

Abstract
Drug delivery to bone is challenging, whereby drug distribution is commonly <1% of injected dose, despite development of several bone-targeted drug delivery systems specific to hydroxyapatite. These bone-targeted drug delivery systems still suffer from poor target cell localization within bone, as at any given time overall bone volume is far greater than acutely remodeled bone volume, which harbors relevant cell targets (osteoclasts or osteoblasts). Thus, there exists a need to target bone-acting drugs specifically to the cells responsible for bone remodeling. To address this need, this study synthesized oligo(ethylene glycol) copolymers based on a peptide with high affinity to tartrate-resistant acid phosphatase (TRAP), an enzyme deposited by osteoclasts during the bone resorption phase of bone remodeling, which provides greater specificity relevant for bone cell drugging. Gradient and random peptide orientations, as well as polymer molecular weights, were investigated. TRAP-targeted, high molecular weight (Mn) random copolymers exhibited superior accumulation in remodeling bone, where fracture accumulation was observed for at least one week and accounted for 14% of tissue distribution. Intermediate and low Mn random copolymer accumulation was lower, indicating residence time depends on Mn. High Mn gradient polymers were cleared, with only 2% fracture accumulation after one week, suggesting TRAP binding depends on peptide density. Peptide density and Mn are easily modified in this versatile targeting platform, which can be applied to a range of bone drug delivery applications.
Keywords: peptide targeting, polymer therapeutics, drug delivery, tartrate-resistant acid phosphatase, oligo(ethylene glycol)
Graphical abstract
1. INTRODUCTION
A critical challenge in drug delivery is achieving therapeutic concentrations to target cell types and tissues while avoiding off-target effects, such as hepatotoxicity and cardiotoxicity.1 Locally injected drugs rapidly disperse from injection sites, while depot-based drug delivery requires invasive surgical placement and retrieval for non-degradable reservoirs.2 Systemic administration is limited by rapid clearance and/or drug degradation, as well as poor target tissue localization.3, 4 Drug delivery systems, including polymer drug conjugates and nanoparticles, increase drug circulation half-life and stability, and a few of these formulations are successful clinically.3, 5 However, drug conjugates require accessible functional groups on drugs for polymer incorporation which, after incorporation, may alter drug pharmacodynamics,6–8 and nanoparticles are prone to uptake and removal by the reticuloendothelial system (RES) prior to distribution to the site of action. Similar to drug conjugates, modification of nanoparticles with hydrophilic polymers such as poly(ethylene glycol) (PEG) or oligo(ethylene glycol) (OEG) improves pharmacokinetic behaviors by enhancing drug solubility and circulation time,9–11 but site-specific delivery remains a challenge.
To realize site-specific drug delivery of both drug conjugates and nanoparticles following systemic administration, active tissue targeting approaches have been developed. These include ligands such as peptides and aptamers with selective affinity to cells or extracellular matrix epitopes within target tissues.12, 13 Multivalent ligand density within drug delivery systems has been shown to significantly influence target affinity14 and, for cell targeting, is essential for receptor clustering.15–18 Targeting ligand presentation, including density and interligand distance (ILD), and other drug delivery system characteristics, such as size and molecular weight, influence biodistribution; for example, large carriers (~100 kDa and greater) exhibit lower target to off-target tissue accumulation.9, 19
Targeted drug delivery approaches are generally applicable to multiple indications, but are especially useful for drug delivery to bone, where accumulation of small-molecule drugs in bone is less than 1% of injected dose.20 There is clear motivation to increase drug delivery to bone, as approximately 6 million bone fractures occur annually in the United States, of which nearly 20% result in non-unions or delayed unions.21 Impaired healing is due to dysfunctions in bone remodeling, including poor vascularization,22–24 overactive osteoclasts,25 and deficient osteoblasts.26–28 A variety of drugs have great potential for overcoming these deficiencies.29 To increase drug delivery to bone, a variety of targeting moieties, including bisphosphonates,30, 31 acidic oligopeptides,32, 33 tetracycline,34, 35 collagen binding peptides,36 and the osteoblast-specific aptamer CH637, have been investigated for their affinity to apatite mineral, organic matrix, and cells in bone.13 Specifically, ovariectomized rodents treated with estradiol-conjugated poly(aspartic acid)38 and tetracycline34 drug delivery systems exhibited significant increases in bone mineral density without off-target effects in the liver or uterus. Additionally, parathyroid hormone (PTH) peptides conjugated to collagen binding domain peptides led to gains in spinal lumbar bone mineral density while avoiding hypercalcemia, which is a major side effect of non-targeted PTH treatment.39 Although these bone-targeting moieties dramatically alter pharmacokinetic profiles of conjugated drugs to favor skeletal delivery, none is selective to the sites of actively remodeling bone matrix, where regenerative cell stimulation is necessary. In bone healing and remodeling, osteoclasts deposit the proteinase tartrate-resistant acid phosphatase (TRAP) onto the bone surface,40 providing a target specific to bone remodeling. A TRAP-binding peptide (TBP) was identified via phage display library to exhibit subnanomolar dissociation constant (KD = 0.1 nM) to TRAP.41 Thus, we sought to incorporate TBP into OEG brush copolymers as a platform from which both drug conjugates and targeted nanoparticles could be developed to provide preferential therapeutic delivery to actively remodeling bone. As ligand density, molecular weight, and the interplay of these characteristics have not been investigated to inform targeting platform design, we developed and characterized a highly controlled polymerization-based approach that is amenable to not only bone targeting but also a range of applications.
2. EXPERIMENTAL SECTION
2.1 Materials
All materials were reagent-grade or higher and used as received unless otherwise indicated. N,N-dimethylformamide (DMF), diethyl ether, acetonitrile, pentane, acetone, chloroform, dichloromethane (DCM), dioxane, petroleum ether, tetrahydrofuran (THF), methanol (MeOH), dimethylsulfoxide (DMSO), Type 3A molecular sieves (8-12 Mesh, Grade 564), Texas Red cadaverine, and N-hydroxysulfosuccinimide (sulfo-NHS) were purchased from Fisher Scientific. N-methyl-2-pyrrolidone (NMP) and lithium chloride (LiCl) were purchased from Acros. Deuterated DMSO (DMSO-d6) and deuterated chloroform (CDCl3) were purchased from Cambridge Laboratories. 0.7-meq g−1-substituted fluorenylmethyloxycarbonyl (Fmoc)-Gly-Wang resin and sodium chloride (NaCl) were purchased from EMD Millipore. Fmoc-protected amino acids were purchased from AAPPTec and Peptides International. Piperazine, N,N-diisopropylethylamine (DIEA), methacrylic anhydride, triisopropylsilane (TIPS), trifluoroacetic acid (TFA), 2-bromoethanol, potassium ethyl xanthogenate, triethylamine (TEA), super activated neutral Grade I aluminum oxide, tris(2-carboxyethyl)phosphine hydrochloride (TCEP), and mouse red blood cell lysis buffer were purchased from Alfa Aesar. O-Benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phophaste (HBTU) was purchased from AnaSpec Inc. 3,6-dioxa-1,8-octanedithiol (DODT) and α-cyano-4-hydroxycinnamic acid (α–CHCA) were purchased from Tokyo Chemical Industry Co. Peptide calibration standards, oligo(ethylene glycol) (OEG) methyl ether methacrylate (300 Da, dehibited with basic alumina), azobisisobutyronitrile (AIBN, recrystallized twice in MeOH), sodium hydroxide (NaOH), isobutylamine, 2,2-dimethoxy-2-phenylacetophenone (DMPA), Tween20, Triton X-100, and powdered paraformaldehyde were purchased from Sigma-Aldrich. Magnesium sulfate and sodium acetate were purchased from J. T. Baker. Sodium bicarbonate (NaHCO3) and hydrochloric acid (HCl) were purchased from Macron. Surface plasmon resonance (SPR) Series S Sensor CM5 Chips with a carboxymethylated dextran matrix, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysuccinimide (NHS), and ethanolamine-hydrochloric acid were purchased from GE Healthcare. Tartrate-resistant acid phosphatase (TRAP) and receptor activator of nuclear factor κB ligand (RANKL) were purchased from R&D Systems. Human bone marrow aspirates were purchased from Lonza. Dulbecco’s phosphate-buffered saline (DPBS), low-glucose 1 g/L Dulbecco’s Modified Eagle Medium (DMEM), penicillin-streptomycin-fungizone (PSF), trypsin-EDTA, Minimum Essential Medium Alpha Medium (α-MEM), GlutaMAX, and non-essential amino acids (NEAA) were purchased from Gibco. Human recombinant basic fibroblast growth factor-2 (bFGF) was purchased from Corning. Fetal bovine serum (FBS) was purchased from Atlanta Biologicals. AlamarBlue solution was purchased from Invitrogen. PE mouse anti-human CD105, FITC mouse anti-human CD90, PE-Cy7 mouse anti-human CD45, APC mouse anti-human CD44, and CompBeads were purchased from BD Biosciences. Cyanine 7 NHS ester was purchased from Lumiprobe. Dialysis tubing was purchased from Spectrum Laboratories. Distilled deionized water (ddH2O) with a resistivity of 18.2 MΩcm−1 was obtained using a Barnstead ultrapure water filtration system. Macrophage colony-stimulating factor (M-CSF) from conditioned medium was a kind gift from Dr. Lianping Xing in the Department of Pathology and Laboratory Medicine at University of Rochester Medical Center.
2.2 Synthesis
2.2.1 Peptides (1)
Tartrate-resistant acid phosphatase (TRAP) binding peptide 1 (TBP: TPLSYLKGLVTVG), derived from the sequence of clone 5 phage that exhibited subnanomolar affinity for TRAP41, and scrambled control peptide (SCP: VPVGTLSYLKLTG) were synthesized at a 0.5 mmol scale on Fmoc-Gly-Wang resin using microwave-assisted solid phase peptide synthesis (CEM Liberty 1) with ultraviolet detection monitoring. Amino acids were prepared at 0.2 M in NMP and deprotected with 5% piperazine in DMF. Coupling was performed with 0.5 M HBTU in DMF (activator) and 2 M DIEA in NMP (activator base). A final deprotect cycle removed the Fmoc group from the final amino acid, exposing a primary amine. 1 was cleaved and deprotected in a solution of 2.5 vol% ddH2O, 2.5 vol% TIPS, and 2.5 vol% DODT in TFA (20 mL per 0.5 mmol of resin) by rotating the mixture at room temperature for 2 h. The solution was filtered and precipitated into ice-cold diethyl ether. 1 was collected via centrifugation (4000 rpm for 10 min), washed thrice with diethyl ether, and dried under vacuum overnight. Correct synthesis was validated using matrix assisted laser desorption/ionization time of flight (MALDI-TOF, Bruker MALDI Autoflex III) with α–CHCA as the matrix (Figure S1A).
2.2.2 Methacrylamide-functionalized peptide (2)
To synthesize methacylamide-functionalized peptide monomer 2, the N-terminus of on-resin 1 was reacted with methacrylic anhydride, as reported previously42 (Scheme 1). Briefly, resin submerged in methacrylic anhydride was microwaved at full power for 3 min and vortexed every 45 s. After cooling, resin was washed with DMF and filtered. 2 was cleaved and validated (Figure S1B) using the same protocol as for 1.
Scheme 1.
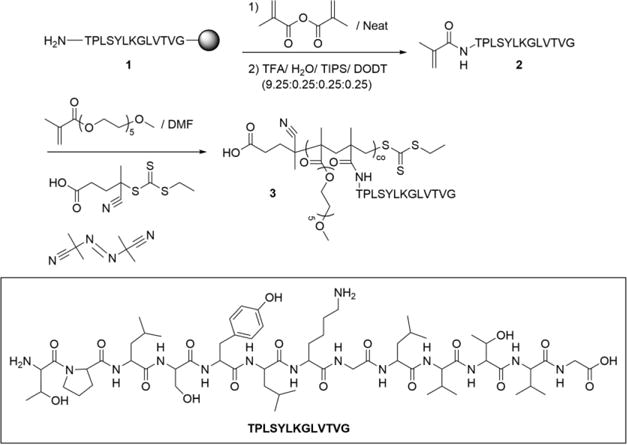
Synthesis and Functionalization of TBP-methacrylamide 2 to Give p(OEG-co-TBP) Gradient Brush Copolymers 3
2.2.3 Gradient peptide-functionalized oligo(ethylene glycol) (OEG) brush copolymer (3)
Reversible addition-fragmentation chain transfer (RAFT) polymerization was used to synthesize p(OEG-co-TBP) and p(OEG-co-SCP) brush copolymers, as illustrated in Scheme 1. OEG methyl ether methacrylate (300 Da) was combined with 2 in DMF (0.5 M) in an 8-mL septa-sealed reaction vessel. 4-cyano-4-(ethylsulfanylthiocarbonyl) sulfanyl pentanoic acid (ECT, synthesized as previously described43 and AIBN were added as the RAFT chain transfer agent (CTA) and initiator, respectively, in a 10:1 CTA:initiator ratio. Monomer:CTA ratios (theoretical degree of polymerization, DP) varied by experiment: DP of 125-600 were used to vary molecular weight, and a DP of 200 was used for reactivity ratio and peptide incorporation experiments. Vials were purged with nitrogen for 30 min and placed in a 60 °C oil bath for 24 h. Polymerization was quenched by opening vials to atmosphere and precipitating in 30 vol% diethyl ether in pentane (50 mL). 3 was collected via centrifugation (1500 rpm for 10 min), then dissolved in acetone (5 mL) and precipitated in diethyl ether (50 mL) thrice. Unreacted 2 was removed by dissolving 3 in a minimal amount of acetone diluted with ddH2O and dialyzing against ddH2O in 6-8 kDa molecular weight cutoff (MWCO) dialysis tubing overnight. The retentate was frozen at −80 °C and lyophilized.
2.2.4 Acrylamide-functionalized peptide (4)
To synthesize acrylamide-functionalized peptide 4, the N-terminus of on-resin 1 was reacted with acryloyl chloride (Scheme 2). Resin was combined with acryloyl chloride (5 eq.) and 2 M DIEA in NMP (5 mL), then incubated at room temperature for 1 h on a mechanical rotator (Labquake). 4 was cleaved and validated (Figure S1C) using the same protocol as for 1.
Scheme 2.
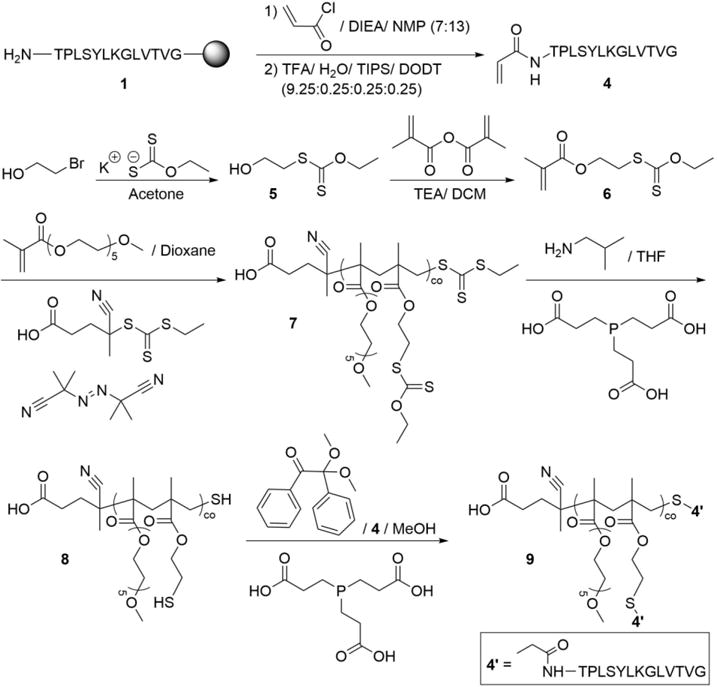
Synthesis and Functionalization of TBP-acrylamide 4 and Xanthate Monomer 6 to Give p(OEG-co-TBP) Random Brush Copolymers 9
2.2.5 S-2-hydroxyethyl-O-ethyl dithiocarbonate monomer (6)
Methacrylated S-alkyl-O-ethyl dithiocarbonate monomer 6 was synthesized according to a protocol modified from Nicolaÿ et al.44, 45 As illustrated in Scheme 2, S-2-hydroxyethyl-O-ethyl dithiocarbonate 5 was first synthesized by adding 2-bromoethanol (26 g) in acetone (80 mL) dropwise over 1 h to a 500-mL roundbottom containing potassium ethyl xanthogenate (40 g) in acetone (200 mL). The reaction was stirred, covered, at room temperature overnight. A yellow liquid was separated from pale yellow solids using a vacuum flask, and the solids were discarded after washing with acetone. The filtrate and washes were combined and rotovapped to dryness. The dried product was dissolved in chloroform (100 mL), washed thrice with saturated NaCl in a separatory funnel, dried with magnesium sulfate, filtered, and rotovapped. Residual chloroform was removed under vacuum overnight (29.52 g, 85% yield). Synthesis of 5 was validated via 1H-nuclear magnetic resonance (1H-NMR) (Figure S2). 1H NMR (400 MHz, CDCl3, 25 °C): δ= 4.65 (q, J = 7.12 Hz, 2 H; CH2), 3.87 ppm (t, J = 6.3 Hz, 2 H; CH2), 3.35 (t, J = 6.3 Hz, 2 H; CH2), 1.41 (t, J = 7.12 Hz, 3 H; CH3).
6 was synthesized by dissolving 5 (29.52 g) and TEA (1.1 eq., 19.79 g) in anhydrous DCM (60 mL) in a 250-mL round bottom flask containing 10 g of activated molecular sieves. To the round bottom flask, methacrylic anhydride (1.1eq., 30.15 g) was added, covered, and gently stirred at room temperature for 3 days. An orange liquid was filtered and washed from the molecular sieves using DCM, then washed in a separatory funnel twice with ddH2O (75 mL each), twice with 2 M HCl (75 mL each), thrice with 2 M NaOH (75 mL each), once with saturated NaHCO3 (75 mL), and once with saturated NaCl (75 mL). The product was dried with magnesium sulfate (10 g), swirled with neutral basic aluminum oxide (50 g), and filtered with a glass fritted funnel. Residual product was washed from the aluminum oxide with DCM and rotovapped to afford a deep yellow liquid (27.08 g, 65% yield). Synthesis was of 6 validated via NMR (Figure S3). 1H NMR (400 MHz, CDCl3, 25 °C): δ= 6.09 (m, J = 1 Hz, 1 H; CH), 5.56 (quint, J = 1.6 Hz, 1 H; CH), 4.63 (q, J = 7.12 Hz, 2 H; CH2), 4.35 ppm (t, J = 6.44 Hz, 2 H; CH2), 3.41 (t, J = 6.44 Hz, 2 H; CH2), 1.91 (dd, J = 1 Hz and J = 1 Hz, 3 H; CH3), 1.40 (t, J = 7.12 Hz, 3 H; CH3).
2.2.6 Random peptide-functionalized OEG brush copolymer precursor polymer (7)
RAFT polymerization was used to synthesize p(OEG-co-6) copolymers, as illustrated in Scheme 2. OEG methyl ether methacrylate (300 Da) was combined with 6 in dioxane (0.5 M) in an 8-mL septa-sealed reaction vessel. ECT and AIBN were added as the RAFT CTA and initiator, respectively, in a 10:1 CTA:initiator ratio. Monomer:CTA ratios (theoretical degree of polymerization, DP) varied by experiment: DP of 100-400 were used to vary molecular weight, and a DP of 200 was used for reactivity ratio and peptide incorporation experiments. Vials were purged with nitrogen for 30 min and placed in a 60 °C oil bath for 24 h. Polymerization was quenched by opening vials to atmosphere and precipitating in petroleum ether. 7 was collected via centrifugation (4000 rpm for 6 min), then dissolved in acetone (5 mL) and precipitated in petroleum ether (50 mL each) thrice. 7 was dried under vacuum overnight before determining incorporation of 6 via NMR (Figure S4).
2.2.7 Random peptide-functionalized OEG copolymer (9)
The thiol group of 6 was deprotected by dissolving 7 in THF (2 mL) in a 20-mL scintillation vial and adding isobutylamine (2 drops) and TCEP (trace, ~20 mg). The solution was stirred for 24 h at room temperature. The deprotected polymer 8 was precipitated in diethyl ether (15 mL) and collected via centrifugation (4000 rpm for 6 min). Ether was decanted, and protecting group removal was confirmed using NMR (Figure S5) and Ellman’s assay. Molecular weight (Mn) and NMR results were used to calculate eq. of thiol per eq. of polymer. 8 was added to a solution of 0.5 w% DMPA, 4 (1 eq. to thiols), and TCEP (1 eq. to thiols, dissolved first in ddH2O) in MeOH, such that the final solution contained 5 w% polymer. The final solution was reacted under long-wavelength UV light (365 nm, 5 mW cm−2) for 1 h and dialyzed against ddH2O using 6-8 kDa MWCO dialysis tubing. After dialysis, water was removed from the retentate by lyophilization.
2.2.8 Fluorescent labeling
Gradient polymers (~10 mg) were dissolved in 2 mL of DMSO (Table S1). Cyanine 7 amine (1 meq. to polymer) and one drop of DIEA were added with stirring at room temperature. After 15 h, reactions were diluted with and dialyzed against ddH2O using 3.5 kDa MWCO dialysis tubing. Water was changed twice a day until clear (~3 days). Polymers were collected through freezing and lyophilization and then reconstituted at 10 mg mL−1 in DPBS. Random polymers (~10 mg) were dissolved in 1 mL of DMSO (Table S2). Texas Red cadaverine, EDC, and sulfo-NHS (1 meq. each to polymer) were added with stirring at room temperature. After 15 h, reactions were diluted with and dialyzed against ddH2O using 6-8 kDa MWCO dialysis tubing. Water was changed twice a day until clear (~3 days). Polymers were collected through freezing and lyophilization and then reconstituted at 10 mg mL−1 in DPBS.
2.3 Characterization
2.3.1 Peptide incorporation
Peptide incorporation was determined using absorbance spectrophotometry.46, 47 3 and 9 were dissolved in ddH2O at 3 mg mL−1 (~100 μM) and measured at λabs of 280 nm (Thermo Scientific Evolution 300), where there is a peak in absorbance due to the tyrosine ring of 1 (Figure S6). Peptide concentration was calculated using an extinction coefficient of 1490 M−1cm−1 (extinction coefficient of tyrosine ring47) to determine mol% peptide incorporation (Figure S7).
2.3.2 Molecular weight (Mn)
Mn and polydispersity were determined via gel permeation chromatography (GPC) (Shimadzu Prominence) using a TSK gel super HM-N column (TOSOH) and a mobile phase of 0.05 M LiCl in DMF at a flow rate of 0.35 mL min−1 (Figure S8). Polymers were dissolved at 5-10 mg mL−1 in mobile phase, filtered with 0.2-μm PTFE syringe filters before injection, and analyzed using a refractive index detector (Wyatt Optilab T-rEX). Absolute Mn was calculated using a light scattering detector (Wyatt miniDAWN TREOS), Astra 6.1 software, and dn dc−1 values (see SI).
2.3.4 Monomer reactivity
Mn, monomer feed, and monomer incorporation were measured over 24 h for 3 and 7 syntheses (Figure S9). For each experiment, 2 g of total monomer and the appropriate ratio of ECT and AIBN were dissolved in 0.5 M solvent in a single 20-mL scintillation vial. Solutions were distributed into 8, 8-mL septa-sealed reaction vessels and purged with N2 for 15 min. Vials were submerged in a 60 °C oil bath, and over the course of 24 h, two vials per polymer per time point were removed and opened to atmosphere to quench the polymerization. A 100 μL sample was pipetted into an NMR tube and combined with 500 μL of DMSO-d6 (for 3) or CDCl3 (for 7). The remaining polymer was precipitated, dialyzed, and characterized as described. Reactivity ratios were determined as previously described.48 For 2 copolymerization with OEG methyl ether methacrylate, r2 = 0 and rOEG = 0.6; for 6 copolymerization with OEG methyl ether methacrylate, r6 = 1.6 and rOEG = 1.6.
2.3.5 Secondary structure of peptides
Peptide secondary structure was analyzed with circular dichroism (AppliedPhotophysics Chirascan). 1, 3, and 9 were dissolved in ddH2O to achieve peptide concentrations of 20 μM, which kept total absorbance at 190 nm under 1.5 au. Four scans per sample ranging from 190 to 260 nm were obtained in 1-nm increments using a 1-cm quartz cuvette at 24 °C. Data were blank subtracted, averaged, and smoothed with a Savitzky-Golay filter with a degree of 1 using MATLAB (Figure S10). To visualize the secondary structure of TBP and SCP, the sequences were entered into the PEP-FOLD2.0 module of Mobyle@RPBS.49
2.4 Mesenchymal stem cell cytocompatibility
Human mesenchymal stem cells (MSCs) were isolated from human bone marrow aspirates as previously described50 and maintained at 37 °C and 5% CO2 in low-glucose (1 g L−1) DMEM supplemented with 1 ng mL−1 bFGF, 10% FBS, and 1% PSF. Cells at passage 2 were seeded in 24-well plates at 8 × 103 cells cm−2 in MSC maintenance medium without bFGF. MSCs were treated 24 h after seeding with MSC maintenance medium supplemented with 1, p(OEG) 3, 7, and 9 at concentrations of 0 (untreated), 0.1, 1.0, and 10.0 μM. A solution of 0.1% Triton X-100 was used as a negative control. After 24 h and 1 week of treatment, media were removed and replaced with 10% alamarBlue in MSC maintenance medium, and MSCs were returned to the incubator for 4 h. A 100 μL sample of medium from each well was taken in triplicate and added to a black well plate. Relative metabolic activity was assessed by measuring fluorescent signal λex = 545 nm, λem = 590 nm) of samples and subtracting fluorescent signal of alamarBlue solution, then normalizing to untreated MSCs (Table S3), as stipulated by ISO-10993-5.51
2.5 Mesenchymal stem cell phenotype
MSCs (passage 2) were seeded at 10,000 cells cm−2 in 24-well plates in MSC maintenance media without bFGF. MSCs were treated 24 h after seeding for 1 week treatment groups, or 6 days after seeding for 24 h treatment groups, with 9 at 1.0 μM in maintenance medium. Media were removed and replaced every other day. To assess alterations in MSC phenotype, cells were prepared for flow cytometry by washing 3x in DPBS, applying 200 μL trypsin-EDTA for 5 minutes at 37 °C, quenching with 300 μL media, and collecting cells by centrifugation (4000 rpm for 5 min). Cells were re-suspended in 110 μL flow buffer (10% FBS in DPBS) and stained with antibodies (PE-CD105, FITC-CD90, PE-Cy7-CD45, and APC-CD44) for 30 minutes on ice. Samples were analyzed via flow cytometry (BD Accuri C6, BD Biosciences) and data were analyzed using FlowJo 10.1 software.
2.6 Osteoclast cytocompatibility
Monocytes were isolated from the spleens of mice via passage through a 40 μm cell strainer, collected via centrifugation (1000 rpm for 5 min), and re-suspended in mouse red blood cell lysis buffer for 10 min at room temperature. Monocytes were subsequently collected via centrifugation and washed in α-MEM supplemented with 10% FBS, 1% PSF, 1% GlutaMAX, and 1% NEAA before re-suspension in medium containing 100 ng mL−1 M-CSF conditioned medium.52 Monocytes were seeded at 2×105 cells well−1 in 96-well plates. After 3 days of incubation at 37 °C and 5% CO2, medium was supplemented with 10 ng mL−1 RANKL to induce osteoclast differentiation. On day 10, osteoclasts were treated with peptides and polymers in differentiation medium with concentrations identical to those used with MSCs. After 24 h, osteoclasts were TRAP-stained, and the entire well was imaged using an EOS Rebel T3i camera (Canon) adapted to the eyepiece of a stereomicroscope (Accu-scope). Blinded viable cell counts were obtained for each well, and each was normalized to untreated osteoclasts, as stipulated by ISO-10993-5.51 Metabolic and DNA analyses were not reported for osteoclasts, as there was a heterogeneous population of osteoclasts, macrophages, and monocytes, and these assays were not representative of osteoclast viability.
2.7 Polymer biodistribution in fractured mice
All animal experiments and care were in accordance with the rules and regulations of and approved by the University of Rochester’s University Committee on Animal Resources. Eight-to-ten week old C57BL/6 mice were bred in-house. Mice were kept in a temperature-controlled environment with a 12-h light/dark cycle and ad libitum access to food and water.
Male and female C57BL/6 mice 8-10 week old were anesthetized with intraperitoneal injections of 60 mg kg−1 of ketamine and 4 mg kg−1 of xylazine and preemptively treated for pain with intraperitoneal injections of 0.1 mg kg−1 buprenorphine. A 1-cm incision was made over the right knee to expose the tibia. A 26 G needle was inserted to the right of the patellar tendon through the intramedullary canal and withdrawn. A 27 G needle was then inserted and withdrawn. A fracture was created mid-diaphysis using a size 11 stainless steel surgical blade, and the 27 G needle was reinserted and cut off at the knee to stabilize the fracture (Figure S11). Size 5 nylon sutures were used to close the incision. The contralateral limb was used as an internal control. Buprenorphine was used for pain management every 12 h for 3 days.
Five days post-surgery, mice were injected intraperitoneally with 100 μL (50 mg mL−1) of labeled polymer or DPBS vehicle on the side contralateral to the fracture. After 24 h, half of the mice were bled to collect plasma for liver enzyme (alanine transaminase (ALT) and aspartate transaminase (AST)) analysis. Mice were then administered 60 mg kg−1 of ketamine and 4 mg kg−1 of xylazine before perfusion with DPBS and anatomized. At 1 week following injection, the remaining mice were perfused and anatomized. The brain, lungs, liver, spleen, kidneys, heart, femurs, and tibias of each mouse were harvested and imaged using the In Vivo Imaging System (IVIS®, PerkinElmer) with λex of 745 nm and λem of 800 nm for cy7-labeled gradient polymers and λex of 570 nm and λem of 620 nm for TexasRed-labeled random polymers. A free draw contour region of interest (ROI) was drawn around individual organs using Living Image software (PerkinElmer). Total radiant efficiency was measured for each organ, summed, and used to calculate percent total radiant efficiency for each organ (Figure S12). Saline controls were used for both gradient and random biodistribution to correct for organ autofluorescence and ensure two different fluorophores did not confound experiments.
2.8 TRAP affinity
Surface plasmon resonance (SPR, GE Healthcare Biacore T200) was used to assess the affinity of 1, 3, and 9 for TRAP. Sensor chips were functionalized with TRAP via EDC/NHS chemistry. Chip surfaces were activated with EDC/NHS for 5 min each using a flow rate of 10 μL min−1. TRAP, at 40 μg mL−1 in pH 5 sodium acetate, was introduced at a flow rate of 10 μL min−1 for 3 min. Ethanolamine-hydrochloric acid was flowed over the surface at a flow rate of 10 μL min−1 for 3 min to quench unreacted carboxysuccinimide groups, yielding ~5000 RU from TRAP-functionalized chip surfaces. Although lower chip functionalization was attempted to enable an evaluation of binding without interference from multivalent effects, realizing 305 RU from TRAP-functionalized surfaces, these chips did not produce measurable signal from soluble TBP, the positive control for binding. To ensure binding was not limited by mass transfer, data were obtained at two flow rates (10 μL min−1 and 30 μL min−1); there was no difference between flow rates, and 30 μL min−1 was used for all subsequent experiments. Polymers were serially diluted to concentrations ranging from 3.125 to 50 μM, and peptides were serially diluted from 25 to 500 μM, using a running buffer of DPBS with 0.01% Tween20. Each concentration was analyzed in duplicate in random order using injections of 100 s and dissociations of 60 s (Figure S13) at a flow rate of 30 μL min−1. Surface regeneration was achieved using a 15 s injection of 100 mM NaOH in ddH2O. Double referencing was achieved by subtracting signal from the EDC/NHS-activated, ethanolamine-hydrochloric acid-quenched chip surface (nonspecific binding) and signal from running buffer (bulk flow). Dissociation constants (KD) were analyzed using BIAcore Evaluation Software (Figure S14), and in-depth analysis was conducted in GraphPad Prism 6 and Microsoft Excel using exported sensorgram data, as explained in SI.
2.9 Statistical Methods
Two-way analysis of variance (ANOVA) with Bonferroni post-hoc testing was used to analyze significant differences (α=0.05) within treatment groups of cytocompatibility assays. One-way ANOVA was used to analyze significant differences (α=0.05) within treatment groups of flow analysis, biodistribution, and liver enzyme assays. Linear regression was used to determine slope and intercept for PAV, SBV, and EDV of SPR binding curves. All statistics were conducted in GraphPad Prism 6.
3. RESULTS AND DISCUSSION
3.1 Design of copolymers
To investigate the effects of multivalent binding on TRAP affinity and bone biodistribution, two reversible addition-fragmentation chain transfer polymerization-mediated reaction schemes were used to form copolymer architectures: a one-step reaction yielding gradient copolymers and a two-step reaction resulting in random copolymers. Gradient copolymers were formed by the compositional drift that occurs during copolymerization of more reactive methacrylate and less reactive methacrylamide monomers.53 Methacrylamide-functionalized TBP 2 was synthesized by functionalizing TBP 1 on-resin with methacrylic anhydride following solid phase peptide synthesis42 (Scheme 1, Figure S1A-B). OEG monomethacrylate (300 Da) and 2 were copolymerized via RAFT to form gradient copolymers 3.
Conversely, random copolymers were formed via statistical conversion of monomers during copolymerization of methacrylate functionalities of similar structure and reactivity.54, 55 A xanthate-based methacrylate monomer 6 was synthesized45 (Scheme 2, Figure S2-3) for copolymerization with OEG monomethacrylate via RAFT to provide a protected thiol available for subsequent peptide conjugation (Figure S4). Acrylamide-functionalized TBP 4 was synthesized by functionalizing 1 on-resin with acryloyl chloride (Figure S1C) and conjugated to deprotected random p(OEG-co-6) brush copolymer 7 (Figure S5) via thiol-ene photoclick chemistry with >99% reaction efficiency to form random copolymers 9. Using these two arrangements in peptide-functionalized polymers, the effect of molecular weight (Mn), which is inherently controlled via RAFT, on biodistribution was analyzed.
3.2 Characterization of copolymers
Peptide incorporation was controlled by monomer feed of 2 and 6 (Figure S6-7). Copolymer Mn exhibited narrow distributions (polydispersity ≤1.1) and were well-controlled by varying the degree of polymerization (DP, i.e. the stoichiometric ratio of monomer and RAFT chain transfer agent) (Figure 1A-B, Figure S8). Mn of 25-100 kDa and 25-125 kDa were achieved by varying DP from 100-600 for 3 and 100-400 for 7, respectively. Mn increased non-linearly over time for 3 (Figure 1C), likely due to high initial 2 incorporation, which has been previously demonstrated as a result of peptide monomer aggregation.56 After 6h, incorporation of 2 reduces precipitously (Figure 1D), which can be attributed to disparate reactivity ratios (see SI). Monomer reactivity mismatch may also explain why higher DP is required to achieve higher Mn for 3.57 In contrast, Mn evolution for 7 was uniform (Figure 1E) due to consistent monomer incorporation (Figure 1F) resulting from similar monomer reactivity (see SI). Thus, 3 exhibited peptide gradients, and 9 exhibited random (uniform) peptide presentation (Figure S9).
Figure 1.
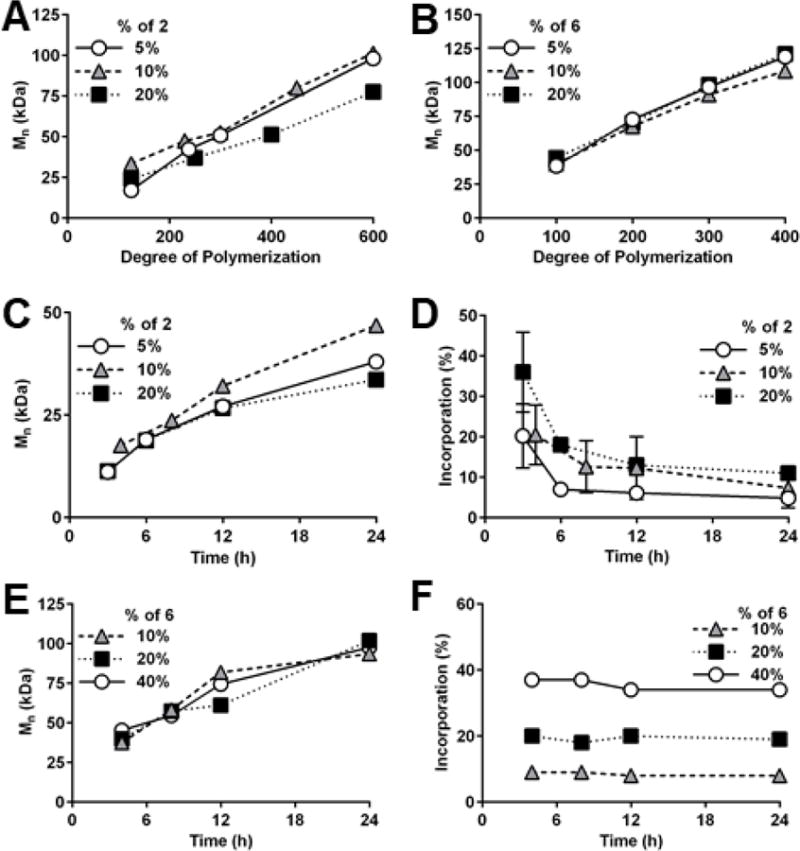
Degree of polymerization controls molecular weight (Mn) of (A) gradient copolymer 3 and (B) random copolymer 7. (C) Mn of 3 increases non-linearly over time. (D) Incorporation of TBP-methacrylamide 2 decreases over time, indicating formation of gradient copolymer 3. (E) Mn of 7 increases linearly over time. (F) Incorporation of xanthate monomer 6 is consistent over time, indicating formation of random copolymer 7.
Peptide secondary structure was evaluated to determine the effects of polymer incorporation and architecture. Unaltered 1 contains mixed secondary structure, with an alpha helix located central to disordered N- and C-termini (Figure S10A).49, 58 This structure was largely preserved in gradient 3 and random 9 copolymers, as the circular dichroism ellipticity ratio of 222/208 nm was consistent and less than 1.25, indicating disordered structures59 (Figure S10B). It is possible the change in ellipticity observed at ~200 nm indicates shorter alpha helix length caused by elongation of the unstructured N-terminus region when juxtaposed to the polymer backbone.60
To ensure cytocompatibility of copolymers, mesenchymal stem cells (MSCs) and osteoclasts, relevant cell types within the bone microenvironment, were exposed to a range of concentrations of peptides 1, gradient copolymers 3, and random copolymers 9. Normalizing to untreated cells, as per ISO 10993-5,51 all treatments resulted in greater than 70% viable MSCs and osteoclasts, the threshold for cytocompatible biomaterials (Figure 2). After 24 hours and 1 week of MSC treatment, there were neither significant differences in viability between treated and untreated MSCs, nor concentration-dependent effects. Similarly for osteoclasts, treatments neither resulted in significantly different viability relative to untreated osteoclasts, nor were dose dependent. Finally, MSC phenotype was conserved with polymer treatment, as there were no significant differences in CD90, CD105, CD44, or CD45 expression relative to untreated controls (Figure S11).
Figure 2.
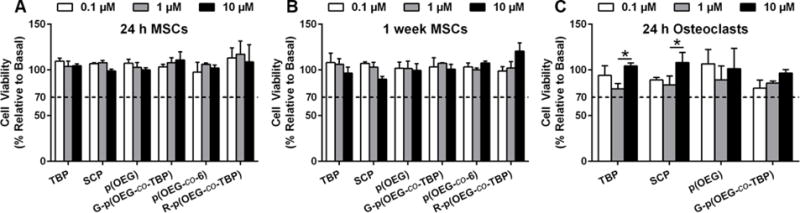
Cytocompatibility of peptides 1, p(OEG), peptide-functionalized gradient copolymers (3), unfunctionalized random copolymers p(OEG-co-6) (7), and peptide-functionalized random copolymers (9). Mesenchymal stem cells (MSCs) were treated for 24 h (A) and 1 week (B), then evaluated using alamarBlue. Osteoclasts were treated for 24 h (C), then stained with TRAP and enumerated. All treatments were significantly greater than 70% viability relative to untreated cells by two-way ANOVA with Bonferroni post-hoc testing, p<0.05. For (C), *p<0.05 between peptide concentrations. Data + SD, n=3.
3.3 Biodistribution of copolymers
The biodistribution of low (L, 10 kDa), intermediate (I, 20 kDa), and high (H, 65 kDa) Mn gradient 3 and random 9 copolymers was investigated. Non-targeting SCP-functionalized gradient and random copolymers of IMn and saline vehicle served as controls to investigate bone targeting specificity. Peptides per polymer chain were normalized (4 per chain) to isolate the effects of Mn on biodistribution (Table S1-2). Five days after tibia fractures (Figure S12), fluorescently labeled polymers were injected intraperitoneally.
After 24 h, only targeted intermediate Mn (TBP-IMn) gradient and targeted high Mn (TBP-HMn) random copolymers resulted in significant accumulation in fractured bone compared to naïve bone. TBP-IMn gradient copolymers exhibited greater accumulation than targeted low Mn (TBP-LMn) and non-targeting scrambled intermediate Mn (SCP-IMn) gradient copolymers (Figure 3A inset). TBP-HMn and TBP-IMn random copolymers exhibited greater accumulation than SCP-IMn random copolymers, and TBP-HMn random copolymers exhibited greater accumulation than TBP-LMn random copolymers (Figure 3B inset). No differences were observed in biodistribution between gradient and random copolymer architectures for any Mn.
Figure 3.
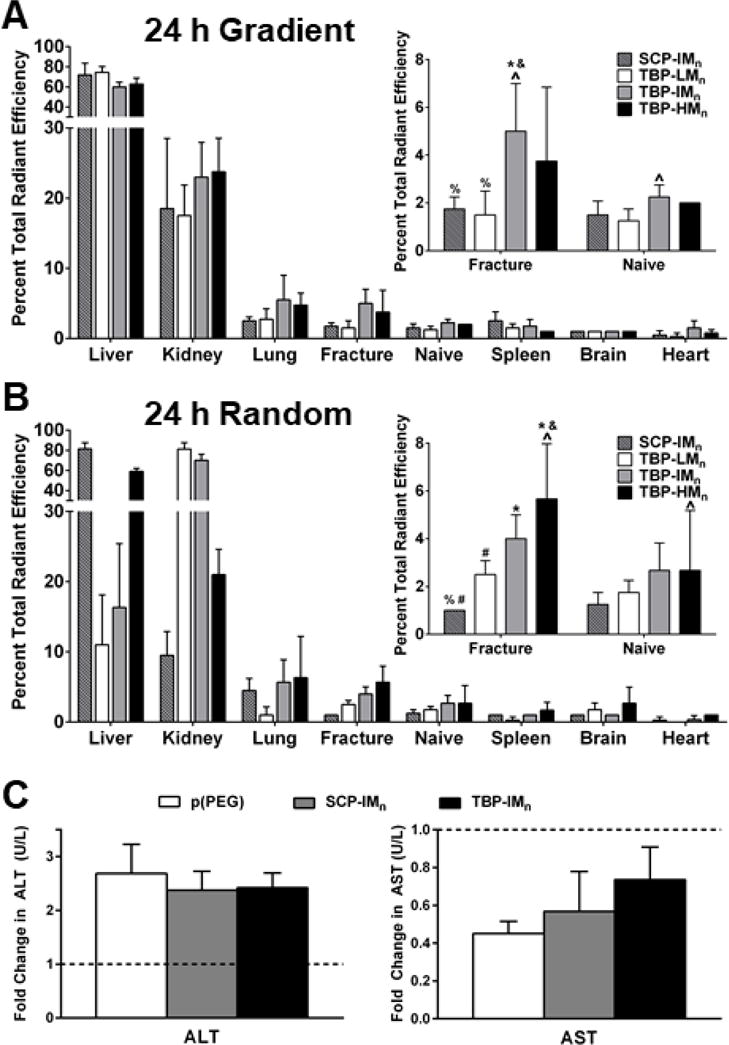
24 h biodistribution of (A) gradient and (B) random copolymers. Significance determined by two-way ANOVA at p<0.05 with * vs SCP-IMn, & vs TBP-LMn, % vs TBP-IMn, # vs TBP-HMn, and ^ between fracture and naïve. Data+SD, n=4. All other organs, see Figure S13. (C) Fold changes (relative to saline vehicle-treated control) in liver enzymes alanine transaminase (ALT) and aspartate transaminase (AST) 24 h after polymer injection are not significantly different by one-way ANOVA (p>0.05) Data+SD, n=3-5 pooled samples.
TBP-LMn, TBP-IMn, and TBP-HMn gradient copolymers showed no significant differences in off-target liver and kidney accumulation relative to SCP-IMn gradient copolymers after 24 h (Figure 3A). Gradient copolymers accumulated preferentially in liver and, to a lesser extent, kidney. Accumulation in liver is likely due to uptake by Kupffer cells.61 Due to lysine residues in peptides, polymers are positively charged at physiological pH. Cationic polymers have been shown to adsorb serum proteins such as opsonins, leading to increased liver accumulation relative to kidney, lung, and spleen, as the liver is the main organ in the RES.62, 63 Despite liver accumulation, there were no significant changes in liver enzymes ALT and AST (Figure 3C), suggesting the polymers were well-tolerated.
The biodistribution profile of SCP-IMn random copolymers was similar to those of gradient copolymers, with the greatest accumulation in liver, followed by kidney (Figure 3B). However, there were significant differences in targeted random copolymer accumulation. TBP-LMn and TBP-IMn random copolymers exhibited significantly lower liver accumulation than TBP-HMn and SCP-IMn random copolymers and lower liver accumulation than comparable gradient copolymers. In contrast, TBP-LMn and TBP-IMn random copolymers exhibited significantly more kidney accumulation than TBP-HMn and SCP-IMn random copolymers, as well as greater kidney accumulation than the corresponding gradient copolymers. This behavior is similar to that of linear dextran polymers, where up to 35% of lower Mn (20-40 kDa) polymers is processed by kidney, and only 5-7% of higher Mn (70-150 kDa) polymers is excreted in urine.64 Nanoparticles, which are commonly used drug delivery platforms accumulate predominantly in liver, lung, and spleen.62 Here, neither gradient nor random copolymers showed appreciable spleen accumulation, likely due to the much smaller hydrodynamic radius of polymer unimers (<5 nm) relative to nanoparticles (>50 nm), and the fracture-to-liver and fracture-to-lung accumulation ratios were significantly greater for targeted copolymers relative to scrambled polymers.
Gradient copolymers were cleared from bone one week after administration (Figure 4A inset). In contrast, there was no change in bone accumulation of random copolymers (Figure 4B inset), suggesting >1 week persistence of targeted polymers and demonstrating a role for ILD in target accumulation. Gradient copolymers likely exhibit ILD of <10 angstroms, but TRAP has a crystal structure measuring 36×42×54 angstroms,65 suggesting multivalent binding is not achievable for gradient copolymers. In contrast, TBP-HMn random copolymers exhibit ILD of ~120 angstroms, which is permissive of multivalent binding. It is possible the smaller ILD of TBP-LMn and TBP-IMn random copolymers, which were ~10 and ~40 angstroms, respectively, were not permissive of multivalent binding due to interference with neighboring ligands and led to lower persistence.66
Figure 4.
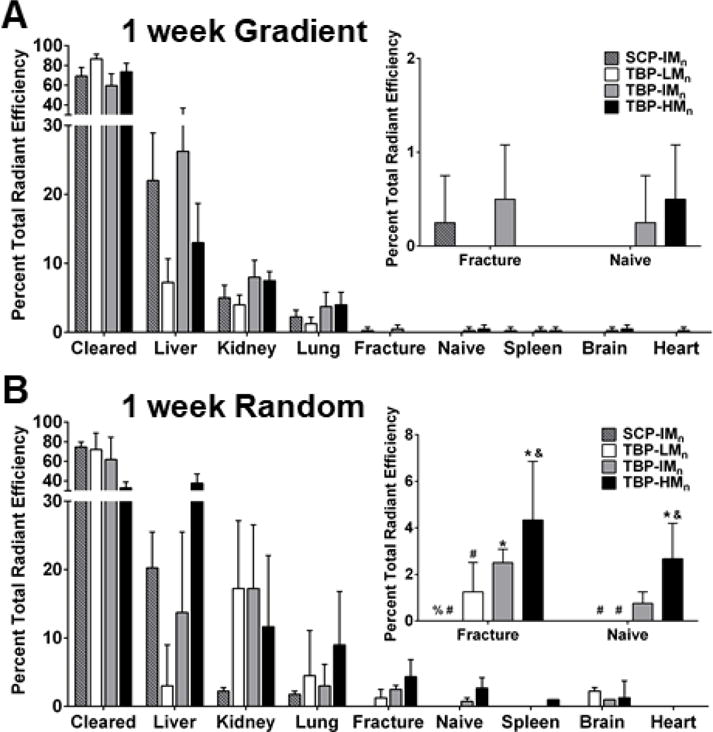
1 week biodistribution of (A) gradient and (B) random copolymers. Statistical symbols are consistent with Figure 3. Data+SD, n=4.
Notably, TBP-HMn random copolymer persistence meets or exceeds other bone targeting systems in literature: the bone accumulation of poly(aspartic acid)-functionalized N-(2-hydroxypropyl)methacrylamide polymers is approximately unchanged between one and three days,67 the concentration of poly(aspartic acid)-conjugated estradiol in bone decreases by two magnitudes within one week,38 and the retention of bisphosphonate-conjugated protein decreases by ~60% in osteoporotic bone68 and ~30% in healthy bone69 between one and three days following injection. While the interactions between hydroxyapatite bone mineral and poly(aspartic acid)70 or bisphosphonates70, 71 are charge-based and dependent on crystallinity,72 the interaction between TBP and TRAP is specific.41 Hydroxyapatite is ubiquitous in bone, and though poly(aspartic acid) may exhibit greater accumulation in areas of high bone turnover,19, 70 TRAP is found only in remodeling bone, giving TBP greater selectivity to fractured bone, which may underpin superior targeting and persistence. With greater persistence, TBP-HMn random copolymers may address the limitations of current non-specific delivery systems. While TRAP is present in naïve bone because of consistent low-level bone remodeling, there is significantly greater TRAP deposition in acutely remodeling fractured bone.73 TBP-HMn random copolymers showed two times greater fracture accumulation relative to naïve bone after 24 h, although the difference is not significant after one week.
Liver accumulation was reduced for the majority of copolymers (Figure 4A-B), which is consistent with bisphosphonate-conjugated albumin and lysozyme.69 Kidney accumulation for TBP-LMn and TBP-IMn random copolymers decreased to levels more similar to those of TBP-HMn and SCP-IMn random copolymers, but more kidney accumulation of TBP-LMn random copolymers was observed relative to TBP-LMn gradient copolymers. Finally, persistence increased with ILD for random copolymers, as TBP-HMn exhibited a lower clearance rate than TBP-LMn random copolymers and TBP-HMn gradient copolymers.
3.4 In Vitro TRAP Affinity
To determine if bone persistence of targeted polymers is related to binding affinity, association of TBP-functionalized polymers and TRAP as a function of multivalency and ILD was investigated by varying number and percent peptide in gradient and random copolymers. Dissociation constants (KD) of peptides and polymers were quantified using Langmuir binding models under the assumption of 1:1 TBP:TRAP binding (Figure S14). Soluble TBP was the positive control for binding and exhibited KD of 2 mM, which was far greater than identified via phage display of TBP (KD of 0.1 nM).41 This dramatic difference is likely due to structural differences between soluble peptides and phage display of peptides. Specifically, the M13 phage displays five copies of peptide on pIII capsid proteins and is connected to the peptide C-terminus (whereas the N-terminus of TBP is conjugated to polymer).74 Thus, a limitation to ligand identification by phage is the potential decrease in affinity when the peptide is chemically synthesized and incorporated into a drug delivery carrier.
For polymers, data exhibited poor fits with binding models, likely due to the complexity of multivalent binding.75, 76 Therefore, alternative analyses were used to compare polymer binding: the peak association value (PAV), the stable binding value (SBV), the end dissociation value (EDV), and the change in value from PAV to SBV (% Drop) (Figure S14).
For gradient copolymers (Figure 5A) used in biodistribution studies, TBP-IMn gradient copolymers exhibited the greatest fractured bone accumulation and the lowest % Drop (−15%, compared to −45% for TBP-LMn, −56% for TBP-HMn, and indeterminable for SCP-IMn), despite having the lowest PAV, SBV, and EDV. Regarding multivalency of gradient copolymers (Figure 5B), PAV, SBV, and EDV linearly increased relative to peptide number per polymer, indicating that greater TBP incorporation led to greater copolymer association to TRAP (Figure 5C, Table S4). However, upon dissociation, % Drop increased with peptide incorporation (Figure 5D). The increase in % Drop suggests that polymer is more loosely bound to TRAP with greater peptide incorporation, potentially due to steric hindrance from neighboring peptides. It is likely that dense peptide packing limits maximal multivalent binding in gradient copolymers, although some degree of multivalent binding still occurs, as soluble TBP control exhibited up to 3.3-fold loss in binding relative to targeted copolymers.66
Figure 5.
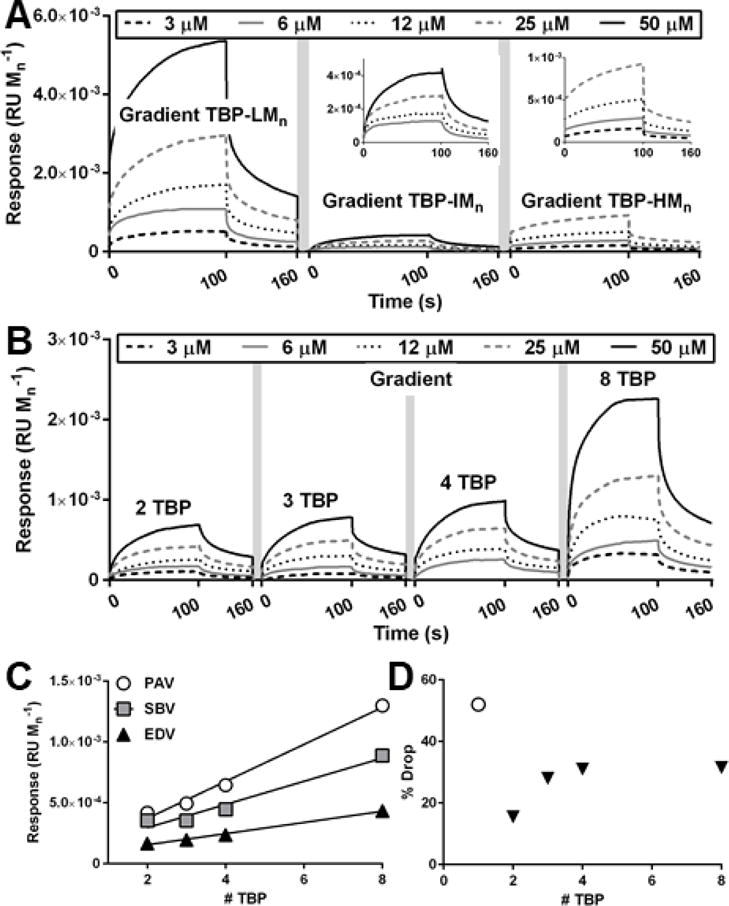
Binding curves and analysis of gradient polymers using surface plasmon resonance (SPR) spectroscopy. Response units are normalized to polymer Mn. (A) Gradient copolymers used in biodistribution studies. (B) Gradient copolymers with increasing number of peptides. (C) Peak association value (PAV), stable binding value (SBV), and end dissociation value (EDV) of polymers in (B). Linear regression, see SI. (D) % Drop of TBP and polymers in (B). White circle is peptide only.
In contrast to gradient copolymers, TBP-LMn, the random copolymer with the lowest fracture biodistribution, exhibited the lowest % Drop (−16%, compared to −31% for TBP-IMn and −38% for TBP-HMn) (Figure 6A). TBP-HMn, the random copolymer with greatest fractured bone accumulation, demonstrated the lowest PAV, SBV, and EDV. To explain this phenomenon and further investigate the interplay of ILD and multivalency, random copolymers were synthesized with various combinations of ILD and peptide incorporation. 10% TBP random copolymers with ILD of ~30 angstroms (Figure S16A) exhibited modest changes in PAV, SBV, and EDV, while 15% TBP random copolymers with ILD ~20 angstroms (Figure 6B) showed robust decreases in PAV, SBV, and EDV with increases in peptide number per polymer (Figure 6D-F, Table S5). However, similar to gradient copolymers, 20% TBP (Figure 6C) and 40% TBP (Figure S16B) random copolymers with smaller ILD of ~10 angstroms exhibited increases in PAV, SBV, and EDV with increases in peptide number per polymer (Table S5). Despite the theoretical increase in binding probability with decreased peptide density and increased ILD,66 increased peptide density enhanced multivalent binding for random copolymers. This paradox may be explained by considering that as peptide number per polymer increases, OEG number per polymer also increases. The water hydration and steric repulsion of OEG prevents protein adsorption.77, 78 Our data suggest that at 85% and greater OEG incorporation, OEG-TRAP repulsive forces dominate. However, at 80% and lower OEG incorporation, TBP-TRAP attractive forces dominate and increase with additional TBP per polymer. Moreover, there was a decreasing relationship between % Drop and number of peptides for 20% and 40% TBP random copolymers (Figure 6G, Table S6), whereby TBP-TRAP interaction allowed less dissociation with additional peptide per polymer. Conversely, there was a positive correlation between % Drop and number of peptides for 15% TBP random copolymers, indicating that additional polymer associated to TRAP was not well bound due to OEG-TRAP repulsion. Interestingly, 10% TBP random copolymers did not follow the same trend, possibly due to small ILD and reduced probability that additional peptide binds without competition.66 These observations explain why TBP-LMn random copolymers exhibited greater in vitro binding than TBP-HMn: TBP-LMn is only 77% OEG, while TBP-HMn is 98% OEG. While TBP-HMn exhibits ILD permissive of multivalent binding to TRAP, TBP-TRAP attraction could not overcome OEG-TRAP repulsion. It is also meaningful to note that in vitro binding analyses do not recapitulate the complex in vivo environment. In opposition to in vitro analysis where polymers are instantaneously exposed to TRAP-functionalized surfaces, in vivo biodistribution is complicated by polymer extravasation from systemic circulation into tissue. High molecular weight polymers exhibit greater circulation times than low molecular weight polymers,79 allowing more opportunities for ligand-target binding. Thus, increased opportunities for binding may have allowed TBP-TRAP attractive forces of TBP-HMn random copolymers to accumulate in bone and remain bound as a result of strong multivalent interactions.
Figure 6.
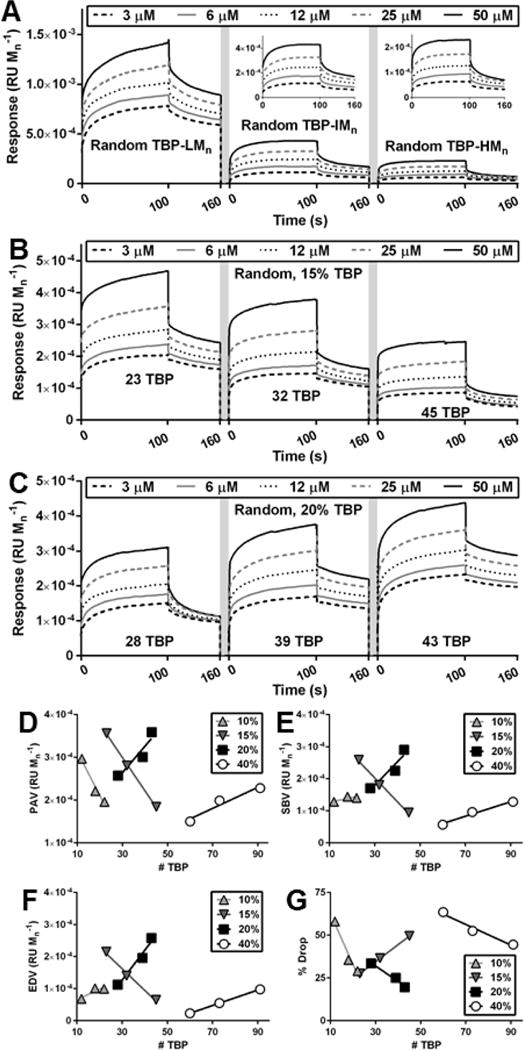
SPR spectroscopy for random copolymers. (A) Random copolymers used in biodistribution studies. (B) 15% random copolymers with increasing number of peptides separated by 6 OEG chains. (C) 20% random copolymers with increasing number of peptides separated by 4 OEG chains. (D) PAV, (E) SBV, (F) EDV, and (G) % Drop of 10%, 15%, 20%, and 40% random copolymers. Linear regression, see SI.
4. CONCLUSIONS
This work demonstrates two versatile copolymerization strategies that yield gradient and random peptide-functionalized polymer architectures to improve the design of drug delivery systems. We exploited heterogeneous comonomer reactivities (e.g. methacrylate and methacrylamide53) to form gradient copolymer architectures and post-polymerization “click” chemistries80, 81 to form random copolymer architectures. TBP, the targeting peptide, retains TRAP binding (Figure 5–6) despite chemical modification and polymer incorporation. While previous groups have used precisely defined systems of monomer feed ratios, solvents, and conversions to avoid compositional drift,82 the described two-step approach avoids tedious experimentation to form homogenously distributed targeting groups. Moreover, this approach may enable new copolymer combinations that were previously unattainable due to reactivity mismatches.
The greatest fracture accumulation was achieved with the targeted high Mn (65 kDa) (TBP-HMn) random copolymer. Since peptide functionalization was consistent, and peritoneal-to-plasma transport of polymers injected intraperitoneally is independent of Mn,64 the differences in bone accumulation are likely due to changes in circulation time and targeting ligand accessibility. Higher Mn polymers avoid rapid renal clearance, leading to longer circulation time and greater delivery to bone via the bloodstream. Binding is then facilitated by targeting peptides with ILD that accommodate target density,66 and stronger binding leads to slower dissociation and enables greater persistence.83, 84 Therefore, careful design of multivalent polymer therapeutics regarding ligand density and Mn is essential for successful targeting.
The benefit of multivalency has been demonstrated in the development of inhibitor peptides,85 vaccines,86, 87 and immunomodulators,88 as well as controlled adhesion domain accessibility on surface-modified scaffolds.18, 89, 90 For example, surfaces modified with fibronectin-derived FN7-1017, 18 and RGD15, 16 must present appropriate ligand density to induce integrin clustering on the surface of cells. Similar to random copolymers, while ligand-receptor binding occurs regardless of peptide arrangement, lower density leads to greater cell motility.15, 18 Analogous to gradient copolymers, greater density increases cell adhesion.16 Binding motif distribution on surfaces,91 as well as nanoparticles,92 is controlled by conjugating peptides to monomers pre-assembly or “clicking” peptides into macrostructures post-assembly. While pre-assembly modification provides greater control over degree of functionalization,93 post-assembly modification provides more homogeneous spatial distribution of targeting moieties.92 Thus, one-step and two-step polymerizations may also be applied to synthesizing polymeric micelle unimers.
Both one-step and two-step polymerizations are amenable to drug incorporation directly. For example, methacrylate/methacrylamide-functionalized drug monomers or “clickable” monomers, such as azide-based monomers, allows for versatile drug incorporation. Importantly, successful bone targeting of quinolone94 and prostaglandin E167 modified acidic oligopeptide polymer conjugates has been achieved while maintaining drug activity, but analysis of drug release from gradient and random polymers is necessary to ensure interference does not occur due to peptide incorporation.95 Alternatively, agonists of bone formation may be loaded into nanoparticles formed from peptide-functionalized polymers96 or nanoparticles modified by peptides post-assembly.97 When combined with these or other osteoanabolic compounds, the described bone-targeting polymer may enhance bone regeneration in fractures and other disease states exhibiting increased osteoclast deposition of TRAP, such as osteoporosis and osteomyelitis.
Supplementary Material
Acknowledgments
In addition to funders, we acknowledge Dr. Joshua Blose (University of Brockport) for assistance with circular dichroism, Dr. Jermaine Jenkins (University of Rochester Medical Center) for surface plasmon resonance expertise, Dr. Lianping Xing (University of Rochester Medical Center) for providing M-CSF conditioned medium, Dr. Matthew Cochran (University of Rochester Medical Center) for flow cytometry expertise, Dr. Roman Eliseev and Dr. Cheryl Ackert-Bicknell (University of Rochester Medical Center) for mice, and Dr. James McGrath (University of Rochester) for equipment use.
Funding Sources
This work was supported by the National Science Foundation (DGE-1419118, CBET-1450987), the National Institutes of Health (R01AR064200, P30ES001247, K12ES019852, P30AR069655, S10RR026542), and the University of Rochester Technology Development Fund.
ABBREVIATIONS
- α–CHCA
α-cyano-4-hydroxycinnamic acid
- α-MEM
Minimum Essential Medium Alpha Medium
- AIBN
azobisisobutyronitrile
- ALT
alanine transaminase
- AST
aspartate transaminase
- DCM
dichloromethane
- ddH2O
distilled deionized water
- DIEA
N,N-diisopropylethylamine
- DMEM
Dulbecco’s Modified Eagle Medium
- DMF
N,N-dimethylformamide
- DMPA
2,2-dimethoxy-2-phenylacetophenone
- DMSO
dimethylsulfoxide
- DODT
3,6-dioxa-1,8-octanedithiol
- DPBS
Dulbecco’s phosphate-buffered saline
- EDC
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
- EDV
end dissociation value
- FBS
fetal bovine serum
- Fmoc
fluorenylmethyloxycarbonyl
- HBTU
O-Benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phophaste
- HCl
hydrochloric acid
- HMn
high molecular weight
- ILD
interligand distance
- IMn
intermediate molecular weight
- IVIS
in vivo imaging system
- KD
dissociation constant
- LiCl
lithium chloride
- LMn
low molecular weight
- M-CSF
macrophage colony-stimulating factor
- MeOH
methanol
- Mn
molecular weight
- MSC
mesenchymal stem cell
- NaCl
sodium chloride
- NaHCO3
sodium bicarbonate
- NaOH
sodium hydroxide
- NEAA
non-essential amino acids
- NHS
N-hydroxysuccinimide
- NMP
N-methyl-2-pyrrolidone
- OEG
oligo(ethylene glycol)
- PAV
peak association value
- PEG
poly(ethylene glycol)
- PSF
penicillin-streptomycin-fungizone
- PTH
parathyroid hormone
- RAFT
reversible addition-fragmentation chain transfer
- RANKL
receptor activator of nuclear factor κB ligand
- RES
reticuloendothelial system
- SBV
stable binding value
- SCP
scrambled control peptide
- SPR
surface plasmon resonance
- sulfo-NHS
N-hydroxysulfosuccinimide
- TBP
TRAP-binding peptide
- TCEP
tris(2-carboxyethyl)phosphine hydrochloride
- TEA
triethylamine
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TIPS
triisopropylsilane
- TRAP
tartrate-resistant acid phosphatase
- XAN
xanthate-based methacrylate monomer
Footnotes
Supporting Information. Characterization spectra and additional experimental information. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Whitebread S, Hamon J, Bojanic D, Urban L. Keynote review: in vitro safety pharmacology profiling: an essential tool for successful drug development. Drug Discov Today. 2005;10(21):1421–33. doi: 10.1016/S1359-6446(05)03632-9. [DOI] [PubMed] [Google Scholar]
- 2.Liechty WB, Kryscio DR, Slaughter BV, Peppas NA. Polymers for drug delivery systems. Annu Rev Chem Biomol Eng. 2010;1:149–73. doi: 10.1146/annurev-chembioeng-073009-100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pisal DS, Kosloski MP, Balu-Iyer SV. Delivery of therapeutic proteins. J Pharm Sci. 2010;99(6):2557–75. doi: 10.1002/jps.22054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knadler MP, Nguyen TH, Campanale K, De Veer MJ, Beals JM, Li S, Hansen R, Siesky A, Michael MD, Porter CJ. Addition of 20-kDa PEG to Insulin Lispro Alters Absorption and Decreases Clearance in Animals. Pharm Res. 2016;33(12):2920–2929. doi: 10.1007/s11095-016-2014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303(5665):1818–22. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 6.Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, Meyer DL, Francisco JA. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10(20):7063–70. doi: 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 7.Bagalkot V, Farokhzad OC, Langer R, Jon S. An aptamer-doxorubicin physical conjugate as a novel targeted drug-delivery platform. Angew Chem Int Ed Engl. 2006;45(48):8149–52. doi: 10.1002/anie.200602251. [DOI] [PubMed] [Google Scholar]
- 8.Santi DV, Schneider EL, Reid R, Robinson L, Ashley GW. Predictable and tunable half-life extension of therapeutic agents by controlled chemical release from macromolecular conjugates. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(16):6211–6. doi: 10.1073/pnas.1117147109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He X, Li L, Su H, Zhou D, Song H, Wang L, Jiang X. Poly(ethylene glycol)-block-poly(epsilon-caprolactone)-and phospholipid-based stealth nanoparticles with enhanced therapeutic efficacy on murine breast cancer by improved intracellular drug delivery. Int J Nanomedicine. 2015;10:1791–804. doi: 10.2147/IJN.S75186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li C, Yu D, Inoue T, Yang DJ, Milas L, Hunter NR, Kim EE, Wallace S. Synthesis and evaluation of water-soluble polyethylene glycol-paclitaxel conjugate as a paclitaxel prodrug. Anticancer Drugs. 1996;7(6):642–8. doi: 10.1097/00001813-199608000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Pan LQ, Wang HB, Lai J, Xu YC, Zhang C, Chen SQ. Site-specific PEGylation of a mutated-cysteine residue and its effect on tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) Biomaterials. 2013;34(36):9115–9123. doi: 10.1016/j.biomaterials.2013.08.020. [DOI] [PubMed] [Google Scholar]
- 12.Larson N, Ghandehari H. Polymeric conjugates for drug delivery. Chem Mater. 2012;24(5):840–853. doi: 10.1021/cm2031569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newman MR, Benoit DS. Local and targeted drug delivery for bone regeneration. Curr Opin Biotechnol. 2016;40:125–32. doi: 10.1016/j.copbio.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stefanick JF, Ashley JD, Kiziltepe T, Bilgicer B. A systematic analysis of peptide linker length and liposomal polyethylene glycol coating on cellular uptake of peptide-targeted liposomes. Acs Nano. 2013;7(4):2935–47. doi: 10.1021/nn305663e. [DOI] [PubMed] [Google Scholar]
- 15.Maheshwari G, Brown G, Lauffenburger DA, Wells A, Griffith LG. Cell adhesion and motility depend on nanoscale RGD clustering. J Cell Sci. 2000;113(Pt 10):1677–86. doi: 10.1242/jcs.113.10.1677. [DOI] [PubMed] [Google Scholar]
- 16.Koo LY, Irvine DJ, Mayes AM, Lauffenburger DA, Griffith LG. Co-regulation of cell adhesion by nanoscale RGD organization and mechanical stimulus. J Cell Sci. 2002;115(Pt 7):1423–33. doi: 10.1242/jcs.115.7.1423. [DOI] [PubMed] [Google Scholar]
- 17.Petrie TA, Raynor JE, Dumbauld DW, Lee TT, Jagtap S, Templeman KL, Collard DM, Garcia AJ. Multivalent integrin-specific ligands enhance tissue healing and biomaterial integration. Sci Transl Med. 2010;2(45):45ra60. doi: 10.1126/scitranslmed.3001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coussen F, Choquet D, Sheetz MP, Erickson HP. Trimers of the fibronectin cell adhesion domain localize to actin filament bundles and undergo rearward translocation. J Cell Sci. 2002;115(Pt 12):2581–90. doi: 10.1242/jcs.115.12.2581. [DOI] [PubMed] [Google Scholar]
- 19.Wang D, Sima M, Mosley RL, Davda JP, Tietze N, Miller SC, Gwilt PR, Kopeckova P, Kopecek J. Pharmacokinetic and biodistribution studies of a bone-targeting drug delivery system based on N-(2-hydroxypropyl)methacrylamide copolymers. Mol Pharmaceut. 2006;3(6):717–725. doi: 10.1021/mp0600539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujisaki J, Tokunaga Y, Takahashi T, Kimura S, Shimojo F, Hata T. Osteotropic drug delivery system (ODDS) based on bisphosphonic prodrug. V. Biological disposition and targeting characteristics of osteotropic estradiol. Biol Pharm Bull. 1997;20(11):1183–7. doi: 10.1248/bpb.20.1183. [DOI] [PubMed] [Google Scholar]
- 21.Bell A, Templeman D, Weinlein JC. Nonunion of the Femur and Tibia: An Update. Orthop Clin North Am. 2016;47(2):365–75. doi: 10.1016/j.ocl.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 22.Cai L, Wang Q, Gu C, Wu J, Wang J, Kang N, Hu J, Xie F, Yan L, Liu X, Cao Y, Xiao R. Vascular and micro-environmental influences on MSC-coral hydroxyapatite construct-based bone tissue engineering. Biomaterials. 2011;32(33):8497–505. doi: 10.1016/j.biomaterials.2011.07.087. [DOI] [PubMed] [Google Scholar]
- 23.Omlor GW, Kleinschmidt K, Gantz S, Speicher A, Guehring T, Richter W. Increased bone formation in a rabbit long-bone defect model after single local and single systemic application of erythropoietin. Acta Orthop. 2016;87(4):425–31. doi: 10.1080/17453674.2016.1198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minkwitz S, Fassbender M, Kronbach Z, Wildemann B. Longitudinal analysis of osteogenic and angiogenic signaling factors in healing models mimicking atrophic and hypertrophic non-unions in rats. PloS one. 2015;10(4):e0124217. doi: 10.1371/journal.pone.0124217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchelli D, Piodi LP, Corradini C, Parravicini L, Verdoia C, Ulivieri FM. Increased serum OPG in atrophic nonunion shaft fractures. J Orthop Traumatol. 2009;10(2):55–8. doi: 10.1007/s10195-009-0047-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mir SA, Azam MQ, Al-Dakheel DA, Acharya S. Healing of Experimentally Created Non-Union of Femur in Rats Using Bone Precursor Cells from Mesenchymal Stem Cells (MSCs) J Stem Cells. 2015;10(2):91–6. [PubMed] [Google Scholar]
- 27.Sunagawa T, Bishop AT, Muramatsu K. Role of conventional and vascularized bone grafts in scaphoid nonunion with avascular necrosis: A canine experimental study. J Hand Surg Am. 2000;25(5):849–59. doi: 10.1053/jhsu.2000.8639. [DOI] [PubMed] [Google Scholar]
- 28.Boyan BD, Caplan AI, Heckman JD, Lennon DP, Ehler W, Schwartz Z. Osteochondral progenitor cells in acute and chronic canine nonunions. J Orthop Res. 1999;17(2):246–55. doi: 10.1002/jor.1100170214. [DOI] [PubMed] [Google Scholar]
- 29.Lo KW, Jiang T, Gagnon KA, Nelson C, Laurencin CT. Small-molecule based musculoskeletal regenerative engineering. Trends Biotechnol. 2014;32(2):74–81. doi: 10.1016/j.tibtech.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gittens SA, Bansal G, Kucharski C, Borden M, Uludag H. Imparting mineral affinity to fetuin by bisphosphonate conjugation: A comparison of three bisphosphonate conjugation schemes. Mol Pharmaceut. 2005;2(5):392–406. doi: 10.1021/mp050017u. [DOI] [PubMed] [Google Scholar]
- 31.Doschak MR, Kucharski CM, Wright JEI, Zernicke RF, Uludag H. Improved Bone Delivery of Osteoprotegerin by Bisphosphonate Conjugation in a Rat Model of Osteoarthritis. Mol Pharmaceut. 2009;6(2):634–640. doi: 10.1021/mp8002368. [DOI] [PubMed] [Google Scholar]
- 32.Ouyang L, Huang WC, He G, Guo L. Bone Targeting Prodrugs Based on Peptide Dendrimers, Synthesis and Hydroxyapatite Binding In Vitro. Lett Org Chem. 2009;6(4):272–277. [Google Scholar]
- 33.Sekido T, Sakura N, Higashi Y, Miya K, Nitta Y, Nomura M, Sawanishi H, Morito K, Masamune Y, Kasugai S, Yokogawa K, Miyamoto K. Novel drug delivery system to bone using acidic oligopeptide: Pharmacokinetic characteristics and pharmacological potential. J Drug Target. 2001;9(2):111–121. doi: 10.3109/10611860108997922. [DOI] [PubMed] [Google Scholar]
- 34.Neale JR, Richter NB, Merten KE, Taylor KG, Singh S, Waite LC, Emery NK, Smith NB, Cai J, Pierce WM. Bone selective effect of an estradiol conjugate with a novel tetracycline-derived bone-targeting agent. Bioorg Med Chem Lett. 2009;19(3):680–683. doi: 10.1016/j.bmcl.2008.12.051. [DOI] [PubMed] [Google Scholar]
- 35.Orme MW, Labroo VM. Synthesis of Beta-Estradiol-3-Benzoate-17-(Succinyl-12a-Tetracycline) - a Potential Bone-Seeking Estrogen. Bioorg Med Chem Lett. 1994;4(11):1375–1380. [Google Scholar]
- 36.Ponnapakkam T, Katikaneni R, Sakon J, Stratford R, Gensure RC. Treating osteoporosis by targeting parathyroid hormone to bone. Drug Discov Today. 2014;19(3):204–8. doi: 10.1016/j.drudis.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang C, Guo B, Wu H, Shao N, Li D, Liu J, Dang L, Wang C, Li H, Li S, Lau WK, Cao Y, Yang Z, Lu C, He X, Au DW, Pan X, Zhang BT, Lu C, Zhang H, Yue K, Qian A, Shang P, Xu J, Xiao L, Bian Z, Tan W, Liang Z, He F, Zhang L, Lu A, Zhang G. Aptamer-functionalized lipid nanoparticles targeting osteoblasts as a novel RNA interference-based bone anabolic strategy. Nature medicine. 2015;21(3):288–94. doi: 10.1038/nm.3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yokogawa K, Miya K, Sekido T, Higashi Y, Nomura M, Fujisawa R, Morito K, Masamune Y, Waki Y, Kasugai S, Miyamoto K. Selective delivery of estradiol to bone by aspartic acid oligopeptide and its effects on ovariectomized mice. Endocrinology. 2001;142(3):1228–1233. doi: 10.1210/endo.142.3.8024. [DOI] [PubMed] [Google Scholar]
- 39.Ponnapakkam T, Katikaneni R, Suda H, Miyata S, Matsushita O, Sakon J, Gensure RC. A single injection of the anabolic bone agent, parathyroid hormone-collagen binding domain (PTH-CBD), results in sustained increases in bone mineral density for up to 12 months in normal female mice. Calcif Tissue Int. 2012;91(3):196–203. doi: 10.1007/s00223-012-9626-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukushima O, Bekker PJ, Gay CV. Ultrastructural localization of tartrate-resistant acid phosphatase (purple acid phosphatase) activity in chicken cartilage and bone. Am J Anat. 1991;191(3):228–36. doi: 10.1002/aja.1001910303. [DOI] [PubMed] [Google Scholar]
- 41.Sheu TJ, Schwarz EM, O’Keefe RJ, Rosier RN, Puzas JE. Use of a phage display technique to identify potential osteoblast binding sites within osteoclast lacunae. J Bone Miner Res. 2002;17(5):915–922. doi: 10.1359/jbmr.2002.17.5.915. [DOI] [PubMed] [Google Scholar]
- 42.Van Hove AH, Wilson BD, Benoit DS. Microwave-assisted functionalization of poly(ethylene glycol) and on-resin peptides for use in chain polymerizations and hydrogel formation. Journal of visualized experiments : JoVE. 2013;(80):e50890. doi: 10.3791/50890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moad G, Chong YK, Postma A, Rizzardo E, Thang SH. Advances in RAFT polymerization: the synthesis of polymers with defined end-groups. Polymer. 2005;46(19):8458–8468. [Google Scholar]
- 44.Le Neindre M, Magny B, Nicolay R. Evaluation of thiocarbonyl and thioester moieties as thiol protecting groups for controlled radical polymerization. Polym Chem-Uk. 2013;4(22):5577–5584. [Google Scholar]
- 45.Nicolay R. Synthesis of Well-Defined Polythiol Copolymers by RAFT Polymerization. Macromolecules. 2012;45(2):821–827. [Google Scholar]
- 46.Stoscheck CM. Quantitation of Protein. Methods in enzymology. 1990;182:50–68. doi: 10.1016/0076-6879(90)82008-p. [DOI] [PubMed] [Google Scholar]
- 47.Anthis NJ, Clore GM. Sequence-specific determination of protein and peptide concentrations by absorbance at 205 nm. Protein Sci. 2013;22(6):851–858. doi: 10.1002/pro.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abdollahi M, Sharifpour M. A new simple procedure to calculate monomer reactivity ratios by using on-line H-1 NMR kinetic experiments: Copolymerization system with greater difference between the monomer reactivity ratios. Polymer. 2007;48(1):25–30. [Google Scholar]
- 49.Shen Y, Maupetit J, Derreumaux P, Tuffery P. Improved PEP-FOLD Approach for Peptide and Miniprotein Structure Prediction. J Chem Theory Comput. 2014;10(10):4745–58. doi: 10.1021/ct500592m. [DOI] [PubMed] [Google Scholar]
- 50.Pittenger MF. Mesenchymal stem cells from adult bone marrow. Methods in molecular biology. 2008;449:27–44. doi: 10.1007/978-1-60327-169-1_2. [DOI] [PubMed] [Google Scholar]
- 51.Tests for in vitro cytotoxicity. Switzerland: 2009. Biological evaluation of medical devices; p. 42. [Google Scholar]
- 52.Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15(8):1477–88. doi: 10.1359/jbmr.2000.15.8.1477. [DOI] [PubMed] [Google Scholar]
- 53.Apostolovic B, Klok HA. Copolymerization Behavior of N-(2-Hydroxypropyl)methacrylamide and a Methacrylated Coiled-Coil Peptide Derivative. Biomacromolecules. 2010;11(7):1891–1895. doi: 10.1021/bm100533g. [DOI] [PubMed] [Google Scholar]
- 54.Manju MVMK, Prasannakumar S, Made Gowda NM, Sherigara BS. Synthesis and Characterization of Copolymers of Methyl Methacrylate and 2-Ethoxyethyl Methacrylate. American Journal of Polymer Science. 2012;2(3):22–27. [Google Scholar]
- 55.Holmberg AL, Karavolias MG, Epps TH. RAFT polymerization and associated reactivity ratios of methacrylate-functionalized mixed bio-oil constituents. Polym Chem-Uk. 2015;6(31):5728–5739. [Google Scholar]
- 56.Murata H, Sanda F, Endo T. Syntheses and radical polymerization behavior of methacrylamides having peptide moieties: Effect of the methylene chain introduced between the methacrylamide and peptide moieties on the polymerizability and polymer structure. Macromolecules. 1997;30(10):2902–2906. [Google Scholar]
- 57.Moad G, Rizzardo E, Thang SH. Radical addition–fragmentation chemistry in polymer synthesis. Polymer. 2008;49(5):1079–1131. [Google Scholar]
- 58.Gokce I, Woody RW, Anderluh G, Lakey JH. Single peptide bonds exhibit poly(pro)II (“random coil”) circular dichroism spectra. J Am Chem Soc. 2005;127(27):9700–1. doi: 10.1021/ja052632x. [DOI] [PubMed] [Google Scholar]
- 59.Wang D, Chen K, Kulp JL, III, Arora PS. Evaluation of biologically relevant short alpha-helices stabilized by a main-chain hydrogen-bond surrogate. J Am Chem Soc. 2006;128(28):9248–56. doi: 10.1021/ja062710w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rodger A, Nordén B. Circular dichroism and linear dichroism. Oxford University Press; Oxford, New York: 1997. p. x, 150. [Google Scholar]
- 61.Moghimi SMFZS. Anatomical and Physiochemical Factors Controlling Nanoparticle Pharmacokinetics. In: Torchilin V, editor. Handbook of Nanobiomedical Research: Fundamentals, Applications and Recent Developments. 2014. pp. 31–44. (Vol. Biology, Safety and Novel Concepts in Nanomedicine). [Google Scholar]
- 62.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–51. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, Agarwal RG, Lam KS. The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles. Biomaterials. 2011;32(13):3435–46. doi: 10.1016/j.biomaterials.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Flessner MF, Dedrick RL, Schultz JS. Exchange of Macromolecules between Peritoneal-Cavity and Plasma. American Journal of Physiology. 1985;248(1):H15–H25. doi: 10.1152/ajpheart.1985.248.1.H15. [DOI] [PubMed] [Google Scholar]
- 65.Lindqvist Y, Johansson E, Kaija H, Vihko P, Schneider G. Three-dimensional structure of a mammalian purple acid phosphatase at 2.2 A resolution with a mu-(hydr)oxo bridged di-iron center. J Mol Biol. 1999;291(1):135–47. doi: 10.1006/jmbi.1999.2962. [DOI] [PubMed] [Google Scholar]
- 66.Hlavacek WS, Posner RG, Perelson AS. Steric effects on multivalent ligand-receptor binding: Exclusion of ligand sites by bound cell surface receptors. Biophys J. 1999;76(6):3031–3043. doi: 10.1016/S0006-3495(99)77456-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pan HZ, Sima M, Miller SC, Kopeckova P, Yang JY, Kopecek J. Efficiency of high molecular weight backbone degradable HPMA copolymer-Prostaglandin E-1 conjugate in promotion of bone formation in ovariectomized rats. Biomaterials. 2013;34(27):6528–6538. doi: 10.1016/j.biomaterials.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uludag H, Gao T, Wohl GR, Kantoci D, Zernicke RF. Bone affinity of a bisphosphonate-conjugated protein in vivo. Biotechnol Prog. 2000;16(6):1115–8. doi: 10.1021/bp000066y. [DOI] [PubMed] [Google Scholar]
- 69.Uludag H, Yang J. Targeting systemically administered proteins to bone by bisphosphonate conjugation. Biotechnol Progr. 2002;18(3):604–611. doi: 10.1021/bp0200447. [DOI] [PubMed] [Google Scholar]
- 70.Wang D, Miller S, Sima M, Kopeckova P, Kopecek J. Synthesis and evaluation of water-soluble polymeric bone-targeted drug delivery systems. Bioconjug Chem. 2003;14(5):853–9. doi: 10.1021/bc034090j. [DOI] [PubMed] [Google Scholar]
- 71.Jahnke W, Henry C. An in vitro assay to measure targeted drug delivery to bone mineral. ChemMedChem. 2010;5(5):770–6. doi: 10.1002/cmdc.201000016. [DOI] [PubMed] [Google Scholar]
- 72.Wang D, Miller SC, Shlyakhtenko LS, Portillo AM, Liu XM, Papangkorn K, Kopekova P, Lyubchenko Y, Higuchi WI, Kopecek J. Osteotropic peptide that differentiates functional domains of the skeleton. Bioconjugate Chem. 2007;18(5):1375–1378. doi: 10.1021/bc7002132. [DOI] [PubMed] [Google Scholar]
- 73.Schell H, Lienau J, Epari DR, Seebeck P, Exner C, Muchow S, Bragulla H, Haas NP, Duda GN. Osteoclastic activity begins early and increases over the course of bone healing. Bone. 2006;38(4):547–54. doi: 10.1016/j.bone.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 74.Kay BK, Kasanov J, Yamabhai M. Screening phage-displayed combinatorial peptide libraries. Methods. 2001;24(3):240–6. doi: 10.1006/meth.2001.1185. [DOI] [PubMed] [Google Scholar]
- 75.Kalinin NL, Ward LD, Winzor DJ. Effects of Solute Multivalence on the Evaluation of Binding Constants by Biosensor Technology - Studies with Concanavalin-a and Interleukin-6 as Partitioning Proteins. Anal Biochem. 1995;228(2):238–244. doi: 10.1006/abio.1995.1345. [DOI] [PubMed] [Google Scholar]
- 76.Muller KM, Arndt KM, Pluckthun A. Model and simulation of multivalent binding to fixed ligands. Anal Biochem. 1998;261(2):149–58. doi: 10.1006/abio.1998.2725. [DOI] [PubMed] [Google Scholar]
- 77.Jin J, Han Y, Zhang C, Liu J, Jiang W, Yin J, Liang H. Effect of grafted PEG chain conformation on albumin and lysozyme adsorption: A combined study using QCM-D and DPI. Colloids Surf B Biointerfaces. 2015;136:838–44. doi: 10.1016/j.colsurfb.2015.10.025. [DOI] [PubMed] [Google Scholar]
- 78.Sun M, Deng J, Tang Z, Wu J, Li D, Chen H, Gao C. A correlation study of protein adsorption and cell behaviors on substrates with different densities of PEG chains. Colloids Surf B Biointerfaces. 2014;122:134–42. doi: 10.1016/j.colsurfb.2014.06.041. [DOI] [PubMed] [Google Scholar]
- 79.Nakaoka R, Tabata Y, Yamaoka T, Ikada Y. Prolongation of the serum half-life period of superoxide dismutase by poly(ethylene glycol) modification. J Control Release. 1997;46(3):253–261. [Google Scholar]
- 80.Singha NK, Gibson MI, Koiry BP, Danial M, Klok HA. Side-chain peptide-synthetic polymer conjugates via tandem “ester-amide/thiol-ene” post-polymerization modification of poly(pentafluorophenyl methacrylate) obtained using ATRP. Biomacromolecules. 2011;12(8):2908–13. doi: 10.1021/bm200469a. [DOI] [PubMed] [Google Scholar]
- 81.Francini N, Purdie L, Alexander C, Mantovani G, Spain SG. Multifunctional poly[N-(2-hydroxypropyl)methacrylamide] copolymers via post-polymerization modification and sequential thiol–ene chemistry. Macromolecules. 2015;48(9):7. [Google Scholar]
- 82.Moraes J, Simionca IM, Ketari H, Klok HA. Avoiding compositional drift during the RAFT copolymerization of N-(2-hydroxypropyl)-methacrylamide and N-acryloxysuccinimide: towards uniform platforms for post-polymerization modification (vol 6, pg 3245, 2015) Polym Chem-Uk. 2015;6(42):7480–7483. [Google Scholar]
- 83.Vauquelin G, Charlton SJ. Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br J Pharmacol. 2010;161(3):488–508. doi: 10.1111/j.1476-5381.2010.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kenanova VE. Engineering of the Fc Region for Improved PK (FcRn Interaction) Vol. 1 Springer Science & Business Media; 2010. [Google Scholar]
- 85.Mourez M, Kane RS, Mogridge J, Metallo S, Deschatelets P, Sellman BR, Whitesides GM, Collier RJ. Designing a polyvalent inhibitor of anthrax toxin. Nat Biotechnol. 2001;19(10):958–61. doi: 10.1038/nbt1001-958. [DOI] [PubMed] [Google Scholar]
- 86.Glaffig M, Palitzsch B, Stergiou N, Schull C, Strassburger D, Schmitt E, Frey H, Kunz H. Enhanced immunogenicity of multivalent MUC1 glycopeptide antitumour vaccines based on hyperbranched polymers. Org Biomol Chem. 2015;13(40):10150–4. doi: 10.1039/c5ob01255d. [DOI] [PubMed] [Google Scholar]
- 87.Wang SK, Liang PH, Astronomo RD, Hsu TL, Hsieh SL, Burton DR, Wong CH. Targeting the carbohydrates on HIV-1: Interaction of oligomannose dendrons with human monoclonal antibody 2G12 and DC-SIGN. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(10):3690–5. doi: 10.1073/pnas.0712326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mandal S, Hammink R, Tel J, Eksteen-Akeroyd ZH, Rowan AE, Blank K, Figdor CG. Polymer-based synthetic dendritic cells for tailoring robust and multifunctional T cell responses. ACS Chem Biol. 2015;10(2):485–92. doi: 10.1021/cb500455g. [DOI] [PubMed] [Google Scholar]
- 89.Benoit DS, Anseth KS. The effect on osteoblast function of colocalized RGD and PHSRN epitopes on PEG surfaces. Biomaterials. 2005;26(25):5209–20. doi: 10.1016/j.biomaterials.2005.01.045. [DOI] [PubMed] [Google Scholar]
- 90.Reyes CD, Petrie TA, Garcia AJ. Mixed extracellular matrix ligands synergistically modulate integrin adhesion and signaling. J Cell Physiol. 2008;217(2):450–8. doi: 10.1002/jcp.21512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41(7):2971–3010. doi: 10.1039/c2cs15344k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Avvakumova S, Colombo M, Tortora P, Prosperi D. Biotechnological approaches toward nanoparticle biofunctionalization. Trends Biotechnol. 2014;32(1):11–20. doi: 10.1016/j.tibtech.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 93.Patil YB, Toti US, Khdair A, Ma L, Panyam J. Single-step surface functionalization of polymeric nanoparticles for targeted drug delivery. Biomaterials. 2009;30(5):859–66. doi: 10.1016/j.biomaterials.2008.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Takahashi T, Yokogawa K, Sakura N, Nomura M, Kobayashi S, Miyamoto K. Bone-targeting of quinolones conjugated with an acidic oligopeptide. Pharm Res. 2008;25(12):2881–8. doi: 10.1007/s11095-008-9605-4. [DOI] [PubMed] [Google Scholar]
- 95.Ding H, Kopeckova P, Kopecek J. Self-association properties of HPMA copolymers containing an amphipathic heptapeptide. J Drug Target. 2007;15(7–8):465–74. doi: 10.1080/10611860701500016. [DOI] [PubMed] [Google Scholar]
- 96.Baranello MP, Bauer L, Benoit DS. Poly(styrene-alt-maleic anhydride)-based diblock copolymer micelles exhibit versatile hydrophobic drug loading, drug-dependent release, and internalization by multidrug resistant ovarian cancer cells. Biomacromolecules. 2014;15(7):2629–41. doi: 10.1021/bm500468d. [DOI] [PubMed] [Google Scholar]
- 97.Wang Y, Newman MR, Ackun-Farmmer M, Baranello MP, Sheu TJ, Puzas JE, Benoit DSW. Fracture-Targeted Delivery of beta-Catenin Agonists via Peptide-Functionalized Nanoparticles Augments Fracture Healing. Acs Nano. 2017;11(9):9445–9458. doi: 10.1021/acsnano.7b05103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.