

Abstract
Patients affected by chronic kidney disease (CKD) exhibit a high risk of cardiovascular mortality that is poorly explained by traditional risk factors. There is a growing awareness about the role of derangement of mineral metabolism that is currently accepted as a trigger and sustainer of cardiovascular disease (CVD) in CKD patients. The synthetic definition of CKD mineral and bone disorder (CKD-MBD) split the concept that the indexes of mineral metabolism extend their effects beyond the bone until the vascular wall and metabolic milieu of CKD patients through complex pathways. A better understanding of the biomarkers and mechanisms of left ventricular hypertrophy, CVD, inflammation, and chronic renal damage may help with the diagnosis and treatment of the systemic impairment that occurs secondary to CKD-MBD, thus slowing the progression of renal and CVD and improving patient survival. Recent insights into fibroblast growth factor (FGF) 23 have led to marked advancement in interpreting data on CVD and CKD progression ascribing to FGF23 a pivotal role in these pathologies independent of its co-receptor klotho and well beyond mineral metabolism. This review article will discuss the current experimental and clinical evidence regarding the role of FGF23 in physiology and pathophysiology of CKD and its associated complications with an emphasis on CVD.
Physiology and Pathophysiology of Fibroblast Growth Factor 23
Primarily secreted by osteocytes, fibroblast growth factor (FGF) 23 is a hormone mainly involved in the regulation of mineral metabolism. In the kidney and the parathyroid glands, FGF23 binds FGF receptor (FGFR)/klotho co-receptor complexes to reduce serum phosphate levels (Fig. 1), inhibit parathyroid hormone (PTH) secretion, and decrease levels of active vitamin D [1]. Specifically in the kidney, FGF23 induces urinary phosphate excretion by decreasing the expression of sodium-phosphate co-transporters in the proximal tubule apparatus [2]. A reduction in active vitamin D levels is achieved by inhibition of 1-α-hydroxylase, which catalyzes the hydroxylation of 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D3 and by stimulation of 24-hydroxylase, which converts 1,25-dihydroxyvitamin D3 to inactive metabolites in the proximal tubule [3]. In the distal tubule, FGF23 has been shown to augment calcium and sodium reabsorption through increased apical expression of epithelial calcium channel TRPV5 and the sodium-chloride co-transporter [4]. Furthermore, FGF23 suppresses the expression of angiotensin converting enzyme-2 in the kidney, thereby leading to an activation of the renin-angiotensin-aldosterone-system (RAAS). Phosphate load, 1,25-dihydroxyvitamin D3, and PTH belong to the main group of physiologic regulators of FGF23 synthesis. However, several additional factors including calcium, the RAAS, oxidative stress, parameters of iron metabolism, and inflammation have been shown to regulate FGF23 production and secretion from osteocytes [5]. Nevertheless, the complete mechanisms behind the production and secretion of FGF23 from osteocytes remain poorly understood, are definitely complex, and also involve a number of local factors such as dentin matrix protein 1 or phosphate regulating endopeptidase homolog X-linked.
Fig. 1.
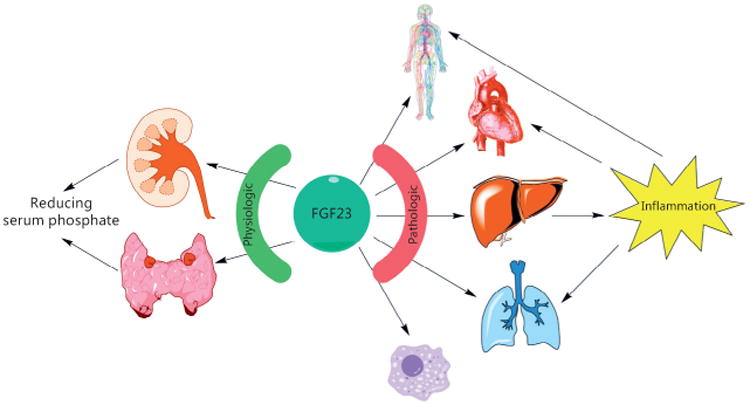
Renal and extrarenal effects of FGF23. In a physiologic state, FGF23 mainly targets the kidney and the parathyroid glands to maintain phosphate homeostasis. In CKD, elevated FGF23 levels might contribute to endothelial dysfunction, cause left ventricular hypertrophy, and promote a chronic inflammatory state. Moreover, FGF23 interferes with the immune system by impairing neutrophil granulocytes. Finally, FGF23 might also account for systemic inflammation observed in COPD. Prolonged chronic inflammation then further accelerates cardiovascular disease.
In patients with chronic kidney disease (CKD), serum levels of FGF23 rise progressively as kidney function declines. This response is mainly a compensatory mechanism to maintain neutral phosphate balance by promoting additional urinary phosphate elimination to counteract the defect in renal excretory capacity. Several large epidemiological studies demonstrated a powerful dose-dependent association between serum levels of FGF23 and higher risk of mortality in end-stage renal disease (ESRD) patients. Moreover, higher FGF23 correlates with increased prevalence of cardiovascular disease (CVD) in general and left ventricular hypertrophy (LVH) in particular among CKD patients. Several in vitro and in vivo studies have been performed to identify a potential causative role of FGF23 in the pathophysiology of abnormal cardiac remodeling in CKD, also known as uremic cardiomyopathy. FGF23 induces hypertrophic growth of cardiac myocytes in vitro. Moreover, rodent models with elevated serum FGF23 levels, either by injection of recombinant FGF23, application of a high phosphate diet or induction of CKD using surgical renal ablation, develop cardiac hypertrophy [6]. Detailed analyses of FGF23-induced signaling events have identified cardiac FGFR4 as the isoform mediating klotho-independent effects of FGF23 on the heart [7]. Subsequent downstream signaling involves the stimulation of the phospholipase Cγ (PLCγ)/calcineurin/nuclear factor of activated T cells (NFAT) pathway, which acts as a potent inducer of cardiac hypertrophy in response to other pathological stimuli. Global deletion of FGFR4 protects mice from the development of high phosphate diet-mediated LVH, whereas systemic blockade of FGFR4 with an isoform-specific blocking antibody or inhibition of calcineurin prevents the development of cardiac hypertrophy in the 5/6 nephrectomy rat model of CKD. Noteworthy, these treatment studies in rats did not affect hypertension or ameliorate kidney function, indicating that high blood pressure or other factors altered by reduced kidney function do not play a major role in the development of LVH at least in this particular animal model of CKD. Finally, knockin mice bearing a gain-of-function mutation of FGFR4, characterized by a single amino acid substitution at position 385 (glycine to arginine), spontaneously develop LVH at 6 months of age [7]. Taken together, these data provide strong evidence of the deleterious effects of FGF23-mediated activation of cardiac FGFR4. Nevertheless, further in vivo studies with cell-type specific deletion of FGFR4 in the presence of elevated FGF23 are needed to confirm whether FGFR4 in cardiac myocytes is required for the development of LVH.
Clinical studies have not only associated FGF23 with cardiovascular injury and LVH, but also with inflammation and elevated serum levels of pro-inflammatory cytokines (Fig. 1). Since the liver expresses high levels of FGFR4, lacks klotho, and plays a decisive role in the acute-phase response and the expression of pro-inflammatory markers, it is intriguing to speculate that FGF23 directly contributes to inflammation in CKD. In a variety of in vitro experiments, Singh et al. [8] demonstrated that FGF23 also activates FGFR4 and subsequent PLCγ/calcineurin/NFAT signaling in hepatocytes, which leads to the production of pro-inflammatory cytokines such as interleukin-6 (IL-6) and C-reactive protein (CRP). Elevated serum FGF23 levels increase hepatic and circulating levels of IL-6 and CRP in wild-type but not in FGFR4 knockout mice. Furthermore, in 5/6 nephrectomized rats, the administration of a FGFR4-blocking antibody reduces the hepatic expression and circulating levels of CRP and IL-6. These findings indicate a novel mechanism for chronic inflammation in CKD and suggest that FGFR4 blockade might not only ameliorate the cardiovascular burden of CKD, but also have therapeutic anti-inflammatory effects.
CKD is also associated with impaired host response and increased susceptibility to infections. Recently, FGF23 has been identified to inhibit chemokine-activated leukocyte arrest on the endothelium. Antibody-mediated neutralization of FGF23 restored leukocyte recruitment and host defense in 5/6 nephrectomized mice with pneumonia, thereby significantly improving the survival in this particular animal model [9]. Mechanistically, FGF23 binds to FGFR2, activates protein kinase A, and inhibits the small GTPase Rap1, thereby counteracting selectin and chemokine-triggered activation of β2-integrin in neutrophil granulocytes [9]. Nevertheless, further mechanistic studies are needed to elucidate the therapeutic potential of targeting FGF23 for the control of inflammatory disorders, especially in the context of CKD (Fig. 1).
Systemic inflammation is not only associated with CKD, but also with a hallmark of other diseases such as chronic obstructive pulmonary disease (COPD), which is associated with increased oxidative stress and significantly increased circulating pro-inflammatory cytokines. Therefore, the question was raised whether FGF23 could serve as a valid biomarker to indicate the aging process in COPD. To date, only a few reports have shown that serum FGF23 levels correlate with smoking [10], and direct effects of FGF23 on the lung have not been described. However, it has been demonstrated that klotho is reduced in COPD patients and protects the alveolar epithelium against oxidative damage [11]. Furthermore, klotho-deficient mice develop widened alveolar spaces, consistent with pulmonary emphysema in the presence of high plasma FGF23 levels [8]. Certainly, future translational studies will assess further potential pathophysiologic roles of FGF23 in a complex scenario such as CKD or COPD.
FGF23 and Clinical Outcomes in CKD and ESRD
In CKD patients, the first striking clinical observation of the potential pathophysiologic role of FGF23 came from a study on incident dialysis patients showing that increased levels of FGF23 associated with increased mortality, independently of the most accredited culprit, serum levels of phosphate. In this study, involving 10,044 patients who were followed up for 1 year, the authors found an impressive monotonic increase in the rate of death, along with increasing FGF23 quartiles, in a nested case–control sample of patients (200 cases per group) [12]. Notably, in these patients, serum levels of FGF23 were up to a thousand times higher than normal. Similar results were evident in the CRIC study involving 3,879 chronic renal failure patients not on dialysis, who were followed for a median of 3.5 years [13]. In this large cohort of patients, higher quartiles of FGF23 were associated with an increased risk of mortality and also of entering renal replacement therapy. By comparison, in this population the median value of FGF23 was only 3-fold higher than normal. Finally, in renal transplantation, higher tertiles of FGF23 associated with increased mortality and worse renal outcome, even though median serum levels of FGF23 were within the normal range [14]. Conceivably, the apparent toxicity associated with serum FGF23 seems to occur for any degree of increment, a finding that raises the question of the underlying pathologic mechanism. Indeed, further data from the CRIC study showed that higher values of FGF23 associated with echocardiographic evidence of LVH, which suggested a possible cardiotoxicity [6].
Interestingly, a recent study evaluated the presence of FGF23 not in bone but in myocardial autopsy samples of deceased chronic renal failure patients. Compared to subjects with normal renal function, the expression of FGF23, FGFR4, and of the calcineurin pathway (involved with myocardial remodeling and hypertrophy) was significantly increased in dialysis patients [15]. Although obtained in a limited number of cases, these results clearly support the link between FGF23 and myocardial uremic toxicity. Furthermore, observational data in conservative chronic renal failure (CRF) analyzing the correlation of serum phosphate and of FGF23 with vascular or cardiac events, clearly suggested that hyperphosphatemia mainly contributes to vascular calcification while FGF23 mainly contributes to myocardial hypertrophy [16].
However, in recent years, a number of other possible links are emerging between FGF23 and its burden of morbidity and mortality through several pathways [17]. In particular, experimental and clinical evidences point to a specific link between FGF23 and inflammation (Fig. 2). Mechanistic studies have demonstrated that FGF23-mediated activation of FGFR4 is capable of enhancing liver synthesis of inflammatory cytokines [8]. These experimental data are in agreement with the observation that, in CKD patients, serum levels of FGF23 and inflammatory markers (IL-6, TNF, CRP, fibrinogen) are positively correlated [18]. Furthermore, a very recent clinical observation in conservative CKD confirmed the association of serum levels of inflammatory biomarkers and FGF23 with the risk of death, but evidenced that their role is independent and additive [19].
Fig. 2.
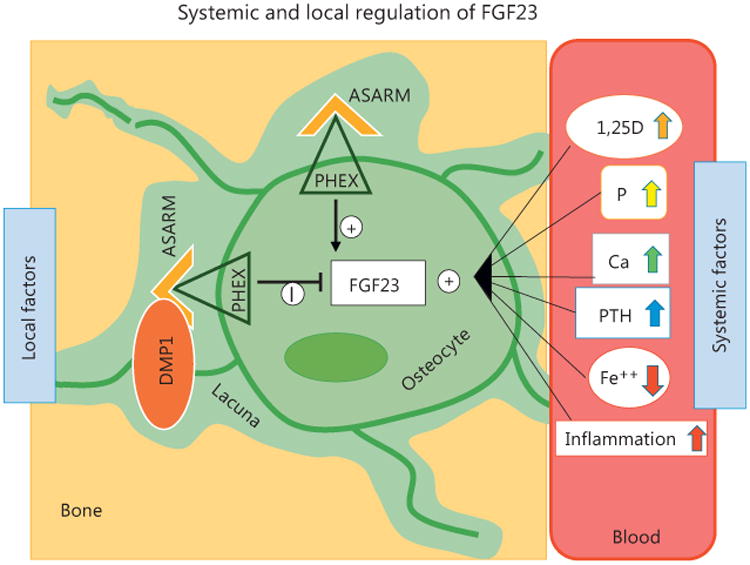
Systemic and local factors regulating osteocyte production of FGF23. Local: DMP1-ASARM binds PHEX and suppresses FGF23 expression; free ASARM-peptide binds PHEX preventing the binding to DMP1 and increased expression of FGF23. Systemic: 1,25D, phosphate, calcium, PTH, and inflammation are positive modulator of FGF23; iron (Fe ++) is a negative modulator of FGF23. ASARM, Acidic Serine Aspartate Rich MEPE-Associated Motif; DMP1, dentin matrix protein 1; PHEX, phosphate regulating endopeptidase homo-log X-linked; 1,25D, 1,25-dihydroxyvitamin D3; P, phosphate; Ca, calcium; PTH, parathyroid hormone; Fe ++, iron.
Another biologic effect experimentally acknowledged to FGF23 is the capacity of impairing immune response [9]. In agreement, a re-evaluation of the HEMO study (originally designed to compare clinical outcomes with different hemodialysis techniques), which included 1,340 hemodialysis patients followed up with repeated biochemical yearly measurements for a median of 3 years, indicate that patients in the highest quartile of FGF23 experienced a higher risk of infectious events, cardiac events, and all-cause mortality [20]. FGF23 has also been shown to associate with atrial fibrillation in CKD. Mehta et al. [21] performed a prospective cohort study on 3,876 individuals demonstrating an independent association of FGF23 with the incidence and prevalence of atrial fibrillation. Mechanistic studies investigating a direct involvement of FGF23 in cardiac arrhythmias however, to date have not been performed. Yet it is likely that the pro-hypertrophic effects of FGF23 promote atrial fibrillation. Fliser et al. [22] assessed FGF23 levels in 227 non-diabetic patients with CKD and followed 177 of the patients prospectively for a median of 53 months to assess the progression of renal disease. In this cohort study, FGF23 independently predicted the progression of CKD after adjustment for covariates such as age, gender, GFR, proteinuria, phosphate, and PTH.
Despite FGF23's powerful correlation with CVD, to date no association has been found between FGF23 and atherosclerosis and arterial calcification. In a study with 1,501 patients, FGF23 did not correlate with coronary artery and thoracic aorta calcium content quantified by computed tomography (CT) [23]. Experimental studies using vascular smooth muscle cells support this hypothesis, since FGF23 also did not induce vascular calcification in vitro [23]. Anemia, a frequent complication seen in patient with CKD and ESRD has also been shown to associate with FGF23. In a cross-sectional observational study of stable CKD patients at stages 3 and 4, high levels of serum FGF23 correlated with low levels of hemoglobin [24]. Certainly, translational studies are needed to elucidate a potential cross-talk between FGF23 and the regulation of iron metabolism and/or the synthesis of erythropoietin.
Due to the growing evidence that FGF23 associates with mortality and morbidity, several studies aimed to reduce FGF23 levels to improve the outcome of CKD patients. Indeed, in a secondary analysis of the EVOLVE trial, the calcimimetic cinacalcet significantly lowered serum FGF23 levels in hemodialysis patients, thereby significantly reducing the rates of cardiovascular events and death. Besides, a novel intravenously administered calcimimetic significantly lowered FGF23 in hemodialysis patients [25]. This study, however, was designed as a non-inferiority trial to target secondary hyperparathyroidism and did not assess clinic outcomes. On the contrary, in a randomized, double blinded, placebo controlled trial, the phosphate binder sevelamer significantly reduced FGF23 in CKD without improving the cardiovascular-related outcomes of interest [26]. Hence, future clinical studies are definitely needed with an intention to reduce FGF23 to clarify if lowering FGF23 improves CVD and overall survival in patients with CKD or ESRD.
FGF23 in Kidney Transplantation
Successful kidney transplantation has been thought to solve the problem of CKD-mineral and bone disorder (CKD-MBD) to a large extent; however, abnormalities in serum calcium, phosphorus, PTH, FGF23, and vitamin D levels are still common in kidney transplant recipients. These conditions are presumably caused by previous bone damage, reduced graft function, CKD-MBD persisting after transplantation or de novo CKD-MBD associated with immunosuppressive therapy.
High levels of PTH, hypophosphatemia, vitamin D deficiency, and hypercalcemia are frequently found in kidney transplant recipients [27, 28]. Interestingly, FGF23 levels decline 3 months after transplantation but remain higher than in CKD patients matched for estimated glomerular filtration rate. Further reductions in FGF23 levels were then repeatedly observed over a longer follow-up period, approximating normal levels 1–3 years after transplantation [28, 29]. In contrast, in a cross-sectional observational study of 279 maintenance kidney recipients with CKD (stages 1–4), Sánchez Fructuoso et al. [30] found that FGF23 levels increased in long-term kidney graft recipients, even in the early stages of CKD, maybe as a result of previous chronic phosphate retention stimulating the secretion of FGF23. These findings support the notion of a persistent (or tertiary) hyperphosphatoninism that in the early post-transplant period mainly reflects previous MBD while in the long-term mirrors a reduction in graft function that is the major determinant of FGF23 serum levels, similar to what is observed in CKD patients.
Nevertheless, only few studies exist investigating the parallel changes of PTH, FGF23 calcium, and phosphate levels in deceased-donor kidney transplantation with a follow-up of >1 year. It is a matter of fact, however, that hyperphosphatoninism, as well as other components of MBD disclose a relapse with the progressive impairment of graft function that follows the progression of chronic allograft deterioration [31, 32].
The impairment of the negative feedback loop between PTH and FGF23, pre-existent in ESRD patients, persists after kidney transplantation. While PTH stimulates FGF23 production, at the same time FGF23 suppresses PTH synthesis acting via the klotho-FGFR1 complex in the parathyroid gland or in the absence of klotho through the NFAT pathway [33].
A decreased expression of the klotho-FGFR1 complex in the parathyroid glands causes this parathyroid resistance to FGF23. Speculatively, the clinical use of calcineurin inhibitors that block calcineurin signaling (NFAT pathway) may further increase the susceptibility to develop, or worsen preexisting hyperparathyroidism (HPTH) in patients with reduced klotho expression such as in kidney transplant recipients (KTRs) [33]. At any rate, decreased FGF23 levels within 1 year after transplantation associated with FGF23 resistance and hyperparathyroidism may account for the restored 1,25-dihydroxyvitamin D3 levels within 3–6 months after transplantation [27].
In kidney transplant recipients, FGF23 has emerged as an important mediator of early hypophosphatemia, and its phosphaturic effect is enforced by persistent hyperparathyroidism and 1,25-dihydroxyvitamin D 3 deficiency. In the long-term, hypophosphatemia and renal phosphate loss are mainly related to persistent hyperparathyroidism. In the first year after transplantation, the faster normalization of FGF23 levels, compared with PTH levels, suggests that FGF23-producing cells may experience a quick reset of their activities in response to the recovery of renal function, all despite persistently elevated PTH levels which stimulate FGF23 production [34]. The long-lasting PTH-related renal phosphate wasting in kidney transplant recipients induces a negative phosphate balance that is appropriately sensed by osteocytes, thus decreasing the FGF23 production in an effort to conserve phosphate, despite the stimulatory effects of high PTH levels (Fig. 3).
Fig. 3.
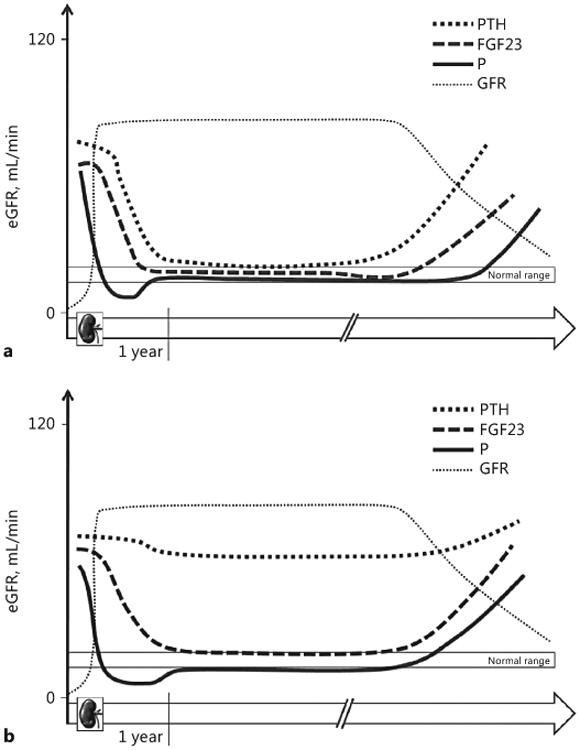
Trends of PTH, FGF23, and phosphate (P) levels at 1 year after renal transplantation and in late period (follow-up). Hyperphosphatoninism in the early post-transplant period mainly reflects previous mineral and bone disorders while in the long-term mirrors graft function and is the main mediator of early hypophosphatemia. In the long term, along with deterioration of graft function, hyperphosphatoninism, and hyperparathyroidism relapse to maintain normophosphatemia and normocalcemia (a). “Persistent” hyperparathyroidism results from pre-existing CKD-MBD with secondary hyperparathyroidism likely complicating post-transplant follow-up with hypophosphatemia and hypercalcemia without affecting the normalization of FGF23. At any rate, hyperphosphatoninism relapses along with the progressive impairment of graft function (b).
Few studies have analyzed the changes in FGF23, PTH, and phosphate levels in living-donor kidney transplants. These studies agree in describing a faster normalization of FGF23 and hypophosphatemia within 1 year after transplantation and a reduced prevalence of hyperparathyroidism [28]. Two factors are held accountable for these findings: the shorter vintage dialysis prior to transplantation, associated with a less severe degree of CKD-MBD and the faster normalization of renal function in living-donor kidney recipients.
Chronically elevated FGF23 levels may be considered ultimately as a maladaptive process in patients with CKD, given the strong associations between higher FGF23 levels and increased risk of LVH, congestive heart failure, CKD progression, and death. To date, clinical studies analyzing the role of FGF23 in transplant patients, however, are still missing.
In a prospective study of stable kidney transplant recipients, elevated FGF23 levels were independently associated with an increased risk of cardiovascular and all-cause mortality and allograft loss [14, 35].
There are several possible mechanisms that may explain this finding. In vitro and in vivo studies have shown that 1,25-dihydroxyvitamin D3 decreases T cell activation and proliferation and inhibits dendritic cell differentiation and maturation, while its supplementation may have beneficial effects on chronic allograft nephropathy [27, 36]. FGF23-mediated suppression of 1,25-dihydroxyvitamin D3 is one possible mechanism through which high FGF23 levels could contribute to allograft loss. In kidney transplant recipients, phosphate depletion, in conjunction with high PTH levels, vitamin D deficiency and chronic steroid use, might worsen skeletal demineralization and contribute directly to fractures, which in turn could increase the risk of mortality. High levels of FGF23 may impair neutrophil recruitment, which additionally could jeopardize antibacterial defense in an already immunocompromised setting [9]. FGF23 has also been shown to cause inflammation and this particular effect may significantly contribute to the inflammatory burden of renal transplant patients [37]. Moreover, chronic elevated FGF23 levels could mimic the known effects of FGF2 inducing glomerulosclerosis and thereby directly contribute to chronic allograft nephropathy and death [7, 28].
Of note, in a retroactive study on deceased pediatric CKD patients, kidney transplantation has been shown to lower cardiac expression of FGF23 and FGFR4 and to normalize klotho protein levels [15].
Finally, an underestimated issue is the effect of immunosuppressive drugs on FGF23 levels, considering their links with vitamin D metabolism and PTH synthesis [27].
Taken together, clearly clinical and mechanistic studies are needed to further elucidate the role of FGF23 in the setting of renal transplantation.
Future Perspectives
FGF23 seems to function as a circulating factor that can directly contribute to cardiac hypertrophy, inflammation, and impaired host response in CKD. Further detailed mechanistic studies are needed to identify a potential causative involvement of FGF23 in further common CKD-related pathophysiologic alterations such as anemia, progression of renal disease, endothelial dysfunction, and atherosclerosis as well other end organ-specific diseases associated with systemic inflammation. Besides, future clinical studies could emphasize the potential differences in gender, ethnicity, and genetic characteristics. Finally, novel laboratory methods have to be developed to reliably assess klotho levels in patients as well as klotho and FGF23 activity in vitro and in experimental in vivo settings. Extending our knowledge of FGF23-FGFR biology in the context of CKD and systemic inflammation will eventually lead to the identification of novel drug targets and the development of pharmacological interventions that ultimately might reduce the burden of cardiovascular injury, decrease systemic inflammation, prevent infections, prolong kidney transplant survival, and ultimately decrease mortality among the millions of CKD patients worldwide.
References
- 1.Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007;117:4003–4008. doi: 10.1172/JCI32409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–568. doi: 10.1172/JCI19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 4.Andrukhova O, Smorodchenko A, Egerbacher M, Streicher C, Zeitz U, Goetz R, et al. FGF23 promotes renal calcium reabsorption through the TRPV5 channel. EMBO J. 2014;33:229–246. doi: 10.1002/embj.201284188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.David V, Martin A, Isakova T, Spaulding C, Qi L, Ramirez V, et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016;89:135–146. doi: 10.1038/ki.2015.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 2015;22:1020–1032. doi: 10.1016/j.cmet.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh S, Grabner A, Yanucil C, Schramm K, Czaya B, Krick S, et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016;90:985–996. doi: 10.1016/j.kint.2016.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rossaint J, Oehmichen J, Van Aken H, Reuter S, Pavenstädt HJ, Meersch M, et al. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J Clin Invest. 2016;126:962–974. doi: 10.1172/JCI83470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vervloet MG, van Zuilen AD, Heijboer AC, ter Wee PM, Bots ML, Blankestijn PJ, et al. Fibroblast growth factor 23 is associated with proteinuria and smoking in chronic kidney disease: an analysis of the MASTERPLAN cohort. BMC Nephrol. 2012;13:20. doi: 10.1186/1471-2369-13-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ravikumar P, Ye J, Zhang J, Pinch SN, Hu MC, Kuro-o M, et al. α-Klotho protects against oxidative damage in pulmonary epithelia. Am J Physiol Lung Cell Mol Physiol. 2014;307:L566–L575. doi: 10.1152/ajplung.00306.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutiérrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359:584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA. 2011;305:2432–2439. doi: 10.1001/jama.2011.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolf M, Molnar MZ, Amaral AP, Czira ME, Rudas A, Ujszaszi A, et al. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J Am Soc Nephrol. 2011;22:956–966. doi: 10.1681/ASN.2010080894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leifheit-Nestler M, Große Siemer R, Flasbart K, Richter B, Kirchhoff F, Ziegler WH, et al. Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol Dial Transplant. 2016;31:1088–1099. doi: 10.1093/ndt/gfv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scialla JJ, Wolf M. Roles of phosphate and fibroblast growth factor 23 in cardiovascular disease. Nat Rev Nephrol. 2014;10:268–278. doi: 10.1038/nrneph.2014.49. [DOI] [PubMed] [Google Scholar]
- 17.Mazzaferro S, Pasquali M, Pirrò G, Rotondi S, Tartaglione L. The bone and the kidney. Arch Biochem Biophys. 2010;503:95–102. doi: 10.1016/j.abb.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 18.Munoz Mendoza J, Isakova T, Ricardo AC, Xie H, Navaneethan SD, Anderson AH, et al. Fibroblast growth factor 23 and Inflammation in CKD. Clin J Am Soc Nephrol. 2012;7:1155–1162. doi: 10.2215/CJN.13281211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munoz Mendoza J, Isakova T, Cai X, Bayes LY, Faul C, Scialla JJ, et al. Inflammation and elevated levels of fibroblast growth factor 23 are independent risk factors for death in chronic kidney disease. Kidney Int. 2017;91:711–719. doi: 10.1016/j.kint.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chonchol M, Greene T, Zhang Y, Hoofnagle AN, Cheung AK. Low vitamin D and high fibroblast growth factor 23 serum levels associate with infectious and cardiac deaths in the HEMO study. J Am Soc Nephrol. 2016;27:227–237. doi: 10.1681/ASN.2014101009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mehta R, Cai X, Lee J, Scialla JJ, Bansal N, Sondheimer JH, et al. Association of fibroblast growth factor 23 with atrial fibrillation in chronic kidney disease, from the chronic renal insufficiency cohort study. JAMA Cardiol. 2016;1:548–556. doi: 10.1001/jamacardio.2016.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fliser D, Kollerits B, Neyer U, Ankerst DP, Lhotta K, Lingenhel A, et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to moderate kidney disease (MMKD) study. J Am Soc Nephrol. 2007;18:2600–2608. doi: 10.1681/ASN.2006080936. [DOI] [PubMed] [Google Scholar]
- 23.Scialla JJ, Lau WL, Reilly MP, Isakova T, Yang HY, Crouthamel MH, et al. Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int. 2013;83:1159–1168. doi: 10.1038/ki.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai MH, Leu JG, Fang YW, Liou HH. High fibroblast growth factor 23 levels associated with low hemoglobin levels in patients with chronic kidney disease stages 3 and 4. Medicine (Baltimore) 2016;95:e3049. doi: 10.1097/MD.0000000000003049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Block GA, Bushinsky DA, Cheng S, Cunningham J, Dehmel B, Drueke TB, et al. Effect of etelcalcetide vs cinacalcet on serum parathyroid hormone in patients receiving hemodialysis with secondary hyperparathyroidism: a randomized clinical trial. JAMA. 2017;317:156–164. doi: 10.1001/jama.2016.19468. [DOI] [PubMed] [Google Scholar]
- 26.Chue CD, Townend JN, Moody WE, Zehnder D, Wall NA, Harper L, et al. Cardiovascular effects of sevelamer in stage 3 CKD. J Am Soc Nephrol. 2013;24:842–852. doi: 10.1681/ASN.2012070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cianciolo G, Galassi A, Capelli I, Angelini ML, La Manna G, Cozzolino M. Vitamin D in kidney transplant recipients: mechanisms and therapy. Am J Nephrol. 2016;43:397–407. doi: 10.1159/000446863. [DOI] [PubMed] [Google Scholar]
- 28.Cianciolo G, Cozzolino M. FGF23 in kidney transplant: the strange case of Doctor Jekyll and Mister Hyde. Clin Kidney J. 2016;9:665–668. doi: 10.1093/ckj/sfw072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evenepoel P, Meijers BK, de Jonge H, Naesens M, Bammens B, Claes K, et al. Recovery of hyperphosphatoninism and renal phosphorus wasting one year after successful renal transplantation. Clin J Am Soc Nephrol. 2008;3:1829–1836. doi: 10.2215/CJN.01310308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sánchez Fructuoso AI, Maestro ML, Pérez-Flores I, Valero R, Rafael S, Veganzones S, et al. Serum level of fibroblast growth factor 23 in maintenance renal transplant patients. Nephrol Dial Transplant. 2012;27:4227–4235. doi: 10.1093/ndt/gfs409. [DOI] [PubMed] [Google Scholar]
- 31.Scolari MP, Cappuccilli ML, Lanci N, La Manna G, Comai G, Persici E, et al. Predictive factors in chronic allograft nephropathy. Transplant Proc. 2005;37:2482–2484. doi: 10.1016/j.transproceed.2005.06.092. [DOI] [PubMed] [Google Scholar]
- 32.Riella LV, Djamali A, Pascual J. Chronic allograft injury: mechanisms and potential treatment targets. Transplant Rev (Orlando) 2016;31:1–9. doi: 10.1016/j.trre.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 33.Olauson H, Lindberg K, Amin R, Sato T, Jia T, Goetz R, et al. Parathyroid-specific deletion of Klotho unravels a novel calcineurin-dependent FGF23 signaling pathway that regulates PTH secretion. PLoS Genet. 2013;9:e1003975. doi: 10.1371/journal.pgen.1003975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wolf M, Weir MR, Kopyt N, Mannon RB, Von Visger J, Deng H, et al. A prospective cohort study of mineral metabolism after kidney transplantation. Transplantation. 2016;100:184–193. doi: 10.1097/TP.0000000000000823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baia LC, Humalda JK, Vervloet MG, Navis G, Bakker SJ, de Borst MH. Fibroblast growth factor 23 and cardiovascular mortality after kidney transplantation. Clin J Am Soc Nephrol. 2013;8:1968–1978. doi: 10.2215/CJN.01880213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cianciolo G, La Manna G, Cappuccilli ML, Lanci N, Della Bella E, Cuna V, et al. VDR expression on circulating endothelial progenitor cells in dialysis patients is modulated by 25(OH)D serum levels and calcitriol therapy. Blood Purif. 2011;32:161–173. doi: 10.1159/000325459. [DOI] [PubMed] [Google Scholar]
- 37.La Manna G, Cappuccilli ML, Cianciolo G, Conte D, Comai G, Carretta E, et al. Cardiovascular disease in kidney transplant recipients: the prognostic value of inflammatory cytokine genotypes. Transplantation. 2010;89:1001–1008. doi: 10.1097/TP.0b013e3181ce243f. [DOI] [PubMed] [Google Scholar]