

Keywords: nerve regeneration, traumatic brain injury, neuroinflammation, nuclear factor kappa B, NLRP3 inflammasome, brain edema, blood-brain barrier, tight junction proteins, apoptosis, neuroprotection, dexmedetomidine, neural regeneration
Abstract
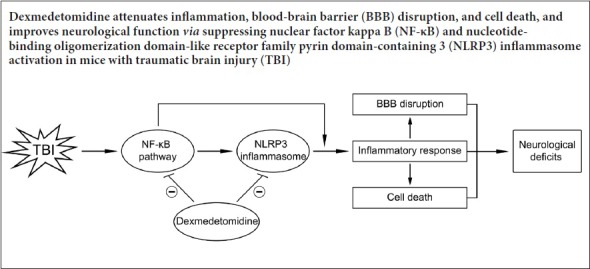
Traumatic brain injury induces potent inflammatory responses that can exacerbate secondary blood-brain barrier (BBB) disruption, neuronal injury, and neurological dysfunction. Dexmedetomidine is a novel α2-adrenergic receptor agonist that exert protective effects in various central nervous system diseases. The present study was designed to investigate the neuroprotective action of dexmedetomidine in a mouse traumatic brain injury model, and to explore the possible mechanisms. Adult male C57BL/6J mice were subjected to controlled cortical impact. After injury, animals received 3 days of consecutive dexmedetomidine therapy (25 µg/kg per day). The modified neurological severity score was used to assess neurological deficits. The rotarod test was used to evaluate accurate motor coordination and balance. Immunofluorescence was used to determine expression of ionized calcium binding adapter molecule-1, myeloperoxidase, and zonula occluden-1 at the injury site. An enzyme linked immunosorbent assay was used to measure the concentration of interleukin-1β (IL-1β), tumor necrosis factor α, and IL-6. The dry-wet weight method was used to measure brain water content. The Evans blue dye extravasation assay was used to measure BBB disruption. Western blot assay was used to measure protein expression of nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3), caspase-1 p20, IL-1β, nuclear factor kappa B (NF-κB) p65, occluding, and zonula occluden-1. Flow cytometry was used to measure cellular apoptosis. Results showed that dexmedetomidine treatment attenuated early neurological dysfunction and brain edema. Further, dexmedetomidine attenuated post-traumatic inflammation, up-regulated tight junction protein expression, and reduced secondary BBB damage and apoptosis. These protective effects were accompanied by down-regulation of the NF-κB and NLRP3 inflammasome pathways. These findings suggest that dexmedetomidine exhibits neuroprotective effects against acute (3 days) post-traumatic inflammatory responses, potentially via suppression of NF-κB and NLRP3 inflammasome activation.
Introduction
The pathophysiological mechanisms of traumatic brain injury (TBI) include primary mechanical damage that leads to a series of secondary injury events, including neuroinflammation, apoptosis, and oxidative stress (Shi et al., 2015). Of these events, post-traumatic neuroinflammation is considered a key factor in secondary brain cascades, which is characterized by glial activation, peripheral inflammatory cell infiltration, and the secretion of inflammatory mediators (Zhang et al., 2013). Although a moderate inflammatory response is essential for reparative and remodeling processes, sustained and excessive inflammation can exacerbate brain edema, blood-brain barrier (BBB) damage, secondary neuronal injury, and subsequent neurological impairment through the secretion of pro-inflammatory mediators such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and IL-6 (Gao et al., 2015; Chen et al., 2017). Previous studies have confirmed that these acute-phase cytokines are regulated via the nuclear factor kappa B (NF-κB) signaling pathway after TBI (Zhu et al., 2014), and that NF-κB expression is rapidly increased (within hours) after brain injury (Hang et al., 2006; Jayakumar et al., 2014). Thus, NF-κB represents a promising target for restricting secondary brain injury after TBI.
In contrast to other acute-phase cytokines, mature and biologically active IL-1β is produced from the inactive precursor pro-IL-1β by cleavage of caspase-1, while caspase-1 activation requires inflammasome-forming nucleotide-binding oligomerization domain-like receptors (McKee and Lukens, 2016). Of these, nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) is the most widely characterized (Liu et al., 2013). NLRP3 can recognize different danger signals, including damage-associated and pathogen-associated molecular pattern molecules, and induce the expression of immune- and inflammation-related genes to defend against infection or tissue damage (Zhou et al., 2016). Through binding of the adaptor protein apoptosis-associated speck-like protein, NLRP3 assembles the NLRP3 inflammasome protein complex in the cytoplasm to mediate caspase-1-dependent processing and release of the pro-inflammatory mediators IL-1β and IL-18 (Zhou et al., 2016; Mamik and Power, 2017). The NLRP3 inflammasome is also involved in the pathogenesis of central nervous system injury, including ischemic and hemorrhagic stroke, and TBI (Lin et al., 2017). Recent studies in animal models and TBI patients have also confirmed up-regulation of NLRP3 inflammasome expression (Liu et al., 2013; Lin et al., 2017). Further, blockade or knockdown of the NLRP3 inflammasome can attenuate inflammatory responses and improve neurological functions in rodent TBI models (Irrera et al., 2017; Ismael et al., 2018). Thus, the NLRP3 inflammasome represents a new potential therapeutic target for TBI.
Dexmedetomidine (DEX) is a highly effective α2-adrenergic receptor agonist that has sedative, analgesic, and anti-anxiety effects, and is extensively used in the intensive care unit and for clinical anesthesia (Fang et al., 2015). In addition, DEX has been shown to reduce cerebral ischemia/reperfusion injury by suppressing inflammation, activating the anti-apoptotic signaling pathways, and inhibiting neuronal autophagy (Akpinar et al., 2016; Luo et al., 2017; Wang et al., 2017). Indeed, in vivo and in vitro studies suggest that DEX has protective effects in TBI (Schoeler et al., 2012; Shen et al., 2017). However, the effects of DEX on the acute inflammatory responses after TBI remain unclear.
The aim of the present study was to examine the protective actions of DEX on acute inflammatory responses, BBB disruption, cellular apoptosis, and neurological function during the acute phase of TBI. Further, we assessed the inhibitory effects of DEX on TBI-induced activation of the NF-κB and NLRP3 inflammasome.
Materials and Methods
Animals
Specific-pathogen-free male C57BL/6J mice aged 8–10 weeks and weighing 20–25 g were purchased from the Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China; License No. SCXK [Jing] 2014-0004). All experimental protocols were approved by the Animal Care and Use Committee of Tianjin Medical University General Hospital (approval No. IRB2016-YX-036). Mice were maintained on a standard environment: controlled temperature and humidity, 12-hour light/dark cycles, and sufficient chow and water. All mice were adapted for 2 weeks before experiments. In total, 115 mice were randomly divided into the sham group (n = 35), TBI + vehicle group (n = 40), and TBI + DEX group (n = 40). The experimental design is shown in Figure 1A.
Figure 1.

Effect of DEX on neurological function of mice following TBI.
(A) Timeline of the experimental design. (B) Schematic representation of the studied area (shaded area: peri-contusional cortex [cortex surrounding the traumatic lesions]). (C) Quantitative analysis of body weight in three individual groups of mice (n = 5 per group). Neurologic deficits were evaluated using the mNSS (D) and rotarod (E) tests (n = 10 per group). Lower scores and longer latency to fall imply better neurological outcomes. All data are expressed as mean ± standard deviation, and were analyzed by one-way analysis of variance followed by Tukey’s multiple comparison post hoc test. ###P < 0.001, vs. sham group; *P < 0.05, vs. TBI + vehicle group. DEX: dexmedetomidine; TBI: traumatic brain injury; mNSS: modified neurological severity score; ELISA: enzyme linked immunosorbent assay; qRT-PCR: quantitative reverse transcription-polymerase chain reaction.
TBI model establishment
A moderate TBI model was established using an electric cortical contusion impactor (Custom Design & Fabrication, Sandston, VA, USA) as we previously reported (Xu et al., 2017). Mice were intraperitoneally anesthetized using chloral hydrate 3.0 mL/kg, and then placed on a stereotaxic apparatus. The coronal and sagittal sutures were fully exposed, and a craniotomy (3.5-mm diameter) was conducted (2-mm posterior from and 2-mm lateral to the bregma) with the entire dura intact. TBI was performed using a 3-mm diameter flat tip with a velocity of 4.5 m/s, a dwell time of 200 ms, and a depth of 2 mm. After restoration of respiration, the scalp incision was closed using intermittent sutures. Animal temperature was monitored and maintained at 37°C during the procedure and until recovery from anesthesia. Mice in the sham group received the same procedures without the cortical contusion impactor.
Drug administration
DEX (Jiangsu Hengrui Medicine Co., Ltd., Jiangsu, China; Drug approval No. H20090248) was dissolved in 0.9% saline. After TBI induction, mice in the TBI + DEX group were injected intraperitoneally with DEX at a dose of 25 µg/kgper day for 3 consecutive days. The DEX dosage and timing were chosen based on previous studies (Liang et al., 2017; Luo et al., 2017). The sham and TBI + vehicle groups received an equal volume (200 μL) of saline with the same schedule.
Modified neurological severity score test
The modified neurological severity score (mNSS) test was performed to assess neurobehavioral deficits (motor, sensory, balance, and reflex) at baseline, and on days 1 and 3 post-injury (Chen et al., 2001). The mNSS test was graded on a scale of 0–18. If a test could not be completed, or the animal did not respond to a test, then the test was scored as 1 point. Therefore, higher scores indicate greater neurological injury.
Rotarod test
The accurate motor coordination and balance abilities of mice were evaluated using the rotarod test at baseline, and on days 1 and 3 post-injury (Xu et al., 2017). One day before surgery, the mice were adapted to the device (RWD Life Science, Shenzhen, China) for 1 minute at low speed (4 revolutions per minute [r/min]). Next, four acceleration tests (from 4–40 r/min over 5 minutes) were performed to record the fall-off time to obtain the baseline values. On days 1 and 3 post-injury, each mouse underwent four acceleration tests with an interval of 30 minutes. The fall-off times were recorded, and the mean value was calculated.
Immunofluorescence
On days 1 and 3 post-injury, brain cryosections (6-μm thick) were blocked in 3% bovine serum albumin at 37°C for 30 minutes, and then incubated with corresponding primary antibodies overnight at 4°C, including rabbit monoclonal anti-ionized calcium binding adapter molecule-1 antibody (Iba-1; microglia marker; 1:500; Wako, Osaka, Japan), rabbit polyclonal anti-myeloperoxidase antibody (MPO; neutrophil marker; 1:100; Abcam, Cambridge, UK), and rabbit polyclonal anti-zonula occluden-1 antibody (ZO-1; tight junction protein ZO-1 marker; 1:100; Invitrogen, Carlsbad, CA, USA). The slides were then incubated at room temperature for 1 hour with an Alexa 488-conjugated donkey anti-rabbit secondary antibody (1:500; Abcam). Finally, nuclei were counterstained with 4′,6-diamidino-2-phenylindole. The slides were visualized under an immunofluorescence microscope (X81; Olympus, Tokyo, Japan) and photographed. The results were analyzed using ImageJ software (Version1.46r; NIH, Bethesda, MD, USA). A total of four slides from each brain, with five random regions of interest surrounding the traumatic lesions (peri-contusional cortex; i.e., cortex surrounding the traumatic lesions; Figure 1B) for each slide, were analyzed. For quantification of MPO-positive neutrophil and Iba-1-positive microglia, positive cells were counted, and data expressed as positive cells/mm2. For quantification of the tight junction protein ZO-1, a thresholding procedure was established to measure the proportion of the ZO-1 immunoreactive area (Shen et al., 2013).
Enzyme-linked immunosorbent assay
Brain samples were collected from the peri-contusional cortex (60 mg; Figure 1B) on days 1 and 3 post-injury. The concentrations of IL-1β, tumor necrosis factor (TNF)-α, and IL-6 in brain homogenates were measured using specific enzyme-linked immunosorbent assay kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Brain water content measurement
The brain water content of mice on day 3 post-injury was measured using the dry-wet weight method (Gao et al., 2015). The lesioned cerebral hemispheres were collected without transcardiac perfusion, and the wet weight was immediately recorded. The tissues were then dried in a 100°C electro-thermostatic blast oven for 24 hours, and the dry weight was measured. The brain water content (%) was calculated with the formula: (wet weight − dry weight)/wet weight × 100%.
Evans blue dye extravasation assay
BBB disruption was quantitatively measured using Evans blue dye extravasation. On day 3 post-injury, 100 μL of 2% Evans blue (Sigma-Aldrich, St. Louis, MO, USA) was administered through the tail vein. After 2 hours of circulation, transcardiac perfusion of phosphate-buffered saline was performed to clear the circulating Evans blue dye. The lesioned cerebral hemisphere was homogenized in formamide and incubated at 60°C for 72 hours. Following centrifugation at 14 000 rpm for 30 minutes, the Evans blue concentration in the resulting supernatant was measured using an optical densitometer (BioTek, Winooski, VT, USA) at 620 nm.
Western blot assay
Brain samples were harvested from the peri-contusional cortex (60 mg; Figure 1B) on days 1 and 3 post-injury, and lysed in radioimmunoprecipitation assay buffer (Beyotime, Jiangsu, China). After denaturing, protein samples (8 μg per lane) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and then transferred to a polyvinylidene difluoride membrane (Roche, South San Francisco, CA, USA). The membrane was blocked with 5% non-fat milk for 2 hours at room temperature, and then incubated at 4°C overnight with the following primary antibodies: mouse monoclonal anti-NLRP3 antibody (NLRP3 inflammasome component NLRP3 protein marker; 1:1000; AdipoGen, San Diego, CA, USA), rabbit polyclonal anti-caspase-1 p20 antibody (NLRP3 inflammasome component activated caspase-1 marker; 1:1000; Millipore, Billerica, MA, USA), rabbit polyclonal anti-IL-1β antibody (NLRP3 inflammasome substrate mature IL-1β marker; 1:1000; Cell Signaling Technology, Danvers, CT, USA), rabbit polyclonal anti-NF-κB p65 antibody (NF-κB signaling pathway marker; 1:2000; Abcam), mouse polyclonal anti-occludin antibody (tight junction protein occludin marker; 1:1000; Invitrogen, Carlsbad, CA, USA), rabbit polyclonal anti-ZO-1 antibody (tight junction protein ZO-1 marker; 1:1000; Invitrogen), and rabbit monoclonal anti-β-actin antibody (1:1000; Cell Signaling Technology). After washing, the blots were incubated with corresponding horseradish peroxidase-conjugated secondary antibodies (1:5000; Beyotime) for 1 hour at room temperature, and then detected with the enhanced chemiluminescence system (Millipore). The band mean optical density was quantified using NIH ImageJ software. β-actin was used as an internal reference.
Flow cytometric analysis for apoptosis
The lesioned hemispheres were harvested on day 3 post-injury, and were homogenized with 40-μm nylon cell strainers. A 5 mL of 30% Percoll solution (Sigma-Aldrich) was used to isolate single cells (700 × g for 5 minutes). After rinsing with phosphate buffered saline, cellular apoptosis was detected using a commercial apoptosis detection kit (BD Bioscience, Franklin Lakes, NJ, USA) according to the manufacturer’s instructions. Flow cytometry was performed and analyzed on an Accuri C6 apparatus (BD Biosciences).
Statistical analysis
Numerical values are presented as mean ± standard deviation. Statistical comparisons were analyzed using one-way analysis of variance followed by Tukey’s multiple comparison post hoc test (SPSS v22.0 statistical software; IBM, Armonk, NY, USA). A P value < 0.05 was considered statistically significant.
Results
DEX administration does not affect body weight after TBI
All mice survived after DEX treatment during the experimental period. There were no differences in body weight between the various groups (Figure 1C; P > 0.05). These data suggest that DEX treatment after TBI was well tolerated.
DEX administration ameliorated early neurological deficits after TBI
To investigate whether DEX promoted recovery of neurological function in the acute phase of TBI, the mNSS test was used to evaluate sensory-motor deficits, while the rotarod test was used to measure motor dysfunction. In the mNSS test, the vehicle-treated mice had higher mNSS scores on days 1 and 3 post-injury compared with sham-injured mice (Figure 1D; P < 0.001). DEX treatment also significantly reduced the mNSS scores on day 3 post-TBI compared with the TBI + vehicle group (Figure 1D; P < 0.05). Similarly, on days 1 and 3 post-injury, the performance of the DEX-treated mice in the rotarod test was better than that of vehicle-treated mice (Figure 1E; P < 0.05 for both).
DEX administration reduced neutrophil infiltration, microglial activation, and pro-inflammatory mediator expression after TBI
Next, we used immunofluorescence to determine whether DEX treatment directly affected the early post-traumatic inflammatory events. TBI induced a robust MPO-positive neutrophil infiltration and Iba-1-positive microglial activation surrounding the traumatic lesions. By contrast, DEX treatment significantly reduced numbers of neutrophils on day 1 post-injury, and numbers of microglia on day 3, post-injury compared with the TBI + vehicle group (Figure 2A–C; P < 0.01 for both). Additionally, morphological analyses showed that TBI induced a marked change in microglial morphology, from a ramified shape to a rounded and enlarged appearance, indicating extensive microglial activation. DEX treatment significantly reduced the microglial cell body and ramification index (Figure 2A).
Figure 2.

Effect of dexmedetomidine (DEX) on neutrophil infiltration, microglial activation, and pro-inflammatory cytokine secretion following traumatic brain injury (TBI).
(A–C) Representative immunofluorescence photomicrographs (A) and quantitative data for myeloperoxidase (MPO)-positive neutrophils on day 1 (B) and ionized calcium binding adapter molecule-1 (Iba-1)-positive microglia on day 3 (C) in the lesioned boundary post-injury (n = 5 per group at each time point). Scale bars: 200 μm. Inset shows positive cells at a high magnification. (D–F) enzyme linked immunosorbent assay results showing the concentration of interleukin-1β (IL-1β; D), tumor necrosis factor-α (TNF-α; E), and IL-6 (F) in the lesioned cerebral cortex on days 1 and 3 post-injury (n = 5 per group). All data are expressed as the mean ± SD, and were analyzed by one-way analysis of variance followed by Tukey’s multiple comparison post hoc test. ###P < 0.001, vs. sham group, *P < 0.05, **P < 0.01, vs. TBI + vehicle group.
IL-1β, TNF-α, and IL-6 are major pro-inflammatory mediators detected in the inflammatory response after TBI (Zhu et al., 2014). Our ELISA data confirmed the upregulation of these pro-inflammatory mediators in the peri-contusional cortex after TBI. By contrast, DEX treatment significantly reduced IL-1β, TNF-α, and IL-6 protein expression on days 1 and 3 post-injury (Figure 2D–F; IL-1β on days 1 and 3 post-injury: P < 0.01; TNF-α and IL-6: on day 1 post-injury: P < 0.01, on day 3 post-injury: P < 0.05).
DEX administration attenuated BBB disruption after TBI
Post-traumatic inflammation was reported to contribute to BBB dysfunction (Abdul-Muneer et al., 2015). Thus, we assessed BBB permeability using the brain water content and Evans blue dye extravasation on day 3 post-injury (peak of post-TBI brain edema). DEX administration caused a significant reduction in the percentage water content in the injured cerebral hemisphere in the TBI + vehicle group (Figure 3A; P < 0.05). Consistent with these results, DEX treatment significantly decreased Evans blue leakage in the injured cerebral cortex compared with the TBI + vehicle group on day 3 post-injury (Figure 3B, C; P < 0.05).
Figure 3.

Effect of dexmedetomidine (DEX) on blood-brain barrier (BBB) permeability, tight junction protein expression, and cellular apoptosis following traumatic brain injury (TBI).
(A) Quantitative data of brain water content in the lesioned cerebral hemisphere on day 3 post-injury (n = 5 per group). (B) Representative images and (C) quantitative data for Evans blue extravasation in the ipsilateral hemisphere on day 3 post-injury (n = 5 per group). (D) Representative immunoblots and mean optical density quantification of occludin (E) and zonula occluden-1 (ZO-1; F) in the lesioned cerebral cortex on day 3 post-injury (n = 5 per group). (G) Representative immunofluorescence photomicrographs and (H) quantitative data for ZO-1 immunoreactivity in the lesioned boundary on day 3 post-injury (n = 5 per group). Scale bar: 200 μm. (I) Gating strategy and (J) quantitative analysis of cellular apoptosis in the lesioned hemispheres on day 3 post-injury (n = 5 per group). All data are expressed as the mean ± SD, and were analyzed by one-way analysis of variance followed by Tukey’s multiple comparison post hoc test. ##P < 0.01, vs. sham group; *P < 0.05, **P < 0.01, vs. TBI + vehicle group.
BBB disruption is accompanied by tight junction protein degradation (Gao et al., 2015). Thus, we assessed the expression and distribution of the tight junction proteins, including ZO-1 and occludin, on day 3 post-injury using western blot assay and immunofluorescence. As shown in Figure 3D–F, western blot results showed that ZO-1 and occludin expression were significantly higher in the TBI + DEX group than that in the TBI + vehicle group on day 3 post-injury (P < 0.05 or P < 0.01, respectively). Additionally, fluorescent staining intensity data confirmed that TBI caused reduced ZO-1 expression in the lesioned boundary, which was reversed by DEX therapy (Figure 3G, H; P < 0.05).
DEX administration reduced cellular apoptosis after TBI
Flow cytometry was used to investigate whether DEX treatment influenced post-TBI cellular apoptosis. DEX treatment significantly reduced cellular apoptosis on day 3 post-injury compared with the vehicle treatment group (Figure 3I, J; P < 0.01).
DEX administration inhibitd NF-κB and NLRP3 inflammasome activation after TBI
To further investigate whether the early neuroprotective actions of DEX were because of inhibition of NF-κB and NLRP3 inflammasome activation, we performed western blot assay on days 1 and 3 post-injury. As shown in Figure 4A–D, TBI induced a significant increase in the expression of total NF-κB p65 and nuclear NF-κB p65, and a decrease in the level of cytoplasmic NF-κB p65, compared with the sham group on days 1 and 3 post-injury (P < 0.001 for all). By contrast, DEX treatment was associated with marked decrease in NF-κB p65 nuclear translocation and total NF-κB p65 expression in the peri-contusional cortex on days 1 and 3 post-injury compared with the TBI + vehicle group (P < 0.05 or P < 0.01, respectively). In addition, the protein levels of NLRP3 inflammasome components, including NLRP3 and caspase-1 p20, and the substrate mature IL-1β, were significantly increased after TBI (Figure 4E–H; P < 0.001 for all). DEX treatment reduced NLRP3, caspase-1 p20, and mature IL-1β expression in the peri-contusional cortex on days 1 and 3 post-injury compared with the vehicle treatment (Figure 4E–H; P < 0.01 or P < 0.001, respectively).
Figure 4.
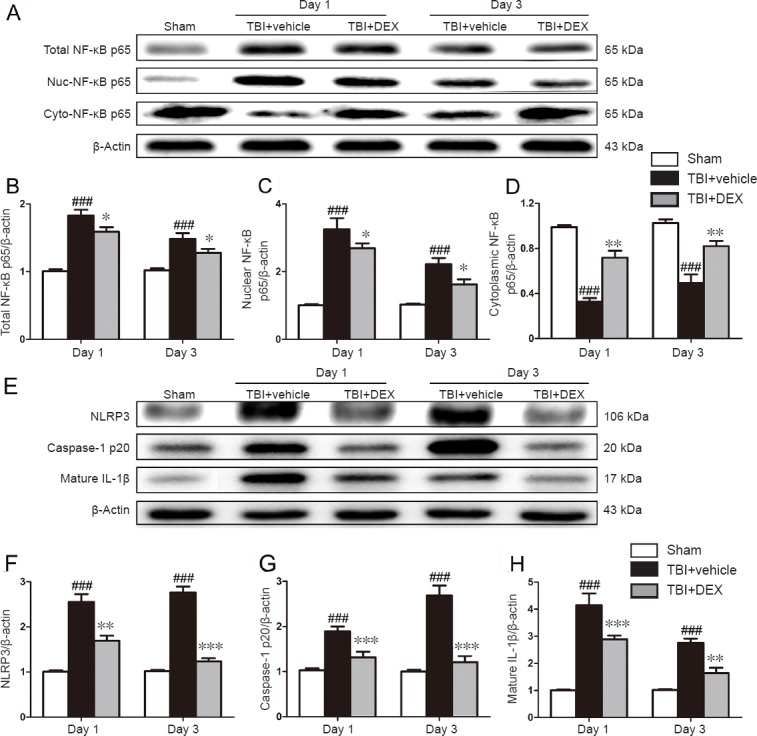
Effect of dexmedetomidine (DEX) on nuclear factor kappa B (NF-κB) and nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation following traumatic brain injury (TBI).
(A) Representative immunoblots and densitometric quantification of total NF-κB p65 (B), nuclear (Nuc) NF-κB p65 (C), and cytoplasmic (Cyto) NF-κB p65 (D) in the lesioned cerebral cortex on days 1 and 3 post-injury (n = 5 per group). (E) Representative immunoblots and densitometric quantification of NLRP3 (F), caspase-1 p20 (G), and mature IL-1β (H) in the lesioned cerebral cortex on days 1 and 3 post-injury (n = 5 per group). All data are expressed as the mean ± SD, and were analyzed by one-way analysis of variance followed by Tukey’s multiple comparison post hoc test. ###P < 0.001, vs. sham group; *P < 0.05, **P < 0.01, ***P < 0.001, vs. TBI + vehicle group.
Discussion
The present study showed that DEX treated attenuated the brain inflammatory responses in the acute phase of TBI via inhibition of NLRP3 inflammasome activation. Our main findings were that DEX treatment: (1) attenuated early neurological dysfunction, (2) reduced neutrophil infiltration, microglial activation, and pro-inflammatory mediator secretion, (3) attenuated TBI-induced BBB damage and cellular apoptosis; and (4) inhibited TBI-induced NF-κB and NLRP3 inflammasome activation.
After TBI, a numerous circulating inflammatory cells (including neutrophils and macrophages) infiltrate the injured sites through the damaged BBB (Kumar and Loane, 2012). Neutrophils are among the first responders to trauma (within hours post-TBI), and are linked to BBB breakdown, brain edema, and neuronal death (McKee and Lukens, 2016). Central nervous system-resident microglia are also activated and undergo marked morphological and behavioral changes. Although moderate microglial activation can clear harmful substances and promote tissue repair, uncontrolled and excessive activation can aggravate brain damage via secretion of inflammatory mediators such as IL-1β (Abdul-Muneer et al., 2015; McKee and Lukens, 2016). In the present study, DEX treatment effectively attenuated neutrophil infiltration and microglial activation, and reduced pro-inflammatory IL-1β, TNF-α, and IL-6 expression.
Accumulating evidence suggests that excessive inflammatory responses also contribute to BBB dysfunction and neuronal damage (Abdul-Muneer et al., 2015; Gao et al., 2015). In the present study, inhibition of inflammation by DEX treatment was accompanied by up-regulation of tight junction proteins, which maintain BBB integrity, and reduced cellular apoptosis. Consistent with these findings, Shen et al. (2017) reported that DEX could reduce brain edema, BBB disruption, and neuronal autophagy via regulation of the phosphatidylinositol-4,5-bisphosphate 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) signaling pathway.
There is increasing evidence for a role of the NF-κB signaling pathway in post-traumatic inflammation, and NF-κB is known to mediate pro-inflammatory cytokine production, and thus augment inflammatory responses (Hang et al., 2006; Jayakumar et al., 2014; Zhu et al., 2014). In the present study, DEX treatment effectively reduced NF-κB p65 translocation from the cytoplasm to the nucleus, and inhibited total NF-κB p65 expression, suggesting that DEX may attenuate post-traumatic inflammatory responses via inhibition of NF-κB activation.
IL-1β is the most important pro-inflammatory mediator in post-traumatic inflammatory responses. IL-1β release peaks within hours after TBI, and causes release of other cytokines, microglial activation, leukocyte recruitment, BBB damage, brain edema, and cellular apoptosis (McKee and Lukens, 2016). Recent evidence also suggests that inflammasome-induced caspase-1 activation is the major pathway for IL-1β production (Ma et al., 2014). The NLRP3 inflammasome is the most widely studied, and is a key mediator in post-traumatic inflammation. Activation of the NLRP3 inflammasome requires two signaling steps (Yang et al., 2018). The first priming signal involves activation of the NF-κB pathway and initiation of transcription and translation of NLRP3 and pro-IL-1β. The second step is regulated by pathogen-associated and damage-associated molecular pattern molecules that trigger the assembly and activation of the NLRP3 inflammasome complex (Zhou et al., 2016). Upon sensing danger stimuli, the NLRP3 inflammasome is assembled and activated to trigger caspase-1 activation, and subsequently the release of pro-inflammatory cytokines IL-1β and IL-18, initiating or amplifying the innate immune response and neuroinflammation following TBI (Mortezaee et al., 2018). NLRP3 inflammasome assembly and caspase-1 activation were confirmed in patients with TBI, and in murine TBI models (Liu et al., 2013; Lin et al., 2017). Inhibition of the NLRP3 inflammasome using gene knockout or small molecule inhibitors was also shown to reduce inflammatory responses, apoptosis, and histopathological injury after TBI (Irrera et al., 2017; Ismael et al., 2018). Further, omega-3 fatty acids and other agents can exert anti-inflammatory roles via inhibition of NLRP3 inflammasome activation (Mortezaee et al., 2018). In the present study, we provide new evidence that DEX treatment can decrease expression of NLRP3, caspase-1 p20, and the subsequent product IL-1β. Given the role of the NF-κB pathway in mediating NLRP3 inflammasome activation (Zhou et al., 2016), our findings suggest that the anti-inflammatory effects of DEX after TBI may be mediated, at least in part, through modulation of NF-κB-mediated NLRP3 activation.
Therapeutic hypothermia has also been shown to attenuate secondary brain injury events, such as inflammation, apoptosis, and free radical generation (Dietrich and Bramlett, 2016). McAdams et al. (McAdams et al., 2015) reported that DEX treatment can decease brain temperature in hypothermic or normal neonatal rats via α2-adrenergic receptor activation. Thus, the protective effects of DEX against TBI may also relate to effects on brain temperature.
There are some limitations of this study. First, we only examined the effects of DEX in early TBI (within 3 days). Thus, the protective function of DEX in long-term TBI remains unclear. Further, the dose of DEX was selected according to previous studies (Liang et al., 2017; Luo et al., 2017), and we did not perform dose-response experiments. Additional studies examining the optimized dose and safety of DEX therapy for TBI, as well as other potential neuroprotective mechanisms (e.g., hypothermia), are required.
In conclusion, DEX treatment attenuated inflammatory responses by limiting the activation of the NF-κB and NLRP3 inflammasome, thereby reducing BBB disruption and cellular apoptosis, and resulting in improved neurobehavioral outcomes in the acute phase of TBI. Thus, DEX may be a promising new therapeutic candidate for patients with TBI.
Additional file (11.2KB, pdf) : Open peer reviewer report 1.
Open peer review report 1
Acknowledgments
The authors are grateful to Li Liu, Wei-Yun Cui, Fang-Lian Chen, and Lei Zhou from the Tianjin Neurological Institute for their excellent technical support.
Footnotes
Conflicts of interest: The authors declare no conflict of interest.
Financial support: This work was supported by the National Natural Science Foundation of China, No. 81330029, 81671380; and the Natural Science Foundation of Tianjin City of China, No. 17JCZDJC35900. The conception, design, execution, and analysis of experiments, as well as the preparation of and decision to publish this manuscript, were made independent of any funding organization.
Institutional review board statement: All protocols were approved by the Animal Care and Use Committee of Tianjin Medical University General Hospital in China (approval No. IRB2016-YX-036). The experiments follow Consensus Author Guidelines for Animal Use, International Association of Veterinary Editors.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Shuichi Hara, Tokyo Medical University, Japan.
Funding: This work was supported by the National Natural Science Foundation of China, No. 81330029, 81671380; and the Natural Science Foundation of Tianjin City of China, No. 17JCZDJC35900.
(Copyedited by Yu J, Li CH, Qiu Y, Song LP, Zhao M)
References
- 1.Abdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol. 2015;51:966–979. doi: 10.1007/s12035-014-8752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akpinar H, Naziroglu M, Ovey IS, Cig B, Akpinar O. The neuroprotective action of dexmedetomidine on apoptosis, calcium entry and oxidative stress in cerebral ischemia-induced rats: Contribution of TRPM2 and TRPV1 channels. Sci Rep. 2016;6:37196. doi: 10.1038/srep37196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen J, Sanberg PR, Li Y, Wang L, Lu M, Willing AE, Sanchez-Ramos J, Chopp M. Intravenous administration of human umbilical cord blood reduces behavioral deficits after stroke in rats. Stroke. 2001;32:2682–2688. doi: 10.1161/hs1101.098367. [DOI] [PubMed] [Google Scholar]
- 4.Chen X, Wu S, Chen C, Xie B, Fang Z, Hu W, Chen J, Fu H, He H. Omega-3 polyunsaturated fatty acid supplementation attenuates microglial-induced inflammation by inhibiting the HMGB1/TLR4/NF-kappaB pathway following experimental traumatic brain injury. J Neuroinflammation. 2017;14:143. doi: 10.1186/s12974-017-0917-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dietrich WD, Bramlett HM. Therapeutic hypothermia and targeted temperature management in traumatic brain injury: Clinical challenges for successful translation. Brain Res. 2016;1640:94–103. doi: 10.1016/j.brainres.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang B, Li XQ, Bi B, Tan WF, Liu G, Zhang Y, Ma H. Dexmedetomidine attenuates blood-spinal cord barrier disruption induced by spinal cord ischemia reperfusion injury in rats. Cell Physiol Biochem. 2015;36:373–383. doi: 10.1159/000430107. [DOI] [PubMed] [Google Scholar]
- 7.Gao W, Zhao Z, Yu G, Zhou Z, Zhou Y, Hu T, Jiang R, Zhang J. VEGI attenuates the inflammatory injury and disruption of blood-brain barrier partly by suppressing the TLR4/NF-kappaB signaling pathway in experimental traumatic brain injury. Brain Res. 2015;1622:230–239. doi: 10.1016/j.brainres.2015.04.035. [DOI] [PubMed] [Google Scholar]
- 8.Hang CH, Chen G, Shi JX, Zhang X, Li JS. Cortical expression of nuclear factor kappaB after human brain contusion. Brain Res. 2006;1109:14–21. doi: 10.1016/j.brainres.2006.06.045. [DOI] [PubMed] [Google Scholar]
- 9.Irrera N, Pizzino G, Calò M, Pallio G, Mannino F, Famà F, Arcoraci V, Fodale V, David A, Francesca C, Minutoli L, Mazzon E, Bramanti P, Squadrito F, Altavilla D, Bitto A. Lack of the Nlrp3 inflammasome improves mice recovery following traumatic brain injury. Front Pharmacol. 2017;8:459. doi: 10.3389/fphar.2017.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ismael S, Nasoohi S, Ishrat T. MCC950, the selective inhibitor of nucleotide oligomerization domain (NOD)-like receptor protein-3 inflammasome, protects mice against traumatic brain injury. J Neurotrauma. 2018;31:1249–1257. doi: 10.1089/neu.2017.5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jayakumar AR, Tong XY, Ruiz-Cordero R, Bregy A, Bethea JR, Bramlett HM, Norenberg MD. Activation of NF-kappaB mediates astrocyte swelling and brain edema in traumatic brain injury. J Neurotrauma. 2014;31:1249–1257. doi: 10.1089/neu.2013.3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 13.Liang H, Liu HZ, Wang HB, Zhong JY, Yang CX, Zhang B. Dexmedetomidine protects against cisplatin-induced acute kidney injury in mice through regulating apoptosis and inflammation. Inflamm Res. 2017;66:399–411. doi: 10.1007/s00011-017-1023-9. [DOI] [PubMed] [Google Scholar]
- 14.Lin C, Chao H, Li Z, Xu X, Liu Y, Bao Z, Hou L, Liu Y, Wang X, You Y, Liu N, Ji J. Omega-3 fatty acids regulate NLRP3 inflammasome activation and prevent behavior deficits after traumatic brain injury. Exp Neurol. 2017;290:115–122. doi: 10.1016/j.expneurol.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 15.Liu HD, Li W, Chen ZR, Hu YC, Zhang DD, Shen W, Zhou ML, Zhu L, Hang CH. Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res. 2013;38:2072–2083. doi: 10.1007/s11064-013-1115-z. [DOI] [PubMed] [Google Scholar]
- 16.Luo C, Ouyang MW, Fang YY, Li SJ, Zhou Q, Fan J, Qin ZS, Tao T. Dexmedetomidine protects mouse brain from ischemia-reperfusion injury via inhibiting neuronal autophagy through up-regulating HIF-1alpha. Front Cell Neurosci. 2017;11:197. doi: 10.3389/fncel.2017.00197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma Q, Chen S, Hu Q, Feng H, Zhang JH, Tang J. NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann Neurol. 2014;75:209–219. doi: 10.1002/ana.24070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mamik MK, Power C. Inflammasomes in neurological diseases: emerging pathogenic and therapeutic concepts. Brain. 2017;140:2273–2285. doi: 10.1093/brain/awx133. [DOI] [PubMed] [Google Scholar]
- 19.McAdams RM, McPherson RJ, Kapur R, Phillips B, Shen DD, Juul SE. Dexmedetomidine reduces cranial temperature in hypothermic neonatal rats. Pediatr Res. 2015;77:772–778. doi: 10.1038/pr.2015.45. [DOI] [PubMed] [Google Scholar]
- 20.McKee CA, Lukens JR. Emerging roles for the immune system in traumatic brain injury. Front Immunol. 2016;7:556. doi: 10.3389/fimmu.2016.00556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mortezaee K, Khanlarkhani N, Beyer C, Zendedel A. Inflammasome: Its role in traumatic brain and spinal cord injury. J Cell Physiol. 2018;233:5160–5169. doi: 10.1002/jcp.26287. [DOI] [PubMed] [Google Scholar]
- 22.Schoeler M, Loetscher PD, Rossaint R, Fahlenkamp AV, Eberhardt G, Rex S, Weis J, Coburn M. Dexmedetomidine is neuroprotective in an in vitro model for traumatic brain injury. BMC Neurol. 2012;12:20. doi: 10.1186/1471-2377-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen M, Wang S, Wen X, Han XR, Wang YJ, Zhou XM, Zhang MH, Wu DM, Lu J, Zheng YL. Dexmedetomidine exerts neuroprotective effect via the activation of the PI3K/Akt/mTOR signaling pathway in rats with traumatic brain injury. Biomed Pharmacother. 2017;95:885–893. doi: 10.1016/j.biopha.2017.08.125. [DOI] [PubMed] [Google Scholar]
- 24.Shen ZY, Zhang J, Song HL, Zheng WP. Bone-marrow mesenchymal stem cells reduce rat intestinal ischemia-reperfusion injury, ZO-1 downregulation and tight junction disruption via a TNF-alpha-regulated mechanism. World J Gastroenterol. 2013;19:3583–3595. doi: 10.3748/wjg.v19.i23.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi H, Wang HL, Pu HJ, Shi YJ, Zhang J, Zhang WT, Wang GH, Hu XM, Leak RK, Chen J, Gao YQ. Ethyl pyruvate protects against blood-brain barrier damage and improves long-term neurological outcomes in a rat model of traumatic brain injury. CNS Neurosci Ther. 2015;21:374–384. doi: 10.1111/cns.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Liu H, Zhang L, Wang G, Zhang M, Yu Y. Neuroprotection of dexmedetomidine against cerebral ischemia-reperfusion injury in rats: involved in inhibition of NF-kappa B and inflammation response. Biomol Ther (Seoul) 2017;25:383–389. doi: 10.4062/biomolther.2015.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu X, Gao W, Cheng S, Yin D, Li F, Wu Y, Sun D, Zhou S, Wang D, Zhang Y, Jiang R, Zhang J. Anti-inflammatory and immunomodulatory mechanisms of atorvastatin in a murine model of traumatic brain injury. J Neuroinflammation. 2017;14:167. doi: 10.1186/s12974-017-0934-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang SJ, Shao GF, Chen JL, Gong J. The NLRP3 inflammasome: an important driver of neuroinflammation in hemorrhagic stroke. Cell Mol Neurobiol. 2018;38:595–603. doi: 10.1007/s10571-017-0526-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang R, Liu Y, Yan K, Chen L, Chen XR, Li P, Chen FF, Jiang XD. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J Neuroinflammation. 2013;10:106. doi: 10.1186/1742-2094-10-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou K, Shi L, Wang Y, Chen S, Zhang J. Recent advances of the NLRP3 inflammasome in central nervous system disorders. J Immunol Res. 2016;2016:9238290. doi: 10.1155/2016/9238290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu HT, Bian C, Yuan JC, Chu WH, Xiang X, Chen F, Wang CS, Feng H, Lin JK. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-kappaB signaling pathway in experimental traumatic brain injury. J Neuroinflammation. 2014;11:59. doi: 10.1186/1742-2094-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Open peer review report 1