

Abstract
Mosaicism due to somatic mutations can cause multiple diseases including cancer, developmental and overgrowth syndromes, neurodevelopmental disorders, autoinflammatory diseases, and atrial fibrillation. With the increased use of next generation sequencing technology, multiple tools have been developed to identify low-frequency variants, specifically from matched tumor-normal tissues in cancer studies. To investigate whether mosaic variants are implicated in congenital heart disease (CHD), we developed a pipeline using the cancer somatic variant caller MuTect to identify mosaic variants in whole-exome sequencing (WES) data from a cohort of parent/affected child trios (n = 715) and a cohort of healthy individuals (n = 416). This is a novel application of the somatic variant caller designed for cancer to WES trio data. We identified two cases with mosaic KMT2D mutations that are likely pathogenic for CHD, but conclude that, overall, mosaicism detectable in peripheral blood or saliva does not account for a significant portion of CHD etiology.
Introduction
Mosaicism, defined as the presence of two or more populations of cells with genetic differences found within one organism, is often due to the acquisition of somatic mutations during development (Taylor et al. 2014). The allelic frequency and anatomical distribution of mosaic mutations depends on developmental timing. Somatic mutations that occur earlier in development can affect multiple tissues, but still occur with a lower alternate allelic fraction (AAF) than de novo germline variants (Biesecker and Spinner 2013). Mosaicism is a naturally occurring consequence of replication errors and is observed in healthy individuals for all types of mutations ranging from single nucleotide variants (SNVs) to large (> 2 Mb) structural changes (Piotrowski et al. 2008). While larger structural changes have been characterized in healthy subjects (Piotrowski et al. 2008), the number of post-zygotic SNVs is unknown. One recent study estimates that between 5 and 7.5% of variants identified as de novo germline are in fact mosaic (Freed and Pevsner 2016; Lim et al. 2017). Mosaicism has been studied in cancer-predisposition traits including retinoblastoma (MIM: 180200), familial adenomatous polyposis (MIM: 175100) and Li–Fraulein syndrome (MIM: 151623) (Aretz et al. 2007; Rushlow et al. 2009; Behjati et al. 2014). Other genetic diseases are associated with mosaicism as well, including Proteus syndrome (MIM: 176920), Ollier disease (MIM: 166000) and autism (Johnston et al. 2011; Pansuriya et al. 2011; Taylor et al. 2014; Freed and Pevsner 2016; Lim et al. 2017).
Mosaic variants are more difficult to detect compared to germline variation due to the defining characteristic of a low alternate allele fraction (AAF < 0.3). Using next generation sequencing (NGS) to identify inherited variation, best practices with GATK HaplotypeCaller (HC) assumes germline heterozygous variants will have an AAF close to 0.5 (McKenna et al. 2010). Thus, mosaic variants are often not detected. As somatic mosaicism is common in tumors, multiple bioinformatics tools have been developed to identify mosaic variants in NGS data with higher sensitivity (Mermel et al. 2011; Larson et al. 2012; Koboldt et al. 2012; Roth et al. 2012; Saunders et al. 2012; Cibulskis et al. 2013; Wang et al. 2013). Tools designed for somatic variation such as MuTect are designed to compare two samples (e.g., tumor to normal) and report all variants regardless of the AAF (Cibulskis et al. 2013).
In this study, we utilized the basic design of MuTect to identify mosaic SNVs from WES trios with high sensitivity and specificity, by designating the child as the “tumor” and each parent as a “normal” sample. We applied our new pipeline to 715 parent/affected child WES trios collected by the Pediatric Cardiac Genomics Consortium (PCGC) to identify mosaic variants that may be causal for congenital heart disease and compared these results to a cohort of healthy individuals.
Methods
Case cohorts
Probands were recruited from 10 centers in the United States and United Kingdom as part of the Congenital Heart Disease Genetic Network study of the PCGC as described previously (Homsy et al. 2015). Seven-hundred and fifteen trios with WES data were included in this study with 19 extracted from saliva and 696 from peripheral whole blood cells. The mean age of probands at the time of enrollment was 7.3 years, the mean maternal age at the time of birth was 30.7 years, and the mean paternal age at the time of birth was 33.0 years. Online Resource Table 1 includes gender, age, parental age at time of birth, DNA sample source and CHD diagnosis for cases.
Control cohort
Control trios were kindly provided by the Simons Foundation Autism Research Initiative Simplex Collection. Simplex families (two unaffected parents, one child with autism spectrum disorder, and one unaffected sibling) underwent whole-exome sequencing using whole blood-derived DNAs (O’Roak et al. 2011; Sanders et al. 2012; Iossifov et al. 2014). We selected 416 trios that were sequenced at the Yale Center for Genome Analysis in order to avoid batch effects between cases and controls. Trios of unaffected siblings and parents served as controls for our study. The mean age of unaffected siblings at time of enrollment was 10.1 years. The mean maternal age at the birth of the sibling was 30.6 years, and the mean paternal age at the birth of the sibling was 32.8 years. Details for the control cohort can be found in Online Resource Table 1.
Exome sequencing
Cases were sequenced at the Yale Center for Genome Analysis as described previously (Homsy et al. 2015). Reference versions hg19/build 37 were used in this study.
Down-sampling of controls
Case trios had a mean depth of coverage (calculated by GATK DepthOfCoverage function) of 60×. Control trios had a mean coverage of 79×. To compare these cohorts, we down-sampled controls trios by 24% using the tool Sambamba (version 0.5.6) because calling of mosaicism is sensitive to read depth. The down-sampled bams had a mean coverage of 60× (Online Resource Fig. 1). The DIR 15 fraction was calculated as the number of WES capture intervals with depth ≥ 15 divided by the number of WES capture intervals with depth ≥ 1 for each sample as reported by the DepthOfCoverage tool from GATK.
The DIR 15 fraction distributions for cases and down-sampled controls (Online Resource Fig. 2) were not significantly different (Mann–Whitney–Wilcoxon test p = 0.776).
Sample identity test
DNA samples were normalized to 5–10 ng/μl, and identity test was performed using iPLEX® Pro Sample ID Panel kit (Cat. No. 25094, Agena Bioscience, San Diego, CA). Multiplex-PCR targeting 44 identity SNPs was carried out in a 5-μl volume on an Applied Biosystems GeneAmp® PCR System 9700 (Thermo Fisher Scientific, Waltham, MA) with the cycling program recommended by the manufacturer’s manual. After PCR, amplicons were treated with 5 U shrimp alkaline phosphatase (SAP; Agena Bioscience, San Diego, CA USA) and followed by adding 2 μl single-base extension (SBE) cocktail (Agena Bioscience, San Diego, CA) to continue the SBE reaction on a GeneAmp® PCR System 9700 with the cycling program recommended by the manufacturer. SBE products were cleaned up using an ion-exchange resin and then spotted on SpectroCHIP arrays (Agena Bioscience, San Diego, CA USA). Raw data were acquired on a MassARRAY® 4 system (Agena Bioscience, San Diego, CA USA) by Matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) analysis. Genotypes for 44 SNPs were generated by MassARRAY Typer software v4.0 (Agena Bioscience, San Diego, CA).
Identification of mosaic variants
Two independent methods were used for variant calling.
1. GATK and HaplotypeCaller
WES data of parent/child trios were aligned using BWA-mem, and variant calling was performed with GATK (version 2.7) (McKenna et al. 2010) and HaplotypeCaller following GATK Best Practices (DePristo et al. 2011; Van Der Auwera et al. 2014). GATK SnpCluster filter was applied with a cluster size of 2 variants within 20 bp. Mosaic variants were defined as de novo variants with an alternate allele fraction (AAF; calculated as the number of alternate allele reads divided by total number of reads at the locus) between 0.15 and 0.35.
2. MuTect somatic variant caller
MuTect somatic variant caller (version 1.1.4) (Cibulskis et al. 2013) from the Broad Institute was used to detect mosaic variants. Proband WES bam files were designated as the “tumor” sample and compared to each parent designated as the “normal” sample. Any variant allele found in the child and not the parent was reported. Variants from the intersection of the child compared to each parent were considered de novo mutations (Online Resource Fig. 3). An important distinction between MuTect and HC is that MuTect does not assume that heterozygous variants have an AAF of 0.5, but rather labels any variant with even one alternate allele present as heterozygous. This allows for higher sensitivity when detecting mosaic variants (Cibulskis et al. 2013). GATK SnpCluster filter was applied as stated above. Filtering for putative mosaic variants was based on sequencing depth in the child, sequencing depth in the parent, AAF in the child and alternate allele depth in the child (Online Resource Table 2). Variants in repetitive regions included in the UCSC repeat masker track were excluded and samples with more than 20 mosaic variants were excluded.
Variant confirmation
Integrated Genomics Viewer (IGV, version 2.3.34) pileup visualization was used as a preliminary method for variant confirmation. Variants were visualized in the proband and parents. Variants were excluded if any of the following aspects were detected: 0 quality reads in child or parents, multiple low-quality alternate alleles in child and/or parents, discordant pairs in child and/or parents.
Digital droplet PCR (ddPCR) was performed to confirm candidate mosaic variants as previously described (Mazaika and Homsy 2014). Variants were considered mosaic with levels of mosaicism (calculated as the ratio of the mutant allele concentration divided by total concentration multiplied by 2) < 0.9. A mosaic level of approximately 1 suggests the variant is heterozygous.
Variant annotation: combined annotation-dependent depletion (CADD)
The publically available tool CADD (version 1.3) (Kircher et al. 2014) was used to annotate mosaic variants detected by both variant callers described above. Based on the literature, we defined C scores ≥ 15 as deleterious for missense mutations and C scores ≥ 30 as deleterious for nonsense and frameshift mutations. The C score is calculated for coding and non-coding variants. CADD was also used to annotate the location and effect of the variant based on the “Consequence” annotation included. Intronic variants included labels “intronic”, “intergenic” and “non_coding change”. Regulatory variants included labels “3Prime_UTR”, “5Prime_UTR”, “Downstream”, “Upstream” and “Regulatory.”
FoxoG scores
The fraction of 8-oxoguanine (FoxoG) scores were calculated to remove variants that are artifacts due to oxidative DNA damage. MuTect2 from the Broad Institute was used to calculate FoxoG scores for the mosaic variants (Costello et al. 2013). FoxoG scores for G>T variants were calculated as the number of alternate alleles on the first read (R1) divided by the number of alternate alleles on R1 and the second read (R2). For C>A variants, FoxoG scores were calculated as the number of alternate alleles on R2 divided by the number of alternate alleles on R1 and R2 (Costello et al. 2013). A score of 1 indicated the alternate alleles were found only on R1 (G>T variants) or R2 (C>A variants) and suggested the variant might be due to DNA damage during the NGS process.
To determine how often a FoxoG score of 1 was seen by chance, we extracted all G>T and C>A inherited heterozygous variants from the 131 samples that had mosaic variants. We filtered these for depth ≥ 10, GQ ≥ 30 and an AAF of 0.4–0.6 to optimize the likelihood that these variants were true and not due to DNA damage. To control for GC content, we annotated each mosaic variant with a GC percentage score from the GC percentage track of the UCSC Genome Browser. We annotated the heterozygous variants with GC percentage scores and required the variant to be within 1 standard deviation of the average GC percentage for mosaic variants. One hundred and thirty-one variants were randomly selected, with one from each sample, and FoxoG scores were calculated as described above. This was replicated 1000 times. A binomial test was used to test for significance between the rate of G>T or C>A variants with a score of 1 found in the mosaic variants compared to the rate of G>T or C>A variants with a FoxoG score of 1 found in inherited heterozygous variants averaged across the 1000 permutations.
Statistical comparison between cases and controls
Enrichment for cases and controls was calculated with a binomial test using R [binom.test(M_PCGC, M_ PCGC + M_SSC, (S_PCGC/(S_PCGC + S_SSC)] where M_PCGC = # of mosaic variants in PCGC cases, M_ SSC = # of mosaic variants in SSC controls, S_PCGC = # of samples in PCGC cases and S_SSC = # of samples in SSC controls. Further enrichment for each variant annotation was calculated using Fisher’s exact test.
Results
Mosaic KMT2D frameshift mutation identified in CHD patient
Identification of de novo variants from WES trios enrolled by the PCGC revealed a frameshift mutation (p.G1722fs) in KMT2D in a subject with hypoplastic left heart syndrome and a double aortic arch (Table 1). Changes in KMT2D are associated with Kabuki syndrome (Liu et al. 2015), which includes CHD, and was clinically suspected in this subject. Twelve induced pluripotent stem cell lines were generated from this individual; four were heterozygous for the mutation, and the other eight were homozygous reference. A PCR-based DNA sample identity assay documented that all of the lines were derived from the same individual, establishing this subject’s KMT2D mutation as mosaic. Review of WES data from the PCGC showed a total read depth of 117 with 39 reads containing the alternate allele. The alternate allele fraction (AAF) for this variant is 0.33 (Table 1). A binomial test confirms the AAF of 0.33 is significantly below (p = 0.0004) the expected AAF (0.5) for heterozygous variants. Finally, mosaicism at 76.8% was confirmed using ddPCR (Online Resource Table 3).
Table 1.
KMT2D de novo variants identified from WES of CHD patients
| Blind-ID | CHD | KMT2D variant | AAF | CADD C Score | Type of mutation | Validation |
|---|---|---|---|---|---|---|
| 1-00596 | HLHSa Double aortic arch Aortic/mitral atresia |
p.G1722 fs | 0.33 | 34 | Mosaic frameshift deletion | iPS clones ddPCR |
| 1-05572 | Hypoplastic left ventricle/mitral valve VSDb |
p.R4198* | 0.53 | 47 | Heterozygous nonsense | ddPCR |
| 1-02566 | Aortic arch hypoplasia VSDb |
p.V5244 fs | 0.55 | 36 | Heterozygous frameshift deletion | ddPCR |
| 1-12480 | HLHSa ASDc |
p.S31* | 0.36 | 35 | Heterozygous nonsense | ddPCR |
| 1-00479 | HLHSa Aortic/mitral atresia |
p.A4576 fs | 0.20 | 35 | Mosaic frameshift deletion | Sanger sequencing |
| 1-10799 | HLHSa | p.Q3607del | 0.25 | 10.72 | Possible mosaic inframe deletion | Unable to confirm in repeat region |
Hypoplastic left heart syndrome
Ventricular septal defect
Atrial septal defect
Five additional subjects were identified as harboring de novo loss-of-function mutations in KMT2D (Homsy et al. 2015) (Table 1). Three had AAFs of 0.53, 0.55 and 0.36 and were confirmed by ddPCR as heterozygous variants (Online Resource Table 3). The fourth KMT2D mutation, a frameshift mutation (p.A4576fs), had an AAF of 0.20 (total depth of 61; Table 1) and was confirmed as mosaic by Sanger sequencing because ddPCR was not possible for this variant. The final KMT2D variant (p.Q3607del) had an AAF of 0.25, but with a total read depth of only eight. It could not be assayed with ddPCR because it was located in a repetitive region. Due to the low read depth of this variant, we concluded that its status vis-à-vis mosaicism is indeterminate.
Development of a robust pipeline to identify mosaic variants from WES data
Having observed somatic mosaicism for at least two KMT2D mutations underlying CHD in our cohort as has also been reported previously in a few cases (Banka et al. 2013), we hypothesized that mosaic mutations might underlie CHD more broadly. In order to explore this hypothesis, we sought to develop a robust high-throughput method to identify mosaic variants from WES data. We began the process of identifying mosaic variants using HaplotypeCaller (HC) to determine how efficiently mosaic variants could be detected using GATK Best Practices, which is implemented robustly for the identification of germline mutations. HC was used to identify de novo variants from a discovery cohort of 427 parent/affected child trios with WES data. We defined putative mosaic variants by an AAF between 0.15 and 0.35 (Fig. 1a). Heterozygous variants are expected to have an AAF of 0.5; however, due to sampling variation, their distribution ranges between 0.35 and 0.75. We used the lower limit of this range to define the maximum AAF for putative mosaic variants. As we are identifying variants in blood/saliva with relation to CHD, we wanted to assure the mosaic variant would be present in the heart as well. To do this, we only considered mosaic variants with a minimum AAF of 0.15, as variants with an AAF > 0.1 are thought to have occurred early enough in development to be present in the blood and affected tissue of different lineage (Zhang et al. 2014; Ju et al. 2017a). In addition, mosaic variants with an AAF lower than 0.15 would increase our rate of false positives due to sequencing errors and variants caused by mutagenic DNA damage, which are enriched for variants with AAFs < 0.15 (Chen et al. 2017). Based on an AAF range of 0.15–0.35, 32 probands from 427 WES trios had one putative mosaic variant each. After visual validation with IGV, 28 of the 32 variants were deemed highly likely to be mosaic (Fig. 1a).
Fig. 1.
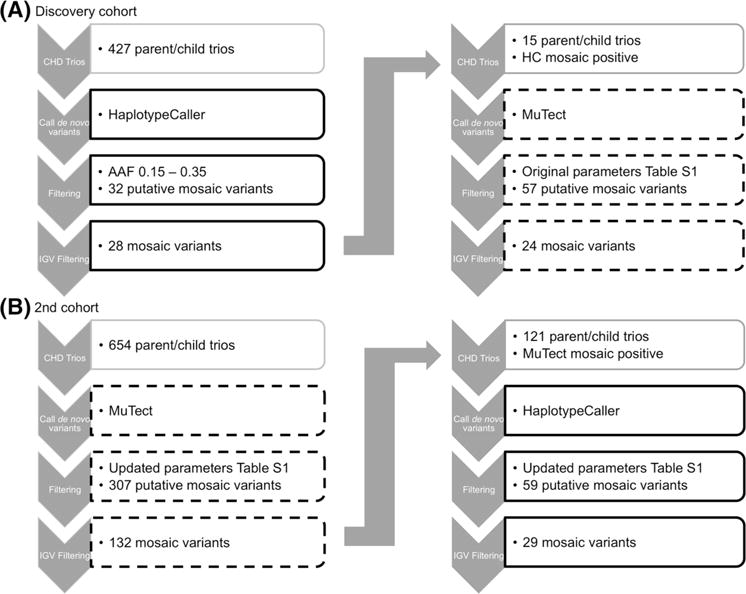
Schematic of mosaic variant identification for two cohorts. Black boxes indicate variant identification by Haplotype Caller. Dashed boxes indicate variant identification by MuTect somatic variant caller. Further details regarding filtering parameters are provided in Online Resource Table 1 and “Methods”
Next, we called mosaic variants in 15 probands with highly likely mosaic variants, identified by the HC, using the MuTect somatic variant caller. We began with a small cohort in order to establish an appropriate pipeline and filtering parameters for this tool (see Methods; Online Resource Table 2). From 15 trios, 57 putative mosaic variants were called, and, following IGV visualization, we identified 24 as high-probability mosaic variants (Fig. 1a). Thirteen of the 24 variants were also identified with HC, and 11 were exclusive to MuTect. HC called two mosaic variants exclusively. ddPCR was used to confirm the presence of the mosaic variants and calculate the level of mosaicism (Online Resource Table 3). ddPCR confirmation revealed that our approach produced a positive predictive value (PPV) of 50% (8/16 mosaic variants; Table 2) in this test cohort. The levels of mosaicism in the discovery cohort ranged from 13 to 87% with a mean of 48% mosaic (Online Resource Table 3).
Table 2.
Discovery cohort ddPCR validation results
| Tool | Number of variants | Mosaic | Heterozygous | Homozygous reference | Undetermined |
|---|---|---|---|---|---|
| Haplotype caller only | 2 | 1 | 0 | 1 | 0 |
| MuTect only | 2 | 2 | 0 | 0 | 0 |
| Haplotype caller and MuTect | 13 | 5 | 1 | 6 | 1 |
| Total | 17 | 8 | 1 | 7 | 1 |
To improve the PPV, we used the allele depth information for the true and false positive mosaic variants to inform the filtering parameters. The data indicated that raising the thresholds for the minimum alternate allele depth in the child and the minimum depth in the parents would be necessary (Online Resource Table 2, Online Resource Fig. 4). In order to achieve higher sensitivity, we used MuTect with the adjusted filtering parameters to identify mosaic variants from 654 trios. From these trios, 132 high-probability mosaic variants were called following IGV visualization (Fig. 1b). HC was run on the MuTect-positive samples and identified only 17 of the high-probability mosaic variants. Twenty-four variants were selected for validation by ddPCR. Variants were selected to represent those identified by both tools and each tool exclusively. Priority was given to exonic variants. ddPCR confirmed 18 of 24 variants as mosaic, raising the PPV to 75% (Table 3). For the second cohort, the level of mosaicism ranged from 30 to 78% with a mean of 47% mosaic (Online Resource Table 3). Final variants are listed in Online Resource Table 4.
Table 3.
Second cohort ddPCR validation results
| Tool | Number of varians | Mosaic | Heterozygous | Homozygous reference | Undetermined |
|---|---|---|---|---|---|
| Haplotype caller only | 5 | 3 | 0 | 0 | 2 |
| MuTect only | 12 | 6 | 1 | 1 | 4 |
| Haplotype caller and MuTect | 15 | 9 | 3 | 1 | 2 |
| Total | 32 | 18 | 4 | 2 | 8 |
To consider the possibility that DNA damage was leading to false positives particularly because these are low-frequency variants, we first checked for the proportions of G>T or C>A variants (Online Resource Fig. 5) (Chen et al. 2017). These changes were not enriched; in fact, C>T variants were seen the most frequently. To consider if the G>T or C>A mosaic variants we identified were due to DNA damage, we used MuTect2 to calculate fraction of 8-oxoguanine (FoxoG) scores (Costello et al. 2013). FoxoG scores represent the percentage of alternate alleles found on reads 1 or 2. Two out of 24 G>T or C>A variants had a FoxoG score of 1 indicating the variant was found exclusively on read 1 or read 2, respectively, and, therefore, could be due to DNA damage. We calculated how often true variants had a FoxoG score of 1 using permutation testing. The average rate of heterozygous variants with a FoxoG score of 1 across 1000 permutations was 0.004. This was significantly lower than the rate of 0.083 observed among the mosaic variants (p = 0.005), further evidence that the mosaic variants with a FoxoG score of 1 in the CHD cohort are false positives.
To test if our pipeline would identify the KMT2D mosaic variants mentioned previously, we used MuTect2 as MuTect1 does not identify indels. From the three KMT2D mosaic variants (two confirmed and one possible), we identified the two confirmed frameshift mosaic variants in samples 1-00596 and 1-00479 with MuTect2, further supporting the robust nature of our pipeline. We did not identify the possible mosaic variant in sample 1-10799, supporting our suspicion that this variant is a false positive.
Mosaicism in healthy subjects
Current estimates of mosaicism are based on DNA mutation rates (Frank 2014). Using MuTect and our pipeline, we quantified mosaicism in the general population. We analyzed WES data for 416 trios comprising unaffected siblings and parents from the Simons Simplex Collection. We used down-sampled bam files (see “Methods”) as the filtering parameters were optimized for a cohort with mean depth of 60×. We identified 83 mosaic variants with MuTect. Based on a PPV of approximately 75%, we predicted there were 62 mosaic variants in this cohort, corresponding to ~ 15% of healthy subjects. The mosaic variants for controls are listed in Online Resource Table 4.
Next, we compared the frequency of mosaicism in CHD cases to healthy subjects. Correcting the number of mosaic variants in CHD cases based on a 75% PPV, 18.7% (125/669) of CHD cases had a mosaic variant compared to 15% (62/416) in healthy subjects, which is not significantly different (binomial test; p = 0.15). Of note, there was no apparent relationship between the rate of mosaic variants and age of the proband in cases or controls (not shown). Using Fisher’s exact test, we tested for further enrichment of mosaic variants that are deleterious based on a CADD C score of 15 or greater for missense mutations; as well as mosaic variants in genes that are highly expressed in the heart based on the top quartile of mean expression levels in E14.5 mice fetal mouse heart (HHE). These categories individually and combined were not enriched in cases compared to controls. In addition there was no enrichment for missense or nonsense/splice variants (Table 4). Lastly, three out of 145 genes previously implicated in CHD, based on Online Mendelian Inheritance of Man (OMIM), were found to harbor mosaic variants in CHD cases (ZEB2, WDR19, TBX20). Although none of the controls harbored a mosaic variant for any of the 145 CHD genes, the difference between cases and controls was not statistically significant (p = 0.55).
Table 4.
Comparison of mosaic variants in CHD trios and controls
| Parameter | Cases (n = 669) | #/Sample | Controls (n = 416) | #/Sample | p value |
|---|---|---|---|---|---|
| Deleterious CADD C score | 23 | 0.034 | 14 | 0.034 | 0.560 |
| High heart expression [HHE] genes | 17 | 0.025 | 5 | 0.013 | 0.339 |
| Deleterious CADD C + HHE | 7 | 0.010 | 3 | 0.007 | 1.000 |
| Missense | 23 | 0.034 | 13 | 0.031 | 0.696 |
| Nonsense/splice | 5 | 0.007 | 3 | 0.007 | 1.000 |
| Synonymous | 17 | 0.025 | 7 | 0.016 | 0.817 |
| Intronic | 47 | 0.070 | 28 | 0.067 | 0.345 |
| Regulatory | 33 | 0.049 | 12 | 0.029 | 0.364 |
Mosaic variants may lead to CHD in a small number of cases
To consider if the mosaic variants that we identified in the CHD probands were causal for their heart anomalies, we investigated the functional impact of the mutation, the affected gene’s expression in the heart and if the affected gene has a role in development based on a literature review. Twenty-three out of 158 mosaic variants affected HHE genes. Thirty-one out of 158 mosaic variants were deemed likely deleterious based on their CADD C scores, of which nine altered HHE genes (Table 4). Of these nine, two variants appeared relevant to CHD. One subject with tetralogy of Fallot (TOF) had a novel p.P117T missense allele in ZEB2, which encodes a zinc finger/homeodomain protein, is not present in the gnomAD database, and is in a gene that is generally constrained (pLi = 1.00; missense constraint metric z = 5.00). Of note, heterozygous ZEB2 mutations underlie Mowat–Wilson syndrome (MIM: 235730), an autosomal dominant trait that includes CHD, although not TOF. Mowat–Wilson syndrome includes characteristic facial features and intellectual disability. There is phenotypic variability with subsets of patients experiencing seizures, hypoplasia of the corpus callosum, Hirschsprung disease and urogenital/renal anomalies (Yamada et al. 2014). Another subject with hypoplastic left heart syndrome had a novel missense variant (p.S254R) in the HHE gene HDAC7, which encodes a histone deacetylase that regulates differentiation of cardio-myocytes and vascular smooth muscle proliferation and was also not observed in the gnomAD database. However, this variant is a G>T substitution and has a FoxoG score of 1, indicating it could be a false positive due to DNA damage. A deleterious variant in a non-HHE gene was detected in a third subject with TOF and atrial/ventricular septal defects. The p.D200G substitution in ADAMTS17 was also not present in the gnomAD database. Homozygous ADAMTS17 mutations cause Weill–Marchesani-like syndrome (MIM: 613195), which can include mitral valve defects. The mosaic variant that we found is not likely sufficient to cause the subject’s CHD, but could be relevant if there was deleterious non-coding variation altering the other ADAMTS17 allele. Of note, the variants detected in two out of the three known CHD genes mentioned above were noncoding and, therefore, unlikely to be causal for CHD, and the third variant was in the gene ZEB2, which is described above.
Discussion
Mosaicism has a well-established role in cancer, and its role in other diseases including developmental disorders is emerging. While mosaicism is recognized as potentially disease-causing, methods to identify somatic variation routinely from large cohorts with NGS data are only now being developed. As documented in this study, we designed a robust pipeline to identify mosaic variants from WES data by altering a cancer-based algorithm used for comparing similar data from paired tumor/non-tumor samples. Moreover, we documented the need for a caller specific for somatic variants as GATK HaplotypeCaller did not identify the majority of the mosaic variants.
We observed mosaic variants in exome sequencing in 15% of healthy subjects. Of note, this estimate included exonic and non-exonic variants from WES data; therefore, this overestimates the frequency of exonic mosaicism and only provided a floor for the frequency of mosaicism genome-wide. Restricting our frequency calculation to only exonic variants, there were 23 exonic mosaic variants observed in 19 subjects. Therefore, 19/416 or 4.6% of healthy subjects have a mosaic exonic variant. This estimate is similar to a recently published one (Freed and Pevsner 2016). Whole-genome sequencing could be used to estimate genome-wide mosaicism, but the typical coverage of 30–40× makes detecting mosaicism, particularly at lower levels, more difficult.
With respect to cardiovascular diseases, relatively little is known about the roles of mosaic variation. The most frequently studied are mosaic aneuploidies such as Turner syndrome (monosomy X) and Down syndrome (trisomy 21), which can include CHDs similar to those observed in children with those aneuploidies inherited through the germline. With respect to SNVs and indels, mosaic mutations altering GJA5, which encodes connexin40, were found in 4 out of 15 patients with atrial fibrillation requiring cardiac resection (Erickson 2010). Mosaicism for a missense SCN5A mutation was recently documented in an infant with long QT syndrome (Priest et al. 2016). With regard to CHD, mosaic frameshift KMT2D mutations underlying Kabuki syndrome have been previously described in three patients (Banka et al. 2013). Our study identified two additional patients with mosaic frameshift mutations in KMT2D that are likely causal for Kabuki syndrome. This confirms that mosaic variation in KMT2D should be considered routinely when trying to identify a causal mutation for Kabuki syndrome, perhaps suggesting enhancing read depth for exome capture kits for this and other genes with increased frequency of mosaicism. It also may suggest that there is a biological driver favoring mosaicism associated with KMT2D loss-of-function mutations.
Considering that germline de novo variants account for about 10% of CHD cases and about 5% of de novo variants are in fact mosaic, we would expect mosaic variants to account for 0.5% of CHD cases. Therefore, in our cohort of 715 trios we would expect to find between 3 and 4 mosaic variants that are causal for CHD if mosaic variants lead to CHD with the same mechanism as germline de novo variants. Although we found one likely causal mosaic variant in ZEB2, we failed to find many other somatic mosaic mutations in our CHD cohort that were likely causal and did not see any enrichment of mosaic variants compared to the control cohort. Therefore, we suggest that mosaic variants that can be identified in blood/saliva at relatively high AAF levels are not present in most individuals with CHD.
There are several limitations to our study. The first is the use of WES data with an average read depth of 60×, as is common for studies of germline genetics. This coverage is significantly lower than the ≥ 100× coverage that is used in many cancer studies (Alioto et al. 2015), and is required to identify low-level mosaic variants. We did not have the ability to identify these low-level variants, therefore CHD relevant mosaic variants may be found at lower levels as disease-causing mosaic variants can be found at levels as low as 1–5% in blood. One report identified three patients with the autoinflammatory disorder cryopryin-associated periodic syndrome with mosaic variants in the CIAS1 gene seen with an AAF of 4% (Saito et al. 2008). Mosaic variants with AAFs as low as 1.8% in blood have also been reported for RB1 mutations, which cause sporadic retinoblastoma (Chen et al. 2014).
A second limitation is the use of saliva to identify mosaic variants that cause disease in the heart. The use of saliva to detect mosaic variants relevant to CHD could be problematic as mosaic variants detected in saliva would need to have occurred earlier in development in order to also affect the heart because the cells in saliva are derived from ectoderm, while cells in the heart are derived from mesoderm. Such early somatic mutational events are plausible as, for example, somatic mutations underlying genetic disorders of neuronal migration in the brain, an ectodermal derivative, have been identified in blood (Poduri et al. 2013). Of note, saliva has been found to be composed of 25% leukocytes that contribute to DNA samples (Endler et al. 1999). This could reduce the robustness of detecting somatic events when using saliva as the source for DNA.
Blood and heart both derive from mesoderm, therefore somatic mutational events causing CHD that occur later in development may still be detectable in blood-derived DNA. However, the use of blood may be limiting when identifying low-level (AAF < 10%) mosaic variants. To appear at low levels, variants must occur later in development, potentially after the differentiation of blood and heart, although this timing is difficult to pinpoint. Currently, the literature supports a minimum AAF of 10% for mosaic variants to be found in multiple tissue types (Ju et al. 2017b). In addition, we did not have corresponding cardiac tissue to confirm the presence of the mosaic variants in the heart. On the other hand, organogenesis includes organized cell migration, and defects in migration during development can lead to CHD (Kurosaka and Kashina 2009). Using a common mesoderm precursor, such as blood, allows us to investigate mosaic mutations that may have affected cell types relevant to cardiogenesis but that ultimately do not constitute the heart. To help strengthen the possibility that the mutations we identified in blood or saliva are also present in the heart, we only considered variants with AAF > 15%.
We compared the rate of CHD probands with mosaic variants with DNA samples extracted from either (saliva n = 19) or blood (n = 696). Forty-two percent of samples from saliva had a mosaic variant compared to 21% of blood samples. Although that difference was not statistically significant (p = 0.07), perhaps due to inadequate power, it might be worth determining in the future if the frequencies are truly different for the two tissues.
The most sensitive approach for detecting CHD-causing somatic mutations that are present only in the heart (or, at least, at highest fraction there) would be to sequence DNA extracted from heart tissues. Whether such somatic events exist and, if they do, whether they cause CHD remain to be proven but could be feasibly studied using cardiac tissues discarded at the time of CHD surgical repairs.
Here, we focused on mosaic variants with a lower AAF limit of 15%. We chose to sacrifice some sensitivity with our approach in order to maintain better specificity, as variants with AAFs < 15% are more likely to arise from sequencing errors or DNA damage. Because somatic mosaic mutations are so frequent in tumors, WES cancer studies generally sequence to far greater depth than is typical for germline genetic studies such as ours so that low AAF calls can, in principle, be called robustly. The recent study of the role of DNA damage during NGS suggests, however, that many putative low-frequency mosaic mutations called in prior cancer studies were susceptible to this source of error (Chen et al. 2017). In contrast, most somatic mutation calls in our study were not G>T or C>A transversions so were highly unlikely to have resulted from DNA damaging during the WES. Among the 24 of those transversions that we identified, only two had the signature of possible DNA damage. Taken as a whole, our pipeline appears to have limited false positive calls due to random errors or DNA damage during the WES process.
Our pipeline was fine-tuned to identify mosaic SNVs, but would not have detected mosaic copy number variations (CNVs) if present. Mosaic CNVs have been described in several diseases including Duchenne muscular dystrophy, hemophilia A and neurofibromatosis type 1 (Notini et al. 2009). Since pathologic CNVs cause about 10% of CHD cases (Glessner et al. 2014), it is plausible to posit that mosaic CNVs might also underlie some proportion of unexplained CHD. Although one study reported no CNVs in heart tissue and peripheral blood from CHD patients (Winberg et al. 2015), the cohort size was small (n = 23), and CNVs were detected by array CGH and FISH, not with sequencing. These limitations leave room for future studies to further explore a possible role for mosaic CNVs in CHD etiology.
Supplementary Material
Acknowledgments
The authors are grateful to the patients and families who participated in this research and team members who supported subject recruitment and sequencing D. Awad, C. Breton, K. Celia, C. Duarte, D. Etwaru, N. Rishman, M. Daspakova, J. Kline, R. Korsin, A. Lanz, E. Marquez, D. Queen, A. Rodriguez, J. Rose, J.K. Sond, D. Warburton, A. Wilpers and R. Yee [Columbia Medical School]; B. McDonough, A. Monafo, J. Stryker [Harvard Medical School]; N. Cross [Yale School of Medicince]; S. M. Edman, J.L. Garbarini, J.E. Tusi, S.H. Woyciechowski (Children’s Hospital of Philadelphia); J. Ellashek and N. Tran (Children’s Hospital of Los Angeles); K. Flack, L. Panesar, N. Taylor (University College London); D. Gruber and N. Stellato (Steve and Alexandra Cohen Children’s Medical Center of New York); D. Guevara, A. Julian, M. MacNeal, C. Mintz (Icahn School of Medicine at Mount Sinai); and E. Taillie (University of Rochester School of Medicine and Dentistry]). We also thank the Simons Foundation for Autism Research for the contribution of control exome trios.
Funding Funding was provided by the National Heart, Blood and Lung Institute from the following grants: U01-HL098147, U01-HL098123, U01-HL098162, U01-HL098153, U01-HL098163, and U01-HL098188.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s0043 9-018-1871-6) contains supplementary material, which is available to authorized users.
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Ethical approval All procedures performed in studies involving human participants were in accordance with the ethical standards of the Institutional Review Boards listed and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Institutional IRBs: Boston Children’s Hospital, Brigham and Women’s Hospital, Great Ormond Street Hospital, Children’s Hospital of Los Angeles, Children’s Hospital of Philadelphia, Columbia University Medical Center, Icahn School of Medicine at Mount Sinai, Rochester School of Medicine and Dentistry, Steven and Alexandra Cohen Children’s Medical Center of New York, and Yale School of Medicine.
Informed consent Informed consent was obtained from all individual participants or their parent/guardian included in this study.
Data availability The PCGC datasets analyzed during the current study are available from dbGAP (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000571.v1.p1). Approved researchers can obtain the SSC population dataset used as controls in this study by applying at SFARI Base (https://base.sfari.org/).
References
- Alioto T, Buchhalter I, Derdak S, et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat Commun. 2015 doi: 10.1038/ncomms10001. https://doi.org/10.1038/ncomms10001. [DOI] [PMC free article] [PubMed]
- Aretz S, Stienen D, Friedrichs N, et al. Somatic APC mosaicism: a frequent cause of familial adenomatous polyposis (FAP) Hum Mutat. 2007;28:985–992. doi: 10.1002/humu.20549. https://doi.org/10.1002/humu.20549. [DOI] [PubMed] [Google Scholar]
- Banka S, Howard E, Bunstone S, et al. MLL2 mosaic mutations and intragenic deletion-duplications in patients with Kabuki syndrome. Clin Genet. 2013;83:467–471. doi: 10.1111/j.1399-0004.2012.01955.x. https://doi.org/10.1111/j.1399-0004.2012.01955.x. [DOI] [PubMed] [Google Scholar]
- Behjati S, Maschietto M, Williams RD, et al. A pathogenic mosaic TP53 mutation in two germ layers detected by next generation sequencing. PLoS ONE. 2014;9:e96531. doi: 10.1371/journal.pone.0096531. https://doi.org/10.1371/journal.pone.0096531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet. 2013;14:307–320. doi: 10.1038/nrg3424. https://doi.org/10.1038/nrg3424. [DOI] [PubMed] [Google Scholar]
- Chen Z, Moran K, Richards-Yutz J, et al. Enhanced sensitivity for detection of low-level germline mosaic RB1 mutations in sporadic retinoblastoma cases using deep semiconductor sequencing. Hum Mutat. 2014;35:384–391. doi: 10.1002/humu.22488. https://doi.org/10.1002/humu.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu P, Evans TC, Ettwiller LM. DNA damage is a pervasive cause of sequencing errors, directly confounding variant identification. Science. 2017;355:752–756. doi: 10.1126/science.aai8690. https://doi.org/10.1101/070334. [DOI] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence M, Carter S, et al. Sensitive deduction of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. https://doi.org/10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello M, Pugh TJ, Fennell TJ, et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013;41:1–12. doi: 10.1093/nar/gks1443. https://doi.org/10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. https://doi.org/10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endler G, Greinix H, Winkler K, et al. Genetic fingerprinting in mouthwashes of patients after allogeneic bone marrow transplantation. Bone Marrow Transplant. 1999;24:95–98. doi: 10.1038/sj.bmt.1701815. https://doi.org/10.1038/sj.bmt.1701815. [DOI] [PubMed] [Google Scholar]
- Erickson RP. Somatic gene mutation and human disease other than cancer: an update. Mutat Res. 2010;705:96–106. doi: 10.1016/j.mrrev.2010.04.002. https://doi.org/10.1016/j.mrrev.2010.04.002. [DOI] [PubMed] [Google Scholar]
- Frank SA. Somatic mosaicism and disease. Curr Biol. 2014;24:R577–R581. doi: 10.1016/j.cub.2014.05.021. https://doi.org/10.1016/j.cub.2014.05.021. [DOI] [PubMed] [Google Scholar]
- Freed D, Pevsner J. The contribution of mosaic variants to autism spectrum disorder. PLoS Genet. 2016;12:1–20. doi: 10.1371/journal.pgen.1006245. https://doi.org/10.1371/journal.pgen.1006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glessner J, Bick AG, Ito K, et al. Increased frequency of de novo copy number variations in congenital heart disease by integrative analysis of SNP array and exome sequence data. Circ Res. 2014 doi: 10.1161/CIRCRESAHA.115.304458. https://doi.org/10.1161/CIRCRESAHA.115.304458. [DOI] [PMC free article] [PubMed]
- Homsy J, Zaidi S, Shen Y, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–1266. doi: 10.1126/science.aac9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O’roak BJ, Sanders SJ, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;13:216–221. doi: 10.1038/nature13908. https://doi.org/10.15154/1149697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JJ, Finn EM, et al. A mosaic activating mutation in AKT1 associated with the proteus syndrome. N Engl J Med. 2011;365(7):611–619. doi: 10.1056/NEJMoa1104017. https://doi.org/10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YS, Martincorena I, Gerstung M, et al. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature. 2017a;543:714–718. doi: 10.1038/nature21703. https://doi.org/10.1038/nature21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YS, Martincorena I, Gerstung M, et al. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature. 2017b;543:714–718. doi: 10.1038/nature21703. https://doi.org/10.1038/nature21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. https://doi.org/10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. https://doi.org/10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaka S, Kashina A. Cell biology of embryonic migration. Birth Defects Res C Embryo Today. 2009 doi: 10.1002/bdrc.20125. https://doi.org/10.1002/bdrc.20125.Cell. [DOI] [PMC free article] [PubMed]
- Larson DE, Harris CC, Chen K, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–317. doi: 10.1093/bioinformatics/btr665. https://doi.org/10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ET, Uddin M, De Rubeis S, et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat Nano Sci. 2017 doi: 10.1038/nn.4598. https://doi.org/10.1038/nn.4598. [DOI] [PMC free article] [PubMed]
- Liu S, Hong X, Shen C, et al. Kabuki syndrome: a Chinese case series and systematic review of the spectrum of mutations. BMC Med Genet. 2015;16:26. doi: 10.1186/s12881-015-0171-4. https://doi.org/10.1186/s12881-015-0171-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazaika E, Homsy J. Digital droplet PCR: CNV analysis and other applications. Curr Protoc Hum Genet. 2014;82:7.24.1–7.24.13. doi: 10.1002/0471142905.hg0724s82. https://doi.org/10.1002/0471142905.hg0724s82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. https://doi.org/10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. https://doi.org/10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notini AJ, Craig JM, White SJ. Copy number variation and mosaicism. Cytogenet Genome Res. 2009;123:270–277. doi: 10.1159/000184717. https://doi.org/10.1159/000184717. [DOI] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–589. doi: 10.1038/ng.835. https://doi.org/10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pansuriya TC, van Eijk R, D’Adamo P, et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet. 2011;43:1256–1261. doi: 10.1038/ng.1004. https://doi.org/10.1038/ng.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski A, Bruder CEG, Andersson R, et al. Somatic mosaicism for copy number variation in differentiated human tissues. Hum Mutat. 2008;29:1118–1124. doi: 10.1002/humu.20815. https://doi.org/10.1002/humu.20815. [DOI] [PubMed] [Google Scholar]
- Poduri A, Evrony GD, Cai X, Walsh CA. Somatic Mutation, Genomic Variation, and Neurological Disease. Science. 2013;341:43–51. doi: 10.1126/science.1237758. https://doi.org/10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priest JR, Gawad C, Kahlig KM, et al. Early somatic mosaicism is a rare cause of long-QT syndrome. Proc Natl Acad Sci. 2016;113:11555–11560. doi: 10.1073/pnas.1607187113. https://doi.org/10.1073/pnas.1607187113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth A, Ding J, Morin R, et al. JointSNVMix: a probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics. 2012;28:907–913. doi: 10.1093/bioinformatics/bts053. https://doi.org/10.1093/bioinformatics/bts053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushlow D, Piovesan B, Zhang K, et al. Detection of mosaic RB1 mutations in families with retinoblastoma. Hum Mutat. 2009;30:842–851. doi: 10.1002/humu.20940. https://doi.org/10.1002/humu.20940. [DOI] [PubMed] [Google Scholar]
- Saito M, Nishikomori R, Kambe N, et al. Disease-associated CIAS1 mutations induce monocyte death, revealing low-level mosaicism in mutation-negative cryopyrin-associated periodic syndrome patients. Blood. 2008;111:2132–2141. doi: 10.1182/blood-2007-06-094201. https://doi.org/10.1182/blood-2007-06-094201. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. https://doi.org/10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WSW, Swamy S, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. https://doi.org/10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- Taylor TH, Gitlin SA, Patrick JL, et al. The origin, mechanisms, incidence and clinical consequences of chromosomal mosaicism in humans. Hum Reprod Update. 2014;20:571–581. doi: 10.1093/humupd/dmu016. https://doi.org/10.1093/humupd/dmu016. [DOI] [PubMed] [Google Scholar]
- Van Der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2014;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. https://doi.org/10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Jia P, Li F, et al. Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers. Genome Med. 2013;5:91. doi: 10.1186/gm495. https://doi.org/10.1186/gm495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winberg J, Berggren H, Malm T, et al. No evidence for mosaic pathogenic copy number variations in cardiac tissue from patients with congenital heart malformations. Eur J Med Genet. 2015;58:129–133. doi: 10.1016/j.ejmg.2015.01.003. https://doi.org/10.1016/j.ejmg.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Yamada Y, Nomura N, Yamada K, et al. The spectrum of Z E B2 mutations causing the Mowat–Wilson syndrome in Japanese populations. Am J Med Genet Part A. 2014 doi: 10.1002/ajmg.a.36551. https://doi.org/10.1002/ajmg.a.36551. [DOI] [PubMed]
- Zhang X, Hill RS, et al. Somatic mutations in cerebral cortical malformations. N Engl Med. 2014;371(8):733–743. doi: 10.1056/NEJMoa1314432. https://doi.org/10.1056/nejmoa1314432. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.