

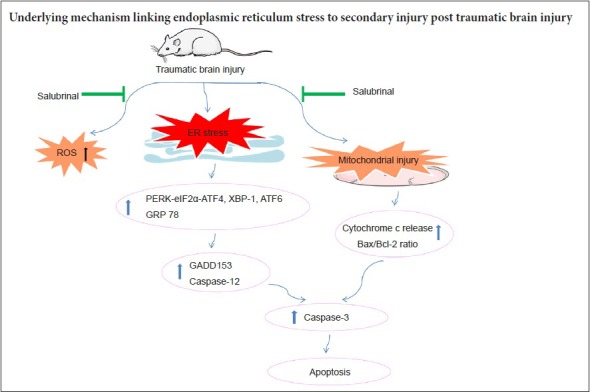
Keywords: nerve regeneration, traumatic brain injury, endoplasmic reticulum stress, apoptosis, mitochondria, reactive oxygen species, unfolded protein response, secondary brain injury, salubrinal, neural regeneration
Abstract
Apoptosis after traumatic brain injury has been shown to be a major factor influencing prognosis and outcome. Endoplasmic reticulum stress may be involved in mitochondrial mediated neuronal apoptosis. Therefore, endoplasmic reticulum stress has become an important mechanism of secondary injury after traumatic brain injury. In this study, a rat model of traumatic brain injury was established by lateral fluid percussion injury. Fluorescence assays were used to measure reactive oxygen species content in the cerebral cortex. Western blot assays were used to determine expression of endoplasmic reticulum stress-related proteins. Hematoxylin-eosin staining was used to detect pathological changes in the cerebral cortex. Transmission electron microscopy was used to measure ultrastructural changes in the endoplasmic reticulum and mitochondria. Our results showed activation of the endoplasmic reticulum stress-related unfolded protein response. Meanwhile, both the endoplasmic reticulum stress response and mitochondrial apoptotic pathway were activated at different stages post-traumatic brain injury. Furthermore, pretreatment with the endoplasmic reticulum stress inhibitor, salubrinal (1 mg/kg), by intraperitoneal injection 30 minutes before injury significantly inhibited the endoplasmic reticulum stress response and reduced apoptosis. Moreover, salubrinal promoted recovery of mitochondrial function and inhibited activation of the mitochondrial apoptotic pathway post-traumatic brain injury. These results suggest that endoplasmic reticulum stress might be a key factor for secondary brain injury post-traumatic brain injury.
Introduction
Traumatic brain injury (TBI) is a major socioeconomic problem and common cause of death and disability worldwide (Langlois et al., 2006; Rao et al., 2015). TBI can cause two types of neural tissue injury. The first is primary injury, which is mainly attributable to the initial physical forces applied to the brain at the moment of impact. Currently, operational treatments are still lacking. The second type is secondary injury, which develops over a period of hours or even days, and involves reactive oxygen species (ROS) generation, vascular abnormalities, endoplasmic reticulum (ER) stress, mitochondrial dysfunction, apoptosis, neuroinflammation, and excitotoxicity. There are many potential targets for intervention during this process (Gajavelli et al., 2015; Cao et al., 2016; Sun et al., 2016; Yang et al., 2017). However, the mechanisms underlying secondary brain injury post-TBI remain to be investigated.
ER stress is characterized by accumulation of unfolded and misfolded ER proteins. There are three major ER stress sensors that mediate the unfolded protein response (UPR) signaling pathway: protein kinase R (PKR)-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol requiring enzyme 1 (IRE1) (Sano and Reed, 2013; Li et al., 2014). Moderate UPR may alleviate cellular dysfunction and increase the chances of survival, however when ER stress is sustained and/or UPR fails to restore ER homeostasis, apoptosis is triggered, which results in cell death (Raghubir et al., 2011). ER stress is an intricate mechanism that interferes with numerous brain damage responses (Yamamoto et al., 2006; Gulyaeva, 2015; Go et al., 2017) during brain ischemic reperfusion injury, neurodegenerative disease, subarachnoid hemorrhage, and epilepsy. Thus, ER stress clearly plays an essential role in determining neuronal fate during various brain injury diseases (Raghubir et al., 2011; Xue et al., 2017). Recent studies have suggested that pathological and behavioral deficits during secondary injury cascades of TBI might contribute to ER stress (Sun et al., 2016; Yin et al., 2017). Nonetheless, more research is needed to explore these mechanisms.
Apoptosis has been confirmed in TBI, and is regarded as a main factor affecting outcome and prognosis post-TBI (Nathoo et al., 2004; Lorente et al., 2015). Apoptotic cell death occurs primarily through intrinsic and extrinsic signaling pathways, and often involves ER stress as well as mitochondrial and membrane-mediated pathways (Jin et al., 2015; Sun et al., 2016). Previous studies have identified numerous crosstalk signaling mechanisms between ER stress and mitochondria-mediated apoptosis (Burton et al., 2017; Plácido et al., 2017; Xue et al., 2017). Indeed, a recent study showed that ER stress may be primarily related to mitochondria-regulated apoptosis (Sophonnithiprasert et al., 2017). In addition, oxidative stress is also a crucial factor in causing apoptosis during secondary injury post-TBI, while prolonged ER stress is well known for inducing oxidative stress (Go et al., 2017). Consequently, the underlying interconnection and regulatory mechanisms of apoptosis during secondary injury post-TBI needs further investigation.
Based on these reasons above, we established the lateral fluid-percussion brain injury model in rats to mimic the insult of TBI and determine whether ER stress is a crucial factor in TBI-induced apoptosis. Our aim was to identify the underlying mechanisms linking ER stress to brain damage, and provide a novel strategy for preventing and treating secondary brain injury post-TBI.
Materials and Methods
Animals
Eighty-four specific-pathogen-free male Sprague-Dawley rats aged 12 weeks and weighing 220–250 g were individually housed under controlled environmental conditions with 12-hour light/dark cycles and unrestricted access to pellet food and water. Rats were purchased from the Experimental Animal Center of the Southern Medical University in China (Certification: SYXK (Yue) 2016-0167). All surgical interventions were performed under anesthesia with a mixture of 13.3% urethane and 0.5% chloralose (0.65 mL/100 g body weight, intraperitoneally) using a standardized protocol established in our laboratory. The experimental procedures were consistent with the ethical requirements established by the Experimental Animal Ethics and Welfare Committee of Guangzhou General Hospital of Guangzhou Military Command of China (No. 2016030095).
Experimental groups and drug administration
The first experiment analyzed ROS production and expression of ER stress- and apoptosis-related proteins in the injured cortex at different time points. Rats were divided into six subgroups including a sham control group and groups sacrificed at 1, 3, 6, 12, and 24 hours post-TBI (n = 6/group). The second experiment was performed to study the effect of the ER stress inhibitor, salubrinal (Sal), post-TBI. Rats were randomized into four groups including sham control, TBI, sham + Sal, and TBI + Sal groups. Salubrinal (1 mg/kg) (Selleck, Houston, TX, USA) was injected intraperitoneally 30 minutes before TBI (Nakka et al., 2010; Zhang et al., 2015) (n = 6/group).
Model establishment of TBI
Surgical procedures were performed as previously described (Chen et al., 2013; Yang et al., 2017). Briefly, anesthetized rats were placed in a stereotaxic frame (Ruiwode, Shenzhen, Guangdong Province, China). After incision of the scalp, the temporal muscles were reflected and a 4.8 mm craniotomy drilled (2.5 mm lateral to the sagittal sinus and centered between bregma and lambda). A hollow female Luer-Lok fitting was placed directly over the dura and rigidly fixed using dental cement. Prior to trauma induction, the female Luer-Lok was connected to the fluid percussion injury device via a transducer (Biomedical Engineering Facility, Medical College of Virginia, Virginia Beach, VA, USA). For TBI, a metal pendulum was released from a pre-selected height, leading to rapid injection of normal saline into the closed cranial cavity. A pulse of increased intracranial pressure of 21–23 ms duration was elicited, and controlled, and recorded by an oscilloscope (Agilent 54622D; MEGAZoom, Munich, Germany). Severity of the injury inflicted was altered by adjusting the amount of force generated by the pendulum. An injury level of severe severity was induced (3.5 ± 0.2 atmospheres) (Chen et al., 2013). Sham animals underwent identical preparatory procedures, including craniotomy, but were not injured.
Isolation of rat cortical neurons
To rapidly isolate cortical neurons from the injured-side of the rat brain, a previously described method with some minor modifications was used (Wang et al., 2013; Yang et al., 2017). Following completion of the TBI procedure, the injured-side cortex was cut into fragments, and the cells dissociated by incubation for 30 minutes at 37°C with 2 mg/mL papain in Dulbecco’s modified Eagle’s minimal essential medium (DMEM). To achieve a pure cell population, the immune adherence method was used. Cell suspensions were poured into anti-neural cell adhesion molecule-coated Petri dishes (Millipore, Boston, MA, USA) and placed on a shaker for one hour. Adhered cells were then collected and trypan blue used to exclude non-viable cells (Yang et al., 2017).
Western blot assay
Protein was extracted from the injured-side cortex using a Total Protein Extraction Kit (Beyotime Institute of Biotechnology, Guangzhou, Guangdong Province, China), following the manufacturer’s protocols. The protein concentration of extracts was determined using an Enhanced Bicinchoninic Acid Protein Assay Kit (Beyotime Institute of Biotechnology). Western blot assay was performed, as described previously (Yang et al., 2017), using primary antibodies against the following proteins: rabbit anti-phospho-PERK, rabbit anti-PERK, rabbit anti-phospho-eukaryotic translation initiation factor 2α (eIF2α), rabbit anti-eIF2α, rabbit anti-spliced X-box binding protein 1 (XBP-1), rabbit anti-78 kDa glucose-regulated protein (GRP78), mouse anti-DNA damage-inducible gene 153 (GADD153), rabbit anti-Bcl-2-associated X protein (Bax) (all Cell Signaling Technology, Danvers, MA, USA), rabbit anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH), rabbit anti-ATF4, rabbit anti-cleaved ATF6, rabbit anti-caspase-12, rabbit anti- B-cell lymphoma 2 (Bcl-2), and mouse anti-cytochrome c (Cyt c) (all Abcam, Cambridge, UK), with each antibody diluted 1:1,000. Horseradish peroxidase-conjugated anti-rabbit/mouse IgG antibody was used as the secondary antibody (1:2000; Abcam) and was incubated at 37°C for 2 hours. Signal was detected using enhanced chemiluminescence substrate (Beyotime Institute of Biotechnology). Quantified grayscales of band intensities were quantified using Image-Quant TL software (GE Healthcare, Piscataway, NJ, USA).
Measurement of ROS
To analyze the kinetics of ROS generation, neurons were isolated post-TBI at different indicated times. ROS was detected using the fluorescent probe, dichloro-dihydro-fluorescein diacetate (DCFH-DA) (Molecular Probes, Carlsbad, CA, USA). Neurons were incubated in the dark with 10 μM DCFH-DA for 30 minutes at 37°C. DCFH-DA oxidized by ROS produces green fluorescent dichlorofluorescein (DCF). The fluorescence intensity generated by ROS probes was analyzed by flow cytometric analysis (Becton Dickinson, Franklin, NJ, USA) (Hiebert et al., 2015).
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential (MMP, ΔΨm) was measured using the fluorescent probe, JC-1 (Molecular Probes). In mitochondria with normal ΔΨm, JC-1 forms red fluorescent aggregates, whereas in damaged, depolarized mitochondria, JC-1 forms green fluorescent monomers. Isolated neurons were incubated in DMEM containing 5 µmol/L JC-1 for 15 minutes at 37°C. Relative fluorescence was subsequently measured by flow cytometry (Becton Dickinson). Data were analyzed using BD FAC Suite software (Wang et al., 2013; Yang et al., 2017).
Histopathological analysis
Rats were anesthetized by intraperitoneal injection of a mixture of 13.3% urethane and 0.5% chloralose (0.65 mL/100 g body weight), and sacrificed. Samples from the injured-side cortex were rapidly excised, sliced into transverse or longitudinal sections, and fixed in 10% neutral-buffered formalin. Tissue was then embedded in paraffin blocks and serial sections stained with hematoxylin and eosin for microscopic evaluation at 200× magnification (Hicks et al., 1996).
Morphological observation
Morphological changes in neuronal ER and mitochondria were observed under transmission electron microscopy. Injured-side cortical tissue was fixed with 2.5% glutaraldehyde and stained with cacodylate-buffered osmium tetroxide (OsO4). Sections were prepared and examined under an electron microscope (Philips CM10; Philips, Eindhoven, The Netherlands) (Wang et al., 2013).
Terminal deoxynucleotidyl transferase-dUTP nick-end labeling (TUNEL) staining
A TUNEL staining kit was used to detect apoptotic cells (Molecular Probes), according to the manufacturer’s instructions. For each specimen, the total number of cells with positive nuclear staining from five microscopic fields (200×) was counted. TUNEL-positive cells were identified, counted, and analyzed by an investigator blinded to experimental treatments. Data are presented as percentage of cells with TUNEL-positive nuclei (Zhou et al., 2014; Hiebert et al., 2015).
Caspase activity assay
Samples from the injured-side cortex were harvested and lysed. Sample lysates were first incubated at −80°C for 30 minutes, and then incubated at 37°C with appropriate caspase substrates using a Quadruple Monochromator Microplate Reader (Infinite M1000; Tecan US, Charlotte, NC, USA). Caspase activity was measured by cleavage of the following fluorogenic peptide substrates: Ac-ATAD-AFC (caspase-12) and Ac-DEVD-AMC (caspase-3). Caspase activity was represented as relative cumulative fluorescence of the kinetic reaction compared with untreated controls (Yi et al., 2017).
Statistical analysis
All data were analyzed for statistical significance using SPSS 13.0 software (SPSS, Chicago, IL, USA). Data are expressed as the mean ± SD. All data were analyzed by one-way analysis of variance combined with Tukey’s multiple-comparisons test. A P value < 0.05 was considered statistically significant.
Results
Elevated ROS production in neurons from the injured cortex post-TBI
ROS production plays a crucial role in neuronal response to brain injury (Abdul-Muneer et al., 2015; Hiebert et al., 2015), therefore we monitored neuronal production of ROS using a cell-permeant fluorescent dye DCFH-DA, which exhibits enhanced fluorescence with ROS generation. We found that neurons immediately showed increased ROS production at one-hour post-TBI. This increase peaked after 6 hours but remained elevated up to 24 hours post-TBI (Figure 1).
Figure 1.

Elevated ROS production in neurons from the injured cortex of rats exposed to TBI induced by lateral fluid percussion.
Neurons were isolated from the injured-side cortex of rat brains at the indicated times (1, 3, 6, 12, and 24 hours) post-TBI. Neurons were labeled with 10 μM DCFH-DA for ROS detection. (A) Mean fluorescence intensity of ROS was analyzed by flow cytometry. (B) Quantification of ROS fluorescence intensity analyzed by flow cytometry. Data are expressed as the mean ± SD (n = 6 per group), and were analyzed by one-way analysis of variance combined with Tukey’s multiple-comparisons test. *P < 0.05, vs. sham group. ROS: Reactive oxygen species; TBI: traumatic brain injury; DCFH-DA: dichloro-dihydro-fluorescein diacetate; h: hour(s).
Elevated ER stress-related UPR in the injured cortex post-TBI
To determine whether TBI can induce ER stress and activate UPR, we detected the main ER stress-related proteins, specifically PERK, phospho-PERK, eIF2α, phospho-eIF2α, ATF4, spliced XBP-1, cleaved ATF6, and GRP78. Our results show that PERK and eIF2α phosphorylation was activated immediately at 1 hour, and peaked at 3 hours post-TBI (Figure 2A–C). Meanwhile, ATF4 expression was induced at 3 hours and peaked at 6 hours post-TBI (Figure 2D). Activation of cleaved ATF6 was observed as early as 1 hour and peaked immediately (3 hours) post-TBI (Figure 2A, F). Expression of spliced XBP-1 was activated at 3 hours post-TBI (Figure 2A, E). GRP78 was induced and peaked at 3 hours post-TBI (Figure 2A, G). Altogether, these results suggest that TBI activates ER stress-related UPR including three UPR transducer pathways: PERK-eIF2α-ATF4, XBP-1, and ATF6.
Figure 2.

Elevated endoplasmic reticulum stress-related unfolded protein response in the injured cortex post-TBI (western blot assay).
The injured-side cortex of rat brain was isolated at the indicated times (1, 3, 6, 12, and 24 hours) post-TBI. (A) Expression of phospho-PERK, PERK, phospho-eIF2a, eIF2a, ATF4, spliced XBP-1, cleaved ATF6, GRP78, and GAPDH were tested by western blot assay. (B–G) Quantified grayscale intensity of phospho-PERK/PERK, phospho-eIF2a/eIF2a, ATF4, spliced XBP-1, cleaved ATF6, and GRP78 expression detected by western blot assay. Data are expressed as the mean ± SD (n = 6 per group), and were analyzed by one-way analysis of variance combined with Tukey’s multiple-comparisons test. *P < 0.05, vs. sham group. TBI: Traumatic brain injury.
Elevated apoptosis-related protein markers in the injured cortex post-TBI
Based on our findings showing that TBI induces ER stress-related UPR signal transduction pathways, we next determined whether TBI can activate the ER stress response apoptotic pathway. We first examined expression of GADD153 and caspase-12, vital mediators of ER stress-induced apoptosis. As shown in Figure 3A and B, both GADD153 and caspase-12 expression were significantly up-regulated at 3 hours, peaked at 6 hours, and remained high up to 24 hours post-TBI (Figure 3A, C). Caspase-12 activity was consistent with these dynamic changes in caspase-12 protein expression (Figure 3D).
Figure 3.
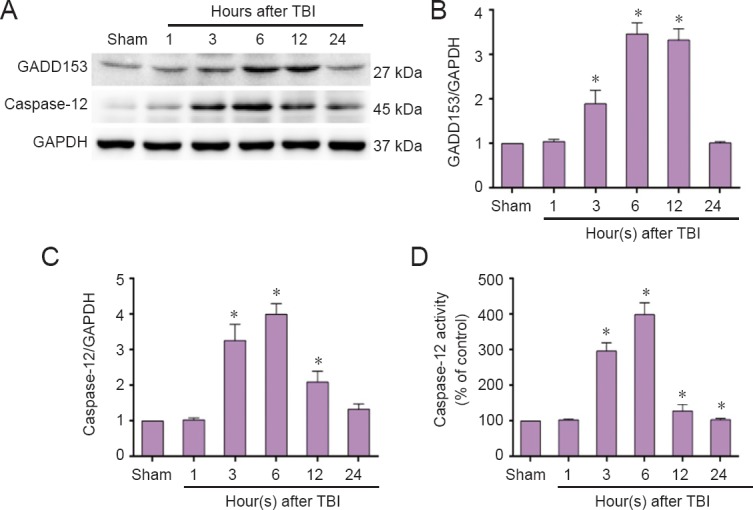
Elevated endoplasmic reticulum stress response pathway-related proteins in the injured cortex post-TBI (western blot assay).
The injured-side cortex of rat brain was isolated at the indicated times (1, 3, 6, 12, and 24 hours) post-TBI. (A) Expression of GADD153 and caspase-12 was detected by western blot assay. (B, C) Quantified grayscale intensity of GADD153 and caspase-12 expression detected by western blot assay. (D) Caspase-12 enzymatic activity was examined in cell lysates using the fluorogenic substrate, Ac-ATAD-AFC. Data are expressed as the mean ± SD (n = 6/group), and were analyzed by one-way analysis of variance combined with Tukey’s multiple-comparisons test. *P < 0.05, vs. sham group. TBI: Traumatic brain injury.
Next, we investigated Cyt c release, change in Bax/Bcl-2 ratio, MMP and caspsase-3 activity. We found a significant increase in Cyt c release from mitochondria to the cytosol, which began at 6 hours and continued for a period of 24 hours post-TBI (Figure 4A, B). Bax abundance showed a similar trend as Cyt c release. Conversely, anti-apoptotic Bcl-2 expression decreased, resulting in an increased Bax/Bcl-2 ratio (Figure 4C, D). As shown in Figure 4E and F, decreased MMP began at 6 hours. Caspase-3 activity significantly increased at 6 hours and peaked at 12 hours post-TBI. Taken together, these findings imply that both the ER stress response and mitochondrial apoptotic pathways are activated post-TBI. The ER stress response apoptotic pathway was activated at the initial stage, while the mitochondrial apoptotic pathway was activated slightly later.
Figure 4.

Increased mitochondrial apoptosis pathway-related proteins in the injured cortex post-TBI (western blot assay).
The injured-side cortex of rat brain was isolated at the indicated times (1, 3, 6, 12, and 24 hours) post-TBI. (A, C) Expression of Cyt c, Bax, and Bcl-2 were detected by western blot assay. (B, D) Quantified grayscale intensity of Cyt c expression and Bax/Bcl-2 ratio detected by western blot. (E) Quantification of mitochondrial membrane potential fluorescence (MMP, ΔΨm) detected by flow cytometry. (F) Caspase-3 enzymatic activity was examined in cell lysates using a fluorogenic substrate, Ac-DEVD-AMC. Data are expressed as the mean ± SD (n = 6/group), and were analyzed by one-way analysis of variance combined with Tukey’s multiple-comparisons test. *P < 0.05, vs. sham group. TBI: Traumatic brain injury; Cyt c: cytochrome c.
Sal alleviated brain damage post-TBI
From our results above, TBI-induced ER stress leads to a peak in the ER stress response apoptotic pathway at 6 hours, with activation of the mitochondrial apoptotic pathway beginning at 6 hours post-TBI. To investigate the effect of inhibited ER stress on activation of both the ER stress response and mitochondrial apoptotic pathways post-TBI, we chose 6 hours post-TBI as an observation time. Microscopy analysis showed that brain tissue in the sham group had clear and dense structures, with normal neuronal structure (Figure 5A, B). In contrast, TBI led to numerous hemorrhagic lesions in the cortex, with swollen neurons exhibiting loose structures, widespread vacuolar changes, and uncentered nuclei (Figure 5C). In contrast, pretreatment with Sal alleviated TBI-induced hemorrhagic lesions and neuronal injury in the brain (Figure 5D).
Figure 5.

Sal alleviates brain damage post-TBI at 6 hours post-TBI (hematoxylin and eosin staining).
Rats were injected intraperitoneally with the endoplasmic reticulum stress inhibitor, Sal (1 mg/kg), 30 minutes before lateral fluid-percussion-induced TBI. (A, B) Brain tissue from the sham group (A) and sham + Sal group (B) shows clear, dense structures with normal neuronal structures. (B) TBI led to numerous hemorrhagic lesions in the cortex (blue arrows), swollen neurons with loose structures, and widespread vacuolar changes (green arrows). (D) Sal alleviated TBI-induced hemorrhagic lesions (blue arrows) and neuronal injury (yellow arrows) in the brain. Scale bars: 200 μm. Sal: salubrinal; TBI: traumatic brain injury.
Sal mitigated both ER and mitochondrial damage in neurons from the injured cortex post-TBI
Morphological changes in neuronal ER and mitochondria were observed by transmission electron microscopy. Both the sham group and sham + Sal group showed normal neurons with normal ER and mitochondrial morphology (Figure 6A, B). In contrast, TBI led to dramatic ultrastructural alterations, including ER swelling and deformation. Mitochondria appeared swollen and irregularly shaped with disrupted and poorly defined cristae (Figure 6B). However, Sal pretreatment resulted in normal ER with markedly improved mitochondrial alterations (Figure 6D).
Figure 6.
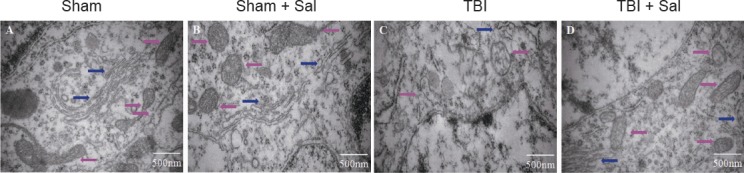
Transmission electron microscopy shows that Sal mitigates endoplasmic reticulum and mitochondrial damage in neurons of the injured cortex post-TBI at 6 hours after surgery (osmium tetroxide staining).
Rats were intraperitoneally injected with the endoplasmic reticulum stress inhibitor, Sal (1 mg/kg), 30 minutes before lateral fluid-percussion-induced TBI. The sham group (A) and sham + Sal group (B) show normal neuronal structures including normal endoplasmic reticulum and mitochondrial morphology. (C) TBI induced dramatic ultrastructural alterations including endoplasmic reticulum swelling and deformation, while mitochondria appeared swollen and irregularly shaped with disrupted and poorly defined cristae. (D) Sal mitigated both endoplasmic reticulum and mitochondrial damage in neurons of the injured cortex post-TBI. Scale bars: 500 nm. Pink arrows represent mitochondria; blue arrows represent endoplasmic reticulum. Sal: Salubrinal; TBI: Traumatic brain injury.
Sal alleviated ROS production, ER stress activation, and apoptosis
To determine whether Sal has an effect on ROS production post-TBI, we assayed ROS levels in neurons. As shown in Figure 7, compared with the TBI group, ROS levels were reduced by Sal pretreatment. Further, Sal induced an obvious increase in the phosphorylation status of eIF2α (Figure 8A, B). Additionally, Sal strongly increased ATF4 and GRP78 expression (Figure 8A, C, D). We next determined whether Sal has an effect on apoptosis-related proteins associated with the ER stress response and mitochondrial apoptotic pathways. As shown in Figure 9, Sal significantly inhibited GADD153 and caspase-12 expression. Further, caspase-12 activity was also inhibited by Sal. Moreover, Sal promoted recovery of MMP, inhibited mitochondrial Cyt c release, decreased the Bax/Bcl-2 ratio, and activated caspase-3 and occurrence of apoptosis post-TBI (Figures 10 and 11). These results suggest that Sal reduces apoptosis by alleviating the ER stress response to exhibit a neuroprotective effect post-TBI. Sal also promoted recovery of ROS homeostasis and mitochondrial function, and inhibited activation of the mitochondrial apoptotic pathway.
Figure 7.

Sal inhibits ROS production at 6 hours post-TBI.
Neurons were isolated from the injured-side cortex of rat brains at 6 hours post-TBI. Neurons were labeled with 10 μM DCFH-DA for ROS detection. (A) Mean fluorescence intensity of ROS was analyzed by flow cytometry. (a–d) Sham group, sham + Sal group, TBI group, and TBI + Sal group, respectively. (B) Quantification of ROS fluorescence intensity by flow cytometry. Data are expressed as the mean ± SD (n = 6/group), and were analyzed by one-way analysis of variance combined with Tukey’s multiple-comparisons test. *P < 0.05, vs. sham group; #P < 0.05, vs. TBI group. Sal: Salubrinal; ROS: reactive oxygen species; TBI: traumatic brain injury; DCFH-DA: dichloro-dihydro-fluorescein diacetate.
Figure 8.
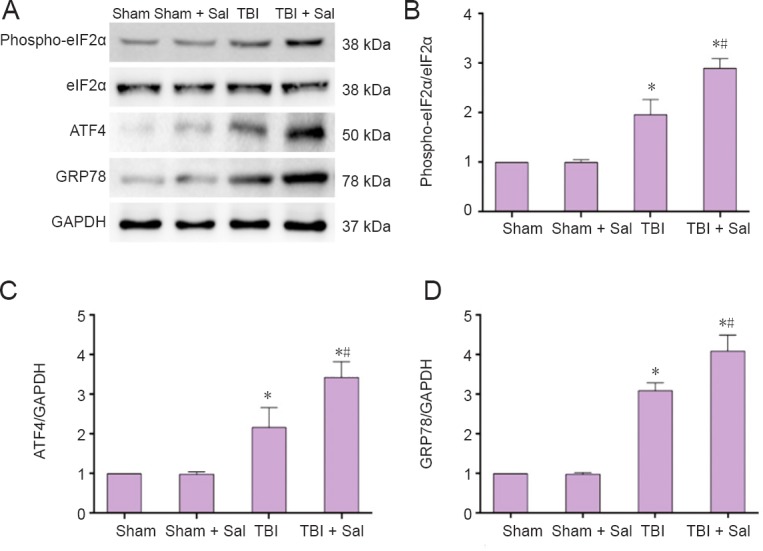
Sal inhibits the endoplasmic reticulum stress-related unfolded protein response post-TBI at 6 hours after surgery.
Rats were intraperitoneally injected with the endoplasmic reticulum stress inhibitor, Sal (1 mg/kg), 30 minutes before lateral fluid-percussion-induced TBI. (A) Expression of phospho-eIF2α, eIF2α, ATF4, GRP78, and GAPDH were detected by western blot assay. (B–D) Quantified grayscale intensity of phospho-eIF2α/eIF2α, ATF4, and GRP78 expression detected by western blot assay. Data are expressed as the mean ± SD (n = 6/group), and were analyzed by one-way analysis of variance combined with Tukey’s multiple-comparisons test. *P < 0.05, vs. sham group; #P < 0.05, vs. TBI group. Sal: Salubrinal; TBI: traumatic brain injury.
Figure 9.
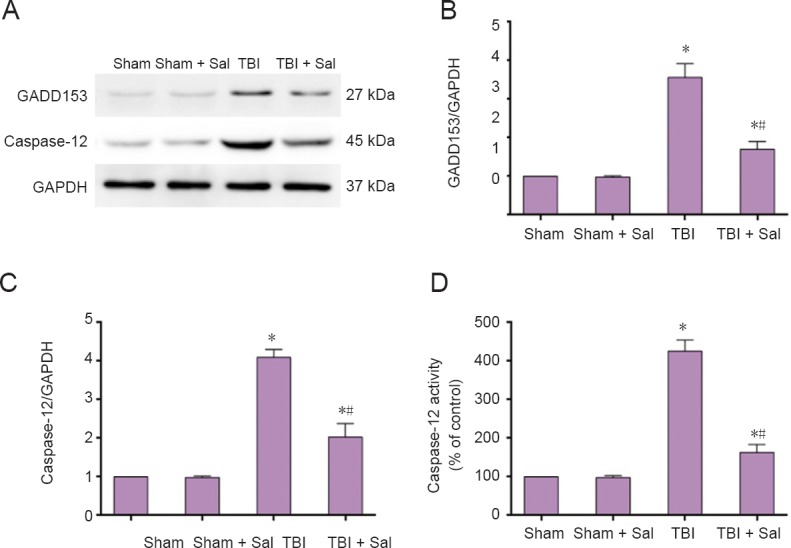
Sal inhibits endoplasmic reticulum stress response pathway-related protein expression post-TBI at 6 hours after surgery.
Rats were intraperitoneally injected with the endoplasmic reticulum stress inhibitor, Sal (1 mg/kg), 30 minutes before lateral fluid-percussion-induced TBI. (A) GADD153 and caspase-12 expression was detected by western blot assay. (B–C) Quantified grayscale intensity of GADD153 and caspase-12 expression detected by western blot assay. (D) Caspase-12 enzymatic activity was measured in cell lysates using the fluorogenic substrate, Ac-ATAD-AFC. Data are expressed as the mean ± SD (n = 6/group), and were analyzed by one-way analysis of variance combined with Tukey’s multiple-comparisons test. *P < 0.05, vs. sham group; #P < 0.05, vs. TBI group. Sal: Salubrinal; TBI: traumatic brain injury.
Figure 10.

Sal prevents mitochondrial apoptosis pathway-related protein expression post-TBI at 6 hours after surgery.
Rats were intraperitoneally injected with the endoplasmic reticulum stress inhibitor, Sal (1 mg/kg), 30 minutes before lateral fluid-percussion-induced TBI. (A, C) Expression of Cyt c, Bax, and Bcl-2 were detected by western blot assay. (B, D) Quantified grayscale intensity of Cyt c expression and Bax/Bcl-2 ratio detected by western blot assay. (E) Quantification of mitochondrial membrane potential fluorescence (ΔΨm) analyzed by flow cytometry. (F) Caspase-3 enzymatic activity was measured in cell lysates using a fluorogenic substrate, Ac-DEVD-AMC. Data are expressed as the mean ± SD (n = 6/group), and were analyzed by one-way analysis of variance combined with Tukey’s multiple-comparisons test. *P < 0.05, vs. sham group; #P < 0.05, vs. TBI group. Sal: Salubrinal; TBI: traumatic brain injury; Cyt c: cytochrome c.
Figure 11.

Sal prevents apoptosis in the injured cortex post-TBI.
Rats were intraperitoneally injected with the endoplasmic reticulum stress inhibitor, Sal (1 mg/kg), 30 minutes before lateral fluid-percussion-induced TBI. After 6 hours, rats were sacrificed. (a, b, c, d) Sham group, sham + Sal group, TBI group, and TBI+ Sal group, respectively. There was no difference between the sham group and sham + Sal group. (A) The cortex stained by TUNEL (blue arrows). Scale bars: 200 μm. (B) Quantification of TUNEL-positive index. Data are expressed as the mean ± SD (n = 6/group), and were analyzed by one-way analysis of variance combined with Tukey’s multiple-comparisons test. *P < 0.05, vs. sham group; #P < 0.05, vs. TBI group. Sal: Salubrinal; TBI: traumatic brain injury; TUNEL: terminal deoxynucleotidyl transferase-dUTP nick-end labeling.
Discussion
Accumulating evidence suggests that the ER is an effector and target of TBI (Begum et al., 2013; Logsdon et al., 2014; Dash et al., 2015). Reducing hippocampal ER stress can improve learning and memory function post-TBI, which is associated with the PERK-P-eIF2α pathway (Dash et al., 2015). Corroboratively, another study found that blast-induced TBI also involves ER stress, with the UPR a potential promising molecular target for relieving neuronal injury (Logsdon et al., 2014). Our present study indicates that major UPR signal transduction pathway initiators (PERK-eIF2α-ATF4, IRE1-XBP-1, and ATF6) are activated post-TBI. More importantly, all these ER stress-related proteins were activated at 3 hours post-TBI, further indicating that ER stress might be an early brain damage event that causes neuronal injury during secondary brain injury post-TBI. Since ER stress can activate extrinsic and intrinsic apoptotic pathways through different molecular transduction mechanisms in response to brain damage, we examined changes in GADD153 and caspase-12 expression. Our results show that GADD153 and caspase-12 levels are progressively increased at 3 hours post-TBI. Taken together, the ER stress response apoptotic pathway is activated at an initial stage (3 hours) post-TBI, implying that inhibition of the ER stress response as early as possible may help reduce secondary brain injury post-TBI.
Overproduction of ROS is a main cause of secondary brain injury post-TBI. The body can naturally produce endogenous antioxidants, suchx as glutathione molecules and superoxide dismutase, to prevent increased ROS production (Hiebert et al., 2015). However, if TBI produces more ROS than these antioxidants, it can result in protein oxidation and damage, peroxidation of cellular and vascular structures, impairment of the mitochondrial electron transport chain, and DNA cleavage (Abdul-Muneer et al., 2015; Hiebert et al., 2015). These mechanisms are sufficient to contribute to early or late apoptosis, neuroinflammation, and immediate cell death. Here, we confirmed that neurons significantly increased ROS production at one hour, and showed this tendency up to 24 hours post-TBI. These findings are consistent with previously published studies on brain damage (Choi et al., 2010; Logsdon et al., 2016). Mitochondria have the vital function of maintaining neuronal homeostasis via a myriad of intricate processes. The main cause of secondary deleterious cascades is neuronal damage, which is also central to mitochondrial function post-TBI (Hiebert et al., 2015). A previous study found that TBI causes neuronal mitochondrial dysfunction, as indicated by promotion of mitochondrial permeability transition pore opening and decreased adenosine triphosphate content post-TBI (Yang et al., 2017). In TBI pathology, pro- and anti-apoptotic Bcl-2 family performance has an impact on neuronal fate. The mitochondrial apoptosis-induced channel is a pathway for Cyt c release, the proposed components of which include oligomeric Bax/Bax. In our present study, Cyt c release from mitochondria into the cytosol began at 6 hours and lasted up to 24 hours post-TBI. Bax/Bcl-2 ratio and caspase-3 activity both showed a similar increase, suggesting that the mitochondrial apoptotic pathway is also activated post-TBI, albeit a bit later than the ER stress response apoptotic pathway, mainly at the late stage of secondary brain injury post-TBI. Compared with other studies, our study shows that both the ER stress response and mitochondrial apoptotic pathways are activated post-TBI. Moreover, activation of the ER stress response apoptotic pathway was earlier than the mitochondrial apoptotic pathway. This may provide new clues for research on the underlying mechanisms triggering secondary brain injury post-TBI.
Sal is a selective inhibitor of eIF2α dephosphorylation. Phosphorylation of eIF2α appears to be cytoprotective during ER stress by inhibiting translation initiation activity of eIF2α, thereby reducing global protein synthesis and leading to reduced ER workload (Matsuoka and Komoike, 2015). In fact, Sal can reduce oxidative stress and neuroinflammation, which are considered common secondary effects of brain injury (Sokka et al., 2007; Begum et al., 2013; Logsdon et al., 2014; Roth et al., 2014; Hiebert et al., 2015). Few studies have focused on the effect of Sal post-TBI. In our study, we administered Sal before TBI to investigate its effects on brain damage post-TBI. We show that Sal significantly enhances eIF2α expression and decreases GADD153 and caspase-12 expression. In addition, ATF4 and GRP78 expression also increased, which is consistent with previous reports showing that enhanced ATF4 or GRP78 levels can alleviate neuronal injury in hypoxic-ischemic brain damage (López-Hernández et al., 2015; Zhang et al., 2015). We also found that Sal can prevent ROS production. More critically, Sal alleviated ultrastructural damage of neuronal mitochondria, promoted recovery of mitochondrial function, and inhibited activation of the mitochondrial apoptotic pathway post-TBI. Therefore, our results suggest that Sal might alleviate the ER stress response via increased P-eIF2α, ATF4, and GRP78 expression as well as reduction of GADD153 expression and inhibition of apoptosis post-TBI. Furthermore, inhibition of ER stress promoted recovery of mitochondrial function and plays a negative role in regulating the mitochondrial apoptotic pathway post-TBI, which previous studies have not addressed. Nonetheless, the mechanism needs further investigation.
In conclusion, ER stress, ROS production, and mitochondrial dysfunction are all involved in secondary brain injury post-TBI, and ER stress is an early event ahead of mitochondrial damage post-TBI. Sal might alleviate the ER stress response through increased P-eIF2α, ATF4, and GRP78 levels and reduced GADD153 expression. This may lead to a decrease in apoptosis post-TBI. Sal pretreatment reduced ROS production and, more importantly, alleviated ultrastructural damage of neuronal mitochondria to promote recovery of mitochondrial function and inhibit activation of the mitochondrial apoptotic pathway post-TBI. This suggests that inhibition of ER stress plays a negative role in regulating the mitochondrial apoptotic pathway post-TBI. Consequently, inhibition of ER stress may be a novel strategy for preventing and treating secondary brain injury post-TBI.
Footnotes
Conflicts of interest: None declared.
Financial support: None.
Institutional review board statement: The study protocol was approved by the Experimental Animal Ethics and Welfare Committee of Guangzhou General Hospital of Guangzhou Military Command of China (Approval number: 2016030095). The experimental procedure followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1985).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer review report:
Reviewer: Jo Anne Stratton, University of Calgary, Canada.
Comments to authors: This manuscript presented a detailed characterization of the time course (1, 3, 6, 12, 24 hours) of ER stress, ROS production, mitochondrial function, and apoptosis following TBI. It then went on to demonstrate that modulating ER stress in vivo reduced mitochondrial dysfunction, ROS production and apoptosis in TBI.
(Copyedited by James R, Hindle A, Yu J, Li CH, Qiu Y, Song LP, Zhao M)
References
- 1.Abdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol. 2015;51:966–979. doi: 10.1007/s12035-014-8752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Begum G, Harvey L, Dixon CE, Sun D. ER stress and effects of DHA as an ER stress inhibitor. Transl Stroke Res. 2013;4:635–642. doi: 10.1007/s12975-013-0282-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burton GJ, Yung HW, Murray AJ. Mitochondrial - Endoplasmic reticulum interactions in the trophoblast: Stress and senescence. Placenta. 2017;52:146–155. doi: 10.1016/j.placenta.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Cao Y, Gao Y, Xu S, Bao J, Lin Y, Luo X, Wang Y, Luo Q, Jiang J, Neale JH, Zhong C. Glutamate carboxypeptidase II gene knockout attenuates oxidative stress and cortical apoptosis after traumatic brain injury. BMC Neurosci. 2016;17:15. doi: 10.1186/s12868-016-0251-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen B, Mutschler M, Yuan Y, Neugebauer E, Huang Q, Maegele M. Superimposed traumatic brain injury modulates vasomotor responses in third-order vessels after hemorrhagic shock. Scand J Trauma Resusc Emerg Med. 2013;21:77. doi: 10.1186/1757-7241-21-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi JH, Choi AY, Yoon H, Choe W, Yoon KS, Ha J, Yeo EJ, Kang I. Baicalein protects HT22 murine hippocampal neuronal cells against endoplasmic reticulum stress-induced apoptosis through inhibition of reactive oxygen species production and CHOP induction. Exp Mol Med. 2010;42:811–822. doi: 10.3858/emm.2010.42.12.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dash PK, Hylin MJ, Hood KN, Orsi SA, Zhao J, Redell JB, Tsvetkov AS, Moore AN. Inhibition of eukaryotic initiation factor 2 alpha phosphatase reduces tissue damage and improves learning and memory after experimental traumatic brain injury. J Neurotrauma. 2015;32:1608–1620. doi: 10.1089/neu.2014.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gajavelli S, Sinha VK, Mazzeo AT, Spurlock MS, Lee SW, Ahmed AI, Yokobori S, Bullock RM. Evidence to support mitochondrial neuroprotection, in severe traumatic brain injury. J Bioenerg Biomembr. 2015;47:133–148. doi: 10.1007/s10863-014-9589-1. [DOI] [PubMed] [Google Scholar]
- 9.Go BS, Kim J, Yang JH, Choe ES. Psychostimulant-induced endoplasmic reticulum stress and neurodegeneration. Mol Neurobiol. 2017;54:4041–4048. doi: 10.1007/s12035-016-9969-0. [DOI] [PubMed] [Google Scholar]
- 10.Gulyaeva NV. Brain ischemia, endoplasmic reticulum stress, and astroglial activation: new insights. J Neurochem. 2015;132:263–265. doi: 10.1111/jnc.13016. [DOI] [PubMed] [Google Scholar]
- 11.Hicks R, Soares H, Smith D, McIntosh T. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol. 1996;91:236–246. doi: 10.1007/s004010050421. [DOI] [PubMed] [Google Scholar]
- 12.Hiebert JB, Shen Q, Thimmesch AR, Pierce JD. Traumatic brain injury and mitochondrial dysfunction. Am J Med Sci. 2015;350:132–138. doi: 10.1097/MAJ.0000000000000506. [DOI] [PubMed] [Google Scholar]
- 13.Jin M, Yang YW, Cheng WP, Lu JK, Hou SY, Dong XH, Liu SY. Serine-threonine protein kinase activation may be an effective target for reducing neuronal apoptosis after spinal cord injury. Neural Regen Res. 2015;10:1830–1835. doi: 10.4103/1673-5374.170313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.López-Hernández B, Ceña V, Posadas I. The endoplasmic reticulum stress and the HIF-1 signalling pathways are involved in the neuronal damage caused by chemical hypoxia. Br J Pharmacol. 2015;172:2838–2851. doi: 10.1111/bph.13095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langlois JA, Rutland-Brown W, Wald MM. The epidemiology and impact of traumatic brain injury: a brief overview. J Head Trauma Rehabil. 2006;21:375–378. doi: 10.1097/00001199-200609000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Li L, Tan H, Gu Z, Liu Z, Geng Y, Liu Y, Tong H, Tang Y, Qiu J, Su L. Heat stress induces apoptosis through a Ca(2)(+)-mediated mitochondrial apoptotic pathway in human umbilical vein endothelial cells. PLoS One. 2014;9:e111083. doi: 10.1371/journal.pone.0111083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Logsdon AF, Lucke-Wold BP, Nguyen L, Matsumoto RR, Turner RC, Rosen CL, Huber JD. Salubrinal reduces oxidative stress, neuroinflammation and impulsive-like behavior in a rodent model of traumatic brain injury. Brain Res. 2016;1643:140–151. doi: 10.1016/j.brainres.2016.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Logsdon AF, Turner RC, Lucke-Wold BP, Robson MJ, Naser ZJ, Smith KE, Matsumoto RR, Huber JD, Rosen CL. Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front Cell Neurosci. 2014;8:421. doi: 10.3389/fncel.2014.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorente L, Martin MM, Argueso M, Ramos L, Sole-Violan J, Riano-Ruiz M, Jimenez A, Borreguero-Leon JM. Serum caspase-3 levels and mortality are associated in patients with severe traumatic brain injury. BMC Neurol. 2015;15:228. doi: 10.1186/s12883-015-0485-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsuoka M, Komoike Y. Experimental evidence shows salubrinal, an eIF2alpha dephosphorylation inhibitor, reduces xenotoxicant-induced cellular damage. Int J Mol Sci. 2015;16:16275–16287. doi: 10.3390/ijms160716275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakka VP, Gusain A, Raghubir R. Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox Res. 2010;17:189–202. doi: 10.1007/s12640-009-9110-5. [DOI] [PubMed] [Google Scholar]
- 22.Nathoo N, Narotam PK, Agrawal DK, Connolly CA, van Dellen JR, Barnett GH, Chetty R. Influence of apoptosis on neurological outcome following traumatic cerebral contusion. J Neurosurg. 2004;101:233–240. doi: 10.3171/jns.2004.101.2.0233. [DOI] [PubMed] [Google Scholar]
- 23.Plácido AI, Pereira CM, Correira SC, Carvalho C, Oliveira CR, Moreira PI. Phosphatase 2A inhibition affects endoplasmic reticulum and mitochondria homeostasis via cytoskeletal alterations in brain endothelial cells. Mol Neurobiol. 2017;54:154–168. doi: 10.1007/s12035-015-9640-1. [DOI] [PubMed] [Google Scholar]
- 24.Raghubir R, Nakka VP, Mehta SL. Endoplasmic reticulum stress in brain damage. Methods Enzymol. 2011;489:259–275. doi: 10.1016/B978-0-12-385116-1.00015-7. [DOI] [PubMed] [Google Scholar]
- 25.Rao W, Zhang L, Peng C, Hui H, Wang K, Su N, Wang L, Dai SH, Yang YF, Chen T, Luo P, Fei Z. Downregulation of STIM2 improves neuronal survival after traumatic brain injury by alleviating calcium overload and mitochondrial dysfunction. Biochim Biophys Acta. 2015;1852:2402–2413. doi: 10.1016/j.bbadis.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 26.Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 2014;505:223–228. doi: 10.1038/nature12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460–3470. doi: 10.1016/j.bbamcr.2013.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci. 2007;27:901–908. doi: 10.1523/JNEUROSCI.4289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sophonnithiprasert T, Mahabusarakam W, Nakamura Y, Watanapokasin R. Goniothalamin induces mitochondria-mediated apoptosis associated with endoplasmic reticulum stress-induced activation of JNK in HeLa cells. Oncol Lett. 2017;13:119–128. doi: 10.3892/ol.2016.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun GZ, Gao FF, Zhao ZM, Sun H, Xu W, Wu LW, He YC. Endoplasmic reticulum stress-induced apoptosis in the penumbra aggravates secondary damage in rats with traumatic brain injury. Neural Regen Res. 2016;11:1260–1266. doi: 10.4103/1673-5374.189190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Song R, Chen Y, Zhao M, Zhao KS. Polydatin--a new mitochondria protector for acute severe hemorrhagic shock treatment. Expert Opin Investig Drugs. 2013;22:169–179. doi: 10.1517/13543784.2013.748033. [DOI] [PubMed] [Google Scholar]
- 32.Xue F, Shi C, Chen Q, Hang W, Xia L, Wu Y, Tao SZ, Zhou J, Shi A, Chen J. Melatonin mediates protective effects against kainic acid-induced neuronal death through safeguarding ER stress and mitochondrial disturbance. Front Mol Neurosci. 2017;10:49. doi: 10.3389/fnmol.2017.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xue LX, Liu HY, Cui Y, Dong Y, Wang JQ, Ji QY, He JT, Yao M, Wang YY, Shao YK, Mang J, Xu ZX. Neuroprotective effects of Activin A on endoplasmic reticulum stress-mediated apoptotic and autophagic PC12 cell death. Neural Regen Res. 2017;12:779–786. doi: 10.4103/1673-5374.206649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto A, Murphy N, Schindler CK, So NK, Stohr S, Taki W, Prehn JH, Henshall DC. Endoplasmic reticulum stress and apoptosis signaling in human temporal lobe epilepsy. J Neuropathol Exp Neurol. 2006;65:217–225. doi: 10.1097/01.jnen.0000202886.22082.2a. [DOI] [PubMed] [Google Scholar]
- 35.Yang H, Gu ZT, Li L, Maegele M, Zhou BY, Li F, Zhao M, Zhao KS. SIRT1 plays a neuroprotective role in traumatic brain injury in rats via inhibiting the p38 MAPK pathway. Acta Pharmacol Sin. 2017;38:168–181. doi: 10.1038/aps.2016.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yi G, Li L, Luo M, He X, Zou Z, Gu Z, Su L. Heat stress induces intestinal injury through lysosome- and mitochondria-dependent pathway in vivo and in vitro. Oncotarget. 2017;8:40741–40755. doi: 10.18632/oncotarget.16580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yin Y, Sun G, Li E, Kiselyov K, Sun D. ER stress and impaired autophagy flux in neuronal degeneration and brain injury. Ageing Res Rev. 2017;34:3–14. doi: 10.1016/j.arr.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, Xie X, Pan H, Wu Z, Lu W, Yang G. Role of endoplasmic reticulum stress in brain damage after cardiopulmonary resuscitation in rats. Shock. 2015;44:65–71. doi: 10.1097/SHK.0000000000000367. [DOI] [PubMed] [Google Scholar]
- 39.Zhou XM, Zhang X, Zhang XS, Zhuang Z, Li W, Sun Q, Li T, Wang CX, Zhu L, Shi JX, Zhou ML. SIRT1 inhibition by sirtinol aggravates brain edema after experimental subarachnoid hemorrhage. J Neurosci Res. 2014;92:714–722. doi: 10.1002/jnr.23359. [DOI] [PubMed] [Google Scholar]