

Abstract
Antibiotics are considered to be the first line of treatment for mild to moderately severe Clostridium difficile infection (CDI) in humans. However, antibiotics are also risk factors for CDI as they decrease colonization resistance against C. difficile by altering the gut microbiota and metabolome. Finding compounds that selectively inhibit different stages of the C. difficile life cycle, while sparing the indigenous gut microbiota is important for the development of alternatives to standard antibiotic treatment. 2-aminoimidazole (2-AI) molecules are known to disrupt bacterial protection mechanisms in antibiotic resistant bacteria such as Pseudomonas aeruginosa, Acinetobacter baumannii, and Staphylococcus aureus, but are yet to be evaluated against C. difficile. A comprehensive small molecule-screening pipeline was developed to investigate how novel small molecules affect different stages of the C. difficile life cycle (growth, toxin, and sporulation) in vitro, and a library of commensal bacteria that are associated with colonization resistance against C. difficile. The initial screening tested the efficacy of eleven 2-AI molecules (compound 1 through 11) against C. difficile R20291 compared to a vancomycin (2 μg/ml) control. Molecules were selected for their ability to inhibit C. difficile growth, toxin activity, and sporulation. Further testing included growth inhibition of other C. difficile strains (CD196, M68, CF5, 630, BI9, M120) belonging to distinct PCR ribotypes, and a commensal panel (Bacteroides fragilis, B. thetaiotaomicron, C. scindens, C. hylemonae, Lactobacillus acidophilus, L. gasseri, Escherichia coli, B. longum subsp. infantis). Three molecules compound 1 and 2, and 3 were microbicidal, whereas compounds 4, 7, 9, and 11 inhibited toxin activity without affecting the growth of C. difficile strains and the commensal microbiota. The antimicrobial and anti-toxin effects of 2-AI molecules need to be further characterized for mode of action and validated in a mouse model of CDI.
Keywords: C. difficile, small molecules, 2-aminoimidazole, growth, toxin, sporulation
Introduction
Clostridium difficile is the leading cause of nosocomial and antibiotic associated infectious diarrhea worldwide. C. difficile causes over 450,000 infections and 29,000 deaths annually in the United States (Lessa et al., 2015; McDonald et al., 2018). The incidence, severity, and recurrence rates have increased markedly with the emergence of epidemic strains, and exposure to classic risk factors such as recent antibiotic use, advanced age, and prior hospitalization (Stabler et al., 2006; Ananthakrishnan, 2011; Loo et al., 2011). In addition, C. difficile is now increasingly being linked to community acquired cases of colitis in individuals not exposed to typical risk factors (CDC, 2008; Gupta and Khanna, 2014; Knetsch et al., 2017; McDonald et al., 2018). The changing epidemiology, and the subsequent challenges in the treatment of this infection has prompted the Centers for Disease Control and Prevention (CDC) to classify C. difficile as an urgent threat to public health (CDC, 2013).
Clostridium difficile infection (CDI) is initiated by spores that are highly resistant to various physical and chemical stressors, enabling them to persist in the environment, and play a key role in disease transmission (Baines et al., 2009; Loo et al., 2011; Deakin et al., 2012; Paredes-Sabja et al., 2014). In the gut, the presence of calcium, glycine, and primary bile acids such as taurocholate sensed by the germinant receptor CspC enables C. difficile spores to germinate into metabolically active vegetative cells (Sorg and Sonenshein, 2008; Francis et al., 2013; Kochan et al., 2017). However, the normal indigenous gut microbiota provides colonization resistance against C. difficile (Theriot et al., 2014; Buffie et al., 2015). Antibiotic mediated disruption of the gut microbiota and metabolome leads to a loss of colonization resistance favoring vegetative cell proliferation, and production of toxins that ultimately mediate disease (Antunes et al., 2011; Theriot et al., 2016). During CDI, C. difficile initiates the sporulation pathway forming metabolically dormant spores there by completing the life cycle. The signals that trigger the onset of sporulation are not well understood, however, substantial evidence supports the link between nutrient limitation or other stress factors with sporulation and virulence (Paredes-Sabja et al., 2014; Nawrocki et al., 2016). Current line of treatment for patients with CDI includes the antibiotics vancomycin, metronidazole, or fidaxomicin, which in approximately 20–30% of the patients is ineffective resulting in recurrence (Cohen et al., 2010; Lessa et al., 2015). The intrinsic damage caused by the current line of antibiotics on the gut microbiota, and its failure to restore colonization resistance is the major limiting factor in the treatment and management of CDI (DuPont, 2011). There are occasional reports of C. difficile having high MIC in vitro to the drugs used for its treatment (Baines et al., 2008; Martin et al., 2008; Snydman et al., 2012), however, to date treatment failures have not been linked to antimicrobial resistance. Considering the ease with which C. difficile spread globally in a short time span (He et al., 2013), coupled with the fact that antibiotics are risk factors, there is growing consensus for drug targets that selectively inhibit C. difficile vegetative cells and or virulence factors, while sparing the indigenous gut microbiota. Compounds that inhibit sporulation would also be beneficial as they would aid in the prevention of transmission and relapse.
Identifying potential drug targets against C. difficile is challenging because of the complex etiology, and the impact of risk factors that lead to the disease (Smits et al., 2016). Traditionally, MIC’s and kill assays were used in initial drug screening pipelines, which focuses only on the growth stage of the C. difficile life cycle. Here we present a comprehensive small molecule pipeline, which evaluates the activity of test compounds on three different stages of the C. difficile life cycle (growth kinetics, toxin activity, and sporulation), and how they impact the growth of C. difficile strains from distinct PCR ribotypes. Additionally, the pipeline evaluates how these small molecules alter the growth of other gut commensals that are associated with colonization resistance against C. difficile. The goal of the in vitro screening strategy described here is to screen and select promising compounds that are able to inhibit one or all of the steps in the C. difficile life cycle. Future work defining the mechanism of action of each compound and validating them in a mouse model of CDI is down stream of this pipeline.
2-aminoimidazole (2-AI) molecules have a unique mechanism of action by targeting two-component systems (TCSs), which are signaling pathways that allow bacteria to respond to environmental signals (antibiotics or quorum sensing molecules) there by inhibiting virulence responses such as antibiotic resistance, toxin secretion, and biofilm formation (Thompson et al., 2012). These processes are important in pathogenesis and survival of the pathogen within the host (Stock et al., 2000; Stephenson and Hoch, 2002; Beier and Gross, 2006). 2-AI molecules have been successfully used for antibiotic potentiation and anti-virulence activities against other antibiotic resistant bacteria such as Pseudomonas aeruginosa, Acinetobacter baumannii, and Staphylococcus aureus, but are yet to be evaluated against C. difficile (Rogers et al., 2010; Brackett et al., 2014; Draughn et al., 2017). C. difficile relies on TCS signaling pathways for toxin production that mediate disease, and sporulation which plays a key role in transmission and recurrence (Underwood et al., 2009; Darkoh et al., 2015, 2016). Therefore, we hypothesized that 2-AI molecules would be able to inhibit different stages of C. difficile life cycle namely toxin activity and sporulation. In this study, we started with eleven 2-AI molecules (compound 1 through 11) in our comprehensive screening pipeline, and tested their ability to inhibit C. difficile growth, toxin activity, and sporulation. Molecules that showed potent activity against C. difficile R20291 were further tested against other C. difficile strains (CD196, CF5, M68, BI9, 630, and M120) belonging to distinct PCR ribotypes, and an eight-member commensal library of bacteria associated with colonization resistance against C. difficile. Compound 1, 2, and 3 were found to inhibit growth kinetics, whereas compounds 4, 7, 9, and 11 inhibited toxin activity without affecting the growth of both C. difficile strains and commensals. Next steps include evaluation of each compound for the mechanism of action, and validation in a mouse model of CDI.
Materials and Methods
Bacterial Strains
Clostridium difficile Strains and Growth Conditions
Clostridium difficile strains selected from a range of PCR ribotypes, including epidemic (R20291 and M68), non-epidemic (CD196, CF5, and 630), current (R20291, M68, and BI9), and a genetically divergent strain (M120) were used in these studies. R20291, CD196, CF5, M68, 630, BI-9, and M120 belongs to ribotypes 027, 027, 017, 017, 012, 001, and 078, respectively. The origin and reference details of the isolates can be obtained from Table 2 of our previous publication (Sebaihia et al., 2006; Stabler et al., 2009; He et al., 2010; Thanissery et al., 2017). All assays using C. difficile were started from spore stocks. Spores were prepared and tested for purity as described previously (Perez et al., 2011; Thanissery et al., 2017). Briefly, individual C. difficile strains were grown anaerobically in 2 ml Columbia broth at 37°C for 12 h and further sub-cultured into 40 ml Clospore media in which it was allowed to sporulate for 5–7 days. Spores were harvested by centrifugation and subjected to 3–5 washes with sterile cold water. Spore stocks were stored at 4°C in sterile water until use. The spores were heat treated (65°C for 20 min) to kill vegetative cells, before enumeration and testing for purity. The viable spores were enumerated on brain heart infusion (BHI, Becton, Dickinson and Company, Sparks, MD, United States) media supplemented with 100 mg/L L-cysteine and 0.1% taurocholate. To ensure purity, spores were plated on BHI media plus 100 mg/L L-cysteine, with and without spore germinant (0.1% taurocholate). The purified spores were further examined under phase contrast microscope in which non-germinated intact spores appeared as phase bright bodies. C. difficile cultures for the assays were prepared by inoculating spores on BHI media supplemented with 100 mg/L L-cysteine and 0.1% taurocholate. The plates were incubated anaerobically overnight at 37°C, and isolated colonies from these plates were used to prepare C. difficile inoculum in BHI broth with 100 mg/L L-cysteine.
Table 2.
Minimum inhibitory concentration of 2-aminoimidazole molecules against C. difficile strain R20291 compared to vancomycin.
| Test compound | MIC (μg/ml) |
|---|---|
| Vancomycin | 0.15–0.31 |
| 1 | 2.5–5 |
| 2 | 5 |
| 3 | 5 |
| 4 | >10 |
| 5 | >10 |
| 6 | >10 |
| 7 | >10 |
| 8 | >10 |
| 9 | >10 |
| 10 | >10 |
| 11 | >10 |
Minimum inhibitory concentration (MIC) was determined by broth microdilution as per modified CLSI guidelines for anaerobes. Data represent mean values from triplicate trials.
Commensal Library Strains and Growth Conditions
Eight different non-C. difficile strains that are members of the healthy human gut microbiota belonging to four dominant bacterial phyla including Bacteroidetes, Firmicutes, Proteobacteria, and Actinobacteria were used to determine MIC’s of various 2-AI molecules. Strain details and sources are shown in Table 1. Bacteroides fragilis NCTC 9343, and Bacteroides thetaiotaomicron VPI-5482 were obtained from Eric Martens (University of Michigan, United States). Clostridium hylemonae TN-271 was obtained from Joson M. Ridlon (University of Illinois Urbana-Champaign, United States). Lactobacillus acidophilus ATCC 700396, Lactobacillus gasseri ATCC 33323, and Bifidobacterium longum subsp. infantis DSM 20090 were obtained from Rodolphe Barrangou (North Carolina State University, United States). Clostridium scindens (ATCC 35704, Cat # 35704) and Escherichia coli (Cat # BAA 2649) were purchased from American Type Culture Collection. All strains were maintained as 15% glycerol stock in -80°C until use. Working stocks of Bacteroides species were prepared in tryptone-yeast extract- glucose (TYG) media (Martens et al., 2008). C. scindens, C. hylemonae, and E. coli were grown in BHI plus 100 mg/L L-cysteine (Barefoot and Klaenhammer, 1983; Ridlon et al., 2010). Lactobacillus acidophilus, and L. gasseri were grown in de Man, Rogosa, and Sharpe broth (MRS, Becton, Dickinson and Company, Sparks, MD, United States), (Barefoot and Klaenhammer, 1983). Bifidobacterium longum subsp. infantis were grown in MRS supplemented with 500 mg/L L-cysteine (Ventura et al., 2003).
Table 1.
Commensal microbiota library.
| Phyla | Bacteria | Strain∗ | Description | Nucleotide accession no. |
|---|---|---|---|---|
| (complete genome)/Reference | ||||
| Bacteroidetes | Bacteroides fragilis | NCTC 9343 | Type strain, appendix abscess | GenBank, CR626927 |
| Bacteroidetes | Bacteroides thetaiotaomicron | VPI-5482 | Type strain, human feces | Xu et al., 2003 |
| Firmicutes | Lactobacillus acidophilus | ATCC 700396/NCFM | Infant feces | Altermann et al., 2005 |
| Firmicutes | Lactobacillus gasseri | ATCC 33323 | Type strain | GenBank, CP000413 |
| Firmicutes | Clostridium scindens | ATCC 35704 | Type strain, human feces | GenBank, ABFY02000000 |
| Firmicutes | Clostridium hylemonae | TN-271 | Type strain, human feces | GenBank, AB023972∗∗ |
| Proteobacteria | Escherichia coli | ATCC BAA 2649 | Not type strain | |
| Actinobacteria | Bifidobacterium longum subsp. infantis | DSM 20090 | Intestine of infants | Mattarelli et al., 2008 |
∗ATCC, American Type Culture Collection; NCTC, National Collection of Type Cultures; DSM, Leibniz-Institute DSMZ-German Collection of Microorganisms and Cell Cultures.
∗∗16S rRNA, partial sequence.
Small Molecule Preparation
2-AI molecules compound 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, and 11 (A kind gift of Agile Sciences Inc., Raleigh, NC, United States, Figure 1) were provided as a 400 μg/mL stock in 10% dimethyl sulfoxide (DMSO, Sigma-Aldrich Co., St. Louis, MO, United States) and were stored at -20°C until use. For all assays, the test compounds were used at a maximum final concentration of 10 μg/mL to ensure efficacy when compared to vancomycin that is currently used for the treatment of CDI (Cohen et al., 2010). Vancomycin (Sigma-Aldrich, St. Louis, MO, United States) was used as a positive control in all assays. Stock solution of vancomycin (8 mg/mL) was diluted in ultrapure water, filter sterilized and stored at 4°C for a week.
FIGURE 1.
2-aminoimidazole chemical structures. The chemical structures of all eleven 2-AI molecules are illustrated in this figure.
Microbroth Dilution for Minimum Inhibitory Concentration Assay
Minimum inhibitory concentration was determined using a modified Clinical and Laboratory Standard Institute (CLSI) broth microdilution method. Test medium used for all Clostridia were BHI with 100 mg/L L-cysteine. Bacteroides were grown in Yeast extract casitone fatty acid medium. Lactobacillus sp. were grown in MRS. The same medium was supplemented with 500 mg/L L-cysteine for growing B. infantis. The inoculum was prepared by the direct colony suspension method. All cell concentrations were adjusted to ∼5 × 105 CFU/mL. An anaerobic environment was maintained at all times using an anaerobic chamber (Coy Industries). An incubation temperature of 37°C was used for all strains. The plates were prepared fresh by making 2-AI molecules or vancomycin dilution stocks in the test media, and adding 90 μL to each well such that the final concentration of the test compounds after the addition of cells (10 μL) ranged from 0.08 to 10 μg/mL. Positive controls included inoculated cells only (in test media to check for media adequacy), and solvent (0.25% DMSO). Uninoculated test media for each strain was used as a negative control to check for sterility. The assay plates were then sealed using a sterile polyester film (VWR, cat # 89134-432) before placing the lid to prevent the panel from dehydrating during incubation. C. difficile, B. fragilis, B. thetaiotaomicron, L. acidophilus, L. gasseri, and E. coli were incubated for 24 h, whereas C. scindens, and C. hylemonae, were allowed to grow for 48 h. MICs were defined as the lowest concentration at which there was no visible growth. The end point optical density at 600 nm (OD600) of the plates was additionally recorded to measure turbidity.
Growth Kinetics Inhibition Assay
The growth inhibition studies of C. difficile were done in a 96-well microtiter plate using previously published methods (Thanissery et al., 2017). All C. difficile strains were cultured overnight at 37°C in pre-reduced BHI plus 100 mg/L L-cysteine broth in an anaerobic chamber. Overnight C. difficile cultures were sub-cultured 1:10 into same media, and allowed to grow for 3 h anaerobically at 37°C. The culture was then diluted in fresh BHI so that the starting OD600 was 0.01. The cell suspension was added in triplicate to a 96-well plate at a final volume of 0.2 ml with the addition of test compound (final concentration: 10 μg/mL), solvent (0.25% DMSO) or vancomycin (final concentration: 2 μg/mL). Each plate contained control wells (without test compounds) and blank wells (without cells). The plates were sealed to ensure anaerobic conditions and passed outside the chamber to measure optical density 600 nm (OD600). The optical density was monitored every 30 min for 10 h, shaking the plate for 90 s before each reading, in a Tecan plate reader. A test plate containing 2-AI or vancomycin in media was run before the assay to measure the optical density and ensure the stability of the compounds over the incubation period. After 24 h, the plates were removed from the plate reader and stored in -80°C until use for measuring toxin activity from the culture supernatants.
Toxin Activity Inhibition Assay
Toxin activity was measured by a Vero cell cytotoxicity assay (Winston et al., 2016; Thanissery et al., 2017). Vero cells were grown and maintained in DMEM media (Gibco Laboratories, 11965-092) with 10% fetal bovine serum (Gibco Laboratories, 16140-071) and 1% Penicillin streptomycin solution (Gibco Laboratories, 15070-063). Cells were incubated with 0.25% trypsin (Gibco Laboratories, 25200-056), washed with 1X DMEM media, and harvested by centrifugation 1,000 RPM for 5 min. Cells were plated at 1 × 104 cells per well in a 96-well flat bottom microtiter plate (Corning, 3596) and incubated overnight at 37°C/5% CO2. Growth inhibition kinetics assay plates were defrosted on ice and then centrifuged at 1,750 RPM for 5 min to pellet vegetative C. difficile. Culture supernatants were collected from each well and serially diluted by 10-fold to a maximum of 10-6 using 1X PBS. Sample dilutions were incubated 1:1 with PBS (for all dilutions) or antitoxin (performed for 10-1 and 10-4 dilutions only, TechLabs, T5000) for 40 min at room temperature. Following incubation, these admixtures were added to the Vero cells. After an overnight incubation at 37°C/5% CO2, plates were viewed under 200× magnification for Vero cell rounding. The cytotoxic titer was defined as the reciprocal of the highest dilution that produced rounding in 80% of Vero cells for each sample. Vero cells treated with purified C. difficile toxins (A and B) and antitoxin (List Biological Labs, 152C and 155C; TechLabs, T5000) were used as controls. A test cytotoxicity assay was run prior to assays to ensure that the 2-AI molecules did not affect the cytoskeleton of Vero cells at the tested concentrations.
Kill Kinetics Assay
Measurement of OD600 Using Plate Reader
Kill kinetics of C. difficile were analyzed on a 96-well plate using a modified growth inhibition assay protocol. Briefly, overnight C. difficile cultures were back-diluted 1:25 into pre-reduced BHI plus 100 mg/L L-cysteine broth and allowed to grow until it reaches mid log (OD600 of 0.45–0.50). The cells were added in triplicates to a 96-well plate at the same volume and concentrations of test compound, solvent, or vancomycin as used in the growth kinetics inhibition assay. Each plate also contained control wells (without test compounds) and blank wells (without cells). The optical density was monitored every 30 min for 12 h, shaking the plate for 90 s before each reading, in a Tecan plate reader.
C. difficile Bacterial Enumeration
Plates were prepared as described here previously for measurement of OD600 using a plate reader. Six hours later, 25 μL aliquots were removed from each treatment, serially diluted 10-fold in phosphate buffered saline (PBS), and plated on BHI plus 100 mg/L L-cysteine and 0.1% taurocholate using a track dilution method (Jett et al., 1997). This method involved plating 10 μL of six dilutions on separate tracks of a single square plate (Genesee Scientific, Cat # 26-275). The dilution plate was then heat treated at 65°C for 20 min to kill all vegetative cells. Following heat treatment, the cells were plated on BHI plus 100 mg/L L-cysteine and 0.1% taurocholate. All plates were incubated at 37°C for 24 h anaerobically. Plates were counted the next day to enumerate total vegetative cells plus spores in the unheated samples, and total spores in the heat-treated samples.
Sporulation Inhibition Assay
The sporulation assay is modified from a method previously described as spore inducing and quantification using heat resistance (Shen et al., 2016). Briefly, R20291 spores were streaked on BHI plates containing 100 mg/L L-cysteine plus 0.1% taurocholate and incubated anaerobically for 24 h. The colonies were sub-cultured into 2 mL BHI plus 100 mg/L L-cysteine and were allowed to grow for 4 to 5 h. The turbid culture was centrifuged for 5 min, and the pellet was resuspended in 70:30 broth [per liter contained 63 g Bacto Peptone, 3.5 g Protease Peptone, 0.7 g NH 4 SO 4, 1.6 g Tris Base, 11.1 g BHI Broth, 1.5 g Yeast Extract, supplemented with 3 mL 10% (w/v) Cysteine] to an OD600 of ∼0.5. Resuspended cultures (195 μL) with or without test compounds (final concentration: 10 μg/mL), vancomycin (2 μg/mL), or solvent (0.25% DMSO) were incubated at 37°C for 24 h anaerobically. The samples after incubation (20 μL) were serially diluted 10-fold, and 4 μL were plated on BHI plates containing 100 mg/L L-cysteine plus 0.1% taurocholate. The dilution plate was passed out of the chamber for heat treatment at 65°C for 20 min. Four μL from each dilution was plated on BHI plates containing 100 mg/L L-cysteine plus 0.1% taurocholate. All plates were incubated anaerobically at 37°C for 24 h. The number of colony forming units (CFUs) were counted on the lowest dilution in which colonies were visible to determine the CFU/mL of total vegetative cells and spores from the unheated samples and spores only from the heat-treated samples.
Statistical Analysis
Statistical tests were performed using Prism version 7.0a for Mac OS X (GraphPad Software, La Jolla, CA, United States). Significance between treatments and solvent control for toxin activity assay (Figure 3B), bacterial enumeration for kill kinetics (Figure 4), and sporulation assay (Figure 5) were calculated by Student’s parametric t-test with Welch’s correction. Statistical significance was set at a p-value of <0.05 for all analyses (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). All assays were done in triplicate.
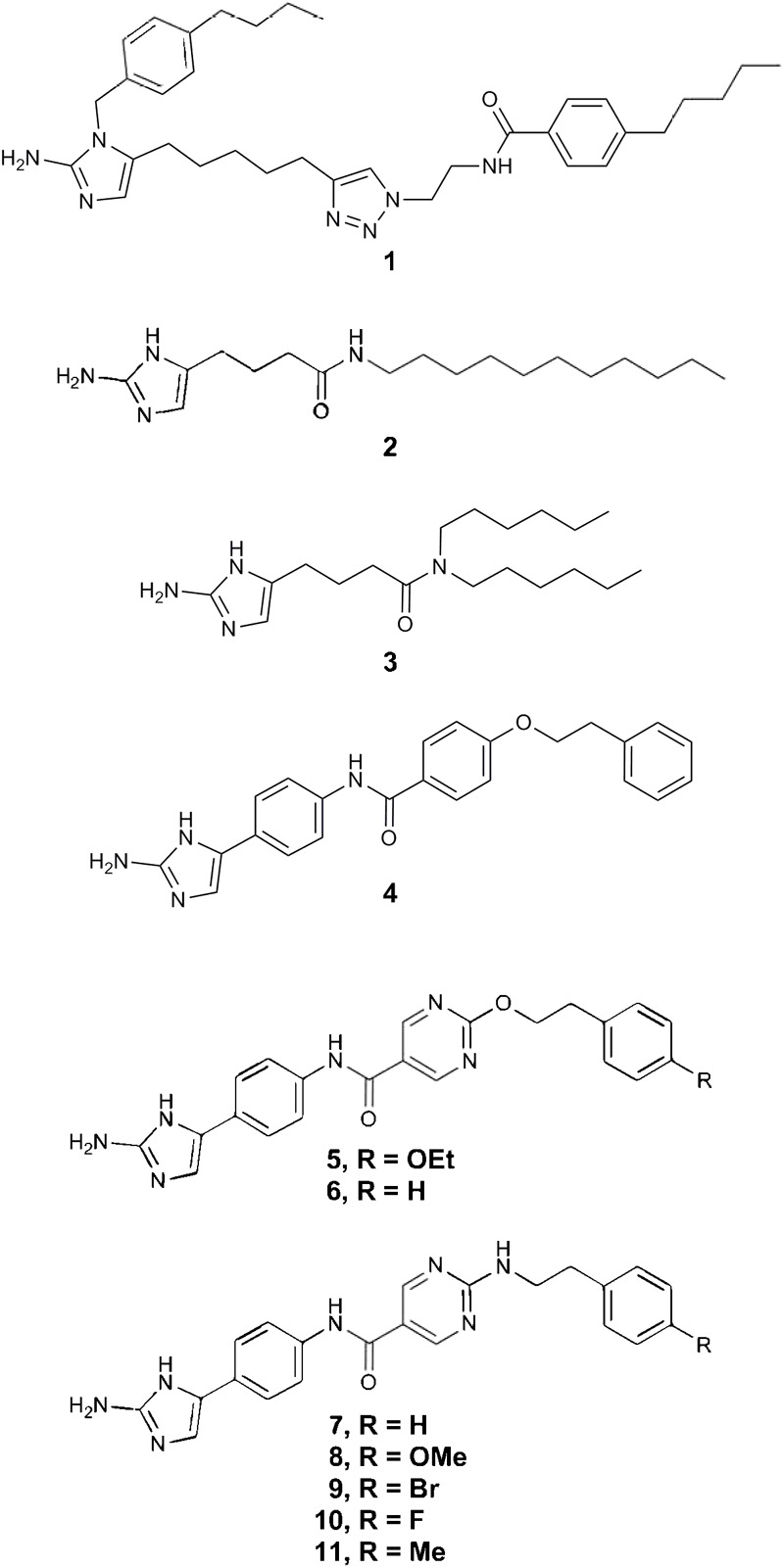
FIGURE 3.
2-aminoimidazole molecules inhibit growth and toxin activity of C. difficile. (A) Inhibition of C. difficile R20291 growth (OD600) in BHI media supplemented with small molecules (Compound 1 through 11) at a concentration of 10 μg/ml, solvent 0.25% DMSO (DMSO), or 2 μg/ml Vancomycin (Vanco). (B) Culture supernatants after 24 h growth inhibition assays were used for Vero cell cytotoxicity assays to measure toxin activity. Toxin titer is expressed as log10 reciprocal dilution toxin per 100 μl of C. difficile culture supernatant. Data presented represents mean ± SEM of triplicate experiments. In (B) statistical significance between positive control (solvent) and treatment groups was determined by Student’s parametric t-test with Welch’s correction (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
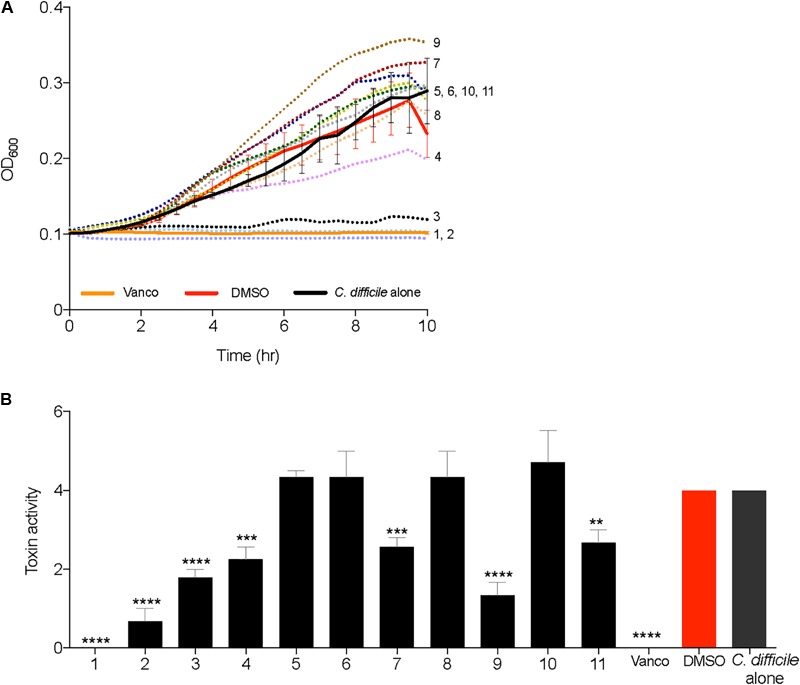
FIGURE 4.
2-aminoimidazole molecules affect C. difficile viability. (A) Killing of C. difficile R20291 growth (OD600) in BHI media supplemented with small molecules at mid log growth phase (Compounds 1 through 11). (B) Total C. difficile R20291 vegetative cells and spores, or spores alone 6 h post exposure to small molecules in (A) (Compounds 1, 2, and 3) at a concentration of 10 μg/ml when compared to solvent 0.25% DMSO (DMSO, positive control), or 2 μg/ml vancomycin (Vanco, negative control). Data presented represent mean ± SEM of triplicate experiments. Statistical significance between positive control (solvent) and treatment groups was determined by Student’s parametric t-test with Welch’s correction (∗p < 0.05, ∗∗p < 0.01).
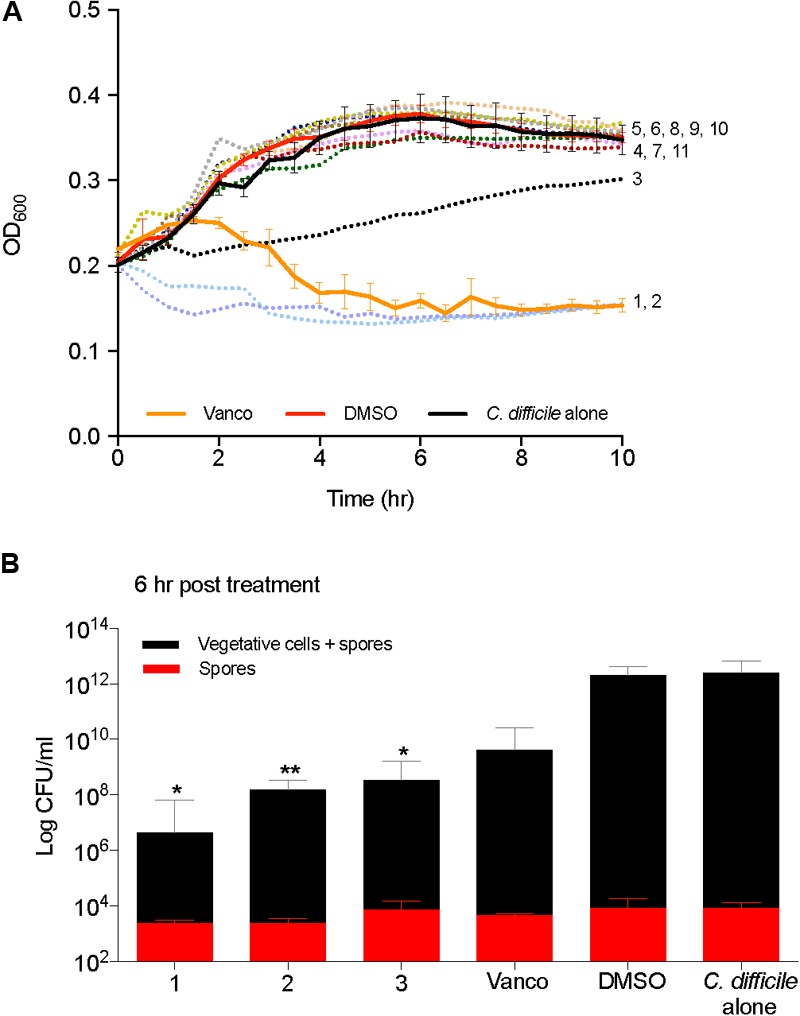
FIGURE 5.
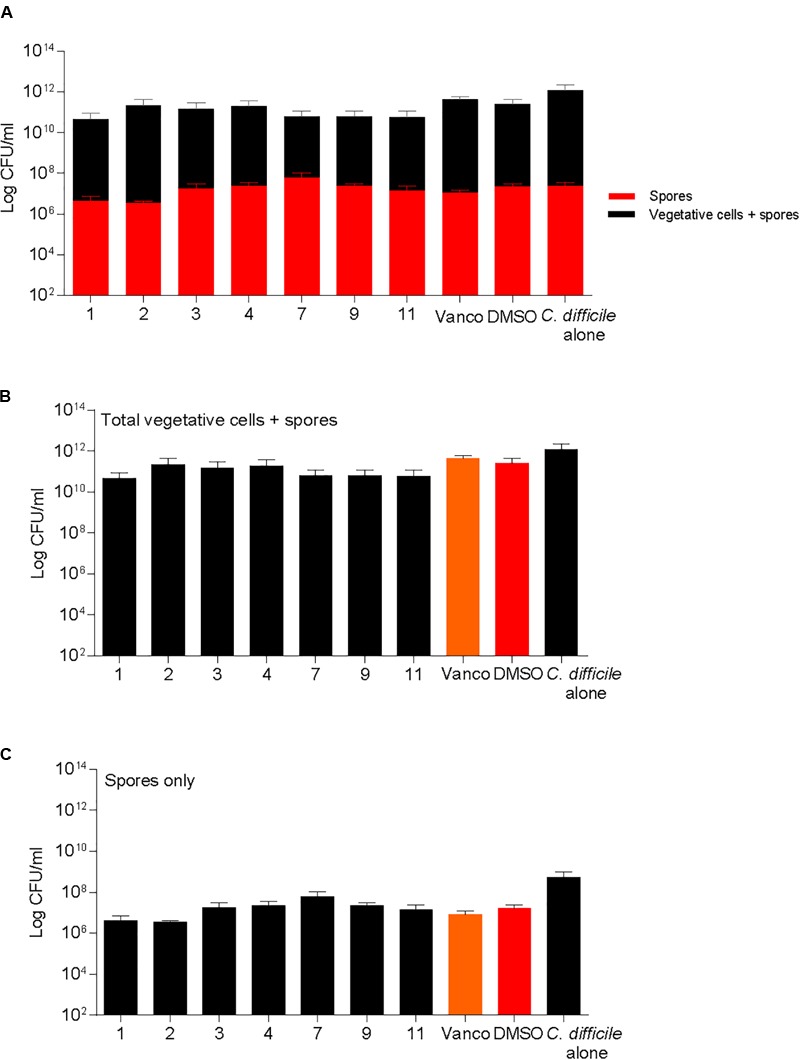
2-aminoimidazole molecules do not alter C. difficile sporulation. Sporulation of C. difficile on 70:30 agar plates after supplementation with 2-AI molecules (Compounds 1, 2, 3, 4, 7, 9, and 11) at a concentration of 10 μg/ml when compared to solvent 0.25% DMSO (DMSO, positive control), or 2 μg/ml vancomycin (Vanco, negative control) after 24 h. Data represents (A) Total vegetative cells and spores, and spores only, (B) Total vegetative cells and spores, (C) Spores only at 24 h post treatment.
Results
Development of a Screening Pipeline to Test Small Molecule Activity Against Different Stages of the C. difficile Life Cycle in Vitro
Figure 2 is an overview of the small molecule-screening pipeline that was developed and implemented in this study. The gray boxes represent the different assays that were used to interrogate how small molecules were able to alter different stages of the C. difficile life cycle including growth, toxin, and sporulation. We also evaluated their activity against other C. difficile strains (CD196, CF5, M68, BI9, 630, M120), and commensals from the gut microbiota (B. fragilis, B. thetaiotaomicron, L. acidophilus, L. gasseri, C. scindens, C. hylemonae, E. coli, and B. longum subsp. infantis) (Table 1). All small molecules begin at the first step, screening on a 96-well plate to determine MICs using a microbroth dilution technique. A MIC of 10 μg/ml was considered an initial cut-off for activity when compared to the reference drug vancomycin, which is currently used to treat patients with CDI. This dose was selected because it was hard to sustain concentrations above 10 μg/ml in animal studies based on previous studies with structurally similar compounds. All molecules along with vancomycin (2 μg/ml) and the solvent (0.25% DMSO) were moved down the pipeline, and assayed for growth kinetics inhibition, toxin activity inhibition, and kill kinetics. Molecules that either inhibited growth and or toxin activity were advanced to the next step in the pipeline. A sporulation induction assay was used to determine if the small molecules were able to alter sporulation. All other molecules were moved to the next step in the pipeline where they were screened for activity against other clinical C. difficile strains, and a commensal microbiota library. Molecules that show promising antimicrobial or anti-toxin activity sparing the commensals in this pipeline will be further evaluated in vivo in a mouse model of CDI.
FIGURE 2.
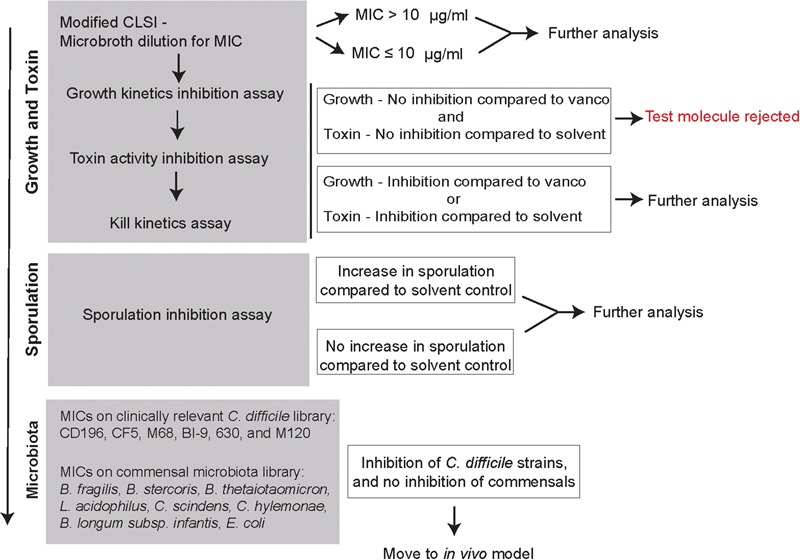
In vitro screening pipeline to evaluate 2-aminoimidazole molecules as potential non-antibiotic therapeutics for C. difficile infection. Flowchart is designed as an in vitro screening pipeline to evaluate small molecules against C. difficile. The gray boxes represent the different protocols used to determine how the small molecules affect different stages of C. difficile life cycle (growth, toxin, and sporulation) and other members of the gut microbiota. Based on the results, at each box a decision is made whether to advance molecules for further screening with the goal of identifying suitable molecules to be advanced to mouse model testing.
2-Aminoimidazole Molecules Alter C. difficile R20291 Growth and Toxin Activity
The MICs for eleven 2-AI molecules with C. difficile are shown in the Table 2. Compounds 1, 2, and 3 were the most active against C. difficile with MICs ranging from 2.5 to 5 μg/ml. C. difficile was not susceptible to all other 2-AI molecules. The control vancomycin had a MIC of 0.15–0.31 μg/ml, and the solvent control (0.25% DMSO) did not inhibit C. difficile.
All 2-AI molecules were moved down the pipeline and tested in a C. difficile growth kinetics inhibition assay at a concentration of 10 μg/ml, along with vancomycin (2 μg/ml), and the solvent (0.25% DMSO). Supplementation of compounds 1, 2, and 3 inhibited the growth of C. difficile and was very similar to the vancomycin control (Figure 3A). There was no change in C. difficile growth kinetics in the presence of all other 2-AI molecules. Toxin activity was measured from culture supernatants of C. difficile supplemented with 2-AI molecules in Figure 3A. Diminished growth correlated with low toxin activity with the addition of compounds 1, 2, and 3 (Figure 3B). Interestingly, growth was unaffected by compounds 4, 7, 9, and 11 yet toxin activity was significantly reduced when compared to the solvent control. The addition of the solvent DMSO to media did not alter C. difficile growth or toxin activity. The cytotoxic activity was neutralized at all dilutions containing the sample and antitoxin confirming that the cell rounding was from C. difficile toxin. All molecules were advanced to the kill kinetics assay, the next step in the pipeline.
Kill kinetics of C. difficile were evaluated by measuring the optical density (OD600) after the addition of 2-AI molecules to cells in mid log growth phase (Figure 4A). Supplementation of vancomycin (2 μg/ml) and solvent (0.25% DMSO) were used as controls. Compounds 1, 2, and 3 (10 μg/ml) altered growth, which was further confirmed by enumerating total colony forming units (CFUs) of vegetative cells and spores at the 6 h time point in Figure 4B. Addition of 2-AI molecules resulted in a significant log reduction in the total number of vegetative cells and spores for compound 1 (5.5 ± 0.57 log), compound 2 (4.0 ± 0.26 log), and compound 3 (3.6 ± 0.27 log) compared to the solvent control. However, no differences were seen in spores. The solvent DMSO did not affect C. difficile kill kinetics like the vancomycin control. 2-AI molecules compound 5, 6, 8, and 10, that did not inhibit growth and toxin activity were rejected at this point in the pipeline. Compounds 1, 2, 3, 4, 7, 9, and 11 were advanced to the next step in the pipeline.
2-Aminoimidazole Molecules Do Not Alter C. difficile R20291 Sporulation
Differences in sporulation were determined by inducing spore formation and quantification of heat resistant spores. Sporulation was unaffected when supplemented with DMSO or compounds 1, 2, 3, 4, 7, 9, and 11 (Figure 5). All molecules tested for sporulation were advanced to next step of screening in the pipeline. Spores were also enumerated in the kill assays described previously and no differences were noticed in the spores recovered in the BHI media both at 6 and 24 h post treatment (Supplementary Figure S1).
2-Aminoimidazole Molecules Affect Other C. difficile Strains Sparing Commensal Members of the Gut Microbiota
Compounds 1, 2, 3, 4, 7, 9, and 11 were screened for MICs against other C. difficile strains and a commensal microbiota library. Other C. difficile strains (CD196, M68, CF5, 630, BI9, and M120) were inhibited by compounds 1, 2, and 3 at a MIC of 2.5–5 μg/ml and 5–10 μg/ml, respectively (Table 3). Vancomycin was inhibitory to all strains at 0.31 μg/ml, except BI9, which was inhibited at 0.16 μg/ml. C. difficile strains were not susceptible to all other 2-AI molecules (compounds 4, 7, 9, and 11) at a concentration of 10 μg/ml.
Table 3.
Minimum inhibitory concentration of 2-aminoimidazole molecules against other C. difficile strains compared to vancomycin.
| C. difficile strain | MIC (μg/mL) |
|||
|---|---|---|---|---|
| Compound 1 | Compound 2 | Compound 3 | Vancomycin | |
| CD196 | 5 | 5 | 5 | 0.31 |
| M68 | 5 | 10 | 10 | 0.31 |
| CF5 | 5 | 5 | 10 | 0.31 |
| 630 | 5 | 5 | 5 | 0.31 |
| BI9 | 2.5–5 | 5 | 5 | 0.16 |
| M120 | 5 | 10 | 10 | 0.31 |
Minimum inhibitory concentration (MIC) was determined by broth microdilution as per modified CLSI guidelines for anaerobes. Data represent mean values from triplicate trials.
Commensal microbes that are associated with a healthy gut microbiota and colonization resistance against C. difficile: B. fragilis, B. thetaiotaomicron, C. scindens, C. hylemonae were also susceptible to compound 1 at a MIC of 5–10 μg/ml (Table 4). In contrast, all other strains (L. acidophilus, L. gasseri, E. coli, B. longum subsp. infantis) remained resistant to compound 1 with a MIC greater than 10 μg/ml. Compound 2 was inhibitory to B. thetaiotaomicron, C. scindens, and C. hylemonae at 10 μg/ml. Interestingly, compound 3, which inhibited C. difficile growth, did not have any effect on the commensals at the tested concentration The commensal panel was resistant to compounds 4, 7, 9, and 11 at a concentration of 10 μg/ml, which also did not inhibit C. difficile strains.
Table 4.
Minimum inhibitory concentration of 2-aminoimidazole molecules on commensal microbiota library.
| Commensal strain | MIC (μg/mL) |
|||
|---|---|---|---|---|
| Compound 1 | Compound 2 | Compound 3 | Vancomycin | |
| B. fragilis | 10 | >10 | >10 | 2.5 |
| B. thetaiotaomicron | 5 | 10 | >10 | 1.25 |
| L. acidophilus | >10 | >10 | >10 | 0.31 |
| L. gasseri | >10 | >10 | >10 | 0.16 |
| C. scindens | 5–10 | 10 | >10 | 0.31 |
| C. hylemonae | 10 | 10 | >10 | 1.25–0.31 |
| E. coli | >10 | >10 | >10 | >10 |
| B. longum subsp. infantis | >10 | >10 | >10 | 0.63 |
Minimum inhibitory concentration (MIC) was determined by broth microdilution as per modified CLSI guidelines for anaerobes. Data represent mean values from triplicate trials.
Discussion
In this study we developed and implemented a small molecule-screening pipeline to screen and select promising compounds that inhibited one or multiple steps in the C. difficile life cycle without altering the growth of a panel of gut commensals associated with colonization resistance. 2-AI molecules that have been successfully used to enhance antibiotic activity and mitigate virulence responses against other insidious pathogens were the first compounds screened through our pipeline. We evaluated eleven 2-AI molecules (compound 1 through 11) for their ability to alter C. difficile growth, toxin, and sporulation, while sparing other members of the gut microbiota. Compounds 1, 2, and 3 were microbicidal and were able to inhibit and kill C. difficile R20291 growth. The antimicrobial activity of compounds 1, 2, and 3 correlated with lower toxin activity. However, there was no difference in the number of spores recovered. Interestingly, compounds 4, 7, 9, and 11 were anti-virulent as they inhibited toxin activity without impacting the growth of C. difficile strains and commensals.
Minimum inhibitory concentrations of all molecules were first evaluated with C. difficile R20291, and then subsequently moved down the pipeline to evaluate how they affected growth kinetics, and virulence factors such as toxin and sporulation. Treatment with compound 1 (5.5 ± 0.57 log), compound 2 (4.0 ± 0.26 log), and compound 3 (3.6 ± 0.27 log) resulted in a higher log reduction of C. difficile vegetative cells and spores then the vancomycin control (2.7 ± 0.50). Based on MIC’s, vancomycin (0.15–0.31 μg/ml) was more potent against C. difficile R20291 compared to compounds 1, 2, and 3 (2.5–5 μg/ml). Similar sensitivity to vancomycin for C. difficile R20291 isolates has been reported (Barbut et al., 2007; Đapa et al., 2013; Brock, 2015). However, different antimicrobial sensitivity testing methods were used making it difficult to compare between studies. Since vancomycin is bacteriostatic to logarithmic phase cultures, it was not surprising that there was a lower log reduction at 2 μg/ml (Levett, 1991; Alam et al., 2015). Several antimicrobials with a range of modes of action are under clinical evaluation for CDI now (Kociolek and Gerding, 2016). Surotomycin is a novel lipopeptide that has antibacterial activity by disrupting the bacterial cell membrane (Mascio et al., 2012). It has potent activity against C. difficile and reduced activity against commensal bacteria (Citron et al., 2012). However, it was not associated with lower recurrence rates in phase III clinical trials (Boix et al., 2017). Cadazolid is another novel oxazolidinone compound which inhibits protein synthesis (Locher et al., 2014a). This compound reduces toxin production and sporulation in vitro in the absence of bacterial killing (Locher et al., 2014b). Ridinilazole a DNA synthesis inhibitor is a novel narrow spectrum antibiotic and has shown promising phase II results (Basseres et al., 2016; Steinebrunner et al., 2018). The mode of action for compounds 1, 2, and 3 screened in our study is unknown, and more studies are needed to explore bactericidal targets including cell wall biosynthesis, DNA replication, and protein synthesis.
Targeting virulence is a therapeutic approach that provides promising opportunities to inhibit pathogenesis in vivo without affecting bacterial growth (Cegelski et al., 2008). Mitigating virulence shifts the advantage to the host since the immune response remains unimpaired by the bacteria. Additionally, the gut microbiota that provide colonization resistance against C. difficile are unaltered, reducing recurrence. Common anti-toxin agents pursued as potential therapeutics for various infectious diseases include inhibitors of toxin transcription factors (Hung et al., 2005), toxin trafficking molecules (Saenz et al., 2007), and the use of toxin neutralizing antibodies (Arnon et al., 2006). Quorum sensing molecules (Hentzer et al., 2003) and bacterial two-component response systems that are central to bacterial virulence are often targeted for anti-virulence effect as well. In our study, compounds 4, 7, 9, and 11 did not affect growth, yet toxin activity decreased significantly compared to the solvent control. This is in line with the mechanism of action of 2-AI molecules that are able to target response regulator protein of bacterial TCS, thereby inhibiting virulence determinants such as antibiotic resistance, toxin secretion, and biofilm formation in other antibiotic resistant bacteria including P. aeruginosa, A. baumannii, and S. aureus (Rogers et al., 2010; Brackett et al., 2014; Draughn et al., 2017). In C. difficile, TCS is a part of the quorum sensing system called accessory gene regulator (agr) system that regulates toxin synthesis (Darkoh et al., 2015). The components of the agr system in strain R20291 includes agrB1 and agrD1 within the agr1 loci, that are responsible for producing the quorum signaling autoinducer peptide, and agrB2D2 and agrC2A2 within agr2 loci that are quorum signal-generation and response genes, respectively (Darkoh et al., 2016). C. difficile also has a Spo0A histidine kinase TCS system that is known to play a key role in both sporulation and toxin production (Underwood et al., 2009). However, the molecular mechanisms that lead to the control of toxin production by Spo0A are found to be strain dependent, and are not well characterized (Darkoh et al., 2015; Martin-Verstraete et al., 2016). Inhibition of any components in the accessory gene regulator pathway, and Spo0A histidine kinase TCS system could result in significant control of the toxin.
Targeting the toxin protein itself rather than bacterial growth to treat CDI is gaining momentum especially after tcdA and tcdB knockouts of toxigenic C. difficile proved to be avirulent in a hamster model (Kuehne et al., 2014). Both toxins are composed of four large domains: putative receptor binding domain, a transmembrane domain, a CPD, and a glucosyltransferase domain, whose conformational changes and the subsequent events leads to cytopathic and cytotoxic effect of the toxins (Pruitt and Lacy, 2012). These domains are potential drug targets for toxin inactivation. Bezlotoxumab an injectable human monoclonal antibody was FDA approved recently for the prevention of recurrent CDI. The antibodies bind to the receptor binding domain of toxin B when given systemically, thereby mitigating the in vivo effects of the toxin (Yang et al., 2015; Wilcox et al., 2017). A viable alternate strategy to target toxins is by using small molecules that could be delivered directly to the site of infection rather than systemic administration. Indeed, a promising bioactive compound, ebselen, which is currently under clinical investigation for unrelated indication was found to inhibit CPD activity in vitro. Ebselen was also validated in a mouse model to bind toxin B, and thereby prevent C. difficile induced clinical pathology (Bender et al., 2015). In another study using a chemical genetics strategy, several small molecules were screened to target potential domains and pathways. This study laid the foundation for identifying first-generation inhibitors of toxin B that mediate CDI (Tam et al., 2015). Antitoxin molecules represent a novel paradigm and could provide the industry with new opportunities in the treatment and management of CDI.
Since 2-AI molecules could potentially affect Spo0A histidine kinase TCS system that controls sporulation, we attempted to measure the inhibitory activity of 2-AI molecules on sporulation induction of mid log C. difficile cells. No differences were observed in the number of spores recovered with or without the addition of 2-AI molecules at a concentration of 10 μg/mL. Compounds 1, 2, and 3 were growth inhibitory at this concentration, however, it is crucial to evaluate if the 2-AI molecules induce stress on the cells resulting in increased spore formation. Fidaxomicin is the only drug currently available that inhibits sporulation when sub inhibitory concentrations are added to early stationary phase cells (Babakhani et al., 2012). Anti-sporulation properties would provide greater effectiveness to control transmission and reduce recurrences of CDI.
Since the gut microbiota plays a key role in providing colonization resistance against C. difficile (Theriot et al., 2014; Buffie et al., 2015), we tested the small molecules against eight different bacterial strains that are members of the healthy human gut microbiota, and six other C. difficile strains from distinct PCR ribotypes. We included members from four of the five dominant phyla of the gut microbiota including Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria (Tremaroli and Backhed, 2012). Firmicutes make up 50–70% of the colonic bacterial community (Frank and Pace, 2008). Members of Firmicutes including L. acidophilus, L. gasseri, C. scindens, C. hylemonae, C. heranonis were added to the panel. B. fragilis and B. sterocis belonging to the phyla Bacteroidetes were added to the panel as they are designated as key stone species in the human gut microbiome (Fisher and Mehta, 2014). Another member of Bacteroidetes added was B. thetaiotaomicron. This commensal is found to antagonize intestinal pathogens through a range of mechanisms (de Sablet et al., 2009; Ferreira et al., 2011; Kamada et al., 2012). B. infantis belonging to the phyla Actinobacteria known to synthesize compounds necessary for functional maturation of enterocytes and host immunity, were also added to the panel (Round and Mazmanian, 2009; Guinane et al., 2011). Compounds 4, 7, 9, and 11 used at a concentration that inhibited C. difficile toxin activity had no effect on the commensal panel. Compounds 1 and 2 were microbicidal to C. difficile, but remained resistant to most of the commensal panel except for Bacteroides and the commensal Clostridia. Compound 3 had narrow spectrum activity against C. difficile, and did not affect growth of the commensal microbiota at a concentration of 10 μg/ml.
Screening novel small molecules against C. difficile rely on MIC assays or growth inhibition assays by measuring optical density in a plate reader. This is not always an accurate readout as exposure of C. difficile to stressors is able to increase sporulation (Wilcox and Fawley, 2000; Fawley et al., 2007). A drop in optical density overtime in a growth inhibition assay does not distinguish between vegetative cell lysis and spore formation. It is also important to evaluate viable counts of vegetative cells and spores to confirm true growth inhibition. In this study, growth was evaluated in multiple assays including a growth kinetics inhibition assay (microbroth dilution technique and OD600 measurement on cells in early log phase), and a kill kinetics assay (OD600 measurement and bacterial enumeration of cells in mid log phase). Another strength of our pipeline is that it takes into consideration other C. difficile strains from distinct ribotypes to ensure there are no differences in susceptibility across strains. Additionally, understanding how these compounds affect other gut commensal bacteria is important for the restoration of colonization resistance in vivo. The pipeline not only allows for quick screening of antimicrobials, but also for anti-virulence agents. The test molecule concentrations selected for screening can be modified based on each molecule.
There are many strengths to using this small-molecule screening pipeline, however, there are some limitations. We did not evaluate the first stage of the C. difficile life cycle, spore germination. However, addition of this assay to the pipeline in the future could be valuable. Although the Vero cell cytotoxicity assay we use in this study is the gold standard for evaluating toxin activity it is semi-quantitative, and other assays such as qRT-PCR and immunoblotting are more quantitative. Another limitation of our toxin assay is that the BHI media used for culturing was supplemented with cysteine, which can reduce toxin expression (Karlsson et al., 2000; Dubois et al., 2016). However, controls using the same media were used for comparison which ensures equal impact across all treatments. The sporulation assay also has limitations as it evaluates sporulation induction when test molecules are added ≥MICs and incubated for 24 h. Therefore, the results of the sporulation assay were not used as a criterion to move the test molecules to the next level of screening. Further testing evaluating sporulation inhibition could be done by adding sub-inhibitory concentrations of test molecules to C. difficile cultures and allowing an extended period of incubation before spore enumeration.
Finally, future studies are needed to characterize the anti-toxin activity and understand the mode of action for these 2-AI compounds. The next step after completing the pipeline is to test the therapeutic properties of 2-AI molecules in a mouse model. Etiology of CDI is complex and a combined approach of drugs inhibiting different stages of C. difficile life cycle are advantageous for the treatment and management of CDI.
Author Contributions
RT, DZ, RD, and CT conceived and designed the experiments. RT performed the experiments. RT and CT performed the analysis. RT, DZ, RD, and CT wrote and edited the manuscript.
Conflict of Interest Statement
CT is a scientific advisor to Locus Biosciences, a company engaged in the development of antimicrobial technologies. DZ and RD are employees of Agile Sciences, Inc., a company engaged in the development of antimicrobial technologies. The other author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the following researchers for providing us with C. difficile and commensal strains used in this study: Trevor Lawley, Joe Sorg, Aimee Shen, Eric Martens, Jason Ridlon, and Rodolphe Barrangou. We also thank David Jung for synthesizing the 2-AI compounds tested.
Abbreviations
- 2-AI
2-aminoimidazole
- CDC
The Centers for Disease Control and Prevention
- CDI
C. difficile infection
- CPD
cysteine protease domain
- MIC
minimum inhibitory concentration
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01206/full#supplementary-material
2-aminoimidazole molecules do not alter C. difficile spores recovered in BHI media at 6 and 24 h post treatment. (A) Total C. difficile R20291 vegetative cells and spores, (B) total vegetative cells and spores, (C) spores only at 6 h and (D) total C. difficile R20291 vegetative cells and spores, (E) total vegetative cells and spores, (F) spores only at 24 h for Compounds 1, 2, and 3 at a concentration of 10 μg/ml when compared to solvent 0.25% DMSO (DMSO, positive control), or 2 μg/ml vancomycin (Vanco, negative control). Data presented represent mean ± SEM of triplicate experiments. Statistical significance between positive control (solvent) and treatment groups was determined by Student’s parametric t-test with Welch’s correction (∗p < 0.05, ∗∗p < 0.01).
References
- Alam M. Z., Wu X., Mascio C., Chesnel L., Hurdle J. G. (2015). Mode of action and bactericidal properties of surotomycin against growing and nongrowing Clostridium difficile. Antimicrob. Agents Chemother. 59 5165–5170. 10.1128/AAC.01087-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altermann E., Russell W. M., Azcarate-Peril M. A., Barrangou R., Buck B. L., McAuliffe O., et al. (2005). Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM. Proc. Natl. Acad. Sci. U.S.A. 102 3906–3912. 10.1073/pnas.0409188102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthakrishnan A. N. (2011). Clostridium difficile infection: epidemiology, risk factors and management. Nat. Rev. Gastroenterol. Hepatol. 8 17–26. 10.1038/nrgastro.2010.190 [DOI] [PubMed] [Google Scholar]
- Antunes L. C. M., Han J., Ferreira R. B., Loliæ P., Borchers C. H., Finlay B. B. (2011). Effect of antibiotic treatment on the intestinal metabolome. Antimicrob. Agents Chemother. 55 1494–1503. 10.1128/AAC.01664-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon S. S., Schechter R., Maslanka S. E., Jewell N. P., Hatheway C. L. (2006). Human botulism immune globulin for the treatment of infant botulism. N. Engl. J. Med. 354 462–471. 10.1056/NEJMoa051926 [DOI] [PubMed] [Google Scholar]
- Babakhani F., Bouillaut L., Gomez A., Sears P., Nguyen L., Sonenshein A. L. (2012). Fidaxomicin inhibits spore production in Clostridium difficile. Clin. Infect. Dis. 55(Suppl. 2) S162–S169. 10.1093/cid/cis453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines S. D., O’Connor R., Freeman J., Fawley W. N., Harmanus C., Mastrantonio P., et al. (2008). Emergence of reduced susceptibility to metronidazole in Clostridium difficile. J. Antimicrob. Chemother. 62 1046–1052. 10.1093/jac/dkn313 [DOI] [PubMed] [Google Scholar]
- Baines S. D., O’Connor R., Saxton K., Freeman J., Wilcox M. H. (2009). Activity of vancomycin against epidemic Clostridium difficile strains in a human gut model. J. Antimicrob. Chemother. 63 520–525. 10.1093/jac/dkn502 [DOI] [PubMed] [Google Scholar]
- Barbut F., Mastrantonio P., Delmée M., Brazier J., Kuijper E., Poxton I. (2007). Prospective study of Clostridium difficile infections in Europe with phenotypic and genotypic characterisation of the isolates. Clin. Microbiol. Infect. 13 1048–1057. [DOI] [PubMed] [Google Scholar]
- Barefoot S. F., Klaenhammer T. R. (1983). Detection and activity of lactacin B, a bacteriocin produced by Lactobacillus acidophilus. Appl. Environ. Microbiol. 45 1808–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basseres E., Endres B. T., Khaleduzzaman M., Miraftabi F., Alam M. J., Vickers R. J., et al. (2016). Impact on toxin production and cell morphology in Clostridium difficile by ridinilazole (SMT19969), a novel treatment for C. difficile infection. J. Antimicrob. Chemother. 71 1245–1251. 10.1093/jac/dkv498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier D., Gross R. (2006). Regulation of bacterial virulence by two-component systems. Curr. Opin. Microbiol. 9 143–152. 10.1016/j.mib.2006.01.005 [DOI] [PubMed] [Google Scholar]
- Bender K. O., Garland M., Ferreyra J. A., Hryckowian A. J., Child M. A., Puri A. W., et al. (2015). A small-molecule antivirulence agent for treating Clostridium difficile infection. Sci. Transl. Med. 7:306ra148 10.1126/scitranslmed.aac9103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boix V., Fedorak R. N., Mullane K. M., Pesant Y., Stoutenburgh U., Jin M., et al. (2017). Primary outcomes from a phase 3, randomized, double-blind, active-controlled trial of surotomycin in subjects with Clostridium difficile infection. Open Forum Infect. Dis. 4:ofw275 10.1093/ofid/ofw275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackett C. M., Melander R. J., An I. H., Krishnamurthy A., Thompson R. J., Cavanagh J., et al. (2014). Small-molecule suppression of beta-lactam resistance in multidrug-resistant gram-negative pathogens. J. Med. Chem. 57 7450–7458. 10.1021/jm501050e [DOI] [PubMed] [Google Scholar]
- Brock T. E. (2015). A Clinical and Molecular Analysis of Clostridium difficile Strains Isolated from Groote Schuur Hospital, Schuur Hospital. Available at: http://hdl.handle.net/11427/19966 [DOI] [PubMed] [Google Scholar]
- Buffie C. G., Bucci V., Stein R. R., McKenney P. T., Ling L., Gobourne A., et al. (2015). Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517 205–208. 10.1038/nature13828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC (2008). Surveillance for community-associated Clostridium difficile–Connecticut, 2006. MMWR Morb. Mortal. Wkly. Rep. 57 340–343. [PubMed] [Google Scholar]
- CDC (2013). Antibiotic Resistance Threats in the United States, 2013 Centres for Disease Control and Prevention, US Department of Health and Human Services. Atlanta, GA: CDC. [Google Scholar]
- Cegelski L., Marshall G. R., Eldridge G. R., Hultgren S. J. (2008). The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol. 6 17–27. 10.1038/nrmicro1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron D. M., Tyrrell K. L., Merriam C. V., Goldstein E. J. (2012). In vitro activities of CB-183,315, vancomycin, and metronidazole against 556 strains of Clostridium difficile, 445 other intestinal anaerobes, and 56 Enterobacteriaceae species. Antimicrob. Agents Chemother. 56 1613–1615. 10.1128/AAC.05655-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. H., Gerding D. N., Johnson S., Kelly C. P., Loo V. G., McDonald L. C., et al. (2010). Clinical practice guidelines for Clostridium difficile infection in adults: update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA). Infect. Control Hosp. Epidemiol. 31 431–455. 10.1086/651706 [DOI] [PubMed] [Google Scholar]
- Đapa T., Leuzzi R., Ng Y. K., Baban S. T., Adamo R., Kuehne S. A., et al. (2013). Multiple factors modulate biofilm formation by the anaerobic pathogen Clostridium difficile. J. Bacteriol. 195 545–555. 10.1128/JB.01980-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darkoh C., DuPont H. L., Norris S. J., Kaplan H. B. (2015). Toxin synthesis by Clostridium difficile is regulated through quorum signaling. mBio 6:e02569-14 10.1128/mBio.02569-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darkoh C., Odo C., DuPont H. L. (2016). Accessory gene regulator-1 locus is essential for virulence and pathogenesis of Clostridium difficile. mBio 7:e01237-16 10.1128/mBio.01237-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sablet T., Chassard C., Bernalier-Donadille A., Vareille M., Gobert A. P., Martin C. (2009). Human microbiota-secreted factors inhibit shiga toxin synthesis by enterohemorrhagic Escherichia coli O157:H7. Infect. Immun. 77 783–790. 10.1128/IAI.01048-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin L. J., Clare S., Fagan R. P., Dawson L. F., Pickard D. J., West M. R., et al. (2012). The Clostridium difficile spo0A gene is a persistence and transmission factor. Infect. Immun. 80 2704–2711. 10.1128/IAI.00147-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draughn G. L., Allen C. L., Routh P. A., Stone M. R., Kirker K. R., Boegli L., et al. (2017). Evaluation of a 2-aminoimidazole variant as adjuvant treatment for dermal bacterial infections. Drug Des. Dev. Ther. 11 153–162. 10.2147/DDDT.S111865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois T., Dancer-Thibonnier M., Monot M., Hamiot A., Bouillaut L., Soutourina O., et al. (2016). Control of Clostridium difficile physiopathology in response to cysteine availability. Infect. Immun. 84 2389–2405. 10.1128/IAI.00121-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPont H. L. (2011). The search for effective treatment of Clostridium difficile infection. N. Engl. J. Med. 364 473–475. 10.1056/NEJMe1013236 [DOI] [PubMed] [Google Scholar]
- Fawley W. N., Underwood S., Freeman J., Baines S. D., Saxton K., Stephenson K., et al. (2007). Efficacy of hospital cleaning agents and germicides against epidemic Clostridium difficile strains. Infect. Control Hosp. Epidemiol. 28 920–925. [DOI] [PubMed] [Google Scholar]
- Ferreira R. B., Gill N., Willing B. P., Antunes L. C., Russell S. L., Croxen M. A., et al. (2011). The intestinal microbiota plays a role in Salmonella-induced colitis independent of pathogen colonization. PLoS One 6:e20338 10.1371/journal.pone.0020338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher C. K., Mehta P. (2014). Identifying keystone species in the human gut microbiome from metagenomic timeseries using sparse linear regression. PLoS One 9:e102451 10.1371/journal.pone.0102451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis M. B., Allen C. A., Sorg J. A. (2013). Muricholic acids inhibit Clostridium difficile spore germination and growth. PLoS One 8:e73653 10.1371/journal.pone.0073653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank D. N., Pace N. R. (2008). Gastrointestinal microbiology enters the metagenomics era. Curr. Opin. Gastroenterol. 24 4–10. 10.1097/MOG.0b013e3282f2b0e8 [DOI] [PubMed] [Google Scholar]
- Guinane C. M., Barrett E., Fitzgerald G. F., van Sinderen D., Ross R. P., Stanton C. (2011). Genome sequence of Bifidobacterium breve DPC 6330, a strain isolated from the human intestine. J. Bacteriol. 193 6799–6800. 10.1128/JB.06196-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A., Khanna S. (2014). Community-acquired Clostridium difficile infection: an increasing public health threat. Infect. Drug Resist. 7 63–72. 10.2147/IDR.S46780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M., Miyajima F., Roberts P., Ellison L., Pickard D. J., Martin M. J., et al. (2013). Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat. Genet. 45 109–113. 10.1038/ng.2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M., Sebaihia M., Lawley T. D., Stabler R. A., Dawson L. F., Martin M. J., et al. (2010). Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc. Natl. Acad. Sci. U.S.A. 107 7527–7532. 10.1073/pnas.0914322107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentzer M., Wu H., Andersen J. B., Riedel K., Rasmussen T. B., Bagge N., et al. (2003). Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 22 3803–3815. 10.1093/emboj/cdg366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung D. T., Shakhnovich E. A., Pierson E., Mekalanos J. J. (2005). Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 310 670–674. 10.1126/science.1116739 [DOI] [PubMed] [Google Scholar]
- Jett B. D., Hatter K. L., Huycke M. M., Gilmore M. S. (1997). Simplified agar plate method for quantifying viable bacteria. Biotechniques 23 648–650. [DOI] [PubMed] [Google Scholar]
- Kamada N., Kim Y. G., Sham H. P., Vallance B. A., Puente J. L., Martens E. C., et al. (2012). Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science 336 1325–1329. 10.1126/science.1222195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson S., Lindberg A., Norin E., Burman L. G., Akerlund T. (2000). Toxins, butyric acid, and other short-chain fatty acids are coordinately expressed and down-regulated by cysteine in Clostridium difficile. Infect. Immun. 68 5881–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knetsch C. W., Kumar N., Forster S. C., Connor T. R., Browne H. P., Harmanus C., et al. (2017). Zoonotic transfer of Clostridium difficile harboring antimicrobial resistance between farm animals and humans. J. Clin. Microbiol. 56:e01384-17 10.1128/JCM.01384-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochan T. J., Somers M. J., Kaiser A. M., Shoshiev M. S., Hagan A. K., Hastie J. L., et al. (2017). Intestinal calcium and bile salts facilitate germination of Clostridium difficile spores. PLoS Pathog. 13:e1006443 10.1371/journal.ppat.1006443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kociolek L. K., Gerding D. N. (2016). Breakthroughs in the treatment and prevention of Clostridium difficile infection. Nat. Rev. Gastroenterol. Hepatol. 13 150–160. 10.1038/nrgastro.2015.220 [DOI] [PubMed] [Google Scholar]
- Kuehne S. A., Collery M. M., Kelly M. L., Cartman S. T., Cockayne A., Minton N. P. (2014). Importance of toxin A, toxin B, and CDT in virulence of an epidemic Clostridium difficile strain. J. Infect. Dis. 209 83–86. 10.1093/infdis/jit426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessa F. C., Mu Y., Bamberg W. M., Beldavs Z. G., Dumyati G. K., Dunn J. R., et al. (2015). Burden of Clostridium difficile infection in the United States. N. Engl. J. Med. 372 825–834. 10.1056/NEJMoa1408913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levett P. (1991). Time-dependent killing of Clostridium difficile by metronidazole and vancomycin. J. Antimicrob. Chemother. 27 55–62. [DOI] [PubMed] [Google Scholar]
- Locher H. H., Caspers P., Bruyere T., Schroeder S., Pfaff P., Knezevic A., et al. (2014a). Investigations of the mode of action and resistance development of cadazolid, a new antibiotic for treatment of Clostridium difficile infections. Antimicrob. Agents Chemother. 58 901–908. 10.1128/AAC.01831-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher H. H., Seiler P., Chen X., Schroeder S., Pfaff P., Enderlin M., et al. (2014b). In vitro and in vivo antibacterial evaluation of cadazolid, a new antibiotic for treatment of Clostridium difficile infections. Antimicrob. Agents Chemother. 58 892–900. 10.1128/AAC.01830-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo V. G., Bourgault A. M., Poirier L., Lamothe F., Michaud S., Turgeon N., et al. (2011). Host and pathogen factors for Clostridium difficile infection and colonization. N. Engl. J. Med. 365 1693–1703. 10.1056/NEJMoa1012413 [DOI] [PubMed] [Google Scholar]
- Martens E. C., Chiang H. C., Gordon J. I. (2008). Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4 447–457. 10.1016/j.chom.2008.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin H., Willey B., Low D. E., Staempfli H. R., McGeer A., Boerlin P., et al. (2008). Characterization of Clostridium difficile strains isolated from patients in Ontario, Canada, from 2004 to 2006. J. Clin. Microbiol. 46 2999–3004. 10.1128/JCM.02437-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Verstraete I., Peltier J., Dupuy B. (2016). The regulatory networks that control Clostridium difficile toxin synthesis. Toxins 8:153 10.3390/toxins8050153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascio C. T., Mortin L. I., Howland K. T., Van Praagh A. D., Zhang S., Arya A., et al. (2012). In vitro and in vivo characterization of CB-183,315, a novel lipopeptide antibiotic for treatment of Clostridium difficile. Antimicrob. Agents Chemother. 56 5023–5030. 10.1128/AAC.00057-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattarelli P., Bonaparte C., Pot B., Biavati B. (2008). Proposal to reclassify the three biotypes of Bifidobacterium longum as three subspecies: Bifidobacterium longum subsp. longum subsp. nov., Bifidobacterium longum subsp. infantis comb. nov. and Bifidobacterium longum subsp. suis comb. nov. Int. J. Syst. Evol. Microbiol. 58(Pt 4) 767–772. 10.1099/ijs.0.65319-0 [DOI] [PubMed] [Google Scholar]
- McDonald L. C., Gerding D. N., Johnson S., Bakken J. S., Carroll K. C., Coffin S. E., et al. (2018). Clinical Practice Guidelines for Clostridium difficile infection in adults and children: 2017 update by the infectious diseases society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis. 66 e1–e48. 10.1093/cid/cix1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki K. L., Edwards A. N., Daou N., Bouillaut L., McBride S. M. (2016). CodY-Dependent regulation of sporulation in Clostridium difficile. J. Bacteriol. 198 2113–2130. 10.1128/JB.00220-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes-Sabja D., Shen A., Sorg J. A. (2014). Clostridium difficile spore biology: sporulation, germination, and spore structural proteins. Trends Microbiol. 22 406–416. 10.1016/j.tim.2014.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez J., Springthorpe V. S., Sattar S. A. (2011). Clospore: a liquid medium for producing high titers of semi-purified spores of Clostridium difficile. J. AOAC Int. 94 618–626. [PubMed] [Google Scholar]
- Pruitt R. N., Lacy D. B. (2012). Toward a structural understanding of Clostridium difficile toxins A and B. Front Cell Infect. Microbiol. 2:28 10.3389/fcimb.2012.00028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon J. M., Kang D. J., Hylemon P. B. (2010). Isolation and characterization of a bile acid inducible 7alpha-dehydroxylating operon in Clostridium hylemonae TN271. Anaerobe 16 137–146. 10.1016/j.anaerobe.2009.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers S. A., Huigens R. W., III, Cavanagh J., Melander C. (2010). Synergistic effects between conventional antibiotics and 2-aminoimidazole-derived antibiofilm agents. Antimicrob. Agents Chemother. 54 2112–2118. 10.1128/AAC.01418-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round J. L., Mazmanian S. K. (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 9 313–323. 10.1038/nri2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz J. B., Doggett T. A., Haslam D. B. (2007). Identification and characterization of small molecules that inhibit intracellular toxin transport. Infect. Immun. 75 4552–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebaihia M., Wren B. W., Mullany P., Fairweather N. F., Minton N., Stabler R., et al. (2006). The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 38 779–786. [DOI] [PubMed] [Google Scholar]
- Shen A., Fimlaid K. A., Pishdadian K. (2016). Inducing and quantifying Clostridium difficile spore formation. Methods Mol. Biol. 1476 129–142. 10.1007/978-1-4939-6361-4_10 [DOI] [PubMed] [Google Scholar]
- Smits W. K., Lyras D., Lacy D. B., Wilcox M. H., Kuijper E. J. (2016). Clostridium difficile infection. Nat. Rev. Dis. Primers 2:16020 10.1038/nrdp.2016.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snydman D. R., Jacobus N. V., McDermott L. A. (2012). Activity of a novel cyclic lipopeptide, CB-183,315, against resistant Clostridium difficile and other Gram-positive aerobic and anaerobic intestinal pathogens. Antimicrob. Agents Chemother. 56 3448–3452. 10.1128/AAC.06257-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg J. A., Sonenshein A. L. (2008). Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol. 190 2505–2512. 10.1128/JB.01765-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabler R. A., Gerding D. N., Songer J. G., Drudy D., Brazier J. S., Trinh H. T., et al. (2006). Comparative phylogenomics of Clostridium difficile reveals clade specificity and microevolution of hypervirulent strains. J. Bacteriol. 188 7297–7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabler R. A., He M., Dawson L., Martin M., Valiente E., Corton C., et al. (2009). Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 10:R102 10.1186/gb-2009-10-9-r102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinebrunner N., Stremmel W., Weiss K. H. (2018). Ridinilazole-a novel antibiotic for treatment of Clostridium difficile infection. J. Thorac. Dis. 10 118–120. 10.21037/jtd.2017.12.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson K., Hoch J. A. (2002). Virulence- and antibiotic resistance-associated two-component signal transduction systems of Gram-positive pathogenic bacteria as targets for antimicrobial therapy. Pharmacol. Ther. 93 293–305. [DOI] [PubMed] [Google Scholar]
- Stock A. M., Robinson V. L., Goudreau P. N. (2000). Two-component signal transduction. Annu. Rev. Biochem. 69 183–215. 10.1146/annurev.biochem.69.1.183 [DOI] [PubMed] [Google Scholar]
- Tam J., Beilhartz G. L., Auger A., Gupta P., Therien A. G., Melnyk R. A. (2015). Small molecule inhibitors of Clostridium difficile toxin B-induced cellular damage. Chem. Biol. 22 175–185. 10.1016/j.chembiol.2014.12.010 [DOI] [PubMed] [Google Scholar]
- Thanissery R., Winston J. A., Theriot C. M. (2017). Inhibition of spore germination, growth, and toxin activity of clinically relevant C. difficile strains by gut microbiota derived secondary bile acids. Anaerobe 45 86–100. 10.1016/j.anaerobe.2017.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot C. M., Bowman A. A., Young V. B. (2016). Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. mSphere 1:e00045-15 10.1128/mSphere.00045-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot C. M., Koenigsknecht M. J., Carlson P. E., Jr., Hatton G. E., Nelson A. M., Li B., et al. (2014). Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 5:3114 10.1038/ncomms4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R. J., Bobay B. G., Stowe S. D., Olson A. L., Peng L., Su Z., et al. (2012). Identification of BfmR, a response regulator involved in biofilm development, as a target for a 2-Aminoimidazole-based antibiofilm agent. Biochemistry 51 9776–9778. 10.1021/bi3015289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremaroli V., Backhed F. (2012). Functional interactions between the gut microbiota and host metabolism. 489 242–249. 10.1038/nature11552 [DOI] [PubMed] [Google Scholar]
- Underwood S., Guan S., Vijayasubhash V., Baines S. D., Graham L., Lewis R. J., et al. (2009). Characterization of the sporulation initiation pathway of Clostridium difficile and its role in toxin production. J. Bacteriol. 191 7296–7305. 10.1128/JB.00882-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura M., Canchaya C., Meylan V., Klaenhammer T. R., Zink R. (2003). Analysis, characterization, and loci of the tuf genes in lactobacillus and bifidobacterium species and their direct application for species identification. Appl. Environ. Microbiol. 69 6908–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox M. H., Fawley W. N. (2000). Hospital disinfectants and spore formation by Clostridium difficile. Lancet 356:1324. [DOI] [PubMed] [Google Scholar]
- Wilcox M. H., Gerding D. N., Poxton I. R., Kelly C., Nathan R., Birch T., et al. (2017). Bezlotoxumab for prevention of recurrent Clostridium difficile infection. N. Engl. J. Med. 376 305–317. 10.1056/NEJMoa1602615 [DOI] [PubMed] [Google Scholar]
- Winston J. A., Thanissery R., Montgomery S. A., Theriot C. M. (2016). Cefoperazone treated mouse model of Clostridium difficile strain R20291: a clinically relevant platform for testing therapeutics. J. Vis. Exp. 10:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Bjursell M. K., Himrod J., Deng S., Carmichael L. K., Chiang H. C., et al. (2003). A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science 299 2074–2076. 10.1126/science.1080029 [DOI] [PubMed] [Google Scholar]
- Yang Z., Ramsey J., Hamza T., Zhang Y., Li S., Yfantis H. G., et al. (2015). Mechanisms of protection against Clostridium difficile infection by the monoclonal antitoxin antibodies actoxumab and bezlotoxumab. Infect. Immun. 83 822–831. 10.1128/IAI.02897-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
2-aminoimidazole molecules do not alter C. difficile spores recovered in BHI media at 6 and 24 h post treatment. (A) Total C. difficile R20291 vegetative cells and spores, (B) total vegetative cells and spores, (C) spores only at 6 h and (D) total C. difficile R20291 vegetative cells and spores, (E) total vegetative cells and spores, (F) spores only at 24 h for Compounds 1, 2, and 3 at a concentration of 10 μg/ml when compared to solvent 0.25% DMSO (DMSO, positive control), or 2 μg/ml vancomycin (Vanco, negative control). Data presented represent mean ± SEM of triplicate experiments. Statistical significance between positive control (solvent) and treatment groups was determined by Student’s parametric t-test with Welch’s correction (∗p < 0.05, ∗∗p < 0.01).