

Abstract
Non-genetic heterogeneity fluctuates over diverse timescales, ranging from hours to months. In specific cases, such variability can profoundly impact the response of cell populations to therapy, in both antibiotic treatments in bacteria and chemotherapy in cancer. It is thus critical to understand the way phenotypes fluctuate in cell populations and the molecular sources of phenotypic diversity. Technical and analytical breakthroughs in the study of single cells have leveraged cellular heterogeneity to gain phenomenological and mechanistic insights of the phenotypic transitions that occur within isogenic cell populations over time. Such an understanding moves forward our ability to design therapeutic strategies with the explicit goal of preventing and controlling the selective expansion and stabilization of drug-tolerant phenotypic states.
Introduction
Surveys of molecular and phenotypic states of single cells have revealed pervasive heterogeneity, both between isogenic cells in a population and within the same cell over time [1]. One potential source of such cell-to-cell variability is at the level of the signals themselves. For instance, individual cells exposed to the same dose of ionizing radiation will effectively receive a variable number of double-stranded DNA breaks [2]. However, even when environmental inputs remain constant, variability in responses can arise as a result of difference in initial states, such as cell cycle or differentiation stage [3]. In addition, cells that populate the same initial state, and are exposed to exactly the same signal, can exhibit different responses due to the stochastic nature of the biochemical reactions that govern the production and degradation of individual molecules [4]. Variability is therefore an intrinsic property of signaling systems that cells (and more recently researchers) have evolved to harness and cope with.
Non-genetic heterogeneity has been identified as a source of molecular and phenotypic diversity that leads to variable responses to treatment within isogenic cell populations. This plasticity has emerged as a predecessor and mediator of the evolution of genetic resistance, which ultimately leads to therapeutic failure [5,6]. Here, we summarize recent evidence of the way non-genetic phenotypic variation contributes to fractional killing in specific experimental systems, from bacteria to cancer cells. Recent technical and analytical developments provide the opportunity to quantitatively understand the dynamics of interconversion between cellular states, both from phenotypic and molecular perspectives. Such an understanding will be critical for designing strategies to optimize therapeutic outcomes that account for the presence of phenotypic heterogeneity and timescales of fluctuations in cellular states.
Cell-to-cell variability limits the success of therapy
It has long been recognized that a small subpopulation of bacterial cells called ‘persisters’ can survive antibiotic treatment under drug concentrations that kill the vast majority of bacteria [7]. Bacterial populations that emerge from the expansion of persister cells after antibiotic removal exhibit similar drug sensitivity to that of the original cell population, arguing that persistence represents a reversible state that is maintained at low frequencies in bacterial populations [8]. Using timelapse microscopy to follow single bacteria over time, Balaban et al. showed that persister cells existed as a small fraction of growth arrested cells in unperturbed bacterial populations [9]. Notably, environmental and mutational perturbations can increase the fraction of persister cells in a population by engaging the stress response pathways that are stochastically activated in exponentially growing populations due to natural variability [10,11]. This suggested that the frequency of phenotypic switching and/or the lifetime of the persister state, could be subject to environmental or evolutionary modulation [12,13].
In striking resemblance to bacterial persistence, drug tolerant phenotypic states have been shown to mediate fractional killing in cancer cell populations in response to targeted therapies. Sharma et al. showed that while the vast majority of lung cancer cells harboring oncogenic EGFR died within a few days upon exposure to the EGFR inhibitor gefitinib, ~0.3% of the cells survived treatment and remained in a largely quiescent state. Eventually, ~20% of drug tolerant cells were able to resume proliferation and could be propagated in the presence of drug concentrations that would be lethal to the drug naïve cell population. The transition from a quiescent to proliferative drug tolerant state was attributed to global changes in histone post-translational modifications and could be blocked by co-treatment with a histone demethylase inhibitor [14]. Interestingly, such transition brought about concomitant changes in the average lifetime of the drug tolerant phenotype: while surviving quiescent cells could re-gained sensitivity within ~9 doublings upon growth in the absence of drug, it took ~90 doublings for the proliferative drug tolerant cells to restore sensitivity [14]. Thus phenotypic heterogeneity was not only present at the level of drug sensitivity vs. tolerance, but also in the relative stability of the tolerant state (Figure 1A).
Figure 1. Non-genetic heterogeneity can pre-exist before, or arise as a result of, treatment leading to fractional killing.
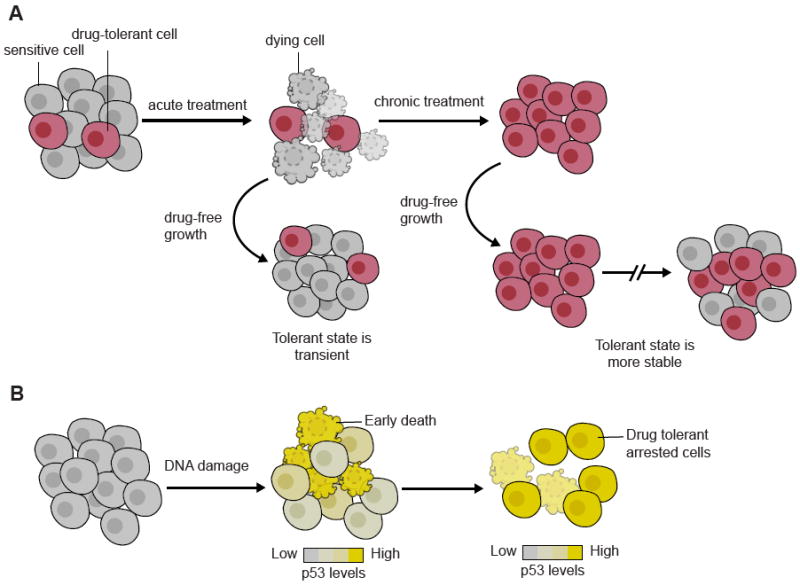
(A) Pre-treatment heterogeneity - Drug tolerant cells exist in a small fraction of drug-naïve populations. Upon treatment, these cells mediate fractional killing. Population heterogeneity is reconstituted rapidly upon drug removal, but recovery of sensitivity takes longer after chronic treatment. This suggests that persistent drug exposure engages a mechanism for the stabilization of drug tolerant states [14,15].
(B) Non-genetic heterogeneity in response to the treatment - Cells exhibit heterogeneous activation of DNA damage signaling upon chemotherapy. Cells with early and fast rate of p53 activation undergo apoptosis. Cells with late and slow p53 activation enter cell cycle arrest and survive even in the context of chronic drug exposure.
Similar results have been reported in the context of BRAFV600E mutant melanoma cells treated with BRAF inhibitor [15]. Shaffer et al. investigated the origin of rare drug resistant colonies that emerged in the presence of BRAFV600E inhibitor [15]. Using long-term live cell imaging they showed that these resulted from the expansion of a small subpopulation of cells that continued cycling normally in the presence of drug. Single molecule RNA fluorescent in situ hybridization (FISH) revealed that heterogeneity in the expression of resistance-associated transcripts preceded drug exposure. In addition, selective expansion of these pre-resistant cells upon treatment was accompanied by a gradual epigenetic reprogramming that transformed transient transcriptional variation in drug naïve cell populations into stable resistance in the course of ~4 weeks (Figure 1A). In an independent study, Fallahi-Sichani et al. discovered a subpopulation of melanoma cells that cycled slowly in the presence of BRAF inhibitor. Drug tolerant cells exhibited a de-differentiated molecular profile, which could be reverted by passaging in the absence of drug and blocked by inhibition of histone modifiers [16]. The activation of proteins involved in developmental plasticity has been linked to resistance in other models [17-19], suggesting that lineage switching could be a widespread mechanism to attain resistance.
In addition to pre-existing stochastic phenotypic variation as a source of drug tolerance [14,15,20], cell-to-cell phenotypic variation can emerge as a direct consequence of the way individual cells respond to treatments. Paek et al. showed that exposure of colon cancer cells to chemotherapy led to heterogeneous activation of the pro-apoptotic tumor suppressor protein p53 and anti-apoptotic proteins. Fractional killing emerged as the result of competition between these antagonistic cellular programs: progressive accumulation of anti-apoptotic proteins gradually increased the threshold in p53 levels required to trigger cell death, limiting apoptosis to cells with early and high rate of p53 induction [21] (Figure 1B). A similar mechanism was identified in the context of TRAIL induced apoptosis, in which the initial rate of caspase-8 activity was shown to distinguish whether a cell underwent apoptosis or survived chronic exposure to the ligand [22]. Thus, heterogeneity in signaling upon perturbations does not only contribute to variability in immediate cellular outcomes, but can also reshape the distribution of phenotypic states in the population and render cells transiently refractory or sensitive to subsequent treatments.
Collectively, these and other studies have revealed a wide diversity in the timescales within which drug tolerant phenotypes fluctuate, ranging from a couple cell generations, when phenotypic variation is due to fluctuations in the levels of proteins directly involved in the response [23], to weeks or months, when phenotypic states become stabilized through engagement of self-reinforcing feedback loops or epigenomic reprogramming [14,15]. Short-lived states can precede the establishment of longer-lived drug tolerant states, with genetic variation ultimately conferring stable resistance [6]. It is thus critical to gain a phenomenological and mechanistic understanding of the rates at which cells enter, exit and stabilize drug tolerant states.
Leveraging uncertainty to advance mechanistic understanding of the transitions between phenotypic states
The recognition of the presence of phenotypic heterogeneity in cell populations prompted the development of experimental paradigms to unmask such variability and to understand the dynamics of diversity-generating processes. Going back to the classical fluctuation analysis developed by Luria and Delbrück’s [24], clonal expansions are powerful tools to unmask heterogeneity in the population that is otherwise missed by population averaging. Beyond clonal expansions, population bottlenecks such as the selective growth of cells expressing specific markers [25] or fractional killing after treatments [14,21,23] generate homogenized cell populations. The dynamics of phenotypic diversification after population bottlenecks hold information about the stability of cellular states even when specific details of the underlying molecular circuits are unknown. While stable phenotypic states persist after prolonged culture, transient states are expected to reconstitute the phenotypic diversity of the original population with a timescale defined by the dynamics of phenotype interconversion (Figure 2A). However, care should be taken when using this rationale to make inferences about the genetic and non-genetic character of phenotypic variation. Non-genetic phenotypes that appear highly stable over limited experimental timescales could in principle be reversible over longer timescales. In addition, differences in growth rates between phenotypic states can contribute to the reconstitution of phenotypic diversity upon drug removal. For instance, ‘drug addiction’ [26,27] – which describes the genetically acquired dependency on drug exposure for proliferation – leads to population-level re-sensitization after drug removal due to competition between resistant and non-resistant cells, and not due to the interconversion between transient phenotypic states. Thus, researchers should be careful when making inferences about single-cell level processes from temporal changes in the diversity of cell populations.
Figure 2. Towards a phenomenological and mechanistic understanding of the dynamics of drug tolerant phenotypic states.
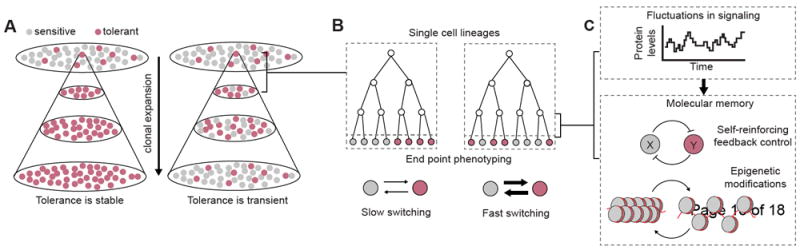
(A) The reconstitution of phenotypic diversity after clonal expansion can provide insights into the stability of drug tolerant states.
(B) Live-cell imaging provides the opportunity to zoom into the process of diversity generation from a single cell. By combining information about lineage relationships and end-point single cell phenotype, it is possible to estimate the rates of phenotypic transitions. Adapted from Hormoz et al. [30].
(C) Understanding the way fluctuations in signaling molecules interact with stabilization mechanisms, such as self-reinforcing feedback loops and epigenetic modifications, is critical for controlling the emergence of drug-tolerant states before and after treatment.
Recent developments in our ability to observe individual cells over time provide the opportunity to characterize, with high temporal resolution, the unfolding of phenotypic diversity in single cells. Using microfluidics and live-cell imaging, Norman and colleagues recorded rare phenotypic switching in individual B. subtilis cells. The waiting-time distributions between switching events revealed insightful features of the mechanisms driving phenotypic transitions: exponential waiting-time distributions indicated memoryless switching where transitions occurred at a constant rate, while non-exponential waiting-time distributions were interpreted to result from the contribution of individual rates in a multistep process [28,29]. Recently, Hormoz and colleagues developed an elegant experimental and analytical strategy that combines end-point phenotyping and single cell lineage relationships to infer transition rates between defined functional states [30,31]. Intuitively, the correlation in cellular states between cells that diverged recently in the lineage are expected to be strong when phenotypes are stable in relation to the cell cycle length, and to decay as state transitions become faster (Figure 2B). Inferences based on kin correlations will be powered by recently developments to infer lineage relationships from fixed cells [32], allowing access to experimental systems that are difficult to address using live imaging. Lastly, synthetic memory devices can be used to record histories of phenotypic transitions within cell populations and to estimate the cumulative probability of switching over time, a valuable approach to understand the dynamics of switching over timescales that are prohibitive for live-cell imaging [33].
The elucidation of molecular mechanisms that mediate the stochastic interconversion between phenotypic states will be critical to account for non-genetic heterogeneity in a clinical setting. Single isogenic cells exposed to the same environment represent independent realizations of very similar dynamical processes. Therefore, the analysis of single cells offers the opportunity to dissect the way variation in specific molecules contributes to phenotypic heterogeneity in an internally controlled setting. As a special case, comparison between sister cells control not only for shared environment, but also for the entire cellular history prior to division [30,34].
Live-fluorescent reporters, recently powered by the development of novel genome engineering technologies [35], allow the detailed quantification of the temporal fluctuations in key signaling proteins before and after treatments, revealing patterns that are hard to infer from fixed timepoint assays [21,22,36]. Complementing this approach, high-throughput assays such as CyTOF [37,38] and single-cell RNA sequencing [39,40] allow the simultaneous profiling multiple molecular species in the same cell. The analysis of these single cell profiles has led to the identification of substructure in cell populations, providing the opportunity to delve deeper into the functional differences of molecularly defined subpopulations of cells. In addition, the density of cells within particular cellular states has been used to estimate the average time cells dwell in such state [41], providing insights into the relative stability of distinct phenotypes. Together, these technological and analytical developments are important milestones towards the overarching goal of understanding the way fluctuations that occur within the timescales of production and degradation of mRNAs and proteins can bring about large phenotypic changes, such as drug tolerance or sensitivity (Figure 2C). Such insights will be pivotal in efforts to predict and control the emergence of phenotypic diversity in pathological cell populations.
Coping with the uncertainty in the response of single cells to therapy
Therapeutic strategies that take into account diversity in drug tolerance within cell populations hold the potential to reduce or forestall the emergence of resistant clones. One promising approach is to homogenize populations through sequential treatments to reduce the burden of phenotypic heterogeneity in the population, as has been proposed to cope with karyotype heterogeneity in yeast and cancer cells [42]. For instance, since cancer cells that are quiescent or in early G1 are more likely to survive chemotherapy [43], synchronized cell cycle re-entry in the population could paradoxically improve the efficacy of acute DNA damaging agents [44]. Targeting global epigenetic modifiers is a promising strategy to prevent the progressive stabilization [14,15] and revert drug tolerant states [20], therefore maintaining sensitivity within cell populations. The identification of vulnerabilities specific to drug tolerant phenotypes could be used to prevent the selective expansion of these subpopulations during therapy. Moreover, genetic alterations can limit the ability of cells to establish drug tolerant quiescent states, such as the impaired entry into cell cycle arrest after DNA damage in p53 mutant cells [45]. Targeting cells that fail to establish a transient drug-tolerant state could selectively ablate the expansion of mutants while sparing their wild-type counterparts, as has been proposed in the context of cyclotherapy [45].
Complementing strategies to reshape heterogeneity in populations, therapies can be designed to specifically account for the diversity and phenotypic dynamics of a given cell population [46]. Resistant states are frequently associated with quiescent or slowly dividing behaviors [14,16,47,48], which stand in contrast to the actively diving cell populations from which drug tolerant cells originate. Theory is particularly suited to optimize therapeutic regimes that take into account tradeoffs and timescales of transitions between cellular states to control the overall growth of cell populations [49-51]. Efforts to fine-tune the frequency and duration of drug exposure are expected to improve the outcomes of therapy by preventing adaptation and off-target effects of chronic drug exposure.
Fractional killing arises from heterogeneity at both the genetic and non-genetic levels [52]. Within non-genetic heterogeneity, different mechanisms can generate drug-tolerant states of different lifetimes. An emerging picture from the study of transient drug-tolerant phenotypic states is that short-lived states can be important for the establishment of longer-lived states. Therefore, a phenomenological and mechanistic understanding of timescales of phenotypic fluctuations is necessary to control the progressive stabilization of drug tolerance and resistance. As considerations of non-genetic heterogeneity are incorporated in the rational design of therapies, it will also be important to understand the extent to cells can modulate the dynamics of phenotypic interconversion to adapt to treatment schedules.
Highlights.
Non-genetic variation gives rise to drug-tolerant transient phenotypic states, imposing a barrier to the success of treatments.
Drug-tolerant phenotypic states fluctuate over diverse timescales and can be stabilized upon chronic drug exposure.
Cell-to-cell variability can be harnesses to understand timescales of phenotypic fluctuations and the molecular underpinnings of entry and exit into drug-tolerant states.
Therapeutic strategies that account for or modulate the rates of phenotypic transitions hold the potential to improve the success of treatments.
Acknowledgments
We thank T. Mitchinson, J. Paulsson and S. Gaudet for helpful feedback and discussions, members of the Lahav laboratory for comments, support, and ideas. This research was supported by NIH grant GM116864. J.R. received support from CONACyT/Fundacion Mexico en Harvard (404476), and Harvard Graduate Merit Fellowship.
Footnotes
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1•.Symmons O, Raj A. What’s Luck Got to Do with It: Single Cells, Multiple Fates, and Biological Nondeterminism. Mol Cell. 2016;62:788–802. doi: 10.1016/j.molcel.2016.05.023. An excellent review that discusses the causes and consequences of heterogeneity in biological systems, with a strong focus on the way distinct input-output relationships can buffer or amplify fluctuations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loewer A, Karanam K, Mock C, Lahav G. The p53 response in single cells is linearly correlated to the number of DNA breaks without a distinct threshold. BMC Biol. 2013;11:114. doi: 10.1186/1741-7007-11-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Battich N, Stoeger T, Pelkmans L. Control of Transcript Variability in Single Mammalian Cells. Cell. 2015;163:1596–1610. doi: 10.1016/j.cell.2015.11.018. [DOI] [PubMed] [Google Scholar]
- 4.Paulsson J. Summing up the noise in gene networks. Nature. 2004;427:415–418. doi: 10.1038/nature02257. [DOI] [PubMed] [Google Scholar]
- 5.Pisco AO, Huang S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: “What does not kill me strengthens me”. Br J Cancer. 2015;112:1725–1732. doi: 10.1038/bjc.2015.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramirez M, Rajaram S, Steininger RJ, Osipchuk D, Roth MA, Morinishi LS, Evans L, Ji W, Hsu C, Thurley K, et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Commun. 2016 doi: 10.1038/ncomms10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balaban NQ. Persistence: Mechanisms for triggering and enhancing phenotypic variability. Curr Opin Genet Dev. 2011;21:768–775. doi: 10.1016/j.gde.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Brauner A, Fridman O, Gefen O, Balaban NQ. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat Rev Microbiol. 2016;14:320–330. doi: 10.1038/nrmicro.2016.34. [DOI] [PubMed] [Google Scholar]
- 9.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science (80-) 2004;305:1622–5. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 10.Radzikowski JL, Vedelaar S, Siegel D, Ortega ÁD, Schmidt A, Heinemann M. Bacterial persistence is an active σS stress response to metabolic flux limitation. Mol Syst Biol. 2016;12:882. doi: 10.15252/msb.20166998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maisonneuve E, Castro-Camargo M, Gerdes K. (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell. 2013;154:1140–1150. doi: 10.1016/j.cell.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 12.Rotem E, Loinger A, Ronin I, Levin-Reisman I, Gabay C, Shoresh N, Biham O, Balaban NQ. Regulation of phenotypic variability by a threshold-based mechanism underlies bacterial persistence. Proc Natl Acad Sci U S A. 2010;107:12541–12546. doi: 10.1073/pnas.1004333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Boxtel C, van Heerden JH, Nordholt N, Schmidt P, Bruggeman FJ. Taking chances and making mistakes: non-genetic phenotypic heterogeneity and its consequences for surviving in dynamic environments. J R Soc Interface. 2017;14 doi: 10.1098/rsif.2017.0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14••.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15••.Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, Beqiri M, Sproesser K, Brafford PA, Xiao M, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431–435. doi: 10.1038/nature22794. These two studies document the stabilization of drug-tolerant states upon chronic treatment of cancer cells with targeted therapies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fallahi-Sichani M, Becker V, Izar B, Baker GJ, Lin J, Boswell SA, Shah P, Rotem A, Garraway LA, Sorger PK. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de-differentiated state. Mol Syst Biol. 2017;13:905. doi: 10.15252/msb.20166796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rothenberg SM, Concannon K, Cullen S, Boulay G, Turke AB, Faber AC, Lockerman EL, Rivera MN, Engelman JA, Maheswaran S, et al. Inhibition of mutant EGFR in lung cancer cells triggers SOX2-FOXO6-dependent survival pathways. Elife. 2015;4:1–25. doi: 10.7554/eLife.06132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen C, Wongvipat J, Ku S, Gao D, Cao Z, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53 - and RB1 -deficient prostate cancer. Science (80-) 2017;355:84–88. doi: 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbé DP, Gomez EC, Wang J, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science (80-) 2017;355:78–83. doi: 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Segerman A, Niklasson M, Haglund C, Bergström T, Jarvius M, Xie Y, Westermark A, Sönmez D, Hermansson A, Kastemar M, et al. Clonal Variation in Drug and Radiation Response among Glioma-Initiating Cells Is Linked to Proneural-Mesenchymal Transition. Cell Rep. 2016;17:2994–3009. doi: 10.1016/j.celrep.2016.11.056. [DOI] [PubMed] [Google Scholar]
- 21••.Paek AL, Liu JC, Loewer A, Forrester WC, Lahav G. Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell. 2016;165:631–642. doi: 10.1016/j.cell.2016.03.025. This study related heterogeneity in the rate of p53 activation in response chemotherapy to fractional induction of apoptosis and the emergence of resistant arrested cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roux J, Hafner M, Bandara S, Sims JJ, Hudson H, Chai D, Sorger PK. Fractional killing arises from cell-to-cell variability in overcoming a caspase activity threshold. Mol Syst Biol. 2015;11:803. doi: 10.15252/msb.20145584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 2009;459:428–432. doi: 10.1038/nature08012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luria S, Delbrück M. Mutations of Bacteria from Virus Sensitivity to Virus Resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011;146:633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 26.Kong X, Kuilman T, Shahrabi A, Boshuizen J, Kemper K, Song J, Niessen HWM, Rozeman EA, Geukes Foppen MH, Blank CU, et al. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature. 2017 doi: 10.1038/nature24037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–255. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Norman TM, Lord ND, Paulsson J, Losick R. Memory and modularity in cell-fate decision making. Nature. 2013;503:481–486. doi: 10.1038/nature12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Norman TM, Lord ND, Paulsson J, Losick R. Stochastic Switching of Cell Fate in Microbes. Annu Rev Microbiol. 2015;69:381–403. doi: 10.1146/annurev-micro-091213-112852. [DOI] [PubMed] [Google Scholar]
- 30••.Hormoz S, Singer ZS, Linton JM, Antebi YE, Shraiman BI, Elowitz MB. Inferring Cell-State Transition Dynamics from Lineage Trees and Endpoint Single-Cell Measurements. Cell Syst. 2016;3:419–433.e8. doi: 10.1016/j.cels.2016.10.015. This study introduces a new experimental and analytical framework to infer rates of state transitions from single cell genealogies and end-point phenotyping. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hormoz S, Desprat N, Shraiman BI. Inferring Epigenetic Dynamics from Kin Correlations. Pnas. 2015 doi: 10.1073/pnas.1504407112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frieda KL, Linton JM, Hormoz S, Choi J, Chow K-HK, Singer ZS, Budde MW, Elowitz MB, Cai L. Synthetic recording and in situ readout of lineage information in single cells. Nature. 2016;541:107–111. doi: 10.1038/nature20777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuan L, Chan GC, Beeler D, Janes L, Spokes KC, Dharaneeswaran H, Mojiri A, Adams WJ, Sciuto T, Garcia-Cardeña G, et al. A role of stochastic phenotype switching in generating mosaic endothelial cell heterogeneity. Nat Commun. 2016;7:10160. doi: 10.1038/ncomms10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feigelman J, Ganscha S, Hastreiter S, Theis FJ. Analysis of Cell Lineage Trees by Exact Bayesian Inference Identifies Negative Autoregulation of Nanog in Mouse Embryonic Stem Cells Report Analysis of Cell Lineage Trees by Exact Bayesian Inference Identifies Negative Autoregulation of Nanog in Mouse Emb. Cell Syst. 2016;3:480–490.e13. doi: 10.1016/j.cels.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 35.Stewart-Ornstein J, Lahav G. Dynamics of CDKN1A in Single Cells Defined by an Endogenous Fluorescent Tagging Toolkit. Cell Rep. 2016;14:1800–1811. doi: 10.1016/j.celrep.2016.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen AA, Geva-Zatorsky N, Eden E, Frenkel-Morgenstern M, Issaeva I, Sigal A, Milo R, Cohen-Saidon C, Liron Y, Kam Z, et al. Dynamic Proteomics of Individual Cancer Cells in Response to a Drug. Science (80-) 2008;322:1511–1516. doi: 10.1126/science.1160165. [DOI] [PubMed] [Google Scholar]
- 37.Krishnaswamy S, Spitzer MH, Mingueneau M, Bendall SC, Litvin O, Stone E, Pe’er D, Nolan GP. Conditional density-based analysis of T cell signaling in single-cell data. Science (80-) 2014;346:1250689–1250689. doi: 10.1126/science.1250689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zunder ER, Lujan E, Goltsev Y, Wernig M, Nolan GP. A continuous molecular roadmap to iPSC reprogramming through progression analysis of single-cell mass cytometry. Cell Stem Cell. 2015;16:323–337. doi: 10.1016/j.stem.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kafri R, Levy J, Ginzberg MB, Oh S, Lahav G, Kirschner MW. Dynamics extracted from fixed cells reveal feedback linking cell growth to cell cycle. Nature. 2013;494:480–483. doi: 10.1038/nature11897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen G, Mulla WA, Kucharavy A, Tsai HJ, Rubinstein B, Conkright J, McCroskey S, Bradford WD, Weems L, Haug JS, et al. Targeting the adaptability of heterogeneous aneuploids. Cell. 2015;160:771–784. doi: 10.1016/j.cell.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryl T, Kuchen EE, Bell E, Shao C, Flórez AF, Mönke G, Gogolin S, Friedrich M, Lamprecht F, Westermann F, et al. Cell-Cycle Position of Single MYC-Driven Cancer Cells Dictates Their Susceptibility to a Chemotherapeutic Drug. Cell Syst. 2017 doi: 10.1016/j.cels.2017.07.005. [DOI] [PubMed] [Google Scholar]
- 44.Overton KW, Spencer SL, Noderer WL, Meyer T, Wang CL. Basal p21 controls population heterogeneity in cycling and quiescent cell cycle states. Proc Natl Acad Sci U S A. 2014;111:E4386–93. doi: 10.1073/pnas.1409797111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rao B, Lain S, Thompson AM. p53-Based cyclotherapy: exploiting the “guardian of the genome” to protect normal cells from cytotoxic therapy. Br J Cancer. 2013;109:2954–8. doi: 10.1038/bjc.2013.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen S hong, Lahav G. Two is better than one; toward a rational design of combinatorial therapy. Curr Opin Struct Biol. 2016;41:145–150. doi: 10.1016/j.sbi.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roesch A, Fukunaga-Kalabis M, Schmidt EC, Zabierowski SE, Brafford PA, Vultur A, Basu D, Gimotty P, Vogt T, Herlyn M. A Temporarily Distinct Subpopulation of Slow-Cycling Melanoma Cells Is Required for Continuous Tumor Growth. Cell. 2010;141:583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levin-Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, Balaban NQ. Antibiotic tolerance facilitates the evolution of resistance. Science (80-) 2017;355:826–830. doi: 10.1126/science.aaj2191. [DOI] [PubMed] [Google Scholar]
- 49.Badri H, Pitter K, Holland EC, Michor F, Leder K. Optimization of radiation dosing schedules for proneural glioblastoma. J Math Biol. 2016;72:1301–1336. doi: 10.1007/s00285-015-0908-x. [DOI] [PubMed] [Google Scholar]
- 50•.Leder K, Pitter K, Laplant Q, Hambardzumyan D, Ross BD, Chan TA, Holland EC, Michor F. Mathematical modeling of pdgf-driven glioblastoma reveals optimized radiation dosing schedules. Cell. 2014;156:603–616. doi: 10.1016/j.cell.2013.12.029. This study combined mathematical modeling and in vivo validation to show that accounting for the dynamics of transitions between resistant and sensitive cellular states can significantly optimize the outcome of radiation therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liao D, Estévez-Salmerón L, Tlsty TD. Generalized principles of stochasticity can be used to control dynamic heterogeneity. Phys Biol. 2012;9:65006. doi: 10.1088/1478-3975/9/6/065006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Almendro V, Marusyk A, Polyak K. Cellular Heterogeneity and Molecular Evolution in Cancer. Annu Rev Pathol Mech Dis. 2013;8:277–302. doi: 10.1146/annurev-pathol-020712-163923. [DOI] [PubMed] [Google Scholar]