

Abstract
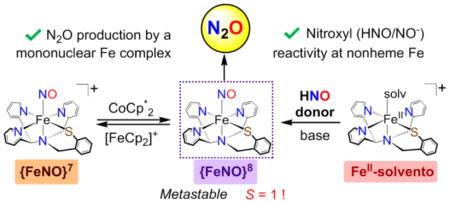
One-electron reduction of [Fe(NO)-(N3PyS)]BF4 (1) leads to the production of the metastable nonheme {FeNO}8 complex, [Fe(NO)(N3PyS)] (3). Complex 3 is a rare example of a high-spin (S = 1) {FeNO}8, and is the first example, to our knowledge, of a mononuclear nonheme {FeNO}8 species that generates N2O. A second, novel route to 3 involves addition of Piloty’s acid, an HNO donor, to an FeII precursor. This work provides possible new insights regarding the mechanism of nitric oxide reductases.
Graphical Abstract
Nitric oxide (NO•) contributes to cellular stress through the secondary production of reactive oxygen and nitrogen species. To combat the toxicity associated with NO•, anaerobic microorganisms have evolved detoxification strategies based on the transition metal mediated reduction of NO• to N2O: 2 NO• + 2 e− + 2 H+ → N2O + H2O. Denitrifying bacteria utilize a mixed heme-nonheme diiron motif to reductively activate NO• (cytochrome c-dependent NOR, cNOR), whereas non-denitrifying bacteria, archaea, and protozoans utilize a nonheme flavodiiron protein (FDP) to catalyze the same reaction (Figure 1).1 Dinuclear Fe active sites can accommodate two Fe-NO units in close proximity, which may promote the key N-N bond formation required for N2O production, and may also help stabilize NO• coupling intermediates (e.g., hyponitrite (ONNO2−)) or oxygen-containing co-products (e.g., O2−, OH−). While both enzymatic1d,2 and biomimetic3 studies have suggested that pre-orientation of two Fe-NO monomers provides an efficient mechanism for N2O production, the example of fungal P450 NO• reductase (P450nor) demonstrates that NO• reduction to N2O can proceed at a mononuclear heme iron center (Figure 1). Alternative mechanisms have been proposed for the diiron active sites of cNOR and FDP that involve N-N bond formation and N2O production occurring at a single nonheme iron center.4
Figure 1.
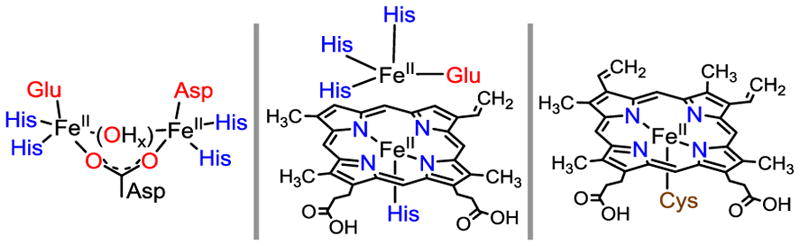
Fe-containing active sites involved in NO reduction. Left: FDPs. Middle: heme-nonheme NORs. Right: P450nor.
Reduction of {FeNO}x (x = 6, 7) species to the reactive {FeNO}8 or {FeN(H)O}8 states is often invoked as a prerequisite for N2O formation in enzymatic systems. There has been considerable interest in the preparation and characterization of {FeNO}8/{FeN(H)O}8 model complexes,5 but their synthesis is challenging because of their instability. A limited number of nonheme {FeNO}8 complexes have been characterized,5a–g and these complexes exhibit low-spin S = 0 ground states,5a–f with one exception of a high-spin S = 1 complex.5g However, there are no reports of a mononuclear {FeNO}8 leading to N2O production in solution.6 In the case of dinuclear Fe complexes, there are only two examples that produce N2O from ({FeNO}7)2 precursors.3a,b Biomimetic systems that not only stabilize the {FeNO}8 unit for spectroscopic interrogation, but also direct reactivity toward N2O production, would be valuable for study.
We previously described the synthesis of a mononuclear {FeNO}7 complex as a model of the NO-bound form of the nonheme iron enzyme cysteine dioxygenase (CDO). This complex, [Fe(NO)(N3PyS)]BF4 (1), prepared from addition of NO•(g) to the FeII precursor [FeII(CH3CN)(N3PyS)]BF4 (2),7 was crystallographically characterized and showed excellent reversibility for both one-electron oxidation and reduction processes by cyclic voltammetry.8 Herein we describe the one-electron reduction chemistry of 1, which leads to the generation of a metastable, nonheme {FeNO}8 complex, [Fe(NO)(N3PyS)] (3). This complex spontaneously generates N2O upon standing in solution at 23 °C and is the first example, to our knowledge, of a mononuclear {FeNO}8 species that reacts to give N2O. Spectroscopic and computational investigations also indicate that complex 3 exhibits a rare high-spin S = 1 ground state. It is also shown that reaction of 2 with the HNO donor Piloty’s acid (P.A.) leads to complex 3, suggesting that a nonheme FeII center could serve as a target for nitroxyl (HNO/NO−) reactivity.
Previous examination of the {FeNO}7 complex 1 by cyclic voltammetry revealed two nicely reversible waves at E1/2 = 0.013 and −1.18 V (vs Fc+/Fc in CH3CN).8a The well-behaved reversibility of the redox couple centered at E1/2 = −1.18 V suggested that the one-electron-reduced {FeNO}8 species may be stable enough for synthesis and characterization by chemical methods. The one-electron reductant decamethylcobaltocene (CoCp*2) has a redox potential of E1/2 = −1.91 V (vs Fc+/Fc in CH3CN),9 indicating that it should be thermodynamically competent to reduce the {FeNO}7 complex. Addition of CoCp*2 (1 equiv) to a dark brown solution of 1 in CH3CN at −40 °C results in the immediate formation of an air- and light-sensitive purple species with absorbance maxima at 520 and 720 nm (ε ≈ 3400 and 2200 M−1 cm−1, respectively), corresponding to the {FeNO}8 complex, [Fe(NO)(N3PyS)] (3) (Scheme 1).
Scheme 1.
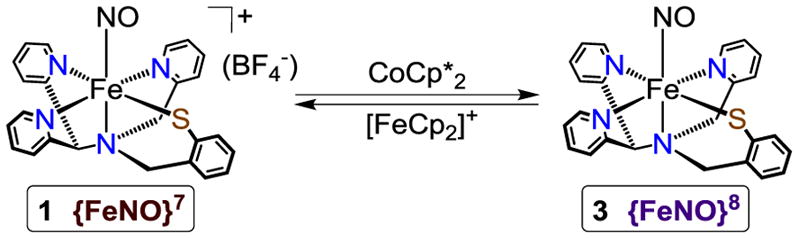
Reversible One-Electron Reduction of [Fe(NO)(N3PyS)]BF4 (1) at −40 °C
Spectral titrations at −40 °C show an isosbestic conversion of 1 to 3, reaching maximal formation of 3 upon addition of 1.0 equiv of reductant (Figure 2). These data confirm the 1:1 stoichiometry of the reduction reaction. Titration of 3 with the one-electron oxidant [FeCp2]+PF6− results in loss of the spectrum for 3 and the quantitative recovery of the spectrum for the {FeNO}7 starting material 1 (Figure S2). The isosbestic point at 470 nm is retained during the re-oxidation, although a second isosbestic point at 334 nm is likely obscured by absorbance from FeCp2. These findings demonstrate that the interconversion of the {FeNO}7 and {FeNO}8 species is both chemically reversible and high-yielding at −40 °C, and provide strong evidence that the Fe(NO)(N3PyS) formulation remains unchanged during reduction and oxidation cycles. The reduced 3 exhibits good stability over several hours under the spectral titration conditions (0.1 mM Fe complex) at −40 °C. However, efforts to concentrate and isolate 3 as a solid at low-temperature have thus far been unsuccessful. Reduction of 1 at 23 °C also leads to complex 3 as seen by UV-vis, but the product has limited stability (t1/2 ≈ 0.5 h at 0.1 mM) at this temperature.
Figure 2.
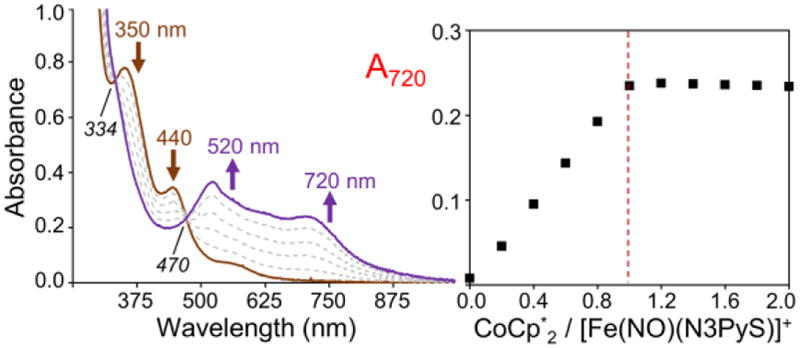
Left: UV-vis spectral titration showing the one-electron reduction of [Fe(NO)(N3PyS)]BF4 (1) (brown line) to [Fe(NO)(N3PyS)] (3) (purple line) upon addition of CoCp*2 (0.2 – 1.0 equiv) in CH3CN/toluene at −40 °C. Right: Plot of absorbance at 720 nm versus CoCp*2/1.
The {FeNO}8 complex was further characterized by NMR and EPR spectroscopies. The 1H-NMR spectrum of 1 in CD3CN/toluene-d8 (95/5 v/v) reveals well-separated, relatively sharp, paramagnetically shifted resonances between −2 and 35 ppm (Figure 3). Upon reduction with 1 equiv of CoCp*2, the characteristic peaks for 1 disappear and are replaced by a new set of paramagnetic signals between −10 and 40 ppm. These new peaks uniformly diminish over two hours at 23 °C, leaving only peaks in the diamagnetic region of the spectrum. These data are consistent with a paramagnetic, one-electron reduced species 3 decaying to diamagnetic products.
Figure 3.
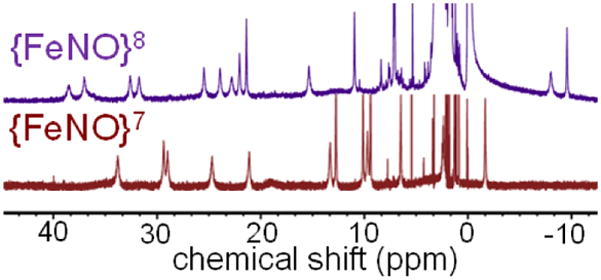
Paramagnetic 1H-NMR spectra of 1 (red) and 3 (purple) in CD3CN/toluene-d8 (95/5 v/v) at 23 °C.
The magnetic moment for 3, prepared from freshly mixed 1 + CoCp*2 in CD3CN/toluene, was measured by the Evans method (see SI) and gave μeff = 2.9 μB, which is in excellent agreement with the theoretical spin-only value for an S = 1 system (2.83 μB). In support of this assignment, DFT calculations yield a triplet (S = 1) ground state for 3, with a singlet (S = 0) state 11.8 kcal mol−1 higher in energy (see SI for details). Mulliken population analysis of triplet 3 reveals spin densities of +3.156 on Fe and −1.154 on NO, suggestive of an electronic structure that is more in line with hs FeI-NO• than hs FeII-3NO−. The EPR spectrum for 1 at 15 K (g = [2.047, 2.007, 1.962]) arises from the S = ½ spin ground state of the {FeNO}7 starting material, and disappears upon one-electron reduction, leaving only a residual EPR signal near g ~ 2 that can be assigned to unreacted CoCp2* (CoII, d7, S = ½) (Figure S5). These spectra indicate that the product 3 is EPR-silent, which, coupled with a paramagnetic 1H-NMR spectrum, is consistent with an S = 1 ground state. Monitoring the slow decay of 3 at 23 °C by EPR shows no new peaks, further indicative of diamagnetic decay products as observed by 1H-NMR.
The {FeNO}8 complex 3 was characterized by low-temperature resonance Raman (RR) spectroscopy and compared to the {FeNO}7 starting material 1. The RR spectra of 1 obtained with 458 nm excitation show a ν(N—O) at 1641 cm−1 and expected 15N18O shifts that matches frequencies previously measured by low-temperature FTIR (Figure 4). We previously showed that 1 exhibits spin-crossover behavior, with a low-spin (S = 1/2) ground state and a thermally accessible high-spin (S = 3/2) excited state.8b The low-spin species exhibited a ν(N—O) at 1641 cm−1 at cryogenic temperatures, whereas the high-spin (S = 3/2) ν(N–O) is observed at 1737 cm−1 above 150 K (CD3CN). The RR spectrum of 3 (Figure 4) (λexc = 647 nm) reveals a prominent, broad band at 1588 cm−1 that shifts to 1518 cm−1 upon substitution with 15N18O; this 70-cm−1 downshift is in agreement with an N-O harmonic oscillator model (calculated Δν = −71 cm−1), confirming the assignment of this band to the N-O stretch of the new {FeNO}8 species. A vibration at 498 cm−1, which also shifts upon isotopic substitution with 15N18O, is tentatively assigned as a ν(Fe—NO) mode (Figure S7). The broadness of the {FeNO}8 signals (half-widths of ~25 cm−1) is noteworthy and may reflect rotational conformers of the nitrosyl ligand. The ν(N–O) frequency of 3 is ~150 cm−1 lower than in the high-spin {FeNO}7 complex 1, and can be compared with the 1618 cm−1 frequency observed by Lehnert and coworkers for the four N-donor ligand (TMG3tren).5g For the few nonheme {FeNO}8 complexes reported to date, a modest decrease in the ν(N–O) frequency has been attributed to reduction of primarily Fe, rather than the nitrosyl ligand.5c,f,g In our case, unrestricted corresponding orbital (UCO) analysis also supports a largely metal-centered reduction of 1 to 3 (Figures S12, S13).
Figure 4.
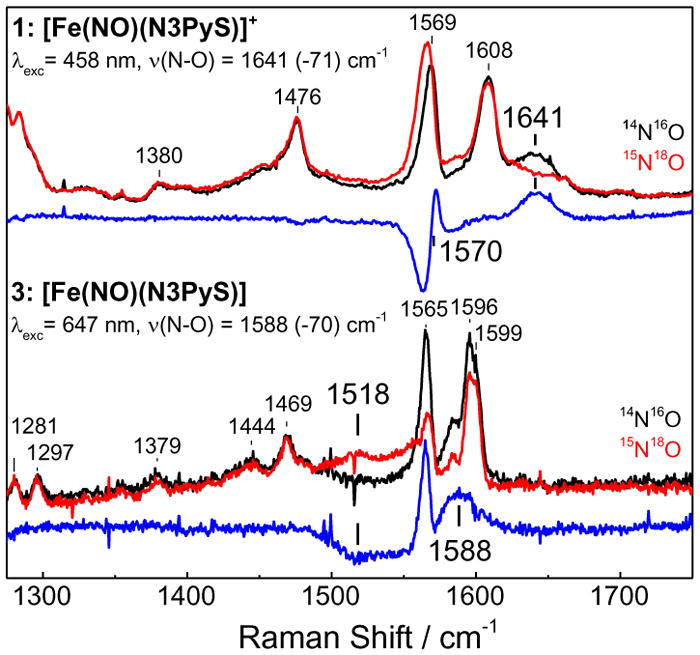
Resonance Raman spectra of 1 (top traces) and 3 (bottom traces) with 14N16O (black), 15N18O (red), and difference spectra (blue) at 110 K.
Solutions of 3 undergo a gradual color change from purple to red-orange at 23 °C, corresponding to slow decay of the UV-vis feature at 720 nm (t1/2 ≈ 0.5 h). This apparent self-decay reaction suggested the possibility of reductive N-N bond formation between two isolated {FeNO}8 monomers, leading to N2O formation. Gas chromatography (GC) analysis of the headspace above the reaction of 1 (0.1 mM) with CoCp*2 (1 equiv) over 20 h showed the formation of N2O in ~54% yield based on an assumed 2:1 stoichiometry for {FeNO}8 self-decay into N2O (Scheme 2). Control experiments established that the {FeNO}7 starting material 1 does not produce any N2O under equivalent conditions, demonstrating the necessity of a reduced {FeNO}8 species for the critical N-N bond forming reaction. Although the final Fe decay products have not been identified, the diamagnetic NMR spectra of final reaction mixtures are consistent with magnetically coupled multi-iron clusters. Thus, complex 1 is a mononuclear nonheme iron complex that mediates N2O formation upon one-electron reduction.
Scheme 2.
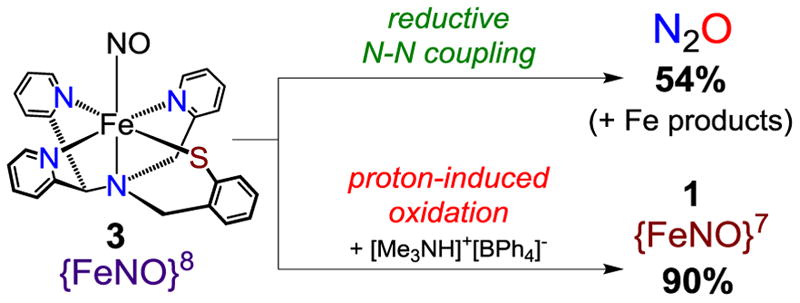
Production of N2O from 3 and Conversion of 3 to 1 by Acid in CH3CN/toluene at 23 °C
It is well known that HNO will rapidly dimerize to give N2O and H2O in aqueous solutions.10 To examine the possibility of N2O production from 3 via protonation from adventitious proton sources (e.g., H2O) and subsequent release of HNO, we reacted freshly generated 3 with the weak acid [Me3NH]+[BPh4]− (1 equiv) at 23 °C. Rather than increasing the yield of N2O, the formation of N2O was greatly suppressed (~4%). This result argues against a mechanism involving release of free HNO from 3 and subsequent N2O formation. Addition of a strong acid such as H+BArF− (1 equiv) also led to almost complete inhibition of N2O production. The reaction of 3 with acids instead led to the one-electron oxidation of 3 to give the {FeNO}7 starting material 1 (confirmed by 1H-NMR and EPR spectroscopies). This reaction is precedented by the proton-induced oxidation of {FeNO}8 complexes supported by sterically unencumbered porphyrins, which give the corresponding {FeNO}7 species upon addition of acid.5h–l
Chemical reduction and bulk electrolysis of {FeNO}7 complexes are the known methods for the generation of {FeNO}8 complexes. However, to the best of our knowledge, there is no report of an {FeNO}8 complex resulting from the reaction of an Fe(II) precursor and a nitroxyl anion (NO−) donor. The HNO donor N-hydroxybenzenesulfonamide (Piloty’s acid (P.A.)), can also furnish NO− under basic conditions.11 We hypothesized that deprotonation of P.A. with 2 equiv of a strong base in the presence of [FeII(N3PyS)(CH3CN)]BF4 (2) would provide access to the {FeNO}8 complex 3 through an FeII + NO− trapping reaction (Figure 5).
Figure 5.
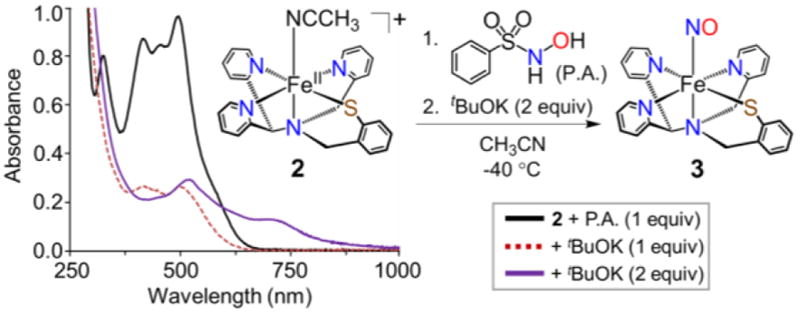
UV-vis spectra showing addition of tBuOK to a pre-mixed solution of 2 and Piloty’s acid in CH3CN at −40 °C.
Mixing of 2 with P.A. (1 equiv) in CH3CN at −40 °C shows no reaction by UV-vis; however, addition of tBuOK/18-crown-6 (1 equiv) triggers the slow decay of the FeII bands at 325, 418, and 493 nm over 1 h (Figure 5) to give a new spectrum with weaker bands at 418 and 501 nm. This new spectrum shows no further change for at least 6 h at −40 °C. However, addition of a second equiv of tBuOK/18-crown-6 results in immediate conversion to the characteristic spectrum of the {FeNO}8 complex 3 (Figure 5, purple trace). These results show that the overall reaction of 2 with P.A. and 2 equiv of base is a net transfer of NO− to the FeII starting material to give 3. The observed intermediate formed after only 1 equiv of base (Figure 5, red trace), could be either an Fe-HNO adduct, or a singly deprotonated FeII-P.A. complex. Future work will focus on characterizing this species. It should be noted that the {FeNO}8 product 3 formed at −40 °C under these conditions has reduced stability (full decay within ~5 min) as compared to one-electron reduction of 1. The lower stability in this case may be due to reaction with one or more of the byproducts of the deprotonation reaction (e.g., K+, tBuOH). Reaction of 2 with P.A. provides a novel approach for the preparation of {FeNO}8 complexes that bypasses {FeNO}7 reduction, and suggests that nonheme FeII sites in biology could be targets for NO− donors.
In summary, a nonheme {FeNO}8 complex was prepared by chemical reduction of an {FeNO}7 precursor and characterized by UV-vis, 1H-NMR, EPR, and RR spectroscopies. This complex exhibits a rare high-spin (S = 1) ground state, and is the first example of a mononuclear nonheme {FeNO}8 species that leads to production of N2O. These observations support the “super-reduced” mechanism for nonheme iron-dependent NO reductases, which requires reduction of {FeNO}7 centers to {FeNO}8 prior to N2O formation. Although this mechanism has been debated for a number of years, up to now there has been no direct characterization of a synthetic {FeNO}8 species that can generate N2O. We have also demonstrated a novel method for forming an {FeNO}8 species from a readily available HNO donor and base. This work suggests that mononuclear nonheme iron centers in biology could be sites for NO reductive activation, as well as for the transport of HNO/NO−.
Supplementary Material
Acknowledgments
The NSF (CHE1566007 to D.P.G.) and NIH (GM074785 to P.M.L.) is gratefully acknowledged for financial support. A.M.C. thanks the Harry and Cleio Greer Fellowship for support. We thank Prof. J.P. Toscano (JHU) and S. Nourian for use of instrumentation (headspace GC). The Kirin cluster at JHU KSAS is thanked for CPU time to A.M.C.
Footnotes
Syntheses, spectroscopy, GC traces, and DFT. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interests.
References
- 1.(a) Kurtz DM., Jr Dalton Trans. 2007:4115–4121. [Google Scholar]; (b) Wasser IM, de Vries S, Moënne-Loccoz P, Schröder I, Karlin KD. Chem Rev. 2002;102:1201. doi: 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]; (c) Watmough NJ, Field SJ, Hughes RJ, Richardson DJ. Biochem Soc Trans. 2009;37:392. doi: 10.1042/BST0370392. [DOI] [PubMed] [Google Scholar]; (d) Sato N, Ishii S, Sugimoto H, Hino T, Fukumori Y, Sako Y, Shiro Y, Tosha T. Proteins. 2014;82:1258. doi: 10.1002/prot.24492. [DOI] [PubMed] [Google Scholar]
- 2.Silaghi-Dumitrescu R, Kurtz DM, Jr, Ljungdahl LG, Lanzilotta WN. Biochemistry. 2005;44:6492–501. doi: 10.1021/bi0473049. [DOI] [PubMed] [Google Scholar]; (b) Hayashi T, Caranto JD, Wampler DA, Kurtz DM, Jr, Moënne-Loccoz P. Biochemistry. 2010;49:7040–9. doi: 10.1021/bi100788y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Jiang Y, Hayashi T, Matsumura H, Do LH, Majumdar A, Lippard SJ, Moënne-Loccoz P. J Am Chem Soc. 2014;136:12524. doi: 10.1021/ja504343t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zheng S, Berto TC, Dahl EW, Hoffman MB, Speelman AL, Lehnert N. J Am Chem Soc. 2013;135:4902. doi: 10.1021/ja309782m. [DOI] [PubMed] [Google Scholar]; (c) Kindermann N, Schober A, Demeshko S, Lehnert N, Meyer F. Inorg Chem. 2016;55:11538–11550. doi: 10.1021/acs.inorgchem.6b02080. [DOI] [PubMed] [Google Scholar]
- 4.(a) Moënne-Loccoz P. Nat Prod Rep. 2007;24:610. doi: 10.1039/b604194a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Timóteo CG, Pereira AS, Martins CE, Naik SG, Duarte AG, Moura JJ, Tavares P, Huynh BH, Moura I. Biochemistry. 2011;50:4251. doi: 10.1021/bi101605p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Duarte AG, Cordas CM, Moura JJ, Moura I. Biochim Biophys Acta. 2014;1837:375. doi: 10.1016/j.bbabio.2014.01.001. [DOI] [PubMed] [Google Scholar]; (d) Blomberg LM, Blomberg MR, Siegbahn PE. J Biol Inorg Chem. 2007;12:79. doi: 10.1007/s00775-006-0166-x. [DOI] [PubMed] [Google Scholar]
- 5.(a) Serres RG, Grapperhaus CA, Bothe E, Bill E, Weyhermüller T, Neese F, Wieghardt K. J Am Chem Soc. 2004;126:5138. doi: 10.1021/ja030645+. [DOI] [PubMed] [Google Scholar]; (b) Montenegro AC, Amorebieta VT, Slep LD, Martín DF, Roncaroli F, Murgida DH, Bari SE, Olabe JA. Angew Chem Int Ed Engl. 2009;48:4213. doi: 10.1002/anie.200806229. [DOI] [PubMed] [Google Scholar]; (c) Patra AK, Dube KS, Sanders BC, Papaefthymiou GC, Conradie J, Ghosh A, Harrop TC. Chem Sci. 2012;3:364. [Google Scholar]; (d) Sanders BC, Patra AK, Harrop TC. J Inorg Biochem. 2013;118:115. doi: 10.1016/j.jinorgbio.2012.08.026. [DOI] [PubMed] [Google Scholar]; (e) Chalkley MJ, Peters JC. Angew Chem Int Ed Engl. 2016;55:11995. doi: 10.1002/anie.201605403. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kupper C, Rees JA, Dechert S, DeBeer S, Meyer F. J Am Chem Soc. 2016;138:7888. doi: 10.1021/jacs.6b00584. [DOI] [PubMed] [Google Scholar]; (g) Speelman AL, Lehnert N. Angew Chem Int Ed Engl. 2013;52:12283. doi: 10.1002/anie.201305291. [DOI] [PubMed] [Google Scholar]; (h) Choi IK, Liu Y, Feng D, Paeng KJ, Ryan MD. Inorg Chem. 1991;30:1832. [Google Scholar]; (i) Pellegrino J, Bari SE, Bikiel DE, Doctorovich F. J Am Chem Soc. 2010;132:989. doi: 10.1021/ja905062w. [DOI] [PubMed] [Google Scholar]; (j) Goodrich LE, Roy S, Alp EE, Zhao J, Hu MY, Lehnert N. Inorg Chem. 2013;52:7766. doi: 10.1021/ic400977h. [DOI] [PubMed] [Google Scholar]; (k) Abucayon EG, Khade RL, Powell DR, Zhang Y, Richter-Addo GB. J Am Chem Soc. 2016;138:104. doi: 10.1021/jacs.5b12008. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Rahman MH, Ryan MD. Inorg Chem. 2017;56:3302. doi: 10.1021/acs.inorgchem.6b02665. [DOI] [PubMed] [Google Scholar]; (m) Hu B, Li J. Angew Chem Int Ed Engl. 2015;54:10579. doi: 10.1002/anie.201505166. [DOI] [PubMed] [Google Scholar]; (n) Kundakarla N, Lindeman S, Rahman MH, Ryan MD. Inorg Chem. 2016;55:2070. doi: 10.1021/acs.inorgchem.5b02384. [DOI] [PubMed] [Google Scholar]
- 6.A site-isolated Fe center in a MOF was shown to mediate reductive disproportionation of NO• to give N2O and NO2−. See: Brozek CK, Miller JT, Stoian SA, Dincă M. J Am Chem Soc. 2015;137:7495. doi: 10.1021/jacs.5b03761.
- 7.McQuilken AC, Jiang Y, Siegler MA, Goldberg DP. J Am Chem Soc. 2012;134:8758. doi: 10.1021/ja302112y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) McQuilken AC, Ha Y, Sutherlin KD, Siegler MA, Hodgson KO, Hedman B, Solomon EI, Jameson GN, Goldberg DP. J Am Chem Soc. 2013;135:14024. doi: 10.1021/ja4064487. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McQuilken AC, Matsumura H, Dürr M, Confer AM, Sheckelton JP, Siegler MA, McQueen TM, Ivanović-Burmazović I, Moënne-Loccoz P, Goldberg DP. J Am Chem Soc. 2016;138:3107. doi: 10.1021/jacs.5b12741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connelly NG, Geiger WE. Chem Rev. 1996;96:877. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 10.Miranda KM. Coord Chem Rev. 2005;249:433. [Google Scholar]
- 11.(a) Switzer CH, Miller TW, Farmer PJ, Fukuto JM. J Inorg Biochem. 2013;118:128. doi: 10.1016/j.jinorgbio.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen J, Campos J, Mercado BQ, Crabtree RH, Balcells D. Organometallics. 2014;33:4417. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.