

Abstract
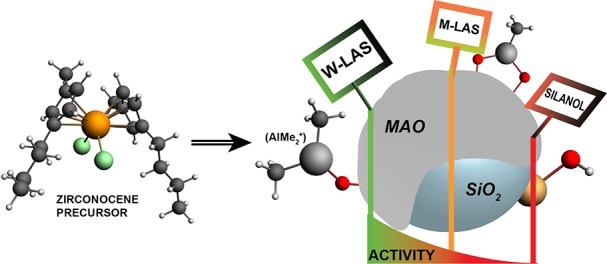
In single-site olefin polymerization catalysis, a large excess of cocatalyst is often required for the generation of highly active catalysts, but the reason for this is unclear. In this work, fundamental insight into the multifaceted role of cocatalyst methylaluminoxane (MAO) in the activation, deactivation, and stabilization of group 4 metallocenes in the immobilized single-site olefin polymerization catalyst was gained. Employing probe molecule FT-IR spectroscopy, it was found that weak Lewis acid sites, inherent to the silica-supported MAO cocatalyst, are the main responsible species for the genesis of active metallocenes for olefin polymerization. These weak Lewis acid sites are the origin of AlMe2+ groups. Deactivation of metallocenes is caused by the presence of silanol groups on the silica support. Interaction of the catalyst precursor with these silanol groups leads to the irreversible formation of inactive metallocenes. Importantly, a high concentration of MAO (14 wt% Al) on the silica support is necessary to keep the metallocenes immobilized, hence preventing metallocene leaching and consequent reactor fouling. Increasing the loading of the MAO cocatalyst leads to larger amounts of AlMe2+, fewer silanol groups, and less metallocene leaching, which all result in higher olefin polymerization activity.
Introduction
The serendipitous discovery of methylaluminoxane (MAO) and the subsequent realization of its superior potential as activator for single-site metallocene catalysts over trimethylaluminum (TMA) sparked the scientific interest in the field of metallocene polymerization catalysis.1,2 Metallocenes typically consist of sandwich complexes composed of a group 4 transition metal and cyclopentadienyl-derived ligands, of which zirconocenes are the most frequently employed.1 The interaction between an inactive metallocene (catalyst precursor) and the MAO cocatalyst, referred to as an activator, gives rise to an unsaturated cationic metallocene species, which is considered the active site in olefin polymerization.3,4
Several activation mechanisms for the standard Cp2ZrCl2/MAO system have been proposed by different research groups.5−9 Interaction of MAO with the dichloride metallocene precursor results in the extraction of the chloride ligands and methylation of the zirconium.10 The cationic metallocene is then stabilized in a complex with the MAO–Cl– species.11 There is, however, no unanimous consensus on the exact species in MAO that are responsible for metallocene activation. This is related to the fact that MAO is a very complex compound, comprising also free TMA. The exact molecular structure of MAO is therefore still not well identified despite extensive research from both experimental12 and theoretical perspectives.13 Another important question remains the apparent necessity for large excesses of MAO for metallocene activation (Al/M ratios of 1000–10 000).1 Coevoet et al. demonstrated that in toluene for low Al/Zr ratios only monomethylation of the metallocene dichloride occurred, while the active cationic metallocene was only formed at high Al/Zr ratios (2000). The use of a polar solvent (CH2Cl2), however, decreased the amount of required MAO by a factor of 20.14,15 Kaminsky and coworkers showed that the Al/Zr ratio can be reduced to a number of 30–50 when placing the metallocene on a support.16,17
The heterogenization of the MAO/metallocene can be done in several ways.1 Metallocenes can be grafted to the support and subsequently be activated by introducing MAO to the system. On the other hand, metallocene precursors can also be directly grafted onto supported MAO activators. The order in which metallocenes and MAO are introduced on the support can significantly influence the final activity due to the formation of different surface species.1 The influence of supporting the catalytic system on the molecular interactions concerning the formation of active metallocenes by MAO, however, has not been accurately and irrefutably reported to the best of our knowledge.
Generally, two kinds of species in the cocatalyst are believed to be responsible for metallocene activation: Lewis acid sites (LAS) and AlMe2+ groups.9,18−23 This has been shown in experimental work on MAO, employing probe-molecule NMR, EPR, and IR spectroscopy by Zakharov and coworkers,18,24−27 and through computational work by Linnolahti and coworkers9,22,28 and Zurek and coworkers.13,29,30 Zakharov et al. stated that the strongest LAS found in silica-supported MAO are the responsible species for the activation of the zirconocene precursor, showing a correlation between the concentration of strong LAS and the activity of the final catalyst.19 Instead, Hirvi et al., Luo and Diefenbach et al., and Thorn and Blakley et al. propose that the main responsible species for the activation of the metallocene precursor must be mobile AlMe2+ groups that are transferred from MAO to the catalyst.21,22,31−36 The modeling study of Ghiotto and coworkers demonstrated both Lewis acid sites and AlMe2+ species to be capable of metallocene activation for olefin polymerization.9 More recent theoretical work by Kuklin and coworkers showed that Lewis acid sites activate metallocenes through direct abstraction of the chloride ligands, directly yielding the cationic zirconocene species, whereas AlMe2+ species activate the metallocene by coordination to the precursor. As a result, [Cp2ZrMe2-μ-AlMe2]+ species are formed, which are considered dormant species of the active cationic species, stabilized with TMA.37 Furthermore, they suggested that the metallocene precursor Cp2ZrMe2 is more likely to be activated by AlMe2+ than Lewis acid sites. However, Lewis acid sites become more active when surrounded by a solvent with a high dielectric constant (polar solvent), indicating that easy transfer and delocalization of charges facilitate metallocene activation.37
In this work, the multifaceted role of methylaluminoxane in the catalytic performance of the silica-supported MAO/zirconocene olefin polymerization catalyst is presented. We have studied a well-defined set of supported activators (MAO/SiO2) and corresponding catalysts (Zr/MAO/SiO2) with increasing MAO loading and constant metallocene loading. Supported activators were prepared through impregnation of a silica support with a 30% MAO solution. Next, the corresponding catalysts were prepared through impregnation of the aforementioned supported activators with the metallocene precursor (bis(1-methyl-3-butylcyclopentadienyl))zirconium dichloride, as illustrated in Scheme 1. The acidic properties of the silica-supported MAO activators were studied with FT-IR spectroscopy using pyridine and CO as probe molecules for Lewis acid sites. The results provide insight into the nature of the active species within the supported MAO that are responsible for metallocene activation, hence explaining the observed changes in catalytic performance in the ethylene–1-hexene olefin copolymerization reaction. As a result, new structural insights into the multifaceted role of MAO in metallocene-based olefin polymerization catalysis were obtained.
Scheme 1. Schematic of the Stepwise Preparation of the Olefin Polymerization Catalyst Material under Study and Research Approach of This Work: the Silica Support (a) Is Impregnated with the Different Loadings of MAO, Yielding the Activators (b); This Is Followed by Impregnation of the Zirconocene Precursor Resulting in the Final Catalysts (c); the Acidity Is Studied with FT-IR Spectroscopy in Combination with Probe Molecules CO and Pyridine; All Results Are Correlated with the Final Catalytic Activity (d), Establishing Structure–Activity Relationships.

Experimental Details
Materials and Synthesis
Catalysts were prepared according to a synthetic protocol comprising three different steps: silica treatment, MAO anchoring, and zirconocene impregnation. All steps in this procedure were carried out under a N2 atmosphere, and all solvents utilized for the synthesis were analytical grade and treated prior to any use in synthesis: Toluene (Fischer Chemical, purity: >99.99%) was degassed through dry nitrogen bubbling and dried employing molecular sieves. n-Pentane (Fischer Chemical, purity: 99%) was dried over calcium hydride. The moisture content was measured by Karl Fischer titration, giving a content level less or equal to 2 ppm. The 30% MAO solution containing approximately 26.2 wt% MAO and 5.2 wt% TMA was stored in a fridge at 255 K in order to prevent gel formation. All synthetic steps were carried out using standard glovebox techniques, and the prepared samples were stored in a N2 glovebox, inside dark and well-sealed containers.
The syntheses yielded a set of samples, consisting of the parent silica (Si-0Al), five supported activators with increasing MAO loading (Si-(6-16)Al), and their corresponding catalysts (Zr/Si-(6-16)Al). In this notation, the number preceding the Al indicates the weight loading of Al, with 6 wt% being the lowest and 16 wt% being the highest loading. Scheme 1 gives a schematic representation of the three types of samples. A reference catalyst (0.27 wt% Zr on the parent silica) without MAO activator was also prepared (Zr/Si-0Al).
Silica Treatment
A commercial amorphous silica (ES767 from PQ), with a surface area of 276 m2/g, a pore volume of 1.56 cm3/g, an average pore width of 19.2 nm, and a mean particle size of approximately 33 μm diameter, was heated at 423 K for 5 h on a fluidized bed under a dry N2 flow to remove moisture.
Supported Activator Synthesis
In a glass round-bottom flask, an MAO solution was slowly added to a silica/toluene slurry (respective weight ratio of 1:5) under gentle mechanical agitation (precursor: Albemarle 30% MAO solution: 26.2 wt% in toluene, 5.2 wt% residual TMA). Subsequently, the whole mixture was heated at toluene reflux temperature (ca. 384 K) for several hours. After cooling down the slurry, a portion of supernatant was analyzed with 1H NMR spectroscopy to determine the residual aluminum content (<2000 ppm). Furthermore, 1H NMR spectroscopy was used to determine the silica OH concentration of the activators after titration of the hydroxyl groups with an excess of an Albemarle TMA solution, 99.4 wt%. The solid was finally filtered on a frit and washed three times with dry n-pentane followed by a drying treatment under vacuum for 1 h at room temperature. Following this procedure, five activators with increasing Al weight loading (6–16 wt%) were prepared.
Catalyst Synthesis
A determined quantity of the metallocene precursor (bis(1-methyl-3-butylcyclopentadienyl)zirconium dichloride) to reach the targeted zirconium content of 0.4 wt% was added to a slurry of the prepared MAO/SiO2 activators in toluene (respective weight ratio of 1:5) in a glass round-bottom flask. The colored slurry was then mechanically stirred for several hours at room temperature. The final supernatant was colorless, indicating that most of the metallocene was anchored to the supported MAO.
Activator and Catalyst Characterization
Inductively Coupled Plasma Atomic Emission Spectroscopy
Inductively coupled plasma atomic emission spectroscopy (ICP-AES) measurements were performed on a PerkinElmer ICP-AES 5300 DV instrument to make an elemental analysis of the samples.
Fourier Transform Infrared Spectroscopy
Fourier transform Infrared (FT-IR) spectra were recorded in transmission mode on a PerkinElmer 2000 instrument with a DTGS detector using 25 scans per spectrum and a resolution of 4 cm–1. The self-supported wafer was positioned in a well-sealed cell that allows switching between vacuum and the probe molecule vapor/gas. All wafers were prepared in a N2 glovebox with a hand-press (PIKE Technologies), resulting in self-supporting wafers (6–13 mg/7 mm diameter) held by a stainless steel collar. All samples were stored and prepared in an inert and dry atmosphere, and therefore no treatment to remove adsorbed water or CO2 was required.
For pyridine FT-IR spectroscopy measurements, pyridine adsorption was allowed for 30 min until equilibrium, with spectra taken every 5 min. Subsequently, vacuum desorption for 45 min followed by a temperature-programmed desorption (TPD) (5 K/min ramp to 823 K) under vacuum was applied, and spectra were taken every 25 K. For CO FT-IR spectroscopy measurements, CO (10% in He, purity 99.9%) was dosed at low temperatures (85 K) and at low pressures (between 1.0 × 10–3 and 1.5 × 10–3 mbar), with spectra being taken after each pulse. The subsequently applied decreasing pressures resulted in desorption.
| 1 |
Equation 1, as proposed by Panchenko et al.,18 derived from Beer’s law applied to dδ (cm–1), was used to determine the concentration of Lewis acid sites, where A (cm–1) represents the integral under the curve delimited by dδ (cm–1). The bands assigned to CO stretching with maxima at 2212 and at 2198 cm–1 and the band assigned to the 19b vibration of pyridine at 1453 cm–1 were employed in this equation. The apparent integral adsorption coefficient A0 (cm μmol–1) for CO at these wavenumbers is 1.1 and 0.95, respectively, and for pyridine this is 2.22. These values were taken from the same work by Panchenko et al. and from the work of Emeis et al.18,38 The mass of the wafer (mg) per cm2 through which the beam is sent (effective cross section) is represented by ρ. Pyridine:LAS and CO:LAS stoichiometries were assumed to be 1:1; that is, only one probe molecule is adsorbed per accessible Lewis acid site.
Integrated areas were evaluated by fitting the raw spectra in BlueprintXAS.39 For the pyridine FT-IR spectra, the spectrum in a vacuum prior to pyridine adsorption was taken as background, and for each spectrum 100 fits were attempted to explore the solution space of all parameters, including peak areas, based on the methodology encoded in this program to generate fits from unbiased start points.40 For CO FT-IR spectra, a holistic model that included a fifth degree polynome, in addition to the CO stretching peaks, was employed. For each spectrum, 500 fits were attempted. In the end, a family of good fits, based on minimum sum of squared errors (SSE) in the regions of interest, and with reasonable baselines, was selected for the estimation of the peak areas along with their uncertainties.
Olefin Polymerization and Characterization
Olefin Polymerization
Ethylene–1-hexene copolymerization took place in a 5 L slurry-phase reactor at 358 K for 1 h employing 60 mg of supported catalyst, 2 L of the isobutane solvent, an ethylene pressure of 8.6 bar, 50 mL of the 1-hexene comonomer, and 6.77 × 10–4 mol of scrubbing agent triisobutylaluminum (TiBA). All catalytic tests were performed in duplicate.
Polymer Characterization
The resulting polymers of the olefin copolymerization reactions were characterized using differential scanning calorimetry (DSC, melting temperature), 13C NMR (short chain branching), gradient columns (polymer density), and size-exclusion chromatography infrared spectroscopy (SEC-IR, molecular weight). Polymer bulk density was determined with the ASTM D1895 method. For polymer density measurements, the ASTM D2839 method was used for the preparation of the samples, and the ASTM D1505 method was used for the actual density measurements.
Differential Scanning Calorimetry
An aluminum pan was placed inside a sample holder and carefully charged with 3–6 mg of resin sample. A lid was manually placed over the pan/sample. Nitrile gloves were worn to protect the sample and standard TA aluminum DSC pans/lids from contamination. The sample holder was transported to the Q2000 crimp press where the pan was sealed. DSC measurements were performed using a TA Instruments DSC Q2000 instrument, heating to 433 K with 10 K/min and cooling to 303 K with 20 K/min. Two heating cycles were performed. The melting temperature measured during second heating cycle is recorded. DSC calibrations were performed for both T0 (sapphire disk) and cell constant (indium) prior to sample analysis.
13C NMR Spectroscopy
Identification of the comonomer branching content of the polyethylene samples was determined using 13C NMR spectroscopy. Measurements were performed on a Bruker Ascend 500 MHz spectrometer utilizing a 5 mm liquid nitrogen cooled Bruker Cryoplatform Prodigy probe along with a variable temperature controller set to 398 K. Samples (approximately 0.1 g) were swollen in a solution (approximately 0.9 g) of 20% 1,4-dichlorobenzene-d4 in 1,2,4-trichlorobenzene in a 5 mm NMR tube at 398 K in a vacuum oven for 30 min. After 30 min the pressure was slowly reduced to ∼100 kPa to remove bubbles and obtain uniform samples. Once homogeneous samples were obtained, the atmospheric pressure was restored. The samples were capped, quickly transferred into the NMR instrument, and allowed to equilibrate for 10 min at 398 K. The experimental acquisition parameters were set as follows: automated tuning, matching, shimming, 90′ pulse angle, 1.6 s acquisition time, 11 s relaxation delay, and a 10 scan loop with sufficient repetitions to acquire a signal-to-noise ratio of greater than 6000 in the final spectrum. Peak assignments and branching content determinations were done according to Randall et al.41
Size-Exclusion Chromatography Infrared Spectroscopy
Samples were prepared by transferring 40 mg of resin to a tared vial and trichlorobenzene (TCB, stabilized with BHT) was added to prepare a 15 mg resin/mL TCB mixture. The mixture was covered and stirred at 433 K for 2 h. An aliquot was dispensed into a tared vial and further diluted to 1.5 mg/mL using TCB. The 1.5 mg/mL solution was covered and stirred at 433 K for 2 h prior to injection for analysis. Analyses were performed on a Freeslate Rapid GPC system equipped with a Polymer Char IR4 detector and a Gilson 305 HPLC pump, using 7.5 mm × 300 nm Varian Plgel 10 μm Mixed-B columns 3 in series. The operating temperature was 423 K at 1.0 mL/min solvent flow rate and 300 mL injection volume with an analysis time of 12 min per sample. Calibration standards polystyrenes in the range 3.050–0.58 kDa were used.
Results and Discussion
A well-defined set of samples comprising the parent silica (Si-0Al), five activators with increasing methylaluminoxane loading (Si-(6-16)Al), and their respective catalysts with a constant zirconocene loading (Zr/Si-(0-16)Al), summarized in Table 1, have been investigated. The parent silica impregnated with the catalyst precursor (Zr/Si-0Al) was included to serve as a reference material for the catalysts.
Table 1. Elemental Composition of the Activators and Catalysts as Determined with ICP-AES (Al wt%, Zr wt%, Al/Zr Molar Ratio) with the Corresponding Catalytic Performance, Fouling Occurrence, and Resulting Polymer Properties (Morphology and Bulk Density) in the Ethylene–1-Hexene Copolymerization Reaction.
| activator (catalyst) | Al (wt%) | Zr (wt%) | Al/Zr molar ratio | activity (kg/mmol (Zr)·h) | fouling (Y/N) | morphology | bulk density (g/cm3) |
|---|---|---|---|---|---|---|---|
| (Zr)/Si-0Al | 0 | 0.26 | |||||
| (Zr)/Si-6Al | 6.3 | 0.42 | 51 | 15.5 | Ya | bad | 0.20 |
| (Zr)/Si-9Al | 8.9 | 0.39 | 77 | 24.7 | Y | bad | 0.17 |
| (Zr)/Si-12Al | 12.2 | 0.42 | 98 | 52.5 | Y | fluffy | 0.26 |
| (Zr)/Si-14Al | 14.0 | 0.36 | 131 | 65.7 | N | OK | 0.31 |
| (Zr)/Si-16Al | 16.4 | 0.42 | 132 | 95.2 | N | OK | 0.44 |
Heavy fouling.
In separate sections, we discuss the catalytic performance of the studied metallocene-based olefin polymerization catalysts in the ethylene–1-hexene copolymerization reaction, including the resulting polymer characteristics. This is followed by a spectroscopic characterization of the activators and corresponding catalysts with FT-IR spectroscopy. Probe molecules CO and pyridine are employed in combination with FT-IR spectroscopy to determine and quantify the Lewis acid sites (LAS) present in the samples before and after zirconocene deposition. The acquired structural information as a function of MAO loading is correlated to the ethylene–1-hexene copolymerization activity of the catalysts, thereby establishing structure–activity relationships for this important catalytic olefin polymerization system.
Catalytic Performances
Table 1 gives the composition of the prepared activator and catalyst materials, as determined with ICP-AES, and their corresponding ethylene–1-hexene copolymerization activity, fouling occurrence, and bulk polymer properties. All catalytic tests were performed in duplicate and showed good reproducibility. Reference material Zr/Si-0Al gives, as expected, no quantifiable activity in the olefin polymerization reaction. A higher zirconium loading for this reference sample could not be achieved due to metallocene leaching from the surface during synthesis.
With increasing MAO content, the activity per mmol of zirconium increases. The produced polymers were analyzed with SEC-IR (Table S1), which indicated similar molecular weight distributions for all produced polymers, corresponding to a molar mass dispersity (DM equals Mw/Mn) of 2.2. Furthermore, DSC analysis (Table S2) indicated similar melting temperatures (393 K) and polymer density (0.926 g/cm3), corresponding to LLDPE grade for all catalysts, regardless of the Al/Zr ratio. On the other hand, an Al/Zr ratio of 130 and higher leads to an improved comonomer incorporation (CH3:1000C, 5–7), as determined with 13C NMR spectroscopy. This indicates that each activated metallocene produces the same quality polymer, regardless of the MAO loading, but the amount of active single-site metallocenes is lower in the catalysts with a low MAO loading despite the excess of aluminum to zirconium centers.
Furthermore, reactor fouling occurs for Al/Zr molar ratios below 130, which is caused by the leaching of metallocene molecules from the surface. The produced polymer from these leached metallocenes becomes a sheet-like plastic that is sticking to the reactor walls on which supported polymer particles can agglomerate. As a consequence, the polymer morphology is nonuniform (bad) with a low bulk density. Therefore, it can be deduced that alongside the activation of metallocene precursors, MAO also functions as an anti-leaching agent. Apparently, a sufficient amount of MAO (Al/Zr 130) is required to keep the metallocenes on the surface and prevent leaching and consequent reactor fouling.
Vibrational Properties of Activators and Catalysts
FT-IR spectroscopy was employed to characterize the structural properties of the silica support, activators, and catalysts. Figure 1 presents the FT-IR spectra (normalized for sample density) at room temperature under vacuum for the parent silica (Si-0Al), activators (Si-(6-16)Al), and corresponding catalysts. All common vibrational bands in Figure 1 can be attributed to the silica support: At low wavenumbers, from 2080 to 1550 cm–1, we can find the combination bands of the silica support network.42,43 Furthermore, in the OH stretching region (3800–3000 cm–1), bands characteristic for silanol groups can be observed: The peak at 3740 cm–1 is attributed to isolated silanol groups with a tail associated with silanol groups localized on inner surfaces (3670 cm–1).44 The broad band centered at 3650 cm–1 is assigned to OH groups retained inside pores (intraglobular),45 and the band at 3535 cm–1 is assigned to ν-OH of H-bonded vicinal silanol groups. The decreasing intensity of this band at 3535 cm–1 with increasing MAO loading characterizes the silica surface dehydroxylation: The interaction with MAO leads to the condensation of the vicinal groups.46,47
Figure 1.

FT-IR spectra of the activators Si-(0-16)Al (a) and the respective catalysts Zr/Si-(0-16)Al (b) in a vacuum at room temperature. Spectra are normalized for sample density and plotted with an offset for clarity.
The silica used for MAO impregnation (Si-0Al) possesses a certain degree of dehydroxylation due to the pretreatment, as can be read in the Experimental Details section (2 mmol of OH per gram). Noteworthy is the observation that FT-IR bands corresponding to isolated terminal silanol groups are visible in the FT-IR spectra for Si-0Al and for the low-loaded activators (Figure 1a), i.e., Si-6Al and Si-9Al, but cannot be detected in the higher loaded activators. This indicates that the terminal silanol groups react with the MAO solution upon impregnation. Bands corresponding to isolated silanol groups are less intense in Zr/Si-0Al and Zr/Si-6Al in comparison with their counter activators (Si-0Al and Si-6Al), and not visible in sample Zr/Si-9Al. This indicates the reaction between the these terminal silanol groups and the zirconocene precursor. The resulting Cp2Zr(X)O(SiO2)n type species can be activated through treatment with excess MAO.1,48 However, in the absence of an MAO treatment after metallocene deposition, as is the case in this work, the reaction between silanol groups and metallocene precursor is reported to negatively affect the ethylene polymerization activity of the catalyst.1,24,49,50
The MAO solutions employed for the impregnation of silica supports are produced through the slow hydrolysis of TMA.28 As a consequence, the MAO solutions contain a small percentage of TMA, which, in this particular case, is ca. 5.2 wt%. Panchenko et al. indicated that it is actually the TMA present in the MAO solution, which reacts with the isolated silanol groups, producing methane (CH4) and Si–O–AlMe2 bonds, whereas MAO itself is merely strongly adsorbed on silica but not chemically grafted.1,18 In this particular case, activators Si-(12-16)Al do not contain isolated silanol groups anymore (in contrast to the lower-loaded activators (Si-(0-9)Al)), which is indicated by the absence of a sharp peak at 3740 cm–1. All isolated silanol groups that were originally present in the parent silica are therefore considered to have reacted with the TMA present in the MAO solution. The amount of TMA in the MAO solution used for the preparation of activator Si-12Al indeed matches the concentration of free silanol groups on the support surface (2 mmol/g).
These results are in accordance with recently published results by Bashir and coworkers, who studied the effect of temperature treatments of the silica support on the catalytic performance of a supported (n-BuCp)2ZrCl2 catalyst. They found that when silica is treated at temperatures higher than 723 K, the impregnated MAO can interact both with isolated silanol groups and siloxane groups, evidenced by the formation of Si–CH3 groups. Since the silica employed in this study was treated at 423 K, the MAO indeed only interacts with the silanol groups, as indicated in Figure 1. According to Bashir and coworkers, higher temperature treatments of the silica lead to the formation of more active sites in the supported MAO on which then, consequently, more metallocenes can anchor. The quality of the resulting polymers, however, does not alter upon different temperature treatments. This is also indicated in our results, since an increase of active species in MAO results in a higher polymerization activity but does not improve the resulting polymer properties.51
With increasing MAO content, there is an appearance and increment of FT-IR bands in the 3050–2750 cm–1 region and around 1430 cm–1, assigned to ν-CH and δ-CH bands, respectively. Since CH bonds are not present in the parent silica, these bands are attributed to ν-CH3 and δ-CH3 vibrations originating from Al–CH3 groups, from increasing the loading of MAO.28 During the synthesis of the activators, these methyl groups in MAO also might interact with the siloxane groups on the silica support, producing Si–CH3 and Si–O–Al–Me2 bonds. However, due to the similarity in atomic mass of aluminum and silicon atoms, it is not possible to distinguish between Al–CH3 and Si–CH3 vibrations, and therefore, it is hard to determine if an interaction between MAO and siloxane groups has taken place.18 The presence of ν-CH bands in the FT-IR spectra of MAO-free Zr/Si-0Al (Figure 1b) at 2965, 2936, 2880, and 2867 cm–1 that were absent for Si-0Al can be ascribed to the CH3 and CH2 stretching vibrations of the methyl and butyl groups of the cyclopentadienyl derivative ligand in the zirconocene precursor. For the MAO-loaded samples, these bands are overshadowed by the CH3 stretching vibrations of the MAO. The cyclopentadienyl ligands of the metallocene, however, can be identified with CH bending modes at 1508, 1470, 1445, and 1384 cm–1 (see Table S3 in the Supporting Information).52,53
Characterization of the Acid Sites by Probe Molecule FT-IR Spectroscopy
The combination of FT-IR spectroscopy with two different probe molecules (pyridine and CO) allowed the study of the acidic properties of the activators and respective catalysts. Pyridine, being a stronger base than CO (pKa = 5.25 for the conjugated acid), is able to interact with strong acid sites and provide a good indication of the overall acidity of the samples.54 Although frequency shifts in FT-IR spectroscopy induced by the coordination of strong bases to Lewis acidic centers are characteristic for particular elements, they are rather insensitive to the local environment. In contrast, the weak base carbon monoxide responds more sensitively to local coordination states and is therefore more informative about the strengths of the different Lewis acid sites in the supported activator.55
In the next sections we will respectively discuss the interactions of the activators and catalysts with pyridine and CO as studied with FT-IR spectroscopy, followed by the quantification of acid sites and the correlation with their respective olefin polymerization activity.
FT-IR Spectroscopy of Pyridine Adsorption at Room Temperature
For all samples (activators and catalysts), pyridine adsorption was allowed for 30 min to reach saturation at ambient temperature and low pressure (10–20 mbar). The resulting spectra are depicted in the Supporting Information along with an extensive discussion (Figures S1 and S2). Since FT-IR bands corresponding to physisorbed, H-bonded, and Lewis acid site coordinated pyridine can overlap in the C–H ring vibration region, the samples underwent vacuum desorption followed by a temperature treatment at 473 K to remove the physisorbed pyridine.56,57 This aided the visualization of chemisorbed pyridine in the studied materials and allowed for a more accurate quantification of the acid sites. Figure 2 depicts the density normalized FT-IR spectra in the C–H region for all activators (a) and catalysts (b) after this desorption treatment.
Figure 2.

FT-IR spectra for the activators (a) and catalysts (b) after pyridine adsorption and consequent desorption treatment at 473 K in a vacuum. The spectra are normalized for sample density and plotted with an offset for clarity. All bands correspond to pyridine adsorbed on Lewis acid sites: 8a at 1620 cm–1 (black ■), 8b at 1576 cm–1 (red ●), 19a at 1493 cm–1 (blue ▼), and 19b at 1453 cm–1 (orange ▲).
Pyridine probed the presence of one type of Lewis acid sites in the studied activators and catalysts, indicated by the four intense bands at 1620, 1576, 1493, and 1453 cm–1. These bands are ascribed to different ring vibrations (8a, 8b, 19a, and 19b) within the same pyridine molecule when coordinated to a Lewis acid site. For pure pyridine, these four ring vibrations are located at 1580, 1570, 1483, and 1439 cm–1. Upon adsorption on acid sites, these modes are perturbed. The stronger the acid site, the stronger is the perturbation of the vibrational modes.58−62 Pyridine allows the distinction between the Lewis acidity of Al3+ with an octahedral (8a: 1614 cm–1) and tetrahedral (8a: 1622 cm–1) coordination, of which the tetrahedral Al3+ expresses a stronger Lewis acidity character compared with the octahedral coordination.59,61,62 In the studied samples, pyridine mainly binds to the stronger tetrahedral Al3+ Lewis acid sites in the studied activators. This does not automatically imply that octahedral Al3+ sites are not present, since calculations by Zurek et al. showed that these are often present in the MAO cages.63 A more likely explanation would be that these sites are not accessible to pyridine. The nature of the Lewis acid sites does not alter upon impregnation of the zirconocene precursor, since the peak positions of pyridine adsorbed on the Lewis acid sites are the same as for the supported activators. The intensity of these peaks, however, is decreased as compared with the supported activators, indicating a loss in the number of accessible acid sites. The quantification of the acid sites is discussed later in this work.
FT-IR Spectroscopy of CO Adsorption at 85 K
As a complementary study, all activators and catalysts were also studied using carbon monoxide adsorption at 85 K. The resulting spectra including discussion can be found in the Supporting Information (Figures S3–S6). In the case of CO adsorption, FT-IR bands corresponding to physisorbed, H-bonded, and Lewis acid site coordinated CO do not overlap. Therefore, spectra taken at saturation, that is, when the bands at 2212 and 2198 cm–1 corresponding to CO stretching adsorbed on Lewis acid sites did not increase upon use of higher CO pressure (around 1 mbar), were employed for band assignment and quantification.18Figure 3 displays these density normalized spectra for all activators (a) and catalysts (b) at saturation. The zoom shows the CO stretching vibration when adsorbed on Lewis acid sites.
Figure 3.

FT-IR spectra for the activators (a) and catalysts (b) after CO adsorption, at 85 K and ca. 1 mbar. The insertions show a magnification of the LAS region for the activators (c) and catalysts (d). The spectra are normalized for sample density and plotted with an offset for clarity. Bands: CO adsorbed on M-LAS at 2212 cm–1 (black ■), W-LAS at 2198 cm–1 (red ●), Si–OH at 2158 cm–1 (blue ▼), O2– at 2142 cm–1 (orange ▲), physisorbed CO at 2136 cm–1 (gray ■), and Zr cationic species at 2153 cm–1 (green ★).
The interaction of CO with the remaining terminal silanol groups of the silica support is characterized with the appearance of a band at 2158 cm–1.18,19,54 With increasing MAO loading, two FT-IR bands at 2142 and 2136 cm–1 grow in intensity, which are ascribed to CO interacting with O2– ions on the MAO surface,64 and liquid physisorbed CO confined in pores, possibly created through multiple layers of MAO.65−68 Neither pyridine nor CO probed the presence of Brønsted acid sites in the studied materials. This was not unexpected, since the hydroxyl groups in the samples are silanol groups. These groups are generally not considered Brønsted acids.69 Moreover, to the best of our knowledge, Brønsted acidity in methylaluminoxane has never been reported in the literature.
With two CO vibrational bands at 2211 and 2198 cm–1, indicating the presence of Lewis acid sites with moderate (M-LAS) and weak (W-LAS) strength, CO differentiates between the presence of two different Lewis acid sites in the studied activators.18 Talsi et al. studied the nature of MAO Lewis acid sites in solution with EPR spectroscopy and concluded that a weak Lewis acid site in MAO consists of a tricoordinated aluminum atom attached to one oxygen atom and two methyl groups (AlOMe2), whereas in the case of moderate LAS the aluminum atom is bound to only one methyl group and two oxygen atoms (AlO2Me). The electron-withdrawing inductive effect of the oxygen atoms makes this aluminum atom a stronger Lewis acid site. Furthermore, Hirvi et al.22 and Luo et al.21 highlighted the importance of the terminal AlMe2+ groups of MAO in the activation of the metallocene precursor. Our results are in agreement with these works; weak Lewis acid sites in silica-supported MAO are terminal Al atoms bonded to two methyl groups and, hence, the origin of mobile AlMe2+ cationic species. The required mobility of these AlMe2+ species within MAO has been reported before.21,23,70 In accordance with the results from pyridine FT-IR spectroscopy, Figure 3 shows that the nature of the LAS does not alter upon zirconocene impregnation. It is important to stress that while pyridine can indeed be used for surface acidity evaluation, CO adsorption at low temperatures studied with FT-IR spectroscopy is more efficient in discriminating between different acid sites.
The coordination number of the aluminum species in MAO responsible for metallocene coordination has been widely researched.22,26,37,71,72 Kuklin and coworkers assigned tricoordinated aluminum to Lewis acid sites and tetracoordinated aluminum species to the origin of mobile AlMe2+ species. The origin of AlMe2+ has also often been ascribed to the presence of TMA, which, as mentioned before, is present in the studied activators. Talsi et al. showed that terminal AlO2Me and AlOMe2 groups form adducts with TMA, which was confirmed by Harlan and coworkers.26,73 Also, Tritto et al. point to TMA adducts as the origin of AlMe2+ species.74 Moreover, the theoretical work of Hirvi et al. calculated that the probability to form the terminal cationic AlMe2+ species is higher when the aluminum atoms are interconnected by bridging methyl species, thus from tetrahedral Al3+ species.22 Luo et al. also proposed that these AlMe2+ species arise from tetrahedral aluminum, where one of the coordination positions is occupied by a heteroatom, such as oxygen,21 in accordance with the EPR work by Talsi et al.26 In connection with our work, CO FT-IR spectroscopy demonstrated the presence of Lewis acid sites with tetrahedral Al3+ species, analogous to the results from pyridine FT-IR.
An additional FT-IR band at 2153 cm–1 (green star in Figure 3b) is visible for the catalysts with higher MAO loadings. This band is assigned to CO adsorbed on the cationic monomethylated zirconocene species.67 No derivative from the zirconocene precursor (bis(1-methyl-3-butylcyclopentadienyl) other than the cationic form is expected to adsorb CO at low temperature and neither does the organometallic complex when grafted to a silica.75 Because of possible overlapping of the band at 2153 cm–1 with the band at 2158 cm–1, it is not possible to deduce from Figure 3 whether catalysts with lower MAO loading do not contain the active monomethylated zirconocene species or whether they are not detected due to a low concentration.
Panchenko and coworkers previously studied the fraction of active metallocene sites on supported metallocene catalysts using the CO insertion reaction into Zr–alkyl bonds with the formation of Zr–acyl derivatives as a probe reaction for the formation of active Zr–alkyl bonds.27,76 This would lead to the formation of bands characterizing acyl (ν-CO 1600–1700 cm–1) and η-acyl (ν-CO 1525–1550 cm–1) complexes of zirconium.75,77,78 The FT-IR spectra of our catalysts upon CO adsorption do not contain any bands in these regions. This is ascribed to the low temperatures (85 K), at which the adsorption experiments were performed. The CO insertion reaction in Zr–alkyl bonds can only take place at elevated temperatures (>273 K).18,76,78
Lewis Acid Site Quantification
For each activator and corresponding catalyst the Lewis acid sites were quantified, which is illustrated in Figure 4. All probe-molecule experiments for the activators (Figure 4a) were carried out in triplicate, and the catalysts (Figure 4b) were measured once. The standard deviation was calculated for each activator as a result of the propagation of uncertainty of the three measurements. The error bars for the catalysts in Figure 4b result from the uncertainties obtained from the mathematical fitting.
Figure 4.

Concentration of the Lewis acid sites vs aluminum content for the activators (a) and catalysts (b) as determined with pyridine FT-IR (blue ◆) and CO FT-IR (moderate (red ●) and weak (black ■)).
A direct comparison between quantitative pyridine and CO FT-IR spectroscopy is not self-evident. Acid sites with an hard character are more likely to interact with pyridine rather than with CO and soft acid sites prefer CO over pyridine. Also, pyridine, being a stronger base, is able to interact with more sites compared to CO. Furthermore, the assumption in the quantification is that probe molecules and acid sites interact in a 1:1 stoichiometry, whereas this is not necessarily true as indicated by studies from Sherborne et al. and Trefz et al.79,80 Therefore, it is better to compare trends rather than absolute numbers when looking at the quantification of the Lewis acid sites.
As demonstrated in Figure 4a, both pyridine and CO clearly indicate a trend of an increasing number of Lewis acid sites with higher MAO loading. It was suspected that an increase in the MAO content would lead to an increase in both moderate and weak LAS. Nonetheless, as can be observed in Figure 4a, with increasing MAO loading, the amount of W-LAS (origin of AlMe2+) increases, but the concentration of M-LAS remains fairly constant. Figure 4b depicts the concentration of Lewis acid sites in the catalysts and indicates that part of the Lewis acid sites is consumed by the metallocene precursor. In particular, with increasing MAO loading, more acid sites contribute to the activation of the metallocene precursor. These conclusions are confirmed by both CO FT-IR and pyridine FT-IR spectroscopy. The error bars derived from the mathematical fitting were too small to be visualized for the pyridine FT-IR results (approximately 4 μmol g–1).
Acidity–Activity Correlation
Figure 5a shows a linear correlation between the concentration of Lewis acid sites in the activators as determined with pyridine FT-IR and activity of the corresponding catalysts. Figure 5b presents the correlation between activity and acidity as determined with CO FT-IR and indicates that it actually is the concentration of weak Lewis acid sites which is linearly correlated with the olefin polymerization activity of the catalysts. The deviating value for Zr/Si-6Al shall be discussed later. This leads to conclude that the immediate interaction between weak LAS and the zirconocene precursor upon impregnation must be essential for the activation process and resulting olefin polymerization activity.
Figure 5.

Ethylene–1-hexene copolymerization activity of the catalysts under study (gram of polyethylene per mole of Zr and per hour) plotted versus the concentration of LAS as determined with pyridine FT-IR spectroscopy, R2 = 0.97 (a), and versus the concentration of weak LAS as determined with CO FT-IR spectroscopy, R2 = 0.99 (b), in the corresponding activators.
Purposely, the acidic properties of the activators, rather than the catalysts, are correlated with the activity, since the essential step in the activation process is the interaction between the zirconocene and the LAS present in the supported activator upon impregnation. This specific interaction determines the loading of active cationic monomethylated zirconocene species and thus the consequent copolymerization activity. Therefore, it is the ratio of weak LAS before zirconocene deposition to the Zr precursor, which determines the activity. On the other hand, the fact that LAS are still present after zirconocene impregnation (Figure 4b) suggests that the activation process may possibly not be completed until exposed to reaction conditions.
Discussion
It can be concluded that weak Lewis acid sites (tetrahedral aluminum with terminal methyl groups) are responsible for metallocene activation in the heterogenized metallocene-based olefin polymerization catalyst. The concentration moderate LAS, on the other hand, was found to be constant and not correlated to the olefin polymerization activity. This is in contrast with a work on a heterogenized metallocene-based olefin polymerization catalyst by Zakharov and coworkers.19 Instead, our results highlight the importance of AlMe2+ species in metallocene activation and resulting catalytic activity in the olefin copolymerization reaction, as proposed by Hirvi et al. and Luo et al.21,22 This is also in agreement with previous theoretical studies by Zurek et al. and Bochmann et al., who showed that only at low Al/Zr ratios the cationic monomethylated metallocene is the main observed active catalyst in the olefin polymerization reaction. With increasing MAO loading, however, the AlMe2+-coordinated metallocene becomes the observed responsible species for olefin polymerization.22,81 This was also the conclusion by Trefz and coworkers, who showed, through electrospray-ionization mass spectrometric studies, that AlMe2+ species are bound in ion pairs to the MAO and become active upon release in a polar solvent (fluorobenzene). These AlMe2+ species were more active than Lewis acid sites in the MAO.70,80 Also, with the use of boron-based activators as molecular models for studying MAO, the formation and reactivity of AlMe2+-bound metallocene dichlorides have also been observed with NMR spectroscopy and crystallography.33,34 Moreover, an important aspect of metallocene activation is the formation of a counteranion stabilizing the cationic metallocene. The resulting ion pair should not be too tight since the steric hindrance inhibits the insertion of the monomer during polymerization. Strong Lewis acid sites create ion pairs that are too tight after chloride abstraction, therefore yielding inactive catalyst species.72 TMA-adducts in MAO activate metallocenes and form loose ion pairs, leading to higher activity in the olefin polymerization reaction.82
The correlation between W-LAS and the activity seems linear in the majority of the studied range, as illustrated in Figure 5. At the lower end the catalyst with the lowest MAO content (Zr/Si-6Al) shows some deviation from this linearity. Zr/Si-6Al shows a significantly higher activity than expected based on the W-LAS concentration. The reason could be related to the first layers of MAO, in other words, the MAO/silica interface. In what follows, we will discuss the consequences of a low MAO loading on the stability of the catalysts.
A computational study by Tymiǹska et al. suggests that the activated cationic metallocene species can interact with the silica surface resulting in extra stabilization.29 This leads to a shift in the equilibrium between the neutral precursor and active cationic metallocene toward the latter. In addition, the silica surface may also lower the energy barrier for the ethylene insertion into the Zr–C bond of inactive species compared with their counterpart homogeneous systems. Thus, catalysts with not enough MAO to fully cover the silica support could exhibit a higher apparent activity because of the possibility of the formation of stabilizing interactions between the active species and the surface. At higher loadings, the support is completely covered with additional MAO layers, and the activity is solely the result of interactions between the MAO and the metallocene species.
Beyond the olefin polymerization activity, the performance of a polymerization catalyst is also defined by a good morphology of the polymer particles and the absence of fouling. Fouling can occur when the MAO/metallocene system leaches from the silica support. Consequently, the leached catalyst performs as a homogeneous polymerization catalyst, which is more active but produces a polymer material with an uncontrollable morphology together with bad (low) polymer bulk density. Zr/Si–6Al, Zr/Si–9Al, and Zr/Si–12Al gave reactor fouling and a bad or improper morphology of the polymer. In this regard, heterogeneous catalysts with a low MAO coverage have a bad stability.
The reactor fouling of low MAO-loaded catalysts showed that the MAO loading is also crucial for its grafting to the silica surface. As reported in the literature, most of the MAO is not directly bonded to silica but strongly adsorbed.1 We propose that the TMA from MAO acts as a linker between the support and the MAO itself by reacting with silanol groups.18 In that way, at low loadings, there is not enough TMA to keep the MAO homogeneously spread over the support. Therefore, the catalysts impregnated on low-loaded MAO/SiO2 samples are more likely to experience leaching during reaction because these catalytic active species are adsorbed on the MAO and hence depend on how well MAO is anchored to the silica support. On the other hand, high MAO loadings would lead to an excess of adsorbed versus grafted MAO, which also would eventually lead to the leaching of the subsequent catalyst.83
Conclusions
The characterization of the silica-supported activator and corresponding metallocene catalyst with increasing loadings of MAO in combination with their catalytic performance allows to draw conclusions on the multifaceted role of cocatalyst methylaluminoxane (MAO) in the heterogenized metallocene-based olefin polymerization catalyst, as illustrated in Scheme 2.
Scheme 2. Schematic of the Main Conclusions from This Work: the Silica Support (a) Impregnated with MAO; (b) Contains AlMe2+ as the Main Responsible Species for Metallocene Activation and Si–OH Groups as the Main Cause for Deactivation; the Reaction between the Metallocene Precursor (c) and Si–OH Groups Yields a Deactivated Metallocene.

Activation of the metallocene is initiated with the complexation of the precursor with weak Lewis acid sites (AlMe2+), followed by the formation of the active cationic metallocene. All (cationic) metallocenes are stabilized on the surface with MAO.
The first role of MAO in the supported metallocene-based olefin polymerization catalyst is to provide AlMe2+ species, originating from weak Lewis acid sites (W-LAS), which are the main responsible species for the activation of the metallocene precursor. All Lewis acid sites in silica-supported MAO originate from tetrahedral Al3+ species. The concentration of M-LAS (moderate Lewis acid sites) for the studied activator samples is low, constant, and uncorrelated with activity. In contrast, the amount of W-LAS is more abundant, increases with MAO content, and has a linear correlation with the ethylene–1-hexene copolymerization activity. The reactivity of these W-LAS mainly relies on the release of AlMe2+ from terminal aluminum centers rather than on their strength as a Lewis acid. These results are in accordance with literature remarking the importance of the AlMe2+ transferable groups.21,22 On the other hand, our results contradict literature that indicate the strong Lewis acid sites as activating species,19 since we show that weak Lewis acid sites are the main responsible species in the MAO/SiO2 activator for the zirconocene activation in a Zr/MAO/SiO2 catalyst.
The second role of MAO in the silica-supported catalyst is to scavenge all surface hydroxyl groups, hence preventing metallocene deactivation. It is proposed that TMA, inherent to commercial MAO solutions, interacts with these silanol groups. In the case of an insufficient amount of TMA, the remaining silanol groups can interact with the metallocene precursor leading to the irreversible formation of deactivated species. In our work, a minimum MAO loading of 12 wt% Al is required to scavenge all silanol groups in the studied silica support.
Third, TMA-titrated silanol groups act as anchors on which multiple layers of MAO can strongly adsorb. A critical amount of MAO (14 wt% Al) is required to stabilize the metallocenes on the surface and prevent them from leaching from the surface. This is necessary to reduce reactor fouling and irregular morphology of the produced polymers.
Acknowledgments
The authors thank Albemarle for financial support and Mario Delgado-Jaime (Utrecht University) for useful discussions about the statistical treatment of the spectral data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol.7b02169.
In the Supporting Information, more details on the polymer characterization and on the activator and catalyst characterization can be found, including the pyridine and CO adsorption spectra for all activators and catalysts, with a more detailed discussion on the peak assignment. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Severn J. R.; Chadwick J. C.; Duchateau R.; Friederichs N. ”Bound but not gagged” - Immobilizing single-site alfa-olefin polymerization catalysts. Chem. Rev. 2005, 105, 4073–4147. 10.1021/cr040670d. [DOI] [PubMed] [Google Scholar]
- Sinn H.; Kaminsky W. Ziegler-Natta Catalysis. Adv. Organomet. Chem. 1980, 18, 99–149. 10.1016/S0065-3055(08)60307-X. [DOI] [Google Scholar]
- Resconi L.; Cavallo L.; Fait A.; Piemontesi F. Selectivity in propene polymerization with metallocene catalysts. Chem. Rev. 2000, 100, 1253–1345. 10.1021/cr9804691. [DOI] [PubMed] [Google Scholar]
- Reddy S. S.; Sivaram S. Homogeneous metallocene-methylaluminoxane catalyst systems for ethylene polymerization. Prog. Polym. Sci. 1995, 20, 309–367. 10.1016/0079-6700(94)00035-Z. [DOI] [Google Scholar]
- Kaminsky W.; Bark A.; Steiger R. Stereospecific polymerization by metallocene aluminoxane catalysts. J. Mol. Catal. 1992, 74, 109–119. 10.1016/0304-5102(92)80228-9. [DOI] [Google Scholar]
- Giannetti E.; Nicoletti G. M.; Mazzocchi R. Homogeneous Ziegler-Natta catalysis. II. Ethylene polymerization by IVB transition metal complexes/methyl aluminoxane catalyst systems. J. Polym. Sci., Polym. Chem. Ed. 1985, 23, 2117–2134. 10.1002/pol.1985.170230805. [DOI] [Google Scholar]
- Cam D.; Giannini U. Concerning the reaction of zirconocene dichloride and methylalumoxane: homogeneous Ziegler-Natta catalytic system for olefin polymerization. Makromol. Chem. 1992, 193, 1049–1055. 10.1002/macp.1992.021930502. [DOI] [Google Scholar]
- Chien J. C. W.; Wang B. Metallocene–methylaluminoxane catalysts for olefin polymerization. I. Trimethylaluminum as coactivator. J. Polym. Sci., Part A: Polym. Chem. 1988, 26, 3089–3102. 10.1002/pola.1988.080261117. [DOI] [Google Scholar]
- Ghiotto F.; Pateraki C.; Tanskanen J.; Severn J. R.; Luehmann N.; Kusmin A.; Stellbrink J.; Linnolahti M.; Bochmann M. Probing the Structure of Methylalumoxane (MAO) by a Combined Chemical, Spectroscopic, Neutron Scattering, and Computational Approach. Organometallics 2013, 32, 3354–3362. 10.1021/om4002878. [DOI] [Google Scholar]
- Coates G. W. Precise control of polyolefin stereochemistry using single-site metal catalysts. Chem. Rev. 2000, 100, 1223–1252. 10.1021/cr990286u. [DOI] [PubMed] [Google Scholar]
- Bochmann M. Kinetic and mechanistic aspects of metallocene polymerisation catalysts. J. Organomet. Chem. 2004, 689, 3982–3998. 10.1016/j.jorganchem.2004.07.006. [DOI] [Google Scholar]
- Zijlstra H. S.; Harder S. Methylalumoxane - History, Production, Properties, and Applications. Eur. J. Inorg. Chem. 2015, 2015, 19–43. 10.1002/ejic.201402978. [DOI] [Google Scholar]
- Zurek E.; Ziegler T. Theoretical studies of the structure and function of MAO (methylaluminoxane). Prog. Polym. Sci. 2004, 29, 107–148. 10.1016/j.progpolymsci.2003.10.003. [DOI] [Google Scholar]
- Coevoet D.; Cramail H.; Deffieux A. U.V./visible spectroscopic study of the rac-Et(Ind)2ZrCl2/MAO olefin polymerization catalytic system, 1. Investigation in toluene. Macromol. Chem. Phys. 1998, 199, 1451–1457. 10.1002/macp.1998.021990725. [DOI] [Google Scholar]
- Coevoet D.; Cramail H.; Deffieux A. U. V./ visible spectroscopic study of the rac-Et(Ind)2ZrCl2/MAO olefin polymerization catalytic system, 2a Investigation in CH2C12. Macromol. Chem. Phys. 1998, 199, 1459–1464. . [DOI] [Google Scholar]
- Kaminsky W.; Külper K.; Brintzinger H. H.; Wild F. R. W. P. Polymerization of Propene and Butene with a Chiral Zirconocene and Methylalumoxane as Cocatalyst. Angew. Chem., Int. Ed. Engl. 1985, 24, 507–508. 10.1002/anie.198505071. [DOI] [Google Scholar]
- Olabisi O.; Atiqullah M.; Kaminsky W. Group 4 metallocenes: supported and unsupported. J. Macromol. Sci., Polym. Rev. 1997, 37, 519–554. 10.1080/15321799708018374. [DOI] [Google Scholar]
- Panchenko V. N.; Semikolenova N. V.; Danilova I. G.; Paukshtis E. A.; Zakharov V. A. IRS study of ethylene polymerization catalyst SiO2/methylaluminoxane/zirconocene. J. Mol. Catal. A: Chem. 1999, 142, 27–37. 10.1016/S1381-1169(98)00275-1. [DOI] [Google Scholar]
- Zakharov V. A.; Panchenko V. N.; Semikolenova N. V.; Danilova I. G.; Paukshtis E. A. IRS study of ethylene polymerization catalyst SiO2/methylaluminoxane/zirconocene. Polym. Bull. 1999, 43, 87–92. 10.1007/s002890050537. [DOI] [Google Scholar]
- Chen E. Y. X.; Marks T. J. Cocatalysts for Metal-Catalyzed Olefin Polymerization: Activators, Activation Processes, and Structure-Activity Relationships. Chem. Rev. 2000, 100, 1391–1434. 10.1021/cr980462j. [DOI] [PubMed] [Google Scholar]
- Luo L.; Sangokoya S. A.; Wu X.; Diefenbach S. P.; Kneale B.. US8354485 B2, 2013.
- Hirvi J. T.; Bochmann M.; Severn J. R.; Linnolahti M. Formation of octameric methylaluminoxanes by hydrolysis of trimethylaluminum and the mechanisms of catalyst activation in single-site a-olefin polymerization catalysis. ChemPhysChem 2014, 15, 2732–2742. 10.1002/cphc.201402298. [DOI] [PubMed] [Google Scholar]
- Imhoff D. W.; Simeral L. S.; Sangokoya S. A.; Peel J. H. Characterization of Methylaluminoxanes and Determination of Trimethylaluminum Using Proton NMR. Organometallics 1998, 17, 1941–1945. 10.1021/om980046p. [DOI] [Google Scholar]
- Moroz B. L.; Semikolenova N. V.; Nosov A. V.; Zakharov V. A.; Nagy S.; O’Reilly N. J. Silica-supported zirconocene catalysts: Preparation, characterization and activity in ethylene polymerization. J. Mol. Catal. A: Chem. 1998, 130, 121–129. 10.1016/S1381-1169(97)00206-9. [DOI] [Google Scholar]
- Bryliakov K. P.; Semikolenova N. V.; Panchenko V. N.; Zakharov V. A.; Brintzinger H. H.; Talsi E. P. Activation of rac-Me2Si(ind)2ZrCl2 by Methylalumoxane Modified by Aluminum Alkyls: An EPR Spin-Probe, 1H NMR, and Polymerization Study. Macromol. Chem. Phys. 2006, 207, 327–335. 10.1002/macp.200500443. [DOI] [Google Scholar]
- Talsi E. P.; Semikolenova N. V.; Panchenko V. N.; Sobolev A. P.; Babushkin D. E.; Shubin A. A.; Zakharov V. A. The metallocene/methylaluminoxane catalysts formation: EPR spin probe study of Lewis acidic sites of methylaluminoxane. J. Mol. Catal. A: Chem. 1999, 139, 131–137. 10.1016/S1381-1169(98)00194-0. [DOI] [Google Scholar]
- Panchenko V. N.; Zakharov V. A.; Paukshtis E. A. Study of the supported zirconocene catalysts by means of UV/Vis and DRIFT spectroscopy. J. Mol. Catal. A: Chem. 2005, 240, 33–39. 10.1016/j.molcata.2005.06.034. [DOI] [Google Scholar]
- Linnolahti M.; Severn J. R.; Pakkanen T. A. Formation of nanotubular methylaluminoxanes and the nature of the active species in single-site alfa-olefin polymerization catalysis. Angew. Chem., Int. Ed. 2008, 47, 9279–9283. 10.1002/anie.200802558. [DOI] [PubMed] [Google Scholar]
- Tymińska N.; Zurek E. DFT-D Investigation of Active and Dormant Methylaluminoxane (MAO) Species Grafted onto a Magnesium Dichloride Cluster: A Model Study of Supported MAO. ACS Catal. 2015, 5, 6989–6998. 10.1021/acscatal.5b01697. [DOI] [Google Scholar]
- Falls Z.; Tyminska N.; Zurek E. The Dynamic Equilibrium Between (AlOMe)n Cages and (AlOMe)n·(AlMe3)m Nanotubes in Methylaluminoxane (MAO): A First-Principles Investigation. Macromolecules 2014, 47, 8556–8569. 10.1021/ma501892v. [DOI] [Google Scholar]
- Luo L.; Diefenbach S. P.. Book of Abstracts, Advances in Polyolefins VIII, Santa Rosa, CA, September 25–28, 2011; American Chemical Society, Division of Polymer Chemistry, Inc.: Blacksburg, VA. [Google Scholar]
- Luo L.; Diefenbach S. P.. Abstr. Papers, 245th ACS National Meeting & Exposition, New Orleans, LA, April 7–11, 2013; CATL-65.
- Thorn M. G.; Diefenbach S. P.. Book of Abstracts, Advances in Polyolefins X, Santa Rosa, CA, September 21–24, 2015; American Chemical Society, Division of Polymer Chemistry, Inc.: Blacksburg, VA. [Google Scholar]
- Thorn M. G.; Marcel L.. SPE International Polyolefins Conference 2016, Global Interdependence, Houston, TX, February 21–24, 2016; SPE South Texas Section, Society of Plastics Engineers: Bethel, CT. [Google Scholar]
- Blakley C. G.; Thorn M. G.; Diefenbach S. P.. Abstr. Papers, 253rd ACS National Meeting & Exposition, San Francisco, CA, April 2–6, 2017; PMSE-176.
- Blakley C. G.; Thorn M. G.. Book of Abstracts, Advances in Polyolefins XI, Santa Rosa, CA, September 24–27, 2017; American Chemical Society, Division of Polymer Chemistry, Inc.: Blacksburg, VA. [Google Scholar]
- Kuklin M. S.; Hirvi J. T.; Bochmann M.; Linnolahti M. Toward Controlling the Metallocene/Methylaluminoxane-Catalyzed Olefin Polymerization Process by a Computational Approach. Organometallics 2015, 34, 3586–3597. 10.1021/acs.organomet.5b00394. [DOI] [Google Scholar]
- Emeis C. Determination of Integrated Molar Extinction Coefficients for Infrared Bands of Pyridine Adsorbed on Solid Acid Catalysts. J. Catal. 1993, 141, 347–354. 10.1006/jcat.1993.1145. [DOI] [Google Scholar]
- Delgado-Jaime M. U.; Mewis C. P.; Kennepohl P. Blueprint XAS: A Matlab-based toolbox for the fitting and analysis of XAS spectra. J. Synchrotron Radiat. 2010, 17, 132–137. 10.1107/S0909049509046561. [DOI] [PubMed] [Google Scholar]
- Delgado-Jaime M. U.; Kennepohl P. Development and exploration of a new methodology for the fitting and analysis of XAS data. J. Synchrotron Radiat. 2010, 17, 119–128. 10.1107/S090904950904655X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall J. C. Characterization of Ethylene-based Polymers. J. Macromol. Sci., Polym. Rev. 1989, 29, 201–317. 10.1080/07366578908055172. [DOI] [Google Scholar]
- Putz A. M.; Putz M. V. Spectral inverse quantum (Spectral-IQ) method for modeling mesoporous systems: Application on Silica films by FTIR. Int. J. Mol. Sci. 2012, 13, 15925–15941. 10.3390/ijms131215925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallas J. P.; Goupil J. M.; Vimont A.; Lavalley J. C.; Gil B.; Gilson J. P.; Miserque O. Quantification of water and silanol species on various silicas by coupling IR spectroscopy and in-situ thermogravimetry. Langmuir 2009, 25, 5825–5834. 10.1021/la802688w. [DOI] [PubMed] [Google Scholar]
- Gallas J. P.; Lavalley J. C.; Burneau A.; Barres O. Comparative study of the surface hydroxyl groups of fumed and precipitated silicas. 4. Infrared study of dehydroxylation by thermal treatments. Langmuir 1991, 7, 1235–1240. 10.1021/la00054a036. [DOI] [Google Scholar]
- Linssen T.; Cassiers K.; Cool P.; Lebedev O.; Whittaker A.; Vansant E. F. Physicochemical and Structural Characterization of Mesoporous Aluminosilicates Synthesized from Leached Saponite with Additional Aluminum Incorporation. Chem. Mater. 2003, 15, 4863–4873. 10.1021/cm031111a. [DOI] [Google Scholar]
- Zhuravlev L. T. The surface chemistry of amorphous silica. Zhuravlev model. Colloids Surf., A 2000, 173, 1–38. 10.1016/S0927-7757(00)00556-2. [DOI] [Google Scholar]
- Vilmin F.; Bottero I.; Travert A.; Malicki N.; Gaboriaud F.; Trivella A.; Thibault-Starzyk F. Reactivity of bis[3-(triethoxysilyl)propyl] tetrasulfide (TESPT) silane coupling agent over hydrated silica: Operando IR spectroscopy and chemometrics study. J. Phys. Chem. C 2014, 118, 4056–4071. 10.1021/jp408600h. [DOI] [Google Scholar]
- Tian J.; Wang S.; Li J.; Collins S. Borane-functionalized oxide supports: development of active supported metallocene catalysts at low aluminoxane loading. J. Mol. Catal. A: Chem. 1999, 144, 137–150. 10.1016/S1381-1169(98)00341-0. [DOI] [Google Scholar]
- Collins S.; Kelly W. M.; Holden D. A. Polymerization of propylene using supported, chiral, ansa-metallocene catalysts: production of polypropylene with narrow molecular weight distributions. Macromolecules 1992, 25, 1780–1785. 10.1021/ma00032a025. [DOI] [Google Scholar]
- Jezequel M.; Dufaud V.; Ruiz-Garcia M. J.; Carrillo-Hermosilla F.; Neugebauer U.; Niccolai G. P.; Lefebvre F.; Bayard F.; Corker J.; Fiddy S.; Evans J.; Broyer J.-P.; Malinge J.; Basset J.-M. Supported metallocene catalysts by surface organometallic chemistry. Synthesis, characterization, and reactivity in ethylene polymerization of oxide-supported Mono– and biscyclopentadienyl zirconium alkyl complexes: establishment of structure/reactivity relationships. J. Am. Chem. Soc. 2001, 123, 3520–3540. 10.1021/ja000682q. [DOI] [PubMed] [Google Scholar]
- Bashir M. A.; Vancompernolle T.; Gauvin R. M.; Delevoye L.; Merle N.; Monteil V.; Taoufik M.; McKenna T. F. L.; Boisson C. Silica/MAO/(n-BuCp)2ZrCl2 catalyst: effect of support dehydroxylation temperature on the grafting of MAO and ethylene polymerization. Catal. Sci. Technol. 2016, 6, 2962–2974. 10.1039/C5CY01285F. [DOI] [Google Scholar]
- Colthup N.; Daly L.; Wiberley S.. Introduction to Infrared and Raman Spectroscopy, 3rd ed.; Academic Press: New York, 1975; pp 257–277. [Google Scholar]
- Dos Santos J. H. Z.; Krug C.; Da Rosa M. B.; Stedile F. C.; Dupont J.; De Camargo Forte M. The effect of silica dehydroxylation temperature on the activity of SiO2-supported zirconocene catalysts. J. Mol. Catal. A: Chem. 1999, 139, 199–207. 10.1016/S1381-1169(98)00173-3. [DOI] [Google Scholar]
- Busca G. Spectroscopic Characterization of the Acid Properties of Metal Oxide Catalysts. Catal. Today 1998, 41, 191–206. 10.1016/S0920-5861(98)00049-2. [DOI] [Google Scholar]
- Paukshtis E.; Shinkarenko V.; Karakchiev L. Spectroscopic investigation of acceptor properties of catalyst surface. Kinet. Katal. 1976, 17, 1029–1034. [Google Scholar]
- Campos J.; López-Serrano J.; Peloso R.; Carmona E. Methyl Complexes of the Transition Metals. Chem. - Eur. J. 2016, 22, 6432–6457. 10.1002/chem.201504483. [DOI] [PubMed] [Google Scholar]
- Madejová J.; Pálková H.; Jankovič L. Near-infrared study of the interaction of pyridine with acid-treated montmorillonite. Vib. Spectrosc. 2015, 76, 22–30. 10.1016/j.vibspec.2014.11.003. [DOI] [Google Scholar]
- Knözinger H. In Handbook of Heterogeneous Catalysis, 2nd ed.; Ertl G., Knözinger H., Schüth F., Weitkamp J., Eds.; Wiley-VCH: Weinheim, 2008; pp 1135–1162. [Google Scholar]
- Zaki M. I.; Hasan M. A.; Al-Sagheer F. A.; Pasupulety L. In situ FTIR spectra of pyridine adsorbed on SiO2-Al2O3, TiO2, ZrO2 and CeO2: General considerations for the identification of acid sites on surfaces of finely divided metal oxides. Colloids Surf., A 2001, 190, 261–274. 10.1016/S0927-7757(01)00690-2. [DOI] [Google Scholar]
- Bagshaw S. A.; Cooney R. P. FTIR surface site analysis of pillared clays using pyridine probe species. Chem. Mater. 1993, 5, 1101–1109. 10.1021/cm00032a013. [DOI] [Google Scholar]
- Barzetti T.; Selli E.; Moscotti D.; Forni L. Pyridine and ammonia as probes for FTIR analysis of solid acid catalysts. J. Chem. Soc., Faraday Trans. 1996, 92, 1401–1407. 10.1039/ft9969201401. [DOI] [Google Scholar]
- Phung T. K.; Herrera C.; Larrubia M. Á.; García-Diéguez M.; Finocchio E.; Alemany L. J.; Busca G. Surface and catalytic properties of some γ-Al2O3 powders. Appl. Catal., A 2014, 483, 41–51. 10.1016/j.apcata.2014.06.020. [DOI] [Google Scholar]
- Zurek E.; Woo T. K.; Firman T. K.; Ziegler T. Modeling the Dynamic Equilibrium between Oligomers of (AlOCH3)n in Methylaluminoxane (MAO). A Theoretical Study Based on a Combined Quantum Mechanical and Statistical Mechanical Approach. Inorg. Chem. 2001, 40, 361–370. 10.1021/ic000845b. [DOI] [PubMed] [Google Scholar]
- Hadjiivanov K.; Penkova A.; Centeno M. A. FTIR indication of CO interaction with O2-ions: A new adsorption form in the gap between chemi- and physisorbed CO. Catal. Commun. 2007, 8, 1715–1718. 10.1016/j.catcom.2007.02.002. [DOI] [Google Scholar]
- Davydov A.Molecular Spectroscopy of Oxide Catalyst Surfaces; John Wiley & Sons: Hoboken, NJ, 2003. [Google Scholar]
- Salla I.; Montanari T.; Salagre P.; Busca G.; Universita D.; Kennedy J. F.; Genova I. Metal Mordenites: Characterization of Multiple Interactions. Phys. Chem. Chem. Phys. 2005, 7, 2526–2533. 10.1039/b502841h. [DOI] [PubMed] [Google Scholar]
- Martra G.; Ocule R.; Marchese L.; Centi G.; Coluccia S. Alkali and alkaline-earth exchanged faujasites: Strength of Lewis base and acid centres and cation site occupancy in Na- and BaY and Na- and BaX zeolites. Catal. Today 2002, 73, 83–93. 10.1016/S0920-5861(01)00521-1. [DOI] [Google Scholar]
- Zecchina A.; Areán C. O. Diatomic molecular probes for mid-IR studies of zeolites. Chem. Soc. Rev. 1996, 25, 187–197. 10.1039/CS9962500187. [DOI] [Google Scholar]
- Tsyganenko A. A.; Storozheva E. N.; Manoilova O. V.; Lesage T.; Daturi M.; Lavalley J. Brønsted acidity of silica silanol groups induced by adsorption of acids. Catal. Lett. 2000, 70, 159–163. 10.1023/A:1018845519727. [DOI] [Google Scholar]
- Trefz T. K.; Henderson M. A.; Wang M. Y.; Collins S.; McIndoe J. S. Mass Spectrometric Characterization of Methylaluminoxane. Organometallics 2013, 32, 3149–3152. 10.1021/om400256f. [DOI] [Google Scholar]
- Luhtanen T. N. P.; Linnolahti M.; Pakkanen T. A. Quantum chemical studies on elementary fragments of three-coordinated methylaluminoxanes. J. Organomet. Chem. 2002, 648, 49–54. 10.1016/S0022-328X(01)01465-6. [DOI] [Google Scholar]
- Atiqullah M.; Anantawaraskul S.; Emwas A. M.; Al-Harthi M. A.; Hussain I.; Ul-Hamid A.; Hossaen A. Silica-supported (nBuCp)2ZrCl2: effect of catalyst active center distribution on ethylene-1-hexene copolymerization. Polym. Int. 2014, 63, 955–972. 10.1002/pi.4587. [DOI] [Google Scholar]
- Harlan C. J.; Bott S. G.; Barron A. R. Three-Coordinate Aluminum Is Not a Prerequisite for Catalytic Activity in the Zirconocene-Alumoxane Polymerization of Ethylene. J. Am. Chem. Soc. 1995, 117, 6465–6474. 10.1021/ja00129a008. [DOI] [Google Scholar]
- Tritto I.; Sacchi M. C.; Locatelli P.; Li S. X. Low-temperature 1H and 13C NMR investigation of trimethylaluminium contained in methylaluminoxane cocatalyst for metallocene-based catalysts in olefin polymerization. Macromol. Chem. Phys. 1996, 197, 1537–1544. 10.1002/macp.1996.021970429. [DOI] [Google Scholar]
- Marsella J. A.; Curtis C. J.; Bercaw J. E.; Caulton K. G. Low-Temperature Infrared Study of d0 Carbonyl Complexes. J. Am. Chem. Soc. 1980, 102, 7244–7246. 10.1021/ja00544a012. [DOI] [Google Scholar]
- Panchenko V. N.; Danilova I. G.; Zakharov V. A.; Paukshtis E. A. An IR-Spectroscopic Study of the State of Zirconium in Supported Zirconocene Catalysts. Kinet. Catal. 2004, 45, 547–553. 10.1023/B:KICA.0000038083.22397.bf. [DOI] [Google Scholar]
- Manriquez J. M.; McAlister D. R.; Sanner R. D.; Bercaw J. E. Reduction of Carbon Mononxide Promoted by Alkyl and Hydride Derivatives of Permethylzirconocene. J. Am. Chem. Soc. 1978, 100, 2716–2724. 10.1021/ja00477a025. [DOI] [Google Scholar]
- Nesterov G. A.; Zakharov V. A.; Volkov V. V.; Myakishev K. Catalysts prepared by the interaction of transition tetrahydroborates with oxide supports: synthesis of surface Ti, Zr, Hf hydrides and their catalytic properties in ethylene polymerization. J. Mol. Catal. 1986, 36, 253–269. 10.1016/0304-5102(86)85082-9. [DOI] [Google Scholar]
- Sherborne G. J.; Chapman M. R.; Blacker A. J.; Bourne R. A.; Chamberlain T. W.; Crossley B. D.; Lucas S. J.; McGowan P. C.; Newton M. A.; Screen T. E. O.; Thompson P.; Willans C. E.; Nguyen B. N. Activation and Deactivation of a Robust Immobilized Cp*Ir-Transfer Hydrogenation Catalyst: A Multielement in Situ X-ray Absorption Spectroscopy Study. J. Am. Chem. Soc. 2015, 137, 4151–4157. 10.1021/ja512868a. [DOI] [PubMed] [Google Scholar]
- Trefz T. K.; Henderson M. A.; Linnolahti M.; Collins S.; McIndoe J. S. Mass Spectrometric Characterization of Methylaluminoxane-Activated Metallocene Complexes. Chem. - Eur. J. 2015, 21, 2980–2991. 10.1002/chem.201405319. [DOI] [PubMed] [Google Scholar]
- Zurek E.; Ziegler T. A theoretical study of the insertion barrier of MAO (methylaluminoxane)-activated, Cp2ZrMe2-catalyzed ethylene polymerization: further evidence for the structural assignment of active and dormant species. Faraday Discuss. 2003, 124, 93–109. 10.1039/b209455j. [DOI] [PubMed] [Google Scholar]
- Guo Y.; He F.; Zhang Z.; Khan A.; Fu Z.; Xu J.; Fan Z. Influence of trimethylaluminum on kinetics of rac-Et(Ind)2ZrCl2/aluminoxane catalyzed ethylene polymerization. J. Organomet. Chem. 2016, 808, 109–116. 10.1016/j.jorganchem.2016.02.024. [DOI] [Google Scholar]
- Haag M. C.; Krug C.; Dupont J.; De Galland G. B.; Dos Santos J. H. Z.; Uozumi T.; Sano T.; Soga K. Effects of Al/Zr ratio on ethylene-propylene copolymerization with supported-zirconocene catalysts. J. Mol. Catal. A: Chem. 2001, 169, 275–287. 10.1016/S1381-1169(01)00045-0. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.