

Abstract
Background: Previous studies have suggested a causative role for agonists of the aromatic hydrocarbon receptor (AhR) in the etiology of breast cancer 1, early-onset (BRCA-1)–silenced breast tumors, for which prospects for treatment remain poor.
Objectives: We investigated the regulation of BRCA1 by the soy isoflavone genistein (GEN) in human estrogen receptor α (ERα)–positive Michigan Cancer Foundation-7 (MCF-7) and ERα-negative sporadic University of Arizona Cell Culture-3199 (UACC-3199) breast cancer cells, respectively, with inducible and constitutively active AhR.
Methods: In MCF-7 cells, we analyzed the dose- and time-dependent effects of GEN and (–)-epigallocatechin-3-gallate (EGCG) control, selected as prototype dietary DNA methyltransferase (DNMT) inhibitors, on BRCA-1 expression after AhR activation with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and in TCDD-washout experiments. We compared the effects of GEN and EGCG on BRCA1 cytosine-phosphate-guanine (CpG) methylation and cell proliferation. Controls for DNA methylation and proliferation were changes in expression of DNMT-1, cyclin D1, and p53, respectively. In UACC-3199 cells, we compared the effects of GEN and α-naphthoflavone (αNF; 7,8-benzoflavone), a synthetic flavone and AhR antagonist, on BRCA1 expression and CpG methylation, cyclin D1, and cell growth. Finally, we examined the effects of GEN and αNF on BRCA1, AhR-inducible cytochrome P450 (CYP)-1A1 (CYP1A1) and CYP1B1, and AhR mRNA expression.
Results: In MCF-7 cells, GEN exerted dose- and time-dependent preventative effects against TCDD-dependent downregulation of BRCA-1. After TCDD washout, GEN rescued BRCA-1 protein expression while reducing DNMT-1 and cyclin D1. GEN and EGCG reduced BRCA1 CpG methylation and cell proliferation associated with increased p53. In UACC-3199 cells, GEN reduced BRCA1 and estrogen receptor-1 (ESR1) CpG methylation, cyclin D1, and cell growth while inducing BRCA-1 and CYP1A1.
Conclusions: Results suggest preventative effects for GEN and EGCG against BRCA1 CpG methylation and downregulation in ERα-positive breast cancer cells with activated AhR. GEN and flavone antagonists of AhR may be useful for reactivation of BRCA1 and ERα via CpG demethylation in ERα-negative breast cancer cells harboring constitutively active AhR.
Keywords: genistein, BRCA1, ESR1, AhR, DNA methylation, epigenetics, breast cancer, flavonoids, prevention
Introduction
The breast cancer 1, early-onset (BRCA1) gene encodes a tumor suppressor protein involved in DNA repair and cell cycle control (1). In women who carry a mutated BRCA1 copy (BRCA1+/−), the silencing of the wild-type allele creates a BRCA-1–deficient phenotype, which is associated with a high probability (∼60–80%) of developing breast cancer (2, 3). On the other hand, sporadic breast cancers, which represent the majority (∼90%) of breast tumor cases, do not have mutations in BRCA1 (BRCA1+/+) but display a “BRCAness” phenotype commonly observed in hereditary BRCA1 tumors. This phenotype includes absent or markedly reduced concentrations of BRCA-1 (4, 5), loss of estrogen receptor α (ERα), and basal-like pathology subtype (6). Therefore, elucidating the nonmutational mechanisms that contribute to silencing of BRCA1 has important implications for the prevention of both hereditary and sporadic breast cancers.
Epigenetics refers to modifications in chromatin structure [i.e., histone posttranslational modifications and DNA cytosine-phosphate-guanine (CpG) methylation] and noncoding RNAs (7). Sporadic breast cancers that have hypermethylated BRCA1 share features with hereditary BRCA1 mutation tumors [i.e., they tend to be triple-negative with reduced or absent expression of ERα, progesterone receptor (PR), and human epidermal growth factor receptor 2] (8). CpG methylation of BRCA1 is associated with reduced BRCA-1 expression in 50–60% of higher-histologic-grade sporadic tumors (9, 10). A high degree of correlation (∼75%) is generally observed between hypermethylation of the BRCA1 and estrogen receptor-1 [ESR1 (ERα)] promoters and reduced expression of BRCA-1 and ERα protein (11, 12), which are invariably associated with resistance to endocrine therapies based on antagonists of the ERα (i.e., tamoxifen) (13). Therefore, main objectives in breast cancer research are to identify the mechanisms linking silencing of BRCA1 to the development of ERα-negative breast cancers, and clarify whether or not opportunities exist for the prevention of these tumors with dietary components.
Agonists of the aromatic hydrocarbon receptor (AhR) are ubiquitous in the environment and include dietary compounds, metabolites of FAs, industrial xenobiotics, and photoproducts generated in the skin from UV radiation (14). Results from our laboratory document that the BRCA1 gene is a target for epigenetic regulation by AhR. In the absence of exogenous ligands, AhR forms a transcription complex with ERα and various cofactors (p300, steroid receptor coactivator-1) (15) contributing to the transcriptional activation of BRCA1 by 17β-estradiol (E2) (16). Conversely, in the presence of agonists, AhR binds to xenobiotic response elements (XRE) with consensus 5′-GCGTG-3′ sequence and harbored in the BRCA1 gene (17), and disrupts transcriptional activation by E2 (18). This repressive effect is coupled to the recruitment of DNA methyltransferase (DNMT) 1 and methyl binding protein (MBD) 2, loss of acetylated histone (AcH) 4 and AcH3K9 (19), and gain of trimethylated H3K9 (H3K9me3) and DNA CpG methylation (20). Recently, we reported that in rodent mammary tissue (21) and human breast tumors (22) with activated AhR, hypermethylation of BRCA1 was associated with reduced BRCA-1 and ERα expression. These cumulative data raised the question of whether or not dietary compounds that possess DNMT and AhR inhibitory properties may protect against CpG hypermethylation of BRCA1 and, ultimately, prevent breast tumorigenesis.
Genistein (GEN), a common dietary isoflavone, exerts antagonistic properties toward DNMT enzymes (23, 24). Evidence that it induces BRCA-1 expression in ERα-positive breast cancer cells suggests potential relevance for this isoflavone in cancer prevention (25). Rodent offspring exposed to GEN in utero, through weaning (26), and prepuberty (27, 28) showed reduced mammary tumorigenesis in adult life. Through the inhibition of DNMT activity, GEN was shown to reactivate the expression of various tumor suppressor genes (i.e., ataxia telangiectasia mutated, adenomatous polyposis coli, phosphatase and tensin homolog) in ERα-positive Michigan Cancer Foundation-7 (MCF-7) and ERα-negative M.D. Anderson Cancer Center-metastatic breast cancer-231 (MDA-MB-231) breast cancer cells (29). Here, we investigated the impact of GEN and (–)-epigallocatechin-3-gallate (EGCG) control on BRCA-1 methylation and expression in a human ERα-positive breast cancer cell line (MCF-7) with inducible AhR and hypomethylated BRCA1. We extended the BRCA-1 expression and DNA methylation studies with GEN and α-naphthoflavone (αNF; 7,8-benzoflavone), a synthetic flavone and AhR antagonist, to a human ERα-negative cell line model University of Arizona Cell Culture-3199 (UACC-3199) of sporadic breast cancer harboring constitutively activated AhR and hypermethylated BRCA1 (4).
Methods
Cell culture
Human MCF-7 and UACC-3199 breast cancer cells were obtained from the American Type Culture Collection and maintained, respectively, in DMEM or Roswell Park Memorial Institute (RPMI) 1640 media (Mediatech) supplemented with 10% fetal calf serum (Hyclone Laboratories). The 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) was supplied by the National Cancer Institute, Division of Cancer Biology, Chemical and Physical Carcinogenesis Branch (NIH), and distributed by Midwest Research Institute (contracts 64 CFR 72090 and 64 CFR 28205). GEN, αNF, EGCG, and E2 were obtained from Sigma-Aldrich and solubilized in stock solutions with ethanol, which was added to DMEM or RPMI as the vehicle control. Cells (passages 3–15) were plated in 6-well plates at a density of 5 × 105 cells/well in Phenol-Red free DMEM (MCF-7) or RPMI (UACC-3199) supplemented with 10% charcoal-stripped fetal calf serum (22). For Western blotting, cells were washed with ice-cold PBS and scraped with cold lysis buffer containing protease inhibitor. For proliferation measurements, cells were washed with ice-cold PBS and counted by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay (Promega), as described previously (30). This assay is based on the conversion of the yellow tretrazolium dye MTT to purple formazan crystals by metabolically active cells. Briefly, 2 × 104 cells were seeded in 96-well tissue culture plates and maintained overnight. Six replicates were assigned to each experimental treatment. After treatment, 15 μL MTT dye solution was added to each well, and the plate was incubated for 4 h at 37°C. Solubilization/stop solution (100 μL) was added for 1 h at room temperature, and the absorbance at 570/650 nm was recorded by using a Synergy HT plate reader (Bio-Tek Instruments).
Western blot analysis
Western blot analyses were performed as previously described (22). Immunoblotting was carried out with antibodies against human BRCA-1 (catalog no. 9010), DNMT-1 (catalog no. 5119), cyclin D1 (catalog no. 92G2), phospho-p53-(Ser20) (catalog no. 9287), and GAPDH (catalog no. 2118) obtained from Cell Signaling Technology, and ERα (catalog no. sc-542) obtained from Santa Cruz Biotechnology. Immunocomplexes were detected by using enhanced chemiluminescence (GE Healthcare Life Sciences). The GAPDH protein was used as an internal control for normalization of protein expression.
Promoter CpG methylation
qPCR analysis of human BRCA1 and ESR1 promoter CpG methylation was performed as described previously (20) with bisulfonated genomic DNA with the use of the following unmethylated (U)- and methylated (M)-specific primers (Sigma-Aldrich):
BRCA1: U-sense: 5′-TTGGTTTTTGTGGTAATGGAAAAGTGT-3′ and U-antisense: 5′-CAAAAAATCTCAACAAACTCACACCA-3′ M-sense: 5′-TGGTAACGGAAAAGCG-3′ and M-antisense 5′-ATCTCAACGAACTCACGC-3′
ESR1: U-sense: 5′-GGATATGGTTTGTATTTTGTTTGT-3′ and U-antisense: 5′-ACAAACAATTCAAAAACTCCAACT-3′ M-sense: 5′-GGTTTTTGAGTTTTTTGTTTTG-3′ and M-antisense: 5′-AACTTACTACTATCCAAATACACCTC-3′
The qPCR was carried out in a volume of 10 μL consisting of the following master mix: 5 μL SYBER Green mix (Life Technologies), 1 μL each of forward and reverse primers, 2 μL nuclease-free water, and 1 μL bisulfonated genomic DNA. Data from qPCR of bisulfonated DNA were presented as ratios of CpG M:U.
mRNA analyses
Total RNA was purified by using an RNeasy Mini Kit as per the manufacturer's instructions (Qiagen) (22). Concentrations and quality of RNA were verified by using the Nanodrop1000 Spectrophotometer (Thermo Scientific). Equal amounts of total RNA (500 ng) were transcribed into cDNA by using the ISCRIPT supermix kit (Bio-Rad Laboratories). Next, cDNA aliquots were analyzed by qPCR with the use of the SYBR Green PCR Reagents kit (Life Technologies). Briefly, reactions were assayed at a final volume of 25 μL consisting of the following master mix: 12.5 μL SYBR Green mix, 1 μL each of forward and reverse primers, 9.5 μL nuclease-free water, and 1 μL cDNA. Amplification of GAPDH mRNA was used for normalization of transcript levels. The primer (Sigma-Aldrich) sequences were as follows—BRCA1: sense, 5′-AGCTCGCTGAGACTTCCTGGA-3′ antisense, 5′-CAATTCAATGTAGACAGACGT-3′ cytochrome P450 (CYP)-1A1 (CYP1A1): sense, 5′-TAACATCGTCTTGGACCTCTTTG-3′ antisense, 5′-GTCGATAGCACCATCAGGGGT-3′ CYP1B1: sense, 5′-AACGTCATGAGTGCCGTGTGT-3′ antisense, 5′-GGCCGGTACGTTCTCCAAATC-3′ AhR: sense, 5′-GAAGCCGGTGCAGAAAACAG-3′ antisense: 5′-GCCGCTTGGAAGGATTTGAC-3′ and control GAPDH: sense, 5′-ACCCACTCCTCCACCTTT-3′ antisense, 5′-CTCTTGTGCTCTTGCTGGG-3′.
Statistical analysis
Densitometry after Western blotting was performed by using Kodak ID Image Analysis Software (Eastman Kodak Company). Statistical analyses were performed by 1-factor ANOVA after assessing data normality by using a Shapiro-Wilk test and variance homogeneity by using Bartlett's test. Post hoc multiple comparisons among all means were conducted by using Tukey's test after main effects and interactions were found to be significant at P ≤ 0.05. Data are presented as means ± SEMs. When ≥3 means were compared, significant differences are highlighted with different letters (a > b > c).
Results
GEN prevents AhR-dependent downregulation of BRCA-1
In control experiments, we first confirmed that at 24-h post-treatment, E2 (10 nM) induced an increase of ∼2.0-fold in BRCA-1 protein expression compared with the vehicle control (Figure 1A, B). This dose of E2 was used throughout this study and was similar to that used previously to investigate regulation of BRCA-1 in human breast cancer cells (16) and detected in women around the human menstrual phase and in patients receiving E2 replacement therapy (31, 32). In contrast, as documented previously (19, 20), an equimolar dose (10 nM) of TCDD did not change basal BRCA-1 concentrations (Figure 1B), but it reduced (∼50%) E2-induced BRCA-1 expression (Figure 1C, D). This dose of TCDD was used throughout this study and approached the concentration found in blood (33, 34) and lipid tissue (35) of women exposed to environmental AhR agonists. Compared with E2 plus TCDD, doses of 0.5 and 1.0 μM GEN counteracted the repressive effects of TCDD on BRCA-1 expression (Figure 1C, D), whereas 2.0 μM GEN had no protective effects. Conversely, GEN at 5, 10, and 20 μM synergized with TCDD to lower BRCA-1 expression to control levels.
FIGURE 1.

GEN prevents repression of BRCA-1 in breast cancer Michigan Cancer Foundation-7 cells with activated aromatic hydrocarbon receptor. Cells were cultured for 72 h in control medium containing E2 (10 nM) or TCDD (10 nM) (A) or E2 + TCDD and various concentrations of GEN (B) as described in Methods. Bands show immunocomplexes for BRCA-1 and the internal standard GAPDH. (C, D) Bars represent means ± SEMs of BRCA-1 expression (fold of control) from 4 (C) and 2 (D) separate experiments performed in duplicate. Labeled means without a common letter differ, P < 0.05. BRCA-1, breast cancer 1, early onset; E2, 17β-estradiol; GEN, genistein; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
In follow-up experiments, we examined the time-dependent effects of GEN at the 1-μM concentration, which approaches the serum concentration of GEN measured in persons with habitual soy intake and is known to induce BRCA-1 expression in breast cancer cells (25). The cotreatment with GEN protected against TCDD-mediated repression of BRCA-1 at 24 h, an effect that persisted at 48 and 72 h (Figure 2A, C). As a positive control for GEN, we used EGCG, which in previous studies was shown to reactivate methylation-silenced genes in cancer cells (36). At equimolar concentrations (1 μM), EGCG counteracted the repressive effects of TCDD and restored BRCA-1 expression to E2 levels by 48 and 72 h (Figure 2B, C).
FIGURE 2.

Time-dependent effects of GEN and EGCG against BRCA-1 downregulation in breast cancer Michigan Cancer Foundation-7 cells with activated aromatic hydrocarbon receptor. Cells were cultured for 24, 48, and 72 h in control medium containing E2 (10 nM), E2 + TCDD (10 nM), and E2 + TCDD and 1 μM GEN (A) or EGCG (B). Bands show immunocomplexes for BRCA-1 and the internal standard GAPDH. In panel C, bars represent means ± SEMs of BRCA-1 expression (fold of E2) from 2 separate experiments performed in duplicate. Labeled means without a common letter differ, P < 0.05. BRCA-1, breast cancer 1, early onset; EGCG, (–)-epigallocatechin-3-gallate; E2, 17β-estradiol; GEN, genistein; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
Growth of MCF-7 cells was induced (∼2.0 fold) by E2 within 72 h. The treatment with TCDD had no effects on cell proliferation, whereas the combination of TCDD plus E2 reduced cell growth by ∼30% (Figure 3A) compared with E2 alone. The cotreatment with GEN plus E2 repressed E2-induced cell growth by ∼50%, irrespective of the presence or absence of TCDD. Inhibitory effects (∼50%) on E2-induced cell growth were also seen for EGCG, whereas the combination of GEN plus EGCG reduced cell growth by ∼80% compared with E2 treatment. As a control for cell proliferation, we examined by Western blotting changes in cyclin D1, whose expression in MCF-7 cotreated with E2 plus TCDD was reduced (∼30%) by GEN (Figure 3B, C). In contrast, GEN and EGCG increased the expression of the tumor suppressor p53 (Figure 4).
FIGURE 3.

GEN and EGCG antagonize E2-induced proliferation of breast cancer Michigan Cancer Foundation-7 cells with activated aromatic hydrocarbon receptor. (A) Cells were cultured for 72 h in control medium or control plus E2 (10 nM), TCDD (10 nM), and E2 + TCDD in the absence or presence of 1 μM GEN or EGCG. Bars represent means ± SEMs of quantitation (fold of control) of proliferation determined by MTT assay from 2 separate experiments with 5 replicates. Panels B and C represent, respectively, immunocomplexes and quantitation (means ± SEMs) from 3 separate experiments performed in duplicate for cyclin D1 and the internal standard GAPDH. Labeled means without a common letter differ, P < 0.05. EGCG, (–)-epigallocatechin-3-gallate; E2, 17β-estradiol; GEN, genistein; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
FIGURE 4.

GEN and EGCG induce p53 expression in breast cancer Michigan Cancer Foundation-7 cells with activated aromatic hydrocarbon receptor. Cells were cultured for 24, 48, and 72 h in control medium containing E2 (10 nM), E2 + TCDD (10 nM), and E2 + TCDD and 1 μM GEN or EGCG. (A) Bands show immunocomplexes for p53 and the internal standard GAPDH. (B) Bars represent means ± SEMs of p53 expression (fold of E2) from 2 separate experiments performed in duplicate. Labeled means without a common letter differ, P < 0.05. EGCG, (–)-epigallocatechin-3-gallate; E2, 17β-estradiol; GEN, genistein; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
GEN reverses AhR-mediated BRCA-1 downregulation
We next asked whether or not GEN could exert reversal effects on BRCA-1 after the removal of AhR agonist. MCF-7 cells were cultured in the presence of E2 or E2 plus TCDD for 24 h. Then, cells were cultured for an additional 24 and 48 h in fresh control medium containing E2 alone or E2 plus GEN or EGCG. The post-treatment with EGCG at 48 h, but not E2 alone or E2 plus EGCG at 24 h, restored BRCA-1 expression to E2 levels (Figure 5A, B). Similarly, the post-treatment with GEN for 24 and 48 h induced (∼0.5–0.7 fold) BRCA-1 expression compared with the E2 control. As previously shown (19), the expression of ERα was not influenced by cotreatment with TCDD plus E2 or GEN plus E2 after washout of TCDD (Figure 5C). Compared with control, the treatment with E2 did not induce cell growth (Figure 5D). We did, however, observe a significant reduction (∼60%) in cell proliferation with GEN (1 μM), irrespective of the presence or absence of E2. We extended the washout studies to longer time periods and found that the post-treatment for 7, 8, and 9 d with E2 plus GEN rescued BRCA-1 expression above E2 alone (Figure 6A, C). Reversal effects on BRCA-1 were also seen for EGCG at days 8 and 9 compared with the E2 control (Figure 6B, C). Overall, these results suggested that post-treatment with GEN and EGCG reversed AhR-dependent downregulation of BRCA-1, albeit with different efficacy (GEN > EGCG).
FIGURE 5.
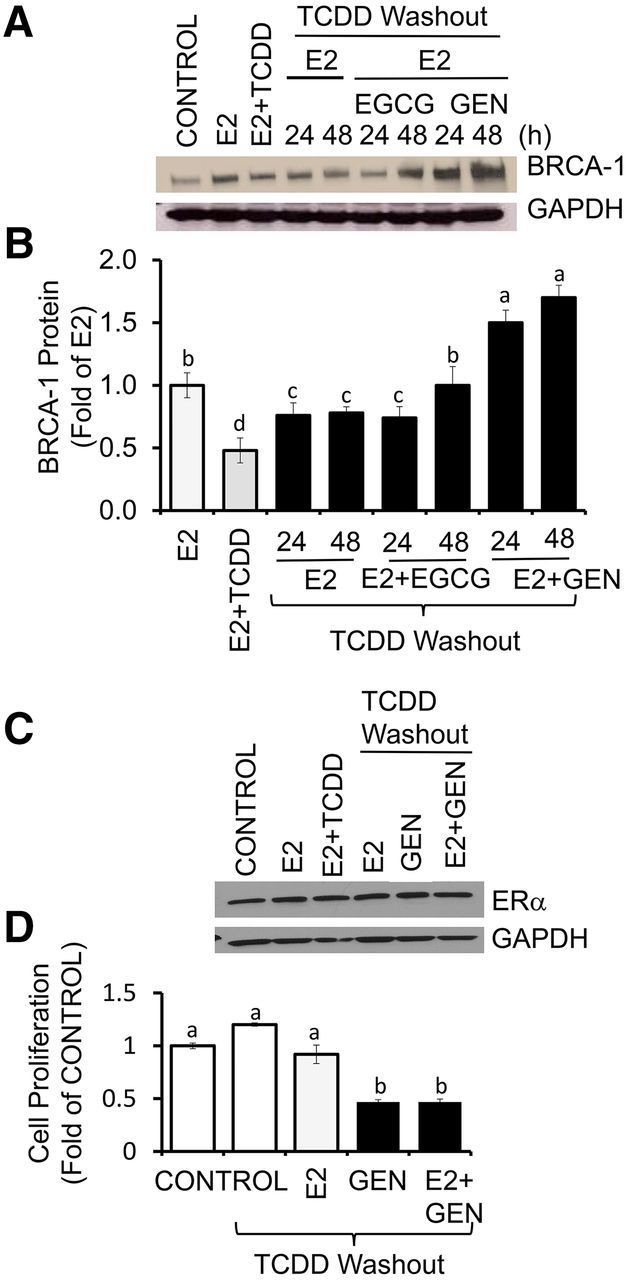
Short-term reversal effects of GEN and EGCG on BRCA-1 expression in breast cancer Michigan Cancer Foundation-7 cells with activated aromatic hydrocarbon receptor. Cells were precultured for 48 h in control medium plus E2 (10 nM) or E2 + TCDD (10 nM). Then, media were washed out and cells were cultured for an additional 24 and 48 h in the presence of E2 alone or E2 + 1 μM GEN or EGCG. Panels A and B show, respectively, bands and quantitation (fold of E2; means ± SEMs) of immunocomplexes for BRCA-1 from 3 independent experiments performed in duplicate. (C) Bands show immunocomplexes for ERα and the internal standard GAPDH. (D) Bars represent means ± SEMs of cell proliferation determined by MTT assay (fold of control) from 2 separate experiments with 6 replicates. Labeled means without a common letter differ, P < 0.05. BRCA-1, breast cancer 1, early onset; EGCG, (–)-epigallocatechin-3-gallate; ERα, estrogen receptor α E2, 17β-estradiol; GEN, genistein; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
FIGURE 6.

Long-term reversal effects of GEN and EGCG on BRCA-1 expression in breast cancer Michigan Cancer Foundation-7 cells with activated aromatic hydrocarbon receptor. Cells were precultured for 48 h in control medium plus E2 (10 nM) or E2 + TCDD (10 nM). Then, media were washed out and cells were cultured for an additional 7, 8, and 9 d in the presence of E2 alone or E2 + 1 μM GEN (A) or EGCG (B). Bands show immunocomplexes for BRCA-1 and the internal standard GAPDH. (C) Bars represent means ± SEMs of BRCA-1 expression (fold of E2) from 3 separate experiments with 3 replicates. Labeled means without a common letter differ, P < 0.05. BRCA-1, breast cancer 1, early onset; EGCG, (–)-epigallocatechin-3-gallate; E2, 17β-estradiol; GEN, genistein; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
GEN counteracts AhR-inducible BRCA1 CpG methylation
DNMT-1 is a maintenance DNA methylation enzyme (7). Therefore, we tested whether or not the BRCA-1 responses to AhR activation and GEN were linked to changes in DNMT-1 expression. We found that after washout of TCDD, the post-treatments of MCF-7 cells with E2 for 24 and 48 h were associated with a 1.8- and 2.8-fold accumulation of DNMT-1, respectively (Figure 7A, B). In contrast, the post-treatment with E2 plus GEN reduced DNMT-1 to control levels. These DNMT-1 changes correlated with induction (1.3-fold) in methylation of a CpG island flanking the BRCA1 transcription start site of exon 1A (Figure 7C, D). Conversely, the post-treatment with GEN lowered BRCA1 CpG methylation compared with E2 control (∼50%) and E2 plus TCDD (∼80%). Similarly, the post-treatment with EGCG reduced BRCA1 methylation compared with the E2 control (∼70%) and E2 plus TCDD (∼90%). These DNA demethylation results were consistent with earlier reports documenting reactivation of tumor suppressor genes by GEN (23, 37) and EGCG (36).
FIGURE 7.

GEN antagonizes DNMT-1 expression and BRCA1 CpG methylation in Michigan Cancer Foundation-7 cells with activated aromatic hydrocarbon receptor. Cells were cultured in control medium for 48 h and after washout of TCDD (10 nM) in the presence of E2 (10 nM) alone or E2 + GEN (1 μM) for 24 and 48 h. (A) Bands show representative immunocomplexes for DNMT-1 and the internal standard GAPDH. (B) Bars represent means ± SEMs of quantitation (fold of control) from 3 independent experiments performed in duplicate of DNMT-1 expression. (C) Location of CpG island flanking exon 1A of the BRCA1 gene and position (arrows) of oligonucleotides for methylation-specific PCR. (D) BRCA1 CpG methylation after pretreatment for 12 h with TCDD (10 nM) and post-treatment for 72 h with E2 (10 nM) + TCDD in the presence or absence of 1 μM GEN or EGCG. Bars represent means ± SEMs (fold of E2) from 3 independent experiments performed in quadruplicate. Labeled means without a common letter differ, P < 0.05. BRCA1, breast cancer 1, early onset; CpG, cytosine-phosphate guanine; DNMT-1, DNA methyltransferase 1; EGCG, (–)-epigallocatechin-3-gallate; E2, 17β-estradiol; GEN, genistein; M:U, methylated/unmethylated; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin.
GEN reverses BRCA1 silencing associated with constitutively active AhR
ERα-negative UACC-3199 cells harbor constitutive hypermethylated BRCA1 (4) and active AhR (22). Compared with control, E2 (10 μM) and GEN at 1 and 5 μM did not influence BRCA-1 expression, which, however, was induced ∼0.4-fold by 10 and 20 μM GEN (Figure 8A, B). As a positive control for AhR inhibition, we used the synthetic flavone αNF (2 μM) (14), which in UACC-3199 cells induced BRCA-1 and ERα expression irrespective of the presence or absence of E2 (Figure 8C, D). Western blots of cell lysates from MCF-7 cells (Figure 8C) provided a positive control for the detection of BRCA-1 and ERα immunocomplexes.
FIGURE 8.

Reversal effects of GEN and αNF on BRCA-1 silencing in UACC-3199 breast cancer cells with constitutively active aromatic hydrocarbon receptor. (A) ERα-negative UACC-3199 cells were cultured for 72 h in control medium and in the presence of E2 (10 nM) or various concentrations of GEN. Bands show representative immunocomplexes for BRCA-1 and the internal standard GAPDH. (B) Bars represent means ± SEMs (fold of control) of BRCA-1 expression from 3 independent experiments (n = 3) performed in duplicate. (C) Bands show immunocomplexes for BRCA-1, ERα, and GAPDH protein in UACC-3199 cells cultured for 72 h in control medium and after treatment with E2, αNF (2 μM), or αNF + E2 and in MCF-7 cells cultured in control medium or in the presence of E2. (D) Bars represent means ± SEMs (fold of control) of BRCA-1 and ERα expression from 3 independent experiments performed in duplicate. Labeled means without a common letter differ, P < 0.05. BRCA-1, breast cancer 1, early onset; ERα, estrogen receptor α E2, 17β-estradiol; GEN, genistein; MCF-7, Michigan Cancer Foundation-7; UACC, University of Arizona Cell Culture; αNF, α-naphthoflavone.
The treatment of UACC-3199 cells for 72 h with E2 and GEN (1 and 10 μM) or αNF (2 μM) (Figure 9A) reduced cell proliferation by ∼50% and 20%, respectively. The antiproliferative effects of GEN (10 μM) and αNF (2 μM) increased to ∼70% and 50%, respectively, in combination with E2. The expression of cyclin D1 (Figure 9B, C) was reduced by αNF (∼40%) and to a larger degree by 10 μM GEN (∼70%), regardless of the absence or presence of E2. Interestingly, although E2 alone reduced growth of UCAA-3199 cells (Figure 9A), it did not elicit measurable changes in cyclin D1 expression compared with the control (Figure 9C).
FIGURE 9.
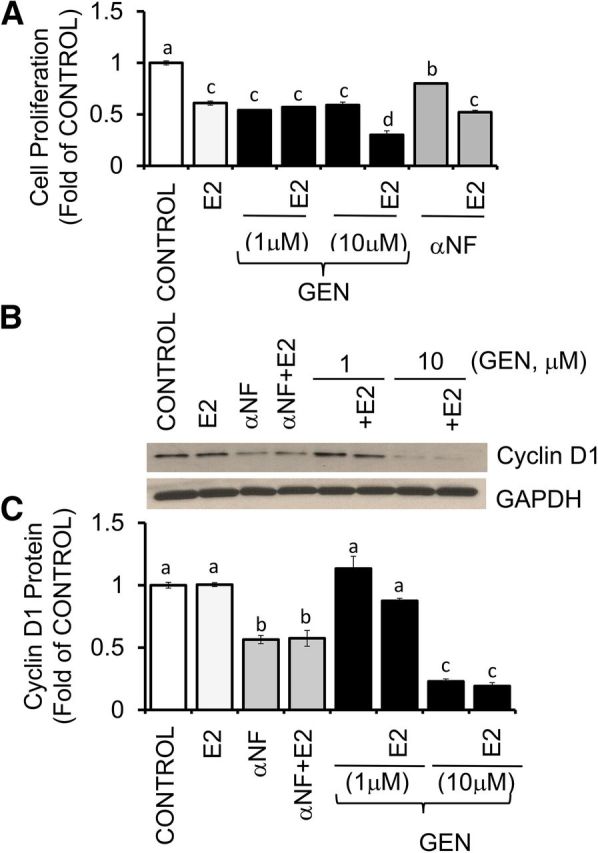
GEN and αNF inhibit cell proliferation in UACC-3199 cells with constitutively active aromatic hydrocarbon receptor. (A) UACC-3199 cells were cultured for 72 h in control medium and in the presence of E2 (10 nM), GEN (1 and 10 μM), and E2 + GEN or αNF (2 μM) and E2 + αNF. Bars represent means ± SEMs of quantitation (fold of control) of proliferation determined by MTT assay from 2 separate experiments with 5 replicates. (B) Bands show representative immunocomplexes for cyclin D1 and the internal standard GAPDH in UACC-3199 cells cultured for 72 h in control medium and after treatment with E2, αNF, E2 + αNF, or E2 + GEN. (C) Bars represent means ± SEMs of cyclin D1 quantitation (fold of control) from 2 independent experiments performed in duplicate. Labeled means without a common letter differ, P < 0.05. E2, 17β-estradiol; GEN, genistein; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; UACC, University of Arizona Cell Culture; αNF, α-naphthoflavone.
The upregulation of BRCA-1 protein by GEN in UACC-3199 cells was paralleled by demethylation of BRCA1 and ESR1 (ERα), as determined by qPCR amplification of bisulfonated DNA (Figure 10A). The treatment with αNF reduced by ∼60% BRCA1 CpG methylation, thus providing a positive control for methylation changes related to AhR. GEN and αNF stimulated BRCA1 mRNA by ∼1.0- and 6.0-fold, respectively, compared with E2 treatment (Figure 10B).
FIGURE 10.

Reversal effects of GEN and αNF on BRCA1 CpG methylation in UACC-3199 cells with constitutively active aromatic hydrocarbon receptor. (A) Bars represent means ± SEMs of quantitation from 2 independent experiments performed in triplicate of BRCA1 and ESR1 promoter CpG methylation (fold of control) in UACC-3199 cells after treatment for 72 h with E2 (10 nM), GEN (1, 5, and 10 μM), or αNF (2 μM). (B) Bars represent means ± SEMs of quantitation of BRCA1 mRNA (fold of E2) by qPCR from 2 independent experiments performed in quadruplicate with GEN (1 μM) and αNF (2 μM) and corrected for GAPDH mRNA as the internal standard. Labeled means without a common letter differ, P < 0.05. BRCA1, breast cancer 1, early onset; CpG, cytosine-phosphate-guanine; ESR1, estrogen receptor-1; E2, 17β-estradiol; GEN, genistein; M:U, methylated/unmethylated; UACC, University of Arizona Cell Culture; αNF, α-naphthoflavone.
GEN and αNF preferentially induce CYP1A1 in breast cancer cells with constitutively active AhR
CYP1A1 and CYP1B1 genes are transcriptional targets for AhR, which is constitutively active in subsets of preclinical and human breast tumors (38, 39). The treatment with GEN alone or in combination with E2 induced (1.2-fold) CYP1A1 mRNA (Figure 11A) but did not affect the expression of CYP1B1 (Figure 11B) or AhR (Figure 10C). As previously shown (22), we found that αNF induced a large increase (∼40-fold) in CYP1A1 (Figure 10A) and a smaller accumulation (1.0- to 1.7-fold) in CYP1B1 (Figure 11B) while lowering (∼50%) AhR (Figure 11C) mRNA. Overall, these cumulative data indicated that in ERα-negative breast epithelial cells with constitutively active AhR, both GEN and αNF stimulated BRCA-1 via CpG demethylation, an effect that was associated with reactivation of ESR1 and preferential activation of CYP1A1 over CYP1B1.
FIGURE 11.
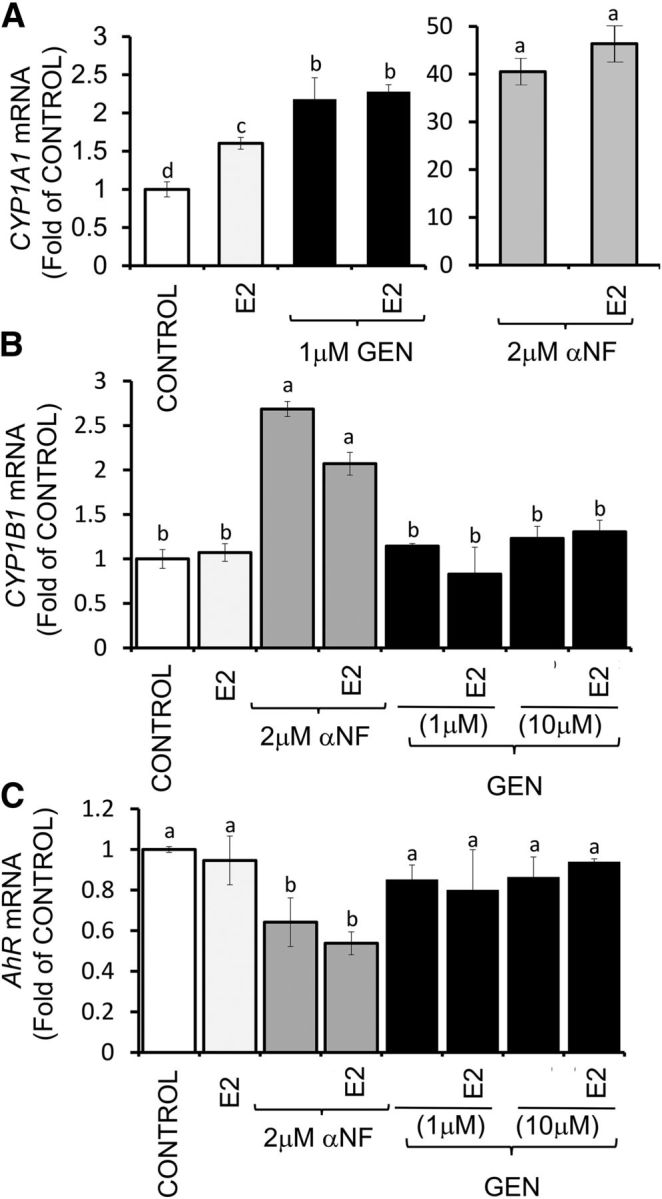
GEN and αNF differentially regulate expression of CYPs and AhR in UACC-3199 cells with constitutively active AhR. UACC-3199 cells were cultured for 72 h in control medium or in the presence of E2 (10 nM) + GEN or αNF. Bars represent means ± SEMs of quantitation (fold of control) of CYP1A1 (A), CYP1B1 (B), and AhR (C) mRNA from 2 independent experiments performed in quadruplicate and corrected for GAPDH mRNA as the internal standard. Labeled means without a common letter differ, P < 0.05. AhR, aromatic hydrocarbon receptor; CYP, cytochrome P450; CYP1A1, cytochrome P450-1A1; CYP1B1, cytochrome P450-1B1; E2, 17β-estradiol; GEN, genistein; UACC, University of Arizona Cell Culture; αNF, α-naphthoflavone.
Discussion
Historically, AhR has been investigated for its role in the transcriptional regulation of genes encoding phase I enzymes (e.g., CYP1A1, CYP1B1). However, studies also proposed a causative role for AhR in the etiology of breast tumorigenesis (39). Our published findings obtained from human breast cell lines (17–20) and rodent mammary tissue (21) indicated that the activation of AhR induced a pattern of BRCA1 methylation around exon 1a that overlapped with that observed in human sporadic breast tumors with reduced BRCA-1 expression (10) and overexpressing AhR (22). Therefore, the first objective of this study was to examine in ERα-positive breast cancer cells with wild-type BRCA1 (4) and a functional AhR pathway (40) the regulation by GEN of BRCA1 expression and CpG methylation. To activate AhR, we used the agonist TCDD because of its long half-life (∼8 y) (33). Therefore, changes in BRCA-1 expression could be analyzed without the confounding effects due to reactive metabolites. We focused on GEN as a cancer preventative because it is the major isoflavone in soy, and its consumption during early life has been linked to reduced breast cancer risk in Asian (41) and North American (42) women. We found that GEN exerted bimodal effects on MCF-7 cells with activated AhR. Doses ranging from 5 to 20 μM amplified the repressive effects of TCDD on BRCA-1 expression. The latter results were supportive of the tumor-promoting effects previously observed for GEN in ERα-dependent (43) and AhR-dependent (44) mammary tumor models. In contrast, the treatment of MCF-7 cells with lower doses of GEN (0.5 and 1.0 μM) counteracted the effects of TCDD and restored BRCA-1 expression to E2 levels. In previous studies, similar concentrations (0.5–1.0 μM) of GEN were shown to stimulate BRCA-1 expression in breast cancer cells (25). We also found that 1.0 μM GEN antagonized cell proliferation, an effect associated with downregulation of cyclin D1 and upregulation of p53. These results were in accord with earlier reports that highlighted the requirement for cyclin D1 in cell proliferation (45) and with other studies that showed that GEN induced G1 arrest in ERα-positive breast cancer cells (46) and protected against AhR-induced mammary tumorigenesis (27, 28).
The presence of putative binding elements for AhR and ERα in the DNMT1 gene (47, 48) may explain, at least in part, the increased DNMT-1 expression observed in MCF-7 cells treated with TCDD and E2. Conversely, GEN reduced DNMT-1 expression and BRCA1 CpG methylation. Repression of DNMT-1 by GEN has been described previously in human breast cancer cells (MCF-7, MDA-MB-231) (29). EGCG provided a positive control for BRCA1 methylation experiments with GEN in MCF-7 cells. It has been shown to reactivate the expression of methylated-silenced tumor suppressor genes (36).
The second objective of this study was to test if GEN could reactivate BRCA1 under conditions of constitutive expression and activation of AhR. For this purpose, we turned to the ERα-negative UACC-3199 cell line, which was derived from a sporadic human breast tumor harboring wild-type, but hypermethylated, BRCA1 (4), and constitutively high levels of AhR (22). GEN doses of 10 and 20 μM induced BRCA-1 expression, whereas lower concentrations (1 and 5 μM) had no effects. These data suggest that higher amounts of GEN may be needed to trigger a BRCA-1 response in ERα-negative breast tumors overexpressing AhR. The CpG demethylation of BRCA1 and ESR1 observed in UACC-3199 cells with 10 μM GEN may be clinically relevant because comparable serum concentrations have been measured in animals (49, 50) and humans (51, 52) for GEN (∼4.5 μM) and for total isoflavones (∼3.0–7.0 μM). Previous studies of isoflavones and catechins showed weak affinity for AhR with half maximal inhibitory concentration (IC50) >50–200 μM (53). Therefore, it is unlikely that GEN and EGCG induced demethylation of BRCA1 through physical interference with AhR. Possibly, the CpG demethylating effects of GEN and EGCG could be due to reduced DNMT-1 expression (29) and activity on BRCA1 (20) and ESR1, as recently reported in ERα-negative breast cancer cells (54).
Previous studies documented the antiproliferative effects of E2 and GEN in ERα-negative breast cancer cells (55). GEN was shown to have greater affinity for ERβ than for ERα, whereas the binding affinities of E2 for ERα and ERβ were equivalent (56). Therefore, E2 and GEN may inhibit the growth of ERα-negative cells by targeting ERβ (57). In support of this idea, agonism and overexpression of ERβ have been shown to attenuate the proliferation of triple-negative breast cancers through cell cycle arrest in the G1 phase (58) and to reduce tumor formation by causing G2 phase arrest (59). Furthermore, CpG demethylation of BRCA1 and ESR1 (ERα) by GEN and αNF in UACC-3199 cells suggested that regimens based on dietary flavonoids may have clinical relevance for therapy of tumors with BRCAness. In support of this inference, we noted that GEN and αNF hampered the expression of cyclin D1 and the growth of UACC-3199 cells. αNF is an AhR antagonist at the BRCA1 gene (60), with IC50 approaching ∼0.4 μM (53), and a potent aromatase inhibitor (61). It shares structural similarity with GEN and the flavonol galangin (3,5,7-trihydroxyflavone; IC50 ∼0.2 μM) (53), which was found to block the proliferation of ERα-negative breast cancer cells overexpressing AhR (62).
Finally, we reported that treatment of UACC-3199 cells with GEN and αNF activated preferentially CYP1A1 with only modest (αNF) or no (GEN) effects on CYP1B1. This selective activation of CYP1A1 may have therapeutic relevance because increased CYP1A1 expression associates with reduced basal AhR activity (63) and increased apoptosis (64). In addition, a reduction in 4-hydroxylation of E2 by αNF, a reaction catalyzed by CYP1B1, was shown to reduce the production of the highly carcinogenic metabolite 4-hydroxy-E2 and mammary tumorigenesis (65). In summary, the results of this study suggest preventative effects for GEN and EGCG against proliferation and AhR-mediated BRCA1 CpG methylation in ERα-positive breast cancer cells. We also presented evidence that GEN and αNF, selected respectively, as a prototype flavone and AhR antagonist, may hold promise for reactivation of BRCA1 in ERα-negative sporadic breast tumors with constitutively activate AhR.
Acknowledgments
The authors' responsibilities were as follows—OIS, AJP, and MGD: conducted the research and data collection; OIS, AJP, MGD, TCD, and DFR: were responsible for the data analysis and interpretation and contributed to the project conception and development; DFR and OIS: wrote the manuscript and had primary responsibility for final content; and all authors: read and approved the final manuscript.
Abbreviations
- AhR
aromatic hydrocarbon receptor
- BRCA1
breast cancer 1, early-onset
- CpG
cytosine-phosphate-guanine
- CYP
cytochrome P450
- DNMT
DNA methyltransferase
- EGCG
(–)-epigallocatechin-3-gallate
- ER
estrogen receptor
- ESR1
estrogen receptor-1
- E2
17β-estradiol
- GEN
genistein
- IC50
half maximal inhibitory concentration
- M
methylated
- MCF-7
Michigan Cancer Foundation-7
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- RPMI
Roswell Park Memorial Institute
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- U
unmethylated
- UACC-3199
University of Arizona Cell Culture-3199
- αNF
α-naphthoflavone
References
- 1. Mullan PB, Quinn JE, Harkin DP.. The role of BRCA1 in transcriptional regulation and cell cycle control. Oncogene 2006;25:5854–63. [DOI] [PubMed] [Google Scholar]
- 2. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. . A strong candidate for the breast and ovarian cancer susceptibility gene BRC. Science 1994;266:66–71. [DOI] [PubMed] [Google Scholar]
- 3. Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, et al. The Breast Cancer Linkage Consortium. Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am J Hum Genet 1998;62:676–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rice JC, Massey-Brown KS, Futscher BW.. Aberrant methylation of the BRCA1 CpG island promoter is associated with decreased BRCA1 mRNA in sporadic breast cancer cells. Oncogene 1998;17:1807–12. [DOI] [PubMed] [Google Scholar]
- 5. Thompson ME, Jensen RA, Obermiller PS, Page DL, Holt JT.. Decreased expression of BRCA1 accelerates growth and is often present during sporadic breast cancer progression. Nat Genet 1995;9:444–50. [DOI] [PubMed] [Google Scholar]
- 6. Lips EH, Mulder L, Oonk A, van der Kolk LE, Hogervorst FB, Imholz AL, Wesseling J, Rodenhuis S, Nederlof PM.. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br J Cancer 2013;108:2172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baylin SB, Jones PA.. A decade of exploring the cancer epigenome—biological and translational implications. Nat Rev Cancer 2011;11:726–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Melchor L, Benítez J.. The complex genetic landscape of familial breast cancer. Hum Genet 2013;132:845–63. [DOI] [PubMed] [Google Scholar]
- 9. Matros E, Wang ZC, Lodeiro G, Miron A, Iglehart JD, Richardson AL.. BRCA1 promoter methylation in sporadic breast tumors: relationship to gene expression profiles. Breast Cancer Res Treat 2005;91:179–86. [DOI] [PubMed] [Google Scholar]
- 10. Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, et al. . Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst 2000;92:564–9. [DOI] [PubMed] [Google Scholar]
- 11. Wei M, Xu J, Dignam J, Nanda R, Sveen L, Fackenthal J, Grushko TA, Olopade OI.. Estrogen receptor alpha, BRCA1, and FANCF promoter methylation occur in distinct subsets of sporadic breast cancers. Breast Cancer Res Treat 2008;111:113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hosey AM, Gorski JJ, Murray MM, Quinn JE, Chung WY, Stewart GE, James CR, Farragher SM, Mulligan JM, Scott AN.. Molecular basis for estrogen receptor alpha deficiency in BRCA1-linked breast cancer. J Natl Cancer Inst 2007;99:1683–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Murphy CG, Moynahan ME.. BRCA gene structure and function in tumor suppression: a repair-centric perspective. Cancer J 2010;16:39–47. [DOI] [PubMed] [Google Scholar]
- 14. Denison MS, Nagy SR.. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu Rev Pharmacol Toxicol 2003;43:309–34. [DOI] [PubMed] [Google Scholar]
- 15. Jeffy BD, Hockings JK, Kemp MQ, Morgan SS, Hager JA, Beliakoff J, Whitesell LJ, Bowden GT, Romagnolo DF.. An estrogen receptor-alpha/p300 complex activates the BRCA-1 promoter at an AP-1 site that binds Jun/Fos transcription factors: repressive effects of p53 on BRCA-1 transcription. Neoplasia 2005;7:873–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Romagnolo D, Annab LA, Thompson TE, Risinger JI, Terry LA, Barrett JC, Afshari CA.. Estrogen upregulation of BRCA1 expression with no effect on localization. Mol Carcinog 1998;22:102–9. [DOI] [PubMed] [Google Scholar]
- 17. Hockings JK, Thorne PA, Kemp MQ, Morgan SS, Selmin O, Romagnolo DF.. The ligand status of the aromatic hydrocarbon receptor modulates transcriptional activation of BRCA-1 promoter by estrogen. Cancer Res 2006;66:2224–32. [DOI] [PubMed] [Google Scholar]
- 18. Hockings JK, Degner SC, Morgan SS, Kemp MQ, Romagnolo DF.. Involvement of a specificity proteins-binding element in regulation of basal and estrogen-induced transcription activity of the BRCA1 gene. Breast Cancer Res 2008;10:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Papoutsis AJ, Lamore SD, Wondrak GT, Selmin OI, Romagnolo DF.. Resveratrol prevents epigenetic silencing of BRCA-1 by the aromatic hydrocarbon receptor in human breast cancer cells. J Nutr 2010;140:1607–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Papoutsis AJ, Borg JL, Selmin OI, Romagnolo DF.. BRCA-1 promoter hypermethylation and silencing induced by the aromatic hydrocarbon receptor-ligand TCDD are prevented by resveratrol in MCF-7 cells. J Nutr Biochem 2012;23:1324–32. [DOI] [PubMed] [Google Scholar]
- 21. Papoutsis AJ, Selmin OI, Borg JL, Romagnolo DF.. Gestational exposure to the AhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin induces BRCA-1 promoter hypermethylation and reduces BRCA-1 expression in mammary tissue of rat offspring: preventive effects of resveratrol. Mol Carcinog 2015;54:261–9. [DOI] [PubMed] [Google Scholar]
- 22. Romagnolo DF, Papoutsis AJ, Laukaitis C, Selmin OI.. Constitutive expression of AhR and BRCA-1 promoter CpG hypermethylation as biomarkers of ERα-negative breast tumorigenesis. BMC Cancer 2015;15:1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Y, Chen H, Hardy TM, Tollefsbol TO.. Epigenetic regulation of multiple tumor-related genes leads to suppression of breast tumorigenesis by dietary genistein. PLoS One 2013;8:e54369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fang M, Chen D, Yang CS.. Dietary polyphenols may affect DNA methylation. J Nutr 2007;137(Suppl):223S–8S. [DOI] [PubMed] [Google Scholar]
- 25. Fan S, Meng Q, Auborn K, Carter T, Rosen EM.. BRCA1 and BRCA2 as molecular targets for phytochemicals indole-3-carbinol and genistein in breast and prostate cancer cells. Br J Cancer 2006;94:407–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Phrakonkham P, Brouland JP, Saad Hel S, Bergès R, Pimpie C, Pocard M, Canivenc-Lavier MC, Perrot-Applanat M.. Dietary exposure in utero and during lactation to a mixture of genistein and an antiandrogen fungicide in a rat mammary carcinogenesis model. Reprod Toxicol 2015;54:101–9. [DOI] [PubMed] [Google Scholar]
- 27. Murrill WB, Brown NM, Zhang JX, Manzolillo PA, Barnes S, Lamartiniere CA.. Prepubertal genistein exposure suppresses mammary cancer and enhances gland differentiation in rats. Carcinogenesis 1996;17:1451–7. [DOI] [PubMed] [Google Scholar]
- 28. de Assis S, Warri A, Benitez C, Helferich W, Hilakivi-Clarke L.. Protective effects of prepubertal genistein exposure on mammary tumorigenesis are dependent on BRCA1 expression. Cancer Prev Res (Phila) 2011;4:1436–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xie Q, Bai Q, Zou LY, Zhang QY, Zhou Y, Chang H, Yi L, Zhu JD, Mi MT.. Genistein inhibits DNA methylation and increases expression of tumor suppressor genes in human breast cancer cells. Genes Chromosomes Cancer 2014;53:422–31. [DOI] [PubMed] [Google Scholar]
- 30. Kemp MQ, Jeffy BD, Romagnolo DF.. Conjugated linoleic acid inhibits cell proliferation through a p53-dependent mechanism: effects on the expression of G1-restriction points in breast and colon cancer cells. J Nutr 2003;133:3670–7. [DOI] [PubMed] [Google Scholar]
- 31. Strecker JR, Lauritzen C, Goessens L.. Plasma concentrations of unconjugated and conjugated estrogens and gonadotrophins following application of various estrogen preparations after oophorectomy and in the menopause. Maturitas 1979;1:183–90. [DOI] [PubMed] [Google Scholar]
- 32. Chuffa LG, Lupi-Júnior LA, Costa AB, Amorim JP, Seiva FR.. The role of sex hormones and steroid receptors on female reproductive cancers. Steroids 2017;118:93–108. [DOI] [PubMed] [Google Scholar]
- 33. Landi MT, Consonni D, Patterson DG Jr, Needham LL, Lucier G, Brambilla P, Cazzaniga MA, Mocarelli P, Pesatori AC, Bertazzi PA, et al. . 2,3,7,8-Tetrachlorodibenzo-p-dioxin plasma levels in Seveso 20 years after the accident. Environ Health Perspect 1998;106:273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eskenazi B, Mocarelli P, Warner M, Needham L, Patterson DG Jr, Samuels S, Turner W, Gerthoux PM, Brambilla P.. Relationship of serum TCDD concentrations and age at exposure of female residents of Seveso, Italy. Environ Health Perspect 2004;112:22–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Patterson DG Jr, Needham LL, Pirkle JL, Roberts DW, Bagby J, Garrett WA, Andrews JS Jr, Falk H, Bernert JT, Sampson EJ, et al. . Correlation between serum and adipose tissue levels of 2,3,7,8-tetrachlorodibenzo-p-dioxin in 50 persons from Missouri. Arch Environ Contam Toxicol 1988;17:139–43. [DOI] [PubMed] [Google Scholar]
- 36. Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, Welsh W, Yang CS.. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res 2003;63:7563–70. [PubMed] [Google Scholar]
- 37. Fang MZ, Chen D, Sun Y, Jin Z, Christman JK, Yang CS.. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, andMGMT genes by genistein and other isoflavones from soy. Clin Cancer Res 2005;11:7033–41. [DOI] [PubMed] [Google Scholar]
- 38. Yang X, Solomon S, Fraser LR, Trombino AF, Liu D, Sonenshein GE, Hestermann EV, Sherr DH.. Constitutive regulation of CYP1B1 by the aryl hydrocarbon receptor (AhR) in pre-malignant and malignant mammary tissue. J Cell Biochem 2008;104:402–17. [DOI] [PubMed] [Google Scholar]
- 39. Schlezinger JJ, Liu D, Farago M, Seldin DC, Belguise K, Sonenshein GE, Sherr DH.. A role for the aryl hydrocarbon receptor in mammary gland tumorigenesis. Biol Chem 2006;387:1175–87. [DOI] [PubMed] [Google Scholar]
- 40. Merchant M, Krishnan V, Safe S.. Mechanism of action of alpha-naphthoflavone as an Ah receptor antagonist in MCF-7 human breast cancer cells. Toxicol Appl Pharmacol 1993;120:179–85. [DOI] [PubMed] [Google Scholar]
- 41. Shu XO, Jin F, Dai Q, Wen W, Potter JD, Kushi LH, Ruan Z, Gao YT, Zheng W.. Soy food intake during adolescence and subsequent risk of breast cancer among Chinese women. Cancer Epidemiol Biomarkers Prev 2001;10:483–8. [PubMed] [Google Scholar]
- 42. Thanos J, Cotterchio M, Boucher BA, Kreiger N, Thompson LU.. Adolescent dietary phytoestrogen intake and breast cancer risk (Canada). Cancer Causes Control 2006;17:1253–61. [DOI] [PubMed] [Google Scholar]
- 43. Hsieh CY, Santel RC, Haslam SZ, Helferich WG.. Estrogenic effects of genistein on the growth of estrogen receptor-positive human breast cancer (MCF-7) cells in vitro and in vivo. Cancer Res 1998;58:3833–8. [PubMed] [Google Scholar]
- 44. Day JK, Besch-Williford C, McMann TR, Hufford MG, Lubahn DB, MacDonald RS.. Dietary genistein increased DMBA-induced mammary adenocarcinoma in wild-type, but not ER alpha KO, mice. Nutr Cancer 2001;39:226–32. [DOI] [PubMed] [Google Scholar]
- 45. Yang K, Hitomi M, Stacey DW.. Variations in cyclin D1 levels through the cell cycle determine the proliferative fate of a cell. Cell Div 2006;1:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tsuboy MS, Marcarini JC, de Souza AO, de Paula NA, Dorta DJ, Mantovani MS, Ribeiro LR.. Genistein at maximal physiologic serum levels induces G0/G1 arrest in MCF-7 and HB4a cells, but not apoptosis. J Med Food 2014;17:218–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aluru N, Kuo E, Helfrich LW, Karchner SI, Linney EA, Pais JE, Franks DG.. Developmental exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin alters DNA methyltransferase (Dnmt) expression in zebrafish (Danio rerio). Toxicol Appl Pharmacol 2015;284:142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shi JF, Li XJ, Si XX, Li AD, Ding HJ, Han X, Sun YJ.. ERα positively regulated DNMT1 expression by binding to the gene promoter region in human breast cancer MCF-7 cells. Biochem Biophys Res Commun 2012;427:47–53. [DOI] [PubMed] [Google Scholar]
- 49. Zhang X, Cook KL, Warri A, Cruz IM, Rosim M, Riskin J, Helferich W, Doerge D, Clarke R, Hilakivi-Clarke L.. Lifetime genistein intake increases the response of mammary tumors to tamoxifen in rats. Clin Cancer Res 2017;23:814–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Supko J, Malspeis L.. Plasma pharmacokinetics of genistein in mice. Int J Oncol 1995;7:847–54. [DOI] [PubMed] [Google Scholar]
- 51. Wiseman H, Casey K, Bowey EA, Duffy R, Davies M, Rowland IR, Lloyd AS, Murray A, Thompson R, Clarke DB.. Influence of 10 wk of soy consumption on plasma concentrations and excretion of isoflavonoids and on gut microflora metabolism in healthy adults. Am J Clin Nutr 2004;80:692–9. [DOI] [PubMed] [Google Scholar]
- 52. Mathey J, Lamothe V, Coxam V, Potier M, Sauvant P, Bennetau-Pelissero C.. Concentrations of isoflavones in plasma and urine of post-menopausal women chronically ingesting high quantities of soy isoflavones. J Pharm Biomed Anal 2006;41:957–65. [DOI] [PubMed] [Google Scholar]
- 53. Ashida H, Fukuda I, Yamashita T, Kanazawa K.. Flavones and flavonols at dietary levels inhibit a transformation of aryl hydrocarbon receptor induced by dioxin. FEBS Lett 2000;476:213–7. [DOI] [PubMed] [Google Scholar]
- 54. Li Y, Meeran SM, Patel SN, Chen H, Hardy TM, Tollefsbol TO.. Epigenetic reactivation of estrogen receptor-α (ERα) by genistein enhances hormonal therapy sensitivity in ERα-negative breast cancer. Mol Cancer 2013;12:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rajah TT, Peine KJ, Du N, Serret CA, Drews NR.. Physiological concentrations of genistein and 17β-estradiol inhibit MDA-MB-231 breast cancer cell growth by increasing BAX/BCL-2 and reducing pERK1/2. Anticancer Res 2012;4:1181–91. [PubMed] [Google Scholar]
- 56. Kuhl H.. Pharmacology of estrogens and progestogens: influence of different routes of administration. Climacteric 2005;8(Suppl 1):3–63. [DOI] [PubMed] [Google Scholar]
- 57. Wisinski KB, Xu W, Tevaarwerk AJ, Saha S, Kim K, Traynor A, Dietrich L, Hegeman R, Patel D, Blank J, et al. . Targeting estrogen receptor beta in a phase 2 study of high-dose estradiol in metastatic triple-negative breast cancer: a Wisconsin Oncology Network study. Clin Breast Cancer 2016;16:256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shanle EK, Zhao Z, Hawse J, Wisinski K, Keles S, Yuan M, Xu W.. Research resource: global identification of estrogen receptor beta target genes in triple negative breast cancer cells. Mol Endocrinol 2013;27:1762–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC.. Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res 2004;64:423–8. [DOI] [PubMed] [Google Scholar]
- 60. Jeffy BD, Chen EJ, Gudas JM, Romagnolo DF.. Disruption of cell cycle kinetics by benzo(a)pyrene: inverse expression patterns of BRCA-1 and p53 in MCF-7 cells arrested in S and G2. Neoplasia 2000;2:460–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kellis JT Jr, Vickery LE.. Inhibition of human estrogen synthetase (aromatase) by flavones. Science 1984;225:1032–4. [DOI] [PubMed] [Google Scholar]
- 62. Murray TJ, Yang X, Sherr DH.. Growth of a human mammary tumor cell line is blocked by galangin, a naturally occurring bioflavonoid, and is accompanied by down-regulation of cyclins D3, E, and A. Breast Cancer Res 2006;8:R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chang CY, Puga A.. Constitutive activation of the aromatic hydrocarbon receptor. Mol Cell Biol 1998;18:525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ciolino HP, Dankwah M, Yeh GC.. Resistance of MCF-7 cells to dimethyl-benz(a)anthracene-induced apoptosis is due to reduced CYP1A1 expression. Int J Oncol 2002;21:385–91. [PubMed] [Google Scholar]
- 65. Mense SM, Singh B, Remotti F, Liu X, Bhat HK.. Vitamin C and alpha-naphthoflavone prevent estrogen-induced mammary tumors and decrease oxidative stress in female ACI rats. Carcinogenesis 2009;30:1202–8. [DOI] [PMC free article] [PubMed] [Google Scholar]