

Abstract
A novel tert-butyl 2-(1-oxoisolndolin-2-yl)acetate derivative is selectively alkylated with propargyl bromide in the presence of lithium hexamethyldisilazide. After removal of the tert-butyl protecting group, the resulting N-isoindolinyl (ethynylalanine) derivative is reacted with a series of azides under ‘click conditions’. The click reactions afford an array of N-isoindolinyl-1,2,3-triazolylalanine derivatives as the free carboxylic adds. Following esterification, the N-isoindolinone protecting group is then transformed into the more easily removable phthaloyl group by selective oxidation at the benzylic position.
Keywords: amino acids, click chemistry, histidine, metal complexes, peptides, triazoles
Graphical Abstract
R1 = t-Bu, R1 = H, R2 = alkyl. benzyl
Click conditions: R2-N3/CuSO4-5H2O/Na ascorbate/THF-H2O, yields, 67–95%, 7 examples, Benzylic oxidation condition: Oxone/KBr/MeCN-H2O, yields, 72–91%, 7 examples.

The synthesis and utilization of unnatural amino adds is a vital component of protein engineering, drug design, and enzyme modification.1 Most frequently, the preparation of many unnatural or otherwise nonproteinogenic amino acids begins with the preparation of so-called glycine equivalents and further manipulation through the installation of side chains.2 In general the term ‘glycine equivalent’ infers a two-carbon fragment whereby the carboxyl group and the amino group bear suitable protection so that the methylene group may be activated as a nucleophile or electrophile to facilitate side-chain installation. In instances where chirality is warranted, the conjunction of side chains may be facilitated by mediation of chiral directing groups, ligands, or adjuvants.3 Unnatural amino acid precursors which have the requisite side chains already in place and contain a set of prochiral carbons, such as amino acrylates, need only to utilize enantioselective reducing agents to complete the scheme.4 In cases where the alkylation of protected glycine derivatives gives the racemic mixtures, the best recourse may be conversion into suitable substrates whereby resolution steps may employ chiral chromatography or enzymes.5 Considering the five-membered nitrogen heterocycles, the metal-complexing properties of the 1,2,3-triazolyl group is notable and has been the focus of development of diverse complexes bearing manganese, iron, nickel, copper, ruthenium, rhodium, iridinium, palladium, and gold for use in catalysis.6 Accordingly, through affixing any of several nitrogen heterocycles with metal-complexing properties to a polypeptide backbone, one may tailor both unique and diverse peptide motifs with catalytic properties.7 One finds many interesting parallels of the 1,2,3-tri-azolyl group to the imidazole group in histidine (Figure 1). In the areas of coordination chemistry, the functional similarities between the two nitrogen heterocycles is noteworthy and has driven the evaluation of the C- or N-substituted triazolyl group.8
Figure 1.

Triazolylalanine vs. histidine residues
For example, functional ized polymers bearing imidazole and triazole residues can effectively coordinate Cu+1 and Cu+2 ions and are being explored as antifouling agents.9 Furthermore, triazole linkers which can be formed from alkynyl amino acid residues and functionalized azides may be used in the fluorogenic labeling of polypeptides.1a,10 Invariably, the most straightforward synthesis of 1,2,3-tri-azoles falls within one major area of 1,3-dipolar cycloaddition chemistry which is the so-called ‘click’ reaction.11 Hence, the formation of the heterocycle only requires the reacting components of an azide and an alkyne and is facilitated under very mild conditions. Our synthesis of triazolyl analogues of histidine starts with the isoindolinone-protected tert-butyl glycine derivative 1 (Scheme 1). We elected to test the use of the isoindolinone group as N-protection of a glycine equivalent due to its increased robustness over the well-known N-phthaloyl group.
Scheme 1.

Preparation of the N-protected ethynylalanine click substrate 3. Reagents and conditions. (a) NaH, tert-butylbromoacetate, THF, 0 °C to r.t., 16 h (97%); (b) LIHMDS, prapargyl bromide, THF, −78 °C to r.t., 3 h, then HCl (84%); (c) K2CO3, MeOH, H2O, 65–70 °C, then 5% HCl (75%).
Furthermore, the employment of a strategic oxidative transformation converting the isoindolinoyl group into the easily removed phthaloyl group enhances or otherwise establishes the effectiveness of this ‘more dormant’ mode of protection. The sodium salt of isoindolinone is generated by treatment with sodium hydride (THF) followed by alkylation with tert-butyl bromoacetate (r.t., 16 h). The resulting N-isoindolinone-protected glycine ester 1 is then C-alkylated with propargyl bromide in the presence of lithium hexamethyldisilazide (−78 °C, THF). The resulting N-isoindolinonoyl ethynylalanine-tert-butyl ester 2 was obtained in 84% yield after purification by silica gel column chromatography.12 The reactions of tert-butyl ester 2 with benzyl-, 4-fluorobenzyl-, or 4-methoxybenzylazides under standard click conditions (CuSO4, ascorbate, THF, H2O) gave less than modest yields of the corresponding click products.
Suspecting that the presence of the tert-butyl ester group was somewhat of a hindrance to the click reaction, it was removed (K2CO3, MeOH, H2O) and replaced with a methyl group (MeOH, acetyl chloride; Scheme 2). Surprisingly, the click reactions with the corresponding methyl ester 3a did not show a marked improvement over similar reaction times and conditions as the tert-butyl substrate 3. However, after removal of the tert-butyl group, the corresponding ethynylalanine isoindolinone-protected free carboxylic acid 3 gave good to excellent yields of the corresponding click products 4–10 (Table 1).13
Scheme 2.

Click reactions of 3 with various azides, esterification to substrates 11–17, and oxidation to the phthaloyl-protected products 18–23. Reagents and conditions: (a) CuSO4-5H2O. Na ascorbate, THF/H2O (2:1), r.t., 16 h (see Table 1); (b) MeOH, acetyl chloride (cat), r.t., 16 h; (c) Oxone, KBr, MeCN/H2O (9:1), 55–60 °C, 16 h.
Table 1.
Click Reactions of Ethynylalanine Analogue 3 with Various Azidesa

| ||
|---|---|---|
| Azide | Product | Yield (%)b |
| N3-C6H13-n | 4 n-hexyl | 86 |
| N3-C6H17-n | 5 n-octyl | 85 |

|
6 cyclopentyl | 67 |
|
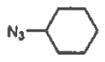
7 cyclohexyl | 63 |

|
8 benzyl | 95 |

|
9 4-fluorobenzyl | 94 |
|
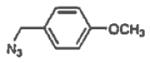
10 4-methoxybenzyl | 84 |
Reagents and conditions: CuSO4-5H2O, Na ascorbate, THF, H2O, r.t., 16 h.
Isolated chemical yield after silica gel column chromatography.
At this point we should note that, in principle, the base-sensitive N-phthaloyl protecting group, as opposed to the present N-isoindolyl protection, would not survive the ester cleavage and immediately suffer hydrolysis. The click products 4–10 were re-esterified with methanol (catalytic acetyl chloride, r.t., 16 h)14 which gave the more soluble corresponding methyl ester substrates 11–17 (64–87%) en route to the benzylic oxidation step. We have reported earlier that the interconversion of the isoindolinone (phthalimidine) group and the phthalimide (N-phthaloyl) group through a selective benzylic oxidation is a useful transformation, albeit with simple N-substituted isoindolinone substrates.15 While our earlier studies of the isoindolinone oxidation utilized an oxochromium(VI)–peroxide system, we now elect to explore a less toxic and more environmentally friendly oxidation system. Although not hitherto reported with nitrogen heterocycles and only with simple alkylarenes, an oxone/KBr-mediated direct benzylic oxidation appeared to be an ideally suited protocol for our purposes.16 Hence, using a modified procedure, treatment of ester substrates 11–17 with Oxone (5 equiv) and KBr (0.5 equiv) in MeCN/H2O (9:1) under slightly elevated temperatures (55–60 °C) gave the corresponding phthalimides 18–23 (72–91%).17 Using the N-benzyltriazolyl alanine product 22 (Table 2), we demonstrate the potential application to triazole-linked peptide units by preparation of N-Boc 24 (see Supporting Information). Removal of the N-phthaloyl group (N2H4, MeOH) of 22 followed by direct installation of the N-Boc group (di-tert-butyl dicarbonate, NaHCO3) gave 24 in 76% overall yield.18a
Table 2.
Esterification of Click Products 4–10 to Methyl Esters 11–17 and Oxidation of 11–17 to Phthalimides 18–23a

| ||
|---|---|---|
| R2 | Yield of 11–17 (%)b | Yield of 18–23 (%)b,c |
| 4 n-hexyl | 11 84 | 18 91 |
| 5 n-octyl | 12 83 | 18 91 |
| 6 cyclopentyl | 13 64 | 20 72 |
| 7 cyclohexyl | 14 63 | 21 76 |
| 8 benzyl | 15 86 | 22 87 |
| 9 4-fluorobenzyl | 16 87 | 23 90 |
| 10 4-methoxybenzyl | 17 87 | NR |
Reagents and conditions: (a) MeOH, acetyl chloride (cat.), r.t., 16 h; (b) Oxone, KBr, MeCN/H2O (9:1), 55–60 °C, 16 h.
Isolated chemical yield after silica gel column chromatography.
NR = no reaction.
In conclusion, we have described the preparation and utilization of a unique amino acid-based, N-isoindolinone-protected intermediate which undergoes base-mediated propargylation to give a protected ethynylalanine derivative. Ester protecting group adjustment then yields a click substrate which reacts with a diverse set of azides to afford the corresponding isoindolinone-protected triazoles. Re-esterification followed by selective benzylic oxidation converts the N-isoindolinone group into the more easily removable phthalimide group. Overall, the processes which involve isoindolinone protection and the benzylic oxidation are novel and will offer considerable synthetic latitude in preparing new amino acids. Separate studies in these laboratories have shown that selective alkylation of the isoindolinone-protected glycine equivalent 2 yields a wide range of unnatural or otherwise non-proteinogenic amino acid derivatives and the results will be reported in due course.
Supplementary Material
Figure 2.

N-Boc-protected benzyltriazole 24
Acknowledgments
Funding Information
This work was financially supported by the NIH/NIDCR through grant 1RO1DEO23206
The measurement of high and low resolution mass spectra by Dr. Michael Walla and the Mass Spectrometry Laboratory, Department of Chemistry and Biochemistry, University of South Carolina is acknowledged. The efforts of Mr. Jarrid Ronnebaum are acknowledged in the preparation of compound 3.
Footnotes
Supporting information for this article is available online at https://doi.org/10.1055/s-0036-1588510.
References and Notes
- 1.Lang K, Chin JW. Chem Rev. 2014;114:4764. doi: 10.1021/cr400355w.Walsh CT, O’Brien RV, Khosla C. Angew Chem Int Ed. 2013;52:7098. doi: 10.1002/anie.201208344.Sorochinsky AE, Acena JL, Moriwaki H, Sato T, Soloshonok VA. Amino Acids. 2013;45:691. doi: 10.1007/s00726-013-1539-4.Perdih A, Dolenc MS. Curr Org Chem. 2011;15:3750.Perdih A, Dolenc MS. Curr Org Chem. 2007;11:801.Calmes M, Daunis J. Amino Acids. 1999;16:215. doi: 10.1007/BF01388170.For example, unnatural amino acids may be used as starting materials for the synthesis of bioactive alkaloids: Singh P, Kumar MSL, Samanta K, Panda G. Tetrahedron. 2017;73:1911.
- 2.Stevenazzi A, Marchini M, Sandrone G, Vergani B, Lattanzio M. Bioorg Med Chem Lett. 2014;24:5349. doi: 10.1016/j.bmcl.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 3.For selected strategies, see: Gnas Y, Glorius F. Synthesis. 2006:1899.O’Donnell MJ. Acc Chem Res. 2004;37:506. doi: 10.1021/ar0300625.O’Donnell MJ, Cooper JT, Mader MM. J Am Chem Soc. 2003;125:2370. doi: 10.1021/ja0298794.Williams RM, Hendrix JA. J Org Chem. 1990;55:3723.Williams RM, Im M-N. J Am Chem Soc. 1991;113:9276.Myers AG, Gleason JL, Yoon T, Kung DW. J Am Chem Soc. 1997;119:656.Myers AG, Schnider P, Kwon S, Kung DW. J Org Chem. 1999;64:3322. doi: 10.1021/jo990341z.
- 4.See, for example: Burk MJ, Feaster JE, Nugent WA, Harlow RL. J Am Chem Soc. 1993;115:10125.Boaz NW, Large SE, Ponasik JA, Jr, Moore MK, Barnette T, Nottingham WD. Org Process Res Dev. 2005;9:472.
- 5.(a) Wang S, Zhou S, Wang J, Nian Y, Kawashima A, Moriwaki H, Acena JL, Soloshonok VA, Liu H. J Org Chem. 2015;80:9817. doi: 10.1021/acs.joc.5b01292. [DOI] [PubMed] [Google Scholar]; (b) Mathew S, Yun H. ACS Catal. 2012;2:993. [Google Scholar]; (c) Winkler M, Klempier N. Anal Bioanal Chem. 2009;393:1789. doi: 10.1007/s00216-008-2564-0. [DOI] [PubMed] [Google Scholar]; (d) Miyazawa T. Amino Acids. 1999;16:191. doi: 10.1007/BF01388169. [DOI] [PubMed] [Google Scholar]; (e) Pugniere M, Domergue N, Castro B, Previero A. Chirality. 1994;6:472. [Google Scholar]
- 6.(a) Zurro M, Mancheno OG. Chem Rec. 2017;17:485. doi: 10.1002/tcr.201600104. [DOI] [PubMed] [Google Scholar]; (b) Huang D, Zhao P, Astruc D. Coord Chem Rev. 2014;272:145. [Google Scholar]
- 7.Lewis JC. Curr Opin Chem Biol. 2015;25:27. doi: 10.1016/j.cbpa.2014.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikeda Y, Kawahara S, Taki M, Kuno A, Hasegawa T, Taira K. Protein Eng. 2003;16:699. doi: 10.1093/protein/gzg084. [DOI] [PubMed] [Google Scholar]
- 9.Trojer MA, Movahedi A, Blanck H, Nyden M. J Chem. 2013:1. [Google Scholar]
- 10.Szychowski J, Mahdavi A, Hodas JJL, Bagert JD, Ngo JT, Landgraf P, Dieterich DC, Schuman EM, Tirrell DA. J Am Chem Soc. 2010;132:18351. doi: 10.1021/ja1083909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Singh MS, Chowdhury S, Koley S. Tetrahedron. 2016;72:5257. [Google Scholar]; (b) Totobenazara J, Burke AJ. Tetrahedron Lett. 2015;56:2853. [Google Scholar]; (c) Majumdar KC, Ray K. Synthesis. 2011:3767. [Google Scholar]; (d) Hein CC, Liu X-M, Wang D. Pharm Res. 2008;25:2216. doi: 10.1007/s11095-008-9616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Meldal M, Tornoe CW. Chem Rev. 2008;108:2952. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]; (f) Moses JE, Moorhouse AD. Chem Soc Rev. 2007;36:1249. doi: 10.1039/b613014n. [DOI] [PubMed] [Google Scholar]
- 12.For an N-isoindolinone-derivatized alanine derivative, see: Golec J, Charifson P, Charrier J-D, Binch H. 20030232846. US. 2003Chem Abstr. 2003:9917h.
- 13.General Procedure for the Synthesis of Click Compounds 4–102-(1-Oxoisoindolin-2-yl)pent-4-ynoic acid (3, 50 mg. 0.22 mmol, 1 equiv) was dissolved in THF followed by the addition of CuSO4-5H2O (5.44 mg. 0.022 mol, 0.1 equiv) and the corresponding azides (0.24 mmol, 1.1 equiv) at r.t. A freshly prepared solution of sodium ascorbate (21.6 mg. 0.11 mmol, 0.5 equiv) in H2O (1 mL) was added to the reaction mixture and stirring was continued (16 h). After completion of reaction, the mixture was concentrated and the resulting crude residue was purified by gravity column chromatography (CHCl3/MeOH, 3:1) to provide pure triazoles 4–10.3-(1-Hexyl-1H-1,2,3-triazol-4-yl)-2-(1-oxaiso-indolin-2-yl)propanoic Acid (4)Off-white solid; 67 mg (86%): mp 138–140 °C; Rf = 0.44 (MeOH/CHCl3, 2.5:7.5). FT-IR (neat): 3383, 2929, 2859, 1705, 1659, 1602, 1398, 1215 cm−1. 1H NMR (400 MHz, DMSO-d6): δ = 7.77 (s, 1 H), 7.61-7.56 (m, 3 H). 7.47-7.43 (m. 1 H), 4.83 (d, J= 9.2 Hz, 1 H), 4.76 (d, J = 17.2 Hz, 1 H), 4.42 (d, J = 17.2 Hz, 1 H). 4.23 (t, J = 6.8 Hz, 2 H), 3.50 (dd, J = 4.0 Hz, 15.6 Hz, 1 H), 3.11 (dd, J = 12.0 Hz, 14.8 Hz, 1 H). 1.70-1.62 (m, 2 H), 1.17-1.10 (m, 4 H), 1.08-1.05 (m, 2 H), 0.80 (t, J = 7.6 Hz, 3 H) ppm. 13C NMR (125 MHz, DMSO-d6): δ = 173.9, 168.2, 145.2, 142.8, 133.3, 131.3, 127.8, 123.6, 122.9, 122.5, 56.5, 49.5, 4.73, 30.9, 30.1, 27.4, 25.8, 22.3, 14.3 ppm. HRMS: m/z calcd for C19H24N4O3, [M + H]: 357, 1921; found: 357.1919.
- 14.General Procedure for the Preparation of Esters 11–17The corresponding click acids 4–10 were dissolved in MeOH (20 mL) followed by the addition of a catalytic amount of acetyl chloride. The reaction mixture was stirred overnight at r.t. After completion of the reaction, the reaction mixture was concentrated, and the oily residue was partitioned between CH2Cl2 (20 mL) and H2O (15 mL). The CH2Cl2 layer was separated, dried over Na2SO4, and then concentrated to provide the corresponding esters 11–17 as clear colorless oils.Methyl 3-(1-Hexyl-1H-1,2,3-triazol-4-yl)-2-(1-oxoisoindolin-2-yl)propanoate (11)Colorless oil; 50 mg (84%); Rf = 0.70 (CHCl3/MeOH, 8:2). FT-IR: 2954, 2930, 1740, 1683, 1469, 1210 cm−1. 1H NMR (400 MHz, CDCl3): δ = 7.74 (d, J = 7.2 Hz, 1 H), 7.49 (t, J = 7.2 Hz, 1 H), 7.41-7.37 (m, 3 H), 535 (dd, J = 4.8 Hz, 10.8 Hz, 1 H), 4.48 (d, J = 2.8 Hz. 2 H). 4.18 (td, J = 1.2 Hz, 7.2 Hz, 14.4 Hz, 2 H), 3.73 (s, 3 H), 3.50-3.44 (m. 2 H), 1.72 (q, J = 7.6 Hz, 2 H), 1.16-1.09 (m. 6 H), 0.78 (t, J = 6.8 Hz, 3 H) ppm. 13C NMR (175 MHz, CDCl3): δ = 170.7, 169.2, 143.2, 141.8, 131.6, 128.0, 123.9, 123,7. 123.2, 121.4, 53.4, 52.7, 50.3, 47.5, 31.0, 30.1, 26.6, 25.9, 22.3, 13.8 ppm. HRMS: m/z calcd for C20H26N4O3 [M + H]: 370.2005; found: 370.2003.
- 15.Luzzio FA, Piatt-Zacherl DP. Tetrahedron Lett. 1999;40:2087. [Google Scholar]
- 16.Moriyama K, Takemura M, Togo H. Org Lett. 2012;14:2414. doi: 10.1021/ol300853z. [DOI] [PubMed] [Google Scholar]
- 17.General Procedure for the Benzylic Oxidation of Isoindolinone-Protected Click Compounds 11–17 Giving Phthalimides 18–23To a solution of isoindolinones 11–17 (1 equiv) in MeCN/H2O (9:1) was added Oxone (5.0 equiv) and KBr (0.5 equiv). The resulting reaction mixture was allowed to reflux (16 h). After completion of the reaction, the reaction mixture was concentrated and the residue was partitioned between H2O (15 mL) and CH2Cl2 (15 mL). The organic layer was separated, dried over anhydrous Na2SO4, and evaporated to obtain a crude residue which was further purified by column chromatography on silica gel (CHCl3/MeOH, 9.5:0.5) to afford pure phthalimides 18–23.2-(13-Dioxolsoindolin-2-yl)-3-(1-hexyl-1H-1,2,3-triazol-4yl)propanoic Acid (18) Colorless oil; 38 mg (91%); Rf = 0.57 (hexane/EtOAc, 1:1). FT-IR: 2925, 2856, 1775, 1744, 1711, 1386, 718, cm−1. 1H NMR (700 MHz, CDCl3): δ = 7.80 (dd, J = 2.8 Hz, 5.6 Hz, 2 H), 7.70 (dd, J = 2.8 Hz, 5.6 Hz, 2 H), 732 (s, 1 H), 5.19 (dd, J = 4.9 Hz, 9.8 Hz, 1 H), 4.22-4.20 (m. 2 H). 3.76 (s, 3 H). 3.69-3.64 (m. 2 H), 1.77-1.74 (m, 2 H), 1.24-1.17 (m, 6 H), 0.83 (t, J = 7.0 Hz. 3 H) ppm. 13C NMR(175 MHz, CDCl3): δ = 169.1, 167.3, 143.1, 134.1, 131.7, 123.5, 121.7, 52.9, 51.7, 50.2, 31.0, 30.1, 25.9, 25.4, 22.3, 13.9 ppm. HRMS: m/z calcd for C20H24N4O4 [M + H) 384.1798; found: 384.1608.
- 18.The spectral properties of 24 were identical with those previously reported: Mindt TL, Schlibi R. J Org Chem. 2007;72:10247. doi: 10.1021/jo702030e.For a unique N-Boc-protected triazolyl alanine methyl ester derivative, see: Boibessot T, Benimelis D, Jean M, Benfodda Z, Meffre P. Synlett. 2016;27:2685.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.