

Abstract
EC1456 is a folate-tubulysin conjugate constructed with an all-D enantiomeric spacer/linker configuration. When tested against folate receptor (FR)-positive cells, EC1456 demonstrated dose-responsive activity with an approximate 1000-fold level of specificity. Treatment of nude mice bearing FR-positive human xenografts (as large as 800 mm3) with non-toxic doses of EC1456 led to cures in 100% of the mice. Combinations of low dose EC1456 with standard of care agents such as platins, taxanes, topotecan and bevacizumab, safely and significantly augmented the growth inhibitory effects of these commonly used agents. When tested against FR-positive human tumor xenograft models having confirmed resistance to a folate-vinca alkaloid (vintafolide), cisplatin or paclitaxel, EC1456 was found to generate partial to curative responses. Taken together, these studies demonstrate that EC1456 has significant anti-proliferative activity against FR-positive tumors, including models which were anticancer drug resistant, thereby justifying a Phase 1 trial of this agent for the treatment of advanced human cancers.
Introduction
The folate receptor (FR) is functionally expressed in high quantities by many primary and metastatic cancers1,2. The vitamin folic acid has been shown to specifically deliver a wide variety of therapeutic- and imaging-based agents to tumors that express the FR protein3,4. Hence, we have been developing folate-targeted small molecule drug conjugates (SMDC’s) to boost the safety and efficacy of oncology agents, resulting in an increased therapeutic advantage5–12.
The tubulysin family of secondary metabolites were originally isolated from the myxobacteria Archangium geophyra and Angiococcus disciformis. A variety of tubulysin analogs13–17 have been synthesized and tested for their activity towards different cancers18–20. These compounds are potent microtubule destabilizing agents with IC50 values in the picomolar range against many cancer cell lines21,22, including those with multidrug resistant properties23.
In spite of the powerful in vitro activity of tubulysins, they have limited in vivo therapeutic activity due to severe toxicity. For example, the natural tubulysin B drug in our hands proved to be inactive against a human cervical cancer tumor model when administered at doses near to or greater than the maximum tolerated dose (MTD)6. For this reason, we believe that tubulysins are perfect candidates to be incorporated into our SMDC delivery system. We have recently described the biological activity of EC03056 and subsequently EC053124, which are folate conjugates of tubulysin B12,25. Here we report on a detailed in vivo investigation of a folate tubulysin conjugate (EC1456) that exploits the stable, water soluble saccharo-peptide spacer already tested in EC0531, with particular emphasis on efficacy towards large subcutaneous tumors, combinations with standard of care agents and activity against relevant drug-resistant tumor models.
Results
EC1456 is the all-D enantiomer of EC0531
All FA-drug conjugates reported to date contain a modular design26. The color-coded modularity of the EC1456 structure is shown in Fig. 1A. This SMDC contains multiple polar carbohydrate segments constructed with novel 1-amino-1-deoxy-glucitolyl-γ-glutamate residues, each separated from the other with d-Glu residues and then terminating with d-Cys. Selection of this saccharopeptidic spacer was based on prior results with other SMDCs showing the need for sterically increasing the hydrophilic spacer region to dis-allow, or significantly reduce non-FR mediated cellular uptake, particularly in highly perfused organs like the liver10,24.
Figure 1.

Chemical structure and in vitro activity of EC1456. (A) Module 1 (in black) is the tumor-targeting ligand, folic acid. Module 2 (in blue) is a hydrophilic saccharo-peptidic spacer. Module 3 (in green) is a bio-cleavable, self-immolative disulfide-based linker system. Module 4 (in red) is the active agent, tubulysin B hydrazide. (B) KB cells were pulsed for 2 h with increasing concentrations of EC1456 in the absence (⚫) or presence of 100 µM folic acid (◼) as a benign competitor.
EC1456’s in vitro activity is dose-dependent and specific for the FR
As shown in Fig. 1B, FR-positive KB cells were found to be highly sensitive to EC1456 with an IC50 of 1.5 nM, a value that is also nearly identical to that measured for its all L-enantiomer, EC053124. This result was important because it confirmed that the activity (i.e. drug release) of a disulfide-based SMDC was not dependent on the stereo-specificity of the spacer-linker moieties. EC1456’s activity was next confirmed to be dependent on FR expression since an excess folic acid (used as a benign competitor ligand) reduced EC1456’s cytotoxicity by ~1000-fold (Fig. 1B).
Anti-tumor activity of EC1456 against large KB tumor xenografts
The activity of EC1456 against the FR-positive parental KB tumor model was assessed by treating mice bearing tumors of increasing sizes with 2 μmol/kg at a three times per week (TIW), 2-week schedule. Mice were divided into three groups and treatments started when the tumors had reached the following range: 224–312 mm3, 386–617 mm3 and 640–821 mm3. As shown in Fig. 2, untreated control mice reached a tumor size of 1500 mm3 by approximately PTI day 19, whereas treatment with EC1456 lead to 100% cures in all groups, regardless of tumor size at the onset of dosing. Importantly, EC1456-treated animals did not lose any significant weight throughout the dosing period and beyond, which is similar to that seen with our previously reported folate-targeted cytotoxic agents5–7.
Figure 2.
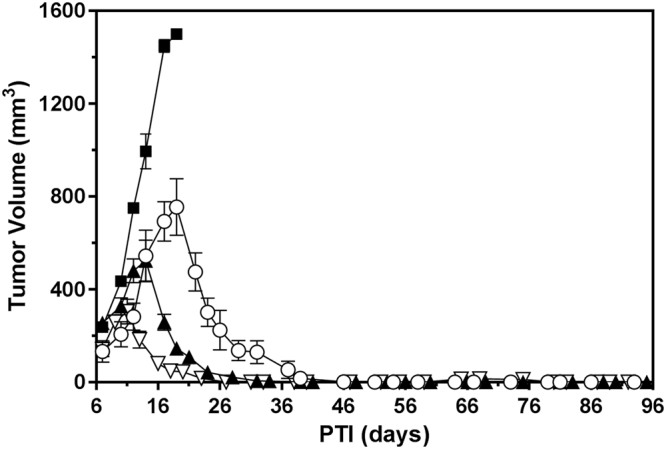
Antitumor effects of EC1456 on various size tumors. KB tumor cells (1 × 106) were inoculated subcutaneously into nu/nu mice and randomized with tumors in various ranges. Mice were treated with EC1456, 2 μmol/kg, TIW x 2 weeks. Tumor volume ranges: (▽) 224–312 mm3; (▲) 386–617 mm3; (○) 640–821 mm3. ◼, untreated control cohort. Each curve shows the average volume of 4–5 tumors.
EC1456 combines synergistically with various clinical anticancer agents
FR-targeted drugs like vintafolide and EC1456 have or are currently being tested clinically against FR-expressing cancers such as ovarian, non-small lung cancer, endometrial and triple-negative breast cancer. The anticancer agents cisplatin, carboplatin, carboplatin/paclitaxel combination, docetaxel, topotecan and bevacizumab are general treatment options for patients with these cancers. In the following studies, EC1456 was combined with these standard agents and evaluated against the FR-expressing KB tumor model for evidence of combined anti-tumor activity.
All of the drug combinations were tested using a lower, less efficacious 1 μmol/kg EC1456 dose level, following either a twice a week (BIW) × 2 week regimen (A) or a TIW × 2 week regimen (B) (see Table 1). When combined with EC1456 on regimen A the DNA crosslinker, cisplatin, produced 100% cures under conditions where single agent EC1456 produced 0 PRs and cisplatin produced only 60% PRs. Carboplatin also combined very well with EC1456 (regimen B) resulting in 100% cures, while EC1456 and carboplatin alone were less effective. At a lower dose of carboplatin (30 mg/kg) and when combined with paclitaxel, significant antitumor activity (80% CRs/20% cures) was observed. Yet, adding EC1456 to this doublet led to 100% cures with no additional toxicity.
Table 1.
Activity and response for each drug combination tested in the KB tumor-nu/nu mouse tumor model.
| Treatment Regimen | PR, % | CR, % | Cures, % |
|---|---|---|---|
| Control mice | 0 | 0 | 0 |
| EC1456 (A: 1 μmol/kg BIW × 2) | 0 | 0 | 0 |
| Cisplatin (3 mg/kg BIW 3 doses) | 60 | 0 | 0 |
| EC1456 (A: 1 μmol/kg BIW × 2) + Cisplatin (3 mg/kg BIW 3 doses) | 0 | 0 | 100 |
| Control mice | 0 | 0 | 0 |
| EC1456 (B: 1 μmol/kg TIW × 2) | 60 | 0 | 0 |
| Carboplatin (50 mg/kg TIW) | 0 | 0 | 0 |
| EC1456 (B: 1 μmol/kg TIW × 2) + Carboplatin (50 mg/kg TIW) | 0 | 0 | 100 |
| Control mice | 0 | 0 | 0 |
| EC1456 (A: 1 μmol/kg BIW × 2) | 20 | 0 | 0 |
| Carboplatin (30 mg/kg BIW × 2) + Paclitaxel (10 mg/kg BIW × 2) | 0 | 80 | 20 |
| EC1456 (A: 1 μmol/kg BIW × 2) +Carboplatin (30 mg/kg BIW × 2) +Paclitaxel (10 mg/kg BIW × 2) | 0 | 0 | 100 |
| Control mice | 0 | 0 | 0 |
| EC1456 (B: 1 μmol/kg TIW × 2) | 0 | 60 | 40 |
| Docetaxel (7 mg/kg BIW 3 doses) | 60 | 0 | 40 |
| EC1456 (B: 1 μmol/kg TIW × 2) + Docetaxel (7 mg/kg BIW 3 doses) | 0 | 0 | 100 |
| Control mice | 0 | 0 | 0 |
| EC1456 (A: 1 μmol/kg BIW × 2) | 20 | 0 | 0 |
| Topotecan (5 mg/kg BIW × 2) | 0 | 0 | 0 |
| EC1456 (A: 1 μmol/kg BIW × 2) + Topotecan (5 mg/kg BIW × 2) | 20 | 40 | 40 |
| Control mice | 0 | 0 | 0 |
| EC1456 (B: 1 μmol/kg TIW × 2) | 60 | 20 | 20 |
| Bevacizumab (5 mg/kg BIW × 2) | 0 | 0 | 0 |
| EC1456 (B: 1 μmol/kg TIW × 2) + Bevacizumab (5 mg/kg BIW × 2) | 0 | 0 | 100 |
All of the drug combinations were tested using a 1 μmol/kg EC1456 dose level, at either a twice a week (BIW) x 2 week regimen (A), or a TIW x 2 week regimen (B). n = 5/cohort. PR, partial response defined as volume regression >50% but with measurable tumor (>2 mm3) remaining at all times; CR, complete response defined as a disappearance of measurable tumor mass (<2 mm3) at some point within 90 days after tumor implantation; cures were defined as CRs without tumor regrowth within the 90-day study time frame.
As single agents both the microtubule inhibitor, docetaxel, and EC1456 (regimen B) produced impressive antitumor activity, with 40% cures/60% PRs and 40% cures/60% CRs, respectively. But when combined, EC1456 + docetaxel worked together to cure 100% of the treated animals, again with no significant change in adverse events.
Significantly greater antitumor effect was also observed when EC1456 was combined with the topoisomerase inhibitor, topotecan. As single agents, topotecan and EC1456 (using the less frequent dosing regimen A) produced minimal antitumor activity as noted by 0% PRs and 20% PRs, respectively. In contrast, this EC1456/topotecan combination produced 40% cures/40% CRs/20% PRs.
EC1456 on regimen B also combined very well with a VEGF inhibitor, bevacizumab, yielding 100% cures, whereas bevacizumab alone or single agent EC1456 produced far lower responses, as noted in Table 1.
Anti-tumor effects of vintafolide and EC1456 on KB-145-55 tumors
The KB-145-55 cell line was created from a parental KB tumor which became resistant to vintafolide (a clinically tested folate-vinca alkaloid conjugate) in a mouse intermittently dosed 43 times with 2 μmol/kg of drug over a 192 day span. The level of KB-145-55’s resistance to vintafolide was assessed by treating the mice 15 days PTI using a normally 100% curative regimen (i.e. 2 μmol/kg given TIW, for 2 weeks;7). As expected and shown in Fig. 3, vintafolide produced a greatly reduced level of antitumor activity (zero CRs and three PRs) in these resistant tumors. This compromised efficacy could not be explained by minor changes in FR copies/cell (6.31 × 106 vs 6.33 × 106 receptors/cell for KB-145-55 and parental KB cells, respectively), tumor growth rate (37 vs. 28 days to reach 1500 mm3 in volume for KB-145-55 and parental KB cells, respectively), or p-glycoprotein (P-gp) expression levels (Fig. 4). In contrast, EC1456 administered using the same dosing regimen had generated a 100% (5/5) complete response rate, of which four of the five animals maintained these CR’s throughout the 90-day duration of the study and were considered cured.
Figure 3.
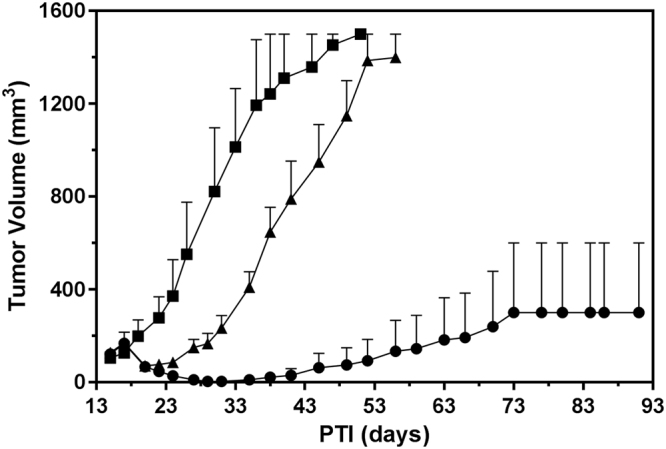
Anti-tumor effects of vintafolide and EC1456 against KB-145–55 tumors. Vintafolide-resistant KB-145–55 tumor cells (1 × 106) were inoculated subcutaneously into nu/nu mice and therapy started on randomized animals with tumors in the 93–173 mm3 range. ■, Control; ▲, vintafolide, 2 μmol/kg, TIW x 2 weeks; ●, EC1456, 2 μmol/kg, TIW × 2 weeks. The EC1456 treatment (●) curve is the result of re-growth of only 1 of 5 tumors after day 35. Each curve shows the average volume of 5 tumors.
Figure 4.
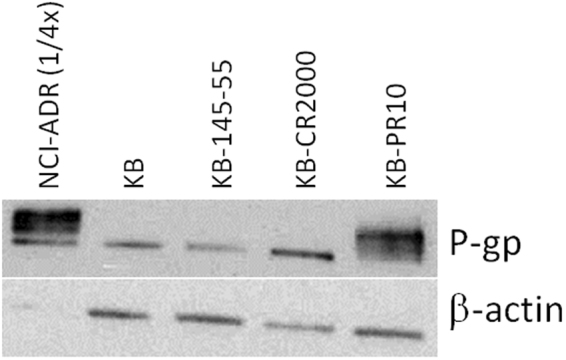
P-glycoprotein (ABCB1, MDR1) expression in vintafolide, cisplatin and paclitaxel resistant cell lines. KB, KB-145–55, KB-CR2000, KB-PR10, and NCI-ADR cell lines were lysed with 100 µL RIPA buffer containing 1:100 Halt phosphatase and protease inhibitor cocktail. Ten μg of each protein lysate (2.5 μg of NCI-ADR) were resolved by SDS-PAGE. P-gp and β-actin were detected with rabbit anti-MDR1 (1:1000) and rabbit anti-β-actin (1:2000) antibodies, respectively. A horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody (1:5000) was used to visualize the signal by an ECL substrate. Western blot image (B) has been cropped for clarity with full blots presented in Figure S1.
Generation of cisplatin and paclitaxel resistant cell lines
Parental KB cells were next used to generate drug-resistant, FR-expressing cell lines through increasing levels of cisplatin or paclitaxel exposure. The cell lines derived through exposure to cisplatin (KB-CR2000, IC50 ~ 10 μM) and paclitaxel (KB-PR10, IC50 ~ 130 nM) both exhibited significant resistance (Table 2) to their corresponding drugs as compared to the parental KB cells (cisplatin IC50 ~ 380 nM, paclitaxel IC50 ~ 3 nM).
Table 2.
In vitro activity (IC50 values) of various agents on KB, KB-CR2000 and KB-PR10 cells.
| Drug/s | KB | KB-CR2000 | KB-PR10 |
|---|---|---|---|
| Cisplatin | 379 nM | 10.8 μM | — |
| Paclitaxel | 3 nM | — | 132 nM |
| Vintafolide | 9 nM | 5 nM | >1 μM |
| Paclitaxel + Verapamil | — | — | 5 nM |
| Vintafolide + Verapamil | — | — | <1 nM |
| EC1456 | 2 nM | 4 nM | 2 nM |
Analysis by western blotting revealed that P-gp expression in the KB-PR10 cells was greatly amplified (~13 fold) in comparison to the parent KB cells or the KB-CR2000 cells (Fig. 4). NCI-ADR cells, which are a known high P-gp expressing cell line, were used as a positive control, albeit at one fourth of the protein concentration as the other cells. This overexpression of P-gp in KB-PR10 cells also caused them to be cross-resistant (IC50 > 1 μM) to vintafolide, which was expected27. The low P-gp expressing KB and KB-CR2000 cells remained sensitive (IC50 of 5–9 nM) to vintafolide. When combined with either paclitaxel or vintafolide, verapamil (a potent inhibitor of P-gp) treatment (10 μM) had restored KB-PR10’s sensitivity to both drugs (IC50s of 1–5 nM). Verapamil alone at 10 μM showed minimal toxicity on KB-PR cells with 90.8 +/− 5.1% of cells remaining viable compared to untreated controls. However, in stark contrast, EC1456 remained potent in all three cell lines (IC50 of 2–4 nM), regardless of their P-gp expression levels (Table 2).
Anti-tumor effects of cisplatin, vintafolide and EC1456 against KB-CR2000 tumors
To build on the results observed in vitro, vintafolide, cisplatin, and EC1456 were dosed in nu/nu mice that had been inoculated subcutaneously with KB-CR2000 cells. The in vivo activity of these three agents against KB-CR2000 tumors was analogous to the in vitro activity of these drugs on KB-CR2000 cells. As expected, cisplatin alone was found to be inactive against the KB-CR2000 tumors (Fig. 5A). However, vintafolide remained highly active against this cisplatin-resistant model yielding 50% CRs/50% cures, and EC1456 at the same dose and schedule produced an even better response with 20% CRs and 80% cures. Changes in FR copies/cell (5.77 × 106 vs. 6.33 × 106 receptors/cell for KB-CR2000 and parental KB cells, respectively) and tumor growth rates (25 vs. 28 days to 1500 mm3 for KB-CR2000s and parental KB cells, respectively) were minimal to have significant effects on drug sensitivities of these tumors.
Figure 5.
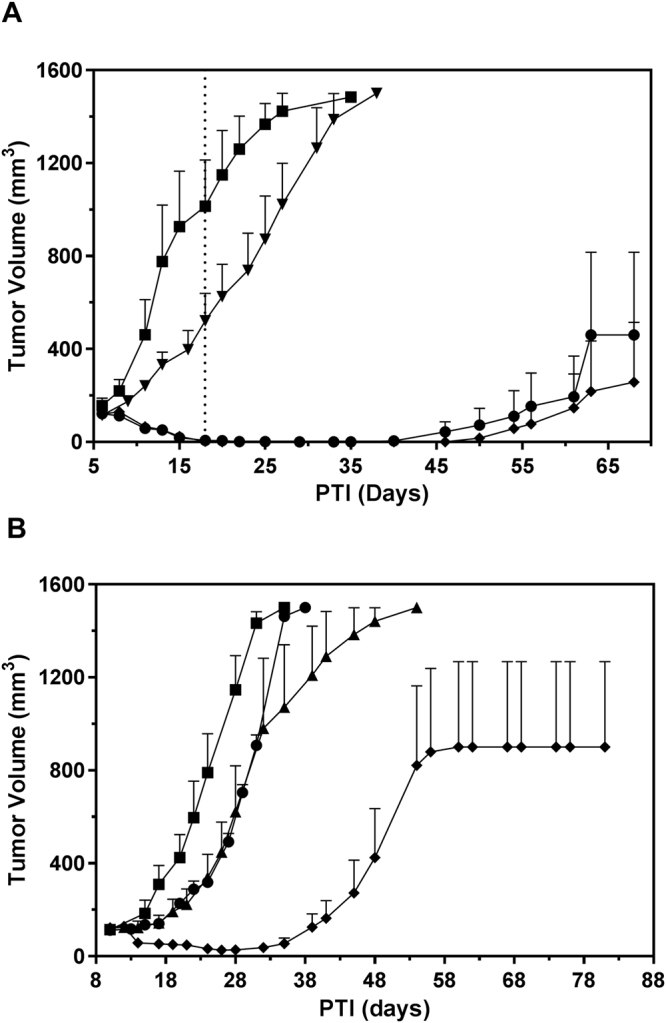
Antitumor effects of cisplatin (A)/paclitaxel (B), vintafolide and EC1456 against cisplatin-resistant KB-CR2000 tumors (A) and paclitaxel resistant KB-PR10 tumors (B). KB-CR2000 (A) and KB-PR10 (B) tumor cells (1 × 106) were inoculated subcutaneously into nu/nu mice and therapy started on randomized animals with tumors in the 99–149 mm3 range. ■, Control; ▼ (A), cisplatin, 3 mg/kg, BIW x 2 weeks; ▲ (B), paclitaxel, 20 mg/kg, TIW x 2 weeks; ●, vintafolide, 2 μmol/kg, TIW x 2 weeks; ◆, EC1456, 2 μmol/kg, TIW x 2 weeks. Each curve shows the average volume of 5 tumors.
Anti-tumor effects of paclitaxel, vintafolide and EC1456 on KB-PR10 tumors
The paclitaxel-resistant cells, KB-PR10, were inoculated subcutaneously into mice and treatment started when tumors were in a range of 99–144 mm3. As shown in Fig. 5B, both single agent paclitaxel and vintafolide had very little effect against the tumors, with 40% PRs and 0% PRs, respectively. On the other hand, single agent EC1456 substantially reduced tumor burden resulting in 60% PRs and 40% cures. FR copies/cell (7.95 × 106 vs. 6.33 × 106 receptors/cell for KB-PR and parental KB cells, respectively) and tumor growth rates (31 vs. 28 days to 1500 mm3 for KB-PR and parental KB cells, respectively) were quite similar to those of KB tumors to have any significant effects on KB-PR drug sensitivities.
Discussion
EC1456 is the third iteration of a folic acid-based SMDC constructed with the potent tubulysin B hydrazide (TubBH) drug linked together with a disulfide bond. The first folate-tubulysin B conjugate reported by us is EC0305, which was constructed with a γGlu-Arg-Asp-Cys spacer connecting the folate moiety to TubBH6. Subsequently, EC0305 was re-engineered with a saccharopeptidic-based hydrophilic spacer (SPS) replacing the peptide spacer. The resulting molecule, called EC0531, thus contained polar carbohydrate (1-amino-1-deoxy-glucitolyl-γ-glutamate) segments, separated by L-Glu residues and terminating with L-Cys24. EC0531 was found to be more active over a wider therapeutic range than EC0305, as supported by a 5.6-fold less free TubBH clearing through the bile following EC0531 injection when compared to EC0305. Within EC1456, we retained the positive pharmacokinetic properties of the SPS spacer system while replacing the L-glu and L-cys amino acids with their D-isoforms. Such stereochemical modifications are known to enhance peptidic biostability because of resistance to proteolytic degradation by most of the endogenous enzymes28. This change in chirality would also likely limit the pericellular proteolysis of EC1456 by membrane associated and secreted proteases in the tumor29.
Following brief cycles of EC1456 therapy, tumors of large sizes were repeatedly observed to totally recede, mostly without recurrence, while producing no noticeable weight loss or adverse events, suggesting that EC1456 may be a useful agent against FR-expressing human cancers. In comparison, untargeted TubBH, regardless of the dose, was not found to produce any antitumor effect in this parental KB tumor model12. This anti-tumor effect against very large s.c. tumors is most probably due to homogeneous EC1456 cellular targeting as previously demonstrated by the uniform fluorescence in the center and periphery of tumors in mice dosed with a small molecule folate-rhodamine conjugate7. ADCs, in contrast, owing to their slow tissue penetrating abilities, often saturate receptors of perivascular cells causing heterogeneous distribution within solid tumors which can impact their overall efficacy30.
Since standard clinical chemotherapy regimens generally use multidrug combinations, we proceeded to determine if EC1456 could improve therapeutic efficacy by combining with clinically approved agents with various mechanisms of action. When low dose EC1456 was combined with the DNA crosslinking agents, cisplatin and carboplatin, enhanced antitumor activity was observed over that produced by the single agents, with some studies yielding 100% cure rates. When EC1456 was added to the often clinically used paclitaxel/carboplatin doublet, it improved the antitumor effect by again yielding 100% cures. EC1456 was also tested in combination with a microtubule inhibitor, docetaxel, the topoisomerase inhibitor, topotecan, and the VEGF inhibitor, bevacizumab. In all cases, the antitumor activity of the EC1456 combinations was far superior to that observed with any of the individual drugs tested alone. In addition, combining EC1456 with these agents did not increase the inherent toxicity above that produced by the untargeted agents, thus resulting in a general increase in the EC1456 combination therapeutic window.
In the past we6 and others23 have shown that tubulysins retain their activity against multidrug resistant cell lines, while vinca alkaloids as a class do not. Our first generation SMDC, vintafolide, is a folic acid-targeted conjugate of a vinca alkaloid. To test if EC1456 would be useful in treating tumors that have developed resistance toward vintafolide, we created a FR-positive cell line KB-145–55, from a tumor which had been made resistant to vintafolide in vivo. We confirmed herein that vintafolide has reduced antitumor activity against KB-145-55 tumors in comparison with the parental KB tumors, whereas EC1456 (dosed similarly) yielded curative activity, likely because of tubulysin being not a good P-gp substrate.
A majority of ovarian cancer patients receive a first-line combination regimen that comprises a taxane and a platinum drug. There is a clear need in advanced ovarian cancer to consider the use of second-line chemotherapeutic options, since most women will ultimately relapse and develop drug-resistant disease31,32. To pre-clinically forecast EC1456’s activity in such patients, paclitaxel and cisplatin-resistant variants of FR-expressing KB cells were selected by continuous in vitro exposure to increasing concentrations of these drugs. When cross resistance was investigated, it was found that the cisplatin-derived resistant line, KB-CR2000, was not cross-resistant to vintafolide. In contrast, the paclitaxel-derived resistant cells, KB-PR10, exhibited significant cross resistance to vintafolide, but not to the tubulysin-containing EC1456. Not surprisingly, the KB-PR10 subline did exhibit a multidrug-resistance phenotype with overexpression of p-glycoprotein, whereas KB-CR2000 did not. Hence verapamil, a P-gp inhibitor, was able to revive KB-PR10’s sensitivity to both paclitaxel and vintafolide. As would be predicted from the cell culture data, the anti-tumor activity of EC1456 was similar to that of vintafolide in the low P-gp expressing KB-CR2000 model, but EC1456 was found to be far superior to vintafolide against the higher P-gp expressing KB-PR10 tumor model. This outcome indicated that the activity of EC1456 was predominantly independent of the levels of p-glycoprotein expression, thereby confirming the previous findings of tubulysin not being a good substrate of this protein23.
While our first generation SMDC, vintafolide, was unable to have much of an effect against some drug-resistant models, the tubulysin-containing EC1456 was able to show curative effects. These results may suggest that EC1456 could be useful in treating tumors that have developed resistance towards some standard chemotherapeutic agents. These exciting preclinical qualities have provided justification for a Phase 1 clinical trial.
Methods
Materials
Clinical vial solutions of vintafolide (formally EC145) and EC1456 were used in all experiments (Endocyte Inc.). Carboplatin, cisplatin and verapamil HCl were purchased from Sigma, St. Louis, MO; docetaxel and bevacizumab were obtained from the Purdue University Pharmacy, West Lafayette, IN; paclitaxel and topotecan were purchased from A.K. Scientific, Mountain View, CA. NCI-ADR cells were a kind gift from Dr. Alberto Gabizon, Shaare Zedek Medical Center, Israel. All other common reagents were purchased from Sigma or other major suppliers.
Chemical characterization of EC1456
1H and 13C NMR were obtained on an Agilent 500 MHz NMR. All experiments were conducted at 25 °C. All spectra were referenced to the DMSO solvent residual signals at 2.5ppm (1H) and 39.50 ppm (13C). Positive electrospray mass spectra were obtained on a high resolution Waters Xevo G2-S TOF mass spectrometer following a reverse-phase separation of the major peak on the Acquity UPLC chromatographic system.
HPLC purity: 96% (XBridge RP18 3.5 µm, 3.0 × 50 mm column at λ = 280 nM); 1H NMR (500 MHz, DMSO-d6/D2O): δ 8.59 (s, 1 H), 8.14 (s, 1 H), 7.56 (d, J = 8.5 Hz, 2 H), 6.95 (d, J = 8.0 Hz, 2 H), 6.61 (d, J = 8.5 Hz, 2 H), 6.57 (d, J = 8.0 Hz, 2 H), 6.16 (d, J = 9.5 Hz, 1 H), 5.68 (d, J = 12.0 Hz, 1 H), 5.22 (d, J = 12.0 Hz, 1 H), 4.45 (s, 2 H), 4.37 (d, J = 9.0 Hz, 2 H), 4.30–4.0 (m, 10 H), 3.60 (m, 3 H), 3.59-3.50(m, 6 H), 3.44 (m, 3 H), 3.43–3.37 (m, 6 H), 3.18 (m, 4 H), 3.05 (m, 3 H), 2.98–2.86 (m, 3 H), 2.78 (d, J = 11.5 Hz, 2 H), 2.68 (m, 2 H), 2.53–2.45 (m, 2 H), 2.40-2.25 (m, 2 H), 2.23-1.98 (series of m, 15 H), 2.06 (s, 3 H), 2.00 (s, 3 H), 1.98-1.62 (series of m, 14 H), 1.62-1.24 (series of m, 9 H), 1.18-1.00 (m, 2 H), 0.98 (d, J = 6.5 Hz, 3 H), 0.92 (d, J = 6.0 Hz, 3 H), 0.78 (d, J = 7.0 Hz, 3 H), 0.74 (d, J = 8.5 Hz, 3 H), 0.73 (d, J = 7.5 Hz, 3 H), 0.62 (d, J = 6.0 Hz, 3 H); 13C NMR (125 MHz, DMSO-d6/D2O): δ 176.77, 176.32, 175.74, 175.42, 174.75, 173.87, 172.68, 172.15, 171.94, 171.84, 173.43, 173.30, 172.79 (2 C), 172.72, 172.46, 170.87, 170.39, 169,30, 166.09, 162.40, 160.70, 156.40, 156.09, 155.71, 154.59, 150.84, 149.63, 149.11, 148.99, 130.44 (2 C), 128.99 (2 C), 128.89, 127.99, 124.97, 122.24, 115.25 (2 C), 111.86 (2 C), 72.17 (3 C), 71.78, 71.74, 71.71, 71.62, 71.59 (2 C), 69.65, 69.57 (2 C), 69.45, 69.34, 68.51, 63.42 (3 C), 63.03, 55.08, 54.05, 53.88, 53.46 (2 C), 53.33, 52.96 (2 C), 52.89, 52.55, 49.77, 46.07, 44.02, 42.85, 42.34 (2 C), 42.29, 39.52, 38.95, 37.43, 35.95, 35.43, 35.38, 34.86, 32.56, 32.36, 32.16, 32.09 (2 C), 31.81, 30.50, 29.95, 28.60, 28.04, 27.78 (2 C), 27.66, 27.00, 25.01, 24.43, 23.04, 20.86, 20.56, 19.64, 18.36, 18.04, 15.64, 13.72, 10.28; HRMS (ESI) (m/z): [M + H]+ calcd. for C110H165N23O45S3, 2625.81; found, 2626.06.
Dose-Dependent FR-Specific Activity of EC1456
Parental KB cells (a human cell line from ATCC containing markers of HeLa cervical cancer origin) were seeded in individual 12-well Falcon plates and allowed to form nearly confluent monolayers overnight in folate-deficient RPMI medium supplemented with 10% fetal bovine serum. Following a detailed published procedure12, a 2 h pulse, 70 h chase assay format was used to evaluate the cytotoxic effects of increasing concentrations of EC1456. Viability was assessed by measuring 3H-thymidine incorporation into trichloroacetic acid precipitable material. Final results were expressed as the percentage of 3H-thymidine incorporation relative to untreated controls.
Creation of an in vivo, vintafolide-resistant EC145-55 model
FR-positive human nasopharyngeal carcinoma KB cells (6.0 × 106 FRs/cell) were used in this study; their integrity was confirmed (Genetica DNA Labs, Cincinnati, Ohio) with cells being similar to reference cells from the ATCC.
A vintafolide-resistant KB tumor model was created after continuous controlled treatment in vivo. For this, ten 6-7 week-old female nu/nu mice maintained on a custom folate deficient diet were each subcutaneously inoculated with 1 × 106 KB cells. These tumors were allowed to grow to 250 mm3 after which these mice were dosed with 2 µmol/kg vintafolide. The tumors were measured three times a week and the decision to dose was based on the tumor volume. This controlled dosing method was used such that the tumor volumes were maintained between 100 mm3 and 300 mm3 as long as possible through intermittent dosing (vide infra). Although several tumors showed resistance to vintafolide, one tumor-bearing mouse, herein referred to as EC145-55, received a total of 43 vintafolide doses. The mouse bearing the EC145-55 tumor received its first dose of vintafolide on PTI (post tumor cell implant) day 17 when the tumor was 272 mm3. The tumor volume oscillation process was maintained by dosing on PTI days 19, 45, 47, 68, 78, 85, 94, 96, 109, 113, 115, 117, 127, 129, 141, 143, 148, 150, 152, 155, 157, 159, 162, 164, 166, 171,173,176, 178, 180, 183, 185, 187, 190, 192, 194, 197, 199, 204, 206, 208, and 211. Following the 43rd dose on PTI day 211, the mouse was euthanized, tumor extracted at a volume of 671 mm3, manually disaggregated and then placed into cell culture for further growth and analysis.
Generation of KB-CR2000 and KB-PR10 cells
Our drug-sensitive FR-expressing KB cell line was used as a parental line to generate additional drug-resistant cell lines through increasing levels of cisplatin and paclitaxel exposure. Cisplatin and paclitaxel stock solutions were made in saline and DMSO respectively; appropriate volumes of the sterile stock solutions were added to cell culture flasks to achieve the desired concentration of drug supplemented media. Drug concentrations were increased when cell death was no longer visible and growth rate had stabilized. Thus, cisplatin-resistant KB cells (KB-CR2000) were selected in vitro through exposure to stepwise-increasing concentrations of cisplatin from 50 to 2000 nM, while paclitaxel-resistant KB cells (KB-PR10) were selected with concentrations from 0.1 to 10 nM paclitaxel, with 0.5- to 1-fold increase at each step of resistance.
Cell growth inhibition studies
Parental KB cells, KB-CR2000 and KB-PR10 cells were maintained in folate-free RPMI medium (FFRPMI) containing 10% heat-inactivated fetal calf serum (HIFCS) at 37 °C in a 5% CO2/95% air-humidified atmosphere with no antibiotics. Exponentially growing cells were seeded in 24-well plates 24 h before treatment with drugs. Cells receiving FR-targeted drugs (vintafolide and EC1456) were pulsed for 2 h at 37 °C, rinsed 4 times with 0.5 mL of medium, and then chased in 1 mL of fresh medium for up to 72 h. Cells treated with non-targeted drugs (cisplatin, paclitaxel and verapamil) were treated continuously for 72 h. Cells were treated with fresh medium containing 3H-thymidine for 2 h at 37 °C, washed with phosphate-buffered saline (PBS) and then treated with ice-cold 5% trichloroacetic acid (TCA). After 15 min, the TCA was aspirated and cells solubilized by the addition of 0.25 N sodium hydroxide for 15 min at room temperature. Each solubilized sample was transferred to scintillation vials containing EcoLume™ scintillation cocktail and counted in a liquid scintillation counter.
In vivoantitumor experiments
Four- to eight-week-old female nu/nu mice (Harlan Sprague Dawley, Inc.), were maintained on a standard 12-h light-dark cycle and fed ad libitum with a low-folate chow (Harlan Teklad diet #TD.01013, Madison, WI) for the duration of dosing and 1 week post dosing schedule. Mice were then switched to Teklad Global 18% Rodent diet (Harlan Teklad diet #2018S) during the monitoring phase of the study. Parental KB, KB-145-55, KB-CR2000 or KB-PR10 cells (1 × 106 per nu/nu mouse) in 100 µL were injected into the subcutis of the dorsal medial area. Mice were divided into groups of 5, and freshly prepared test articles were injected through the lateral tail vein under sterile conditions in a volume of 200 μL of PBS. Intravenous treatments typically initiated on day 15 PTI when KB-145-55 tumors were approximately 93 to 173 mm3 in volume, on day 10 PTI when the KB-PR10 tumors were about 96 to 162 mm3, and on day 6 PTI when the KB-CR2000 were around 99 to 149 mm3. In combination experiments, EC1456 was administered 2-3 h ahead of the combined drug, when given on the same day.
The mice in the control groups received no treatment. Growth of each subcutaneous tumor was followed by measuring the tumor 3 times per week during treatment and twice per week thereafter, until a maximum volume of 1500 mm3 was reached. Tumors were measured in 2 perpendicular directions using Vernier calipers, and their volumes were calculated as V = 0.5 × L × W2, where L = measurement of longest axis in mm and W = measurement of axis perpendicular to L in mm. As a general measure of gross toxicity, changes in body weights were determined on the same schedule as tumor volume measurements. Survival of animals was monitored daily. Animals that were moribund (or unable to reach food or water) were euthanized by CO2 asphyxiation. All animal housing, care, and procedures were followed according to Purdue Animal Care and Use Committee (PACUC)-approved animal care and use protocols.
Individual tumor response endpoints were reported in terms of tumor volume change. A partial response (PR) was defined as volume regression >50% but with measurable tumor (>2 mm3) remaining at all times. Complete response (CR) was defined as a disappearance of measurable tumor mass (<2 mm3) at some point within 90 days after tumor implantation. Cures were defined as CRs without tumor regrowth within the 90-day study time frame.
Analysis of P-gp expression
Confluent parental KB, KB-145-55, KB-CR2000, KB-PR10, and NCI-ADR cell lines were cultured in 6 well plates in FFRPMI-1640 with 5% HIFCS with their respective drug concentration, if required (KB-CR2000: 2000 nM cisplatin, KB-PR10: 10 nM paclitaxel), for 24 h. The cells were lysed with 100 µL RIPA buffer containing 1:100 Halt phosphatase and protease inhibitor cocktail for 5 minutes at room temperature with gentle agitation. Protein concentrations were determined by BCA assay, and 10 μg of lysate were resolved by SDS-PAGE. P-gp and β-actin were detected with rabbit anti-MDR1 antibody (1:1000, Cell Signaling) and rabbit anti-β-actin (1:2000, Rockland Immunochemicals), respectively. A horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody (1:5000, Jackson Immunoresearch) was used to visualize the signal by an ECL substrate (Pierce). Quantitative comparison of pgp expression in these cell lines was done by determining the intensities of the pgp bands with ImageJ.
Data Availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
Electronic supplementary material
Author Contributions
Synthesis and characterization of EC1456 was done by P.K. and H.S. with guidance from I.V. The in vitro experiments were performed by R.D., C.D. and M.V. The in vivo experiments were performed by A.B. and M.N. J.R. and C.L. guided the experiments and wrote the main manuscript text. All authors reviewed the manuscript.
Competing Interests
The authors declare that they are shareholders of Endocyte, Inc.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-27320-5.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.O’Shannessy DJ, Somers EB, Smale R, Fu YS. Expression of folate receptor-alpha (FRA) in gynecologic malignancies and its relationship to the tumor type. Int J Gynecol Pathol. 2013;32:258–268. doi: 10.1097/PGP.0b013e3182774562. [DOI] [PubMed] [Google Scholar]
- 2.O’Shannessy DJ, et al. Folate receptor alpha expression in lung cancer: diagnostic and prognostic significance. Oncotarget. 2012;3:414–425. doi: 10.18632/oncotarget.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Assaraf YG, Leamon CP, Reddy JA. The folate receptor as a rational therapeutic target for personalized cancer treatment. Drug Resist Updat. 2014;17:89–95. doi: 10.1016/j.drup.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Vlahov IR, Leamon CP. Engineering folate-drug conjugates to target cancer: from chemistry to clinic. Bioconjug Chem. 2012;23:1357–1369. doi: 10.1021/bc2005522. [DOI] [PubMed] [Google Scholar]
- 5.Reddy JA, et al. Rational combination therapy of vintafolide (EC145) with commonly used chemotherapeutic drugs. Clin Cancer Res. 2014;20:2104–2114. doi: 10.1158/1078-0432.CCR-13-2423. [DOI] [PubMed] [Google Scholar]
- 6.Reddy JA, et al. In vivo structural activity and optimization studies of folate-tubulysin conjugates. Mol Pharm. 2009;6:1518–1525. doi: 10.1021/mp900086w. [DOI] [PubMed] [Google Scholar]
- 7.Reddy JA, et al. Preclinical evaluation of EC145, a folate-vinca alkaloid conjugate. Cancer Res. 2007;67:4434–4442. doi: 10.1158/0008-5472.CAN-07-0033. [DOI] [PubMed] [Google Scholar]
- 8.Naumann RW, et al. PRECEDENT: a randomized phase II trial comparing vintafolide (EC145) and pegylated liposomal doxorubicin (PLD) in combination versus PLD alone in patients with platinum-resistant ovarian cancer. J Clin Oncol. 2013;31:4400–4406. doi: 10.1200/JCO.2013.49.7685. [DOI] [PubMed] [Google Scholar]
- 9.Vlahov IR, et al. Acid mediated formation of an N-acyliminium ion from tubulysins: a new methodology for the synthesis of natural tubulysins and their analogs. Bioorg Med Chem Lett. 2011;21:6778–6781. doi: 10.1016/j.bmcl.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 10.Leamon CP, et al. Reducing undesirable hepatic clearance of a tumor-targeted vinca alkaloid via novel saccharopeptidic modifications. J Pharmacol Exp Ther. 2011;336:336–343. doi: 10.1124/jpet.110.175109. [DOI] [PubMed] [Google Scholar]
- 11.Vlahov IR, et al. Carbohydrate-based synthetic approach to control toxicity profiles of folate-drug conjugates. J Org Chem. 2010;75:3685–3691. doi: 10.1021/jo100448q. [DOI] [PubMed] [Google Scholar]
- 12.Leamon CP, et al. Folate targeting enables durable and specific antitumor responses from a therapeutically null tubulysin B analogue. Cancer Res. 2008;68:9839–9844. doi: 10.1158/0008-5472.CAN-08-2341. [DOI] [PubMed] [Google Scholar]
- 13.Nicolaou KC, et al. Total Synthesis and Biological Evaluation of Natural and Designed Tubulysins. J Am Chem Soc. 2016;138:1698–1708. doi: 10.1021/jacs.5b12557. [DOI] [PubMed] [Google Scholar]
- 14.Murray BC, Peterson MT, Fecik RA. Chemistry and biology of tubulysins: antimitotic tetrapeptides with activity against drug resistant cancers. Nat Prod Rep. 2015;32:654–662. doi: 10.1039/C4NP00036F. [DOI] [PubMed] [Google Scholar]
- 15.Xiangming X, Friestad GK, Lei Y. Recent advances in the synthesis of tubulysins. Mini Rev Med Chem. 2013;13:1572–1578. doi: 10.2174/13895575113139990063. [DOI] [PubMed] [Google Scholar]
- 16.Shibue T, et al. Total syntheses of tubulysins. Chemistry. 2010;16:11678–11688. doi: 10.1002/chem.201000963. [DOI] [PubMed] [Google Scholar]
- 17.Pando O, et al. First total synthesis of tubulysin B. Org Lett. 2009;11:5567–5569. doi: 10.1021/ol902320w. [DOI] [PubMed] [Google Scholar]
- 18.Shankar SP, et al. Synthesis and structure-activity relationship studies of novel tubulysin U analogues–effect on cytotoxicity of structural variations in the tubuvaline fragment. Org Biomol Chem. 2013;11:2273–2287. doi: 10.1039/c3ob27111k. [DOI] [PubMed] [Google Scholar]
- 19.Balasubramanian R, Raghavan B, Begaye A, Sackett DL, Fecik RA. Total synthesis and biological evaluation of tubulysin U, tubulysin V, and their analogues. J Med Chem. 2009;52:238–240. doi: 10.1021/jm8013579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patterson AW, Peltier HM, Ellman JA. Expedient synthesis of N-methyl tubulysin analogues with high cytotoxicity. J Org Chem. 2008;73:4362–4369. doi: 10.1021/jo800384x. [DOI] [PubMed] [Google Scholar]
- 21.Sasse F, Steinmetz H, Heil J, Hofle G, Reichenbach H. Tubulysins, new cytostatic peptides from myxobacteria acting on microtubuli. Production, isolation, physico-chemical and biological properties. J Antibiot (Tokyo) 2000;53:879–885. doi: 10.7164/antibiotics.53.879. [DOI] [PubMed] [Google Scholar]
- 22.Steinmetz H, et al. Isolation, crystal and solution structure determination, and biosynthesis of tubulysins–powerful inhibitors of tubulin polymerization from myxobacteria. Angew Chem Int Ed Engl. 2004;43:4888–4892. doi: 10.1002/anie.200460147. [DOI] [PubMed] [Google Scholar]
- 23.Kaur G, et al. Biological evaluation of tubulysin A: a potential anticancer and antiangiogenic natural product. Biochem J. 2006;396:235–242. doi: 10.1042/BJ20051735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leamon CP, et al. Enhancing the therapeutic range of a targeted small-molecule tubulysin conjugate for folate receptor-based cancer therapy. Cancer Chemother Pharmacol. 2017;79:1151–1160. doi: 10.1007/s00280-017-3311-z. [DOI] [PubMed] [Google Scholar]
- 25.Vlahov IR, Wang Y, Kleindl PJ, Leamon CP. Design and regioselective synthesis of a new generation of targeted chemotherapeutics. Part II: Folic acid conjugates of tubulysins and their hydrazides. Bioorg Med Chem Lett. 2008;18:4558–4561. doi: 10.1016/j.bmcl.2008.07.041. [DOI] [PubMed] [Google Scholar]
- 26.Leamon CP, Jackman AL. Exploitation of the folate receptor in the management of cancer and inflammatory disease. Vitam Horm. 2008;79:203–233. doi: 10.1016/S0083-6729(08)00407-X. [DOI] [PubMed] [Google Scholar]
- 27.Guertin AD, et al. High Levels of Expression of P-glycoprotein/Multidrug Resistance Protein Result in Resistance to Vintafolide. Mol Cancer Ther. 2016;15:1998–2008. doi: 10.1158/1535-7163.MCT-15-0950. [DOI] [PubMed] [Google Scholar]
- 28.Feng Z, Xu B. Inspiration from the mirror: D-amino acid containing peptides in biomedical approaches. Biomol Concepts. 2016;7:179–187. doi: 10.1515/bmc-2015-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sevenich L, Joyce JA. Pericellular proteolysis in cancer. Genes Dev. 2014;28:2331–2347. doi: 10.1101/gad.250647.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rhoden JJ, Wittrup KD. Dose dependence of intratumoral perivascular distribution of monoclonal antibodies. J Pharm Sci. 2012;101:860–867. doi: 10.1002/jps.22801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Della Pepa C, et al. Ovarian cancer standard of care: are there real alternatives? Chin J Cancer. 2015;34:17–27. doi: 10.5732/cjc.014.10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markman M. Current standards of care for chemotherapy of optimally cytoreduced advanced epithelial ovarian cancer. Gynecol Oncol. 2013;131:241–245. doi: 10.1016/j.ygyno.2013.05.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available from the corresponding author on reasonable request.