

Abstract
Myelin-associated inhibitory factors within the central nervous system (CNS) are considered to be one of the main obstacles for axonal regeneration following disease or injury. The nogo receptor 1 (NgR1) has been well documented to play a key role in limiting axonal regrowth in the injured and diseased mammalian CNS. However, the role of nogo receptor in immune cell activation during CNS inflammation is yet to be mechanistically elucidated. Microglia/macrophages are immune cells that are regarded as pathogenic contributors to inflammatory demyelinating lesions in multiple sclerosis (MS). In this study, the animal model of MS, experimental autoimmune encephalomyelitis (EAE) was induced in ngr1+/+ and ngr1–/– female mice following injection with the myelin oligodendrocyte glycoprotein (MOG35–55) peptide. A fate-map analysis of microglia/macrophages was performed throughout spinal cord sections of EAE-induced mice at clinical scores of 0, 1, 2 and 3, respectively (increasing locomotor disability) from both genotypes, using the CD11b and Iba1 cell markers. Western immunoblotting using lysates from isolated spinal cord microglia/macrophages, along with immunohistochemistry and flow cytometric analysis, was performed to demonstrate the expression of nogo receptor and its two homologs during EAE progression. Myelin protein engulfment during EAE progression in ngr1+/+ and ngr1–/– mice was demonstrated by western immunblotting of lysates from isolated spinal cord microglia/macrophages, detecting levels of Nogo-A and MOG. The numbers of M1 and M2 microglia/macrophage phenotypes present in the spinal cords of EAE-induced ngr1+/+ and ngr1–/– mice, were assessed by flow cytometric analysis using CD38 and Erg-2 markers. A significant difference in microglia/macrophage numbers between ngr1+/+ and ngr1–/– mice was identified during the progression of the clinical symptoms of EAE, in the white versus gray matter regions of the spinal cord. This difference was unrelated to the expression of NgR on these macrophage/microglial cells. We have identified that as EAE progresses, the phagocytic activity of microglia/macrophages with myelin debris, in ngr1–/– mice, was enhanced. Moreover, we show a modulation from a predominant M1-pathogenic to the M2-neurotrophic cell phenotype in the ngr1–/– mice during EAE progression. These findings suggest that CNS-specific macrophages and microglia of ngr1–/– mice may exhibit an enhanced capacity to clear inhibitory molecules that are sequestered in inflammatory lesions.
Keywords: microglia, experimental autoimmune encephalomyelitis, nogo receptor, myelin-associated inhibitory factors (MAIFs), Nogo A, neural regeneration
Introduction
Microglia are innate immune cells of the central nervous system (CNS), considered to exacerbate neuroinflammation, in chronic multiple sclerosis (MS) lesions (Streit et al., 2004). MS is a severe CNS disorder (Cudrici et al., 2006), characterized by extensive demyelination, with axonal damage or loss (Petratos et al., 2012; Singh et al., 2013b; Lee et al., 2014). It is well documented that CNS myelin-associated inhibitory molecules are considered major molecular barriers for axonal regeneration (Lee and Petratos, 2013). The inhibitory components of myelin include Nogo-A, oligodendrocyte-myelin glycoprotein (OMgp) as well as myelin-associated glycoprotein (MAG) (Lee and Petratos, 2013). They are mainly found on the myelin sheath and interact directly with nogo receptor 1 (NgR1) expressed on axons (Neumann et al., 2009). During neuroinflammation or physical injury within the CNS, demyelination generates myelin debris enriched with myelin-associated inhibitors to axonal regeneration. Thus, the clearance of myelin debris may be an endogenous attempt at establishing a diseased tissue environment that promotes neural repair leading to neurological improvement (Neumann et al., 2009). In fact, it has been reported that there is a robust association between the efficiency of remyelination and the success of myelin debris elimination (Neumann et al., 2009). In addition, a number of studies have found that insufficient myelin clearance leads to a lack of axonal regeneration as seen in the damaged CNS (David and Lacroix, 2003; Oudega and Xu, 2006; Neumann et al., 2009). Current evidence implies that the existence of myelin-associated inhibitory factors also disturbs oligodendrocyte precursor cell differentiation into mature oligodendrocytes throughout the remyelination process (Kotter et al., 2005; Sozmen et al., 2016).
Histological studies have shown that NgR and its co-receptor Troy/Lingo-1 are also expressed on macrophage/microglia in demyelinating MS lesions (Satoh et al., 2007; Yan et al., 2012). These data raise an interesting question of whether the expression of NgR on microglia might be a key factor in the regulation of inflammation following CNS injury. Importantly, our laboratory has defined that limiting neurodegeneration during EAE progression can be achieved through abrogating the NgR1 signal transduction mechanism (Petratos et al., 2012). NgR1-dependent axonal pathology has been defined as the mechanism governing EAE and potentially MS pathology (Petratos et al., 2012). Furthermore, recent evidence suggests that microglia/macrophages may express NgR (Satoh et al., 2005; Fang et al., 2015; Liu et al., 2015), and thus blockade of the signal transduction mechanisms in our NgR1-null mice, during the progression of EAE, may regulate innate immune mechanisms of disease induction and propagation.
Two homologs of NgR exist, defined as NgRH1 and NgRH2 (NgR2 and NgR3 respectively) but their biological relevance in disease is yet not to be fully elucidated (Pignot et al., 2003). Similar to NgR, these homologs contain a C-terminal GPI signal sequence and eight leucine-rich repeats (LRR) surrounded by a leucine-rich repeat N-terminus (LRRNT) and a leucine-rich repeat C-terminus (LRRCT) (Pignot et al., 2003). NgR2 is a receptor protein that binds MAG with high affinity and can probably inhibit neurite outgrowth (Domeniconi et al., 2002). MAG is a well-known inhibitor of axonal growth during development and regeneration (Filbin, 2003; Sicotte et al., 2003; Venkatesh et al., 2005), but blocking this inhibitor is not enough to promote axonal regrowth within the injured spinal cord of mice (Venkatesh et al., 2005). NgR3 has been identified as a receptor for chondroitin sulfate proteoglycans (CSPGs) (Dickendesher et al., 2012).
In the current study, we aimed to identify a functional role for NgR in microglial activity during inflammatory demyelination and axonal damage that manifests during the clinical progression of EAE. We investigated the expression of NgR within microglia/macrophage populations during the progression of EAE. We identified that NgR1-dependent microglial/macrophage activity did not govern the dynamics of these cell populations during EAE progression.
Materials and Methods
Animals
Both ngr1–/–(Kim et al., 2004) and their ngr1+/+ littermates, were bred in-house and maintained at the Alfred Medical Research and Education Precinct (AMREP) animal facility. The AMREP Animal Ethics Committee (AMREP AEC#E/1532/2015/M) approved the use of these animals for experimentation, in accordance with the National Health and Medical Research Council of Australia code for the care and use of animals for scientific purposes.
MOG35-55 induction of EAE and evaluation of clinical progression
The induction of EAE was performed to demonstrate the number and role of macrophages/microglia expressing NgR during the disease course. Sixty-eight ngr1+/+ and 51 ngr1–/– female mice, weighing 18–22 g, aged 8–12 weeks were utilized in this study. They were subcutaneously injected in the lower flanks with either 200 μg MOG35–55 (Purar Chemical, Shanghai, China) emulsified in 200 μL of Complete Freund’s Adjuvant (CFA; Sigma-Aldrich, St. Louis, MO, USA) accompanied with 4 μL/mL of Mycobacterium tuberculosis or CFA alone. The immunized mice were then injected intraperitoneally (i.p.) with two doses of 350 μg pertussis toxin (List Biological Laboratories, Inc., Campbell, CA, USA), on days 1 and 3 post-induction (dpi), with MOG35–55. The mice were checked daily and scored for their disease progression as follows: pre-onset (score 0, usually observed throughout 1–12 dpi), onset (score 1; usually observed between 10–18 dpi), peak stage of the disease (score 2, usually between 16–26 dpi) and chronic stage of the disease (score ≥ 3, usually between 22–30 dpi). Mice were then killed through CO2 inhalation at different stages of the disease and either transcardially perfused with 4% paraformaldehyde (PFA) for immunohistochemical analysis to be performed on spinal cords, or the same freshly collected CNS tissues for macrophage/microglia immunopanning or protein extraction for western immunoblotting.
Immunofluorescent labeling and confocal microscopy
Immunohistochemistry was performed to identify and enumerate microglia/macrophage cells, which populate lesion and non-lesion areas of the spinal cord with and without EAE induction. CNS tissues from EAE-induced mice were examined for the presence of either CD11b- and Iba1-positive microglia/macrophage cells, along with the colocalization of either NgR or NgR3. The tissue sections were incubated for 1 hour at 60°C, followed by paraffin dewaxing. After incubation, the sections were incubated in the proteinase K (PK) (Qiagen, Hilden, Germany), then washed with phosphate-buffered saline (PBS). Tissues were then post-fixed with 4% paraformaldehyde for 30 minutes, and then washed with PBS. The slides were blocked overnight at 4°C in blocking buffer (5% fetal bovine serum (FBS), 5% normal goat serum, 0.1% Triton-X 100 in PBS) for all EAE tissues. Then, the slides were incubated for 2 hours at room temperature with the primary antibodies polyclonal rabbit anti-CD11b (1:100) (Abcam, Cambridge, United Kingdom) or polyclonal rabbit anti-Iba1 (1:500) (Wako Chemicals USA, Richmond, VA, USA). Sections were then further incubated with a monoclonal anti-NgR antibody (1:100) (R&D Systems, Minneapolis, MN, USA). After the slides were washed with PBS, the secondary antibody anti-rabbit 555 (1:200) (Alexafluor 555 conjugated, anti-rabbit IgG (H+L), Life technologies, Carlsbad, CA, USA) was used for both CD11b and Iba1, while anti-rat 488 (1:200) (Alexafluor 488 conjugated goat anti-rat IgG, Life technologies, Carlsbad, CA, USA) was used as a secondary antibody for anti-NgR. Next, the slides were washed with PBS and finally incubated with 4′,6-diamidino-2-phenylindole (DAPI) at a dilution of 1:2000. The slides were analyzed under an upright Nikon A1r confocal microscope (Nikon, Tokyo, Japan) and the images were captured.
Semi-quantitative analysis of CD11b-positive microglia/macrophage cells along with NgR within white and grey matter spinal cord
The immunostained EAE-induced mouse CNS sections stained with the anti-CD11b antibody were then processed for semi-quantitative analysis of the number of microglia/macrophages, which expressed NgR in grey and white matter using MetaMorph™ software (Molecular devices, Silicon Valley, CA, USA). The areas of white and grey matter, which had NgR-positive microglia/macrophage cells, were estimated per unit area (μm2). Approximately 500 sections (> 50 μm apart) were analysed (from n = 3–12 mice/clinical score, 3–7 sections/mouse and 3–7 images per section).
The data were analyzed using GraphPad Prism 6 software (GraphPad Software, La Jolla, CA, USA), in order to compare the numbers of either single CD11b-positive microglia/macrophages or double labeled NgR-positive microglia/macrophages in both genotypes across different clinical scores.
Western blot analysis
To further investigate the expression of NgR on activated microglia, western blot analysis was performed on microglial cell lysates. The cells were firstly isolated from the spinal cord of naïve ngr1+/+, ngr1–/– mice and EAE-induced ngr1+/+ and ngr1–/– mice after being immuno-panned using a polyclonal Iba1 antibody. Microglia were re-suspended in a cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA) along with PhosSTOP™ phosphatase inhibitor cocktail (Roche Applied Science, Penzberg, Germany) and protease inhibitor cocktail (Sigma-Aldrich). Homogenates were centrifuged at 13,000 × g for 15 minutes and protein concentrations were estimated, using the bicinchoninic acid (BCA) protein assay reagent kit (Life technologies, Carlsbad, CA, USA).
Cell and tissue lysates (10 μg of protein) were run on 4–12% Bis-Tris gel (Life technologies). The membranes from the previously defined lysates were blocked with 5% skim milk in Tris-buffered saline with 0.1% Tween-20 TBST and then incubated overnight at 4°C with the primary antibodies that included polyclonal anti-NgR (Merck Millipore, Darmstadt, Germany) (1:1000), polyclonal rabbit anti-NgR2 (1:200) (Santa Cruz Biotechnology, Dallas, TX, USA) and polyclonal rabbit anti-NgR3 (1:200) (Santa Cruz Biotechnology) antibodies, all diluted in the same blocking buffer. In addition, isolated microglia were also probed with different primary antibodies, such as monoclonal anti-myelin oligodendrocyte glycoprotein (MOG) (1:5000) (Merck Millipore, Darmstadt, Germany), polyclonal rabbit anti-myelin basic protein (MBP) (1:20,000)(Merck Millipore), polyclonal rabbit anti-contactin-associated protein (Caspr) (1:10,000) (Abcam, Cambridge, United Kingdom) and polyclonal rabbit anti-Nogo-A (1:4000) (Merck Millipore) antibodies, to analyze the microglial behavior and activity within demyelination and axonal damage during the progression of EAE disease. The membranes were then washed in TBST, and HRP-conjugated secondary antibodies for each primary antibody were used that included anti-rabbit or anti-mouse or anti-goat (all diluted at 1:10,000) IgG (Merck Millipore) for 2 hours at room temperature. The membranes were washed with TBST and incubated with Luminata ECL chemiluminescence (Merck Millipore), developed, and imaged using ChemiDoc™ Touch Gel Imaging System (Bio-Rad, Hercules, CA, USA). Protein loading levels were analyzed by further staining of these membranes using monoclonal anti-alpha-tubulin (1:15,000) (Merck Millipore) or monoclonal anti-beta-actin (1:20,000) (Merck Millipore) incubated in skim milk blocking buffer that consisted of 0.01% w/v sodium azide. Four to six individual samples were analyzed for all tissue and cell lysate experimental paradigms then individual protein band optical densities measured by Image Lab v5.2.1 (Bio-Rad) for statistical purposes.
Isolation of macrophages/microglia using immunopanning
An immunopanning method was adopted to isolate spinal cord macrophages/microglia from ngr1+/+ and ngr1–/– mice with or without EAE induction, using the polyclonal anti-Iba-1 antibody (Wako Chemicals USA, Richmond, VA, USA).
Preparation of panning plate
Two petri dishes (100 mm × 15 mm) were incubated overnight at 4°C with 2 mL of Tris buffer (50 mM, pH 9.5) and 150 μL of anti-rabbit IgG1 was used as the capture antibody. After three washes with 1× PBS, the dishes were incubated with 2 mL of polyclonal rabbit anti-Iba-1 antibody (Wako Chemicals USA) for 2 hours at room temperature.
Preparation of mouse spinal cord cell suspension
The spinal cord was dissected from the ngr1+/+ and ngr1–/– mice and transferred into 15 mL tubes with 8 mL Earle’s balanced salt solution (EBSS). After the fragments had settled, the EBSS was removed. The tissue was enzymatically dissociated to make a single-cell suspension. The tissue was incubated at 37°C for 30 minutes with papain at a final concentration of 20 U/mL (Worthington Biochemical Corp., Lakewood, NJ, USA). Next, the tissue was washed with 4 mL EBSS, followed by an ovomucoid solution in BSA (10 mg/mL) (Sigma) and 100 μL DNAse (0.005%). The cells were triturated until a homogenous solution was obtained.
Panning
The dissociated cells were then re-suspended in 2 mL panning buffer (0.02% BSA, 12.5 U/mL in dPBS) and filtered through a 40 μm mesh. Subsequently, the single cell suspension was sequentially panned on anti-Iba-1 antibody coated panning plates and incubated for 45 minutes in a 10% CO2 incubator at 37°C. Then, the purified cells were removed by 1 mL of trypsin solution (12 mL) (0.25% Trypsin EDTA (1.2 mL), Ca2+ Mg2+- free EBSS (10.8 mL). The trypsin was neutralized with 10% FBS in DMEM, then washed by adding 2 mL DMEM with PBS to the panning dish, with continuous swirling to gather more cells. Finally, the cells were gently dislodged by pipetting and harvested by centrifugation at 220 × g for 15 minutes.
Analysis of isolated cells by fluorescence-activated cell sorting (FACS)
The cells were re-suspended in 2 mL FACS wash buffer and then filtered through a 40 μm mesh. Viable cells were estimated and the cells were then added to 1 mL of FACS wash and centrifuged for 3 minutes at 220 × g and 4°C. After removing the supernatant, the cells were incubated for 30 minutes on ice with either polyclonal rabbit anti-CD11b antibody (1:10) or polyclonal rabbit anti-Iba-1 (1:10) and polyclonal rabbit anti-NgR antibody (1:10), polyclonal rabbit anti-NgR2 and polyclonal rabbit anti-NgR3 antibodies (1:100) as primary antibodies, followed by 30 minute incubation on ice with anti-rabbit 647 (1:200), anti-rat 488 (Alexafluor 488 goat anti-rat IgG (1:200) and anti-goat 488 (1:200), Life Technologies) as secondary antibodies. The cells were also counterstained with DAPI (1:2000). Finally, the cell population was analyzed by using an FACS Canto II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Post-flow cytometric analysis was performed on FlowLogic (Inivai Technologies, Mentone, Australia).
Phenotyping of microglia/macrophages
To identify the biological properties and function of microglia/macrophages during neuroinflammation, cells were isolated from spinal cords of EAE-induced ngr1+/+ and ngr1–/– mice using the immunopanning method. The cells were then immuno-stained with anti-rat CD38-FITC monoclonal antibody (1:50) (BD Biosciences) to define M1 phenotype and anti-rat Egr-2-PE monoclonal antibody (1:20) to detect M2 phenotype, along with their isotype controls including RatIgG2a, kappa Isotype Control FITC (1:200) (BD Biosciences), and Rat IgG2a, kappa Isotype Control PE (1:200) (BD Biosciences). The immuno-stained live cells were analyzed using Canto II flow cytometer (BD Biosciences), and then post-analysis was performed on FlowLogic (Inivai Technologies, Mentone, Australia).
Statistics
Data were analyzed using GraphPad Prism v6.05 software (GraphPad software, La Jolla, CA, USA). Data were represented as the mean ± standard error of the mean (SEM) and standard errors were calculated for each group of naïve or diseased mice at different clinical scores of EAE, followed by a one-way analysis of variance (ANOVA) analysis with a Tukey’s post-hoc test. A one-way ANOVA was used to determine statistical significance (P < 0.05) at a 95% confidence level for semi-quantitative analysis and a one-tailed Student’s t-test was performed for optical density levels acquired from immunoblotting analysis as well as FACS data. Two-way ANOVA and Friedman’s non-parametric repeated measures were used for analysis of differences in the clinical progression of EAE between different genotypes (ngr1+/+ and ngr1–/– mice).
Results
NgR expression on microglia/macrophages throughout spinal cord sections of naïve and EAE-induced ngr1+/+ or ngr1–/– mice
To assess the NgR expression among microglia/macrophage populations present throughout the CNS during neuroinflammation and axo-myelin degeneration, immunofluorescence was performed on mouse spinal cord and optic nerve tissues. Mouse microglia/macrophages were immunostained using two specific markers to these reactive cells, CD11b antibody and Iba-1. In the ngr1+/+ mice without disease, NgR was expressed within both CD11b- and Iba1-positive cells throughout the grey and white matter tracts (Additional Figure 1A, B (1.2MB, tif) ). During the chronic stage of EAE disease (clinical score 3), we were able to identify immunolabeled microglia/macrophages expressing NgR, which exhibited an activated amoeboid morphology (Additional Figure 1A, B (1.2MB, tif) ). These findings confirmed that NgR is expressed within microglia/macrophages throughout the spinal cord at different stages of disease, displaying various active cell morphologies following EAE induction.
Microglial cells expressing NgR in the spinal cord of EAE-induced ngr1+/+andngr1–/– mice during inflammatory demyelinating pathology.
(A) CD11b-positive microglial/macrophage cells were co-labeled with the anti-NgRantibody within the spinal cord, during the progression of EAE in ngr1+/+mice. (B)Microglial/macrophage cells were also immunostained with Iba-1 for further confirmation of their activation, along with anti-NgR, demonstrating the elevationin the numbers of these cells within spinal cord inflammatory lesions in ngr1+/+mice. (C) The ngr1–/–micethat developed EAE were also immunolabeled for both CD11b and NgR, utilizing the same antibodies as for ngr1+/+mouse tissue. NgR expression could still be seen in large numbers of these macrophage/microglial cells in the spinal cord sections of EAE-induced ngr1–/–mice. (D) The Iba-1-positive macrophage/microglial cells, immunopositive for NgR, were present in the spinal cords of ngr1–/–mice during the progression of their EAE. Scale bar: 50 μm.
Throughout the spinal cord of ngr1–/– mice that were immunized with MOG35–55, we identified NgR-immunoreactivity within the CD11b-positive microglia/macrophages in clusters of DAPI+ cells (Additional Figure 1C (1.2MB, tif) ). Similarly, anti-NgR- and Iba1-labeled microglia/macrophages were demonstrated to be present within white and grey matter in the spinal cord sections of ngr1–/– mice obtained from EAE-induced mice (Additional Figure 1D (1.2MB, tif) ). The detection of NgR-immunolabeling in the ngr1–/– mice may suggest that the anti-NgR antibodies are non-specific for NgR1 detection.
Mapping microglial/macrophage numbers throughout the spinal cord of ngr1+/+ and ngr1–/– mice during the progression of EAE
Only ~50% of the EAE-induced ngr1–/– mice exhibited disease symptoms (Figure 1A and Table 1), therefore we only included those mice that progressed with EAE for immunohistochemical analysis to obtain a direct comparison with wild type littermate mice at the same stage of disease. We mapped the activated microglia/macrophage populations that expressed NgR within the spinal cord grey and white matter to illustrate their relevance during the disease progression in particular as the cell populations relate to neurodegeneration. It was clear that ngr1–/– mice had a reduced incidence of EAE, delay in onset (if EAE was successfully induced) and exhibited reduced severity of symptoms (Figure 1A and Table 1).
Figure 1.
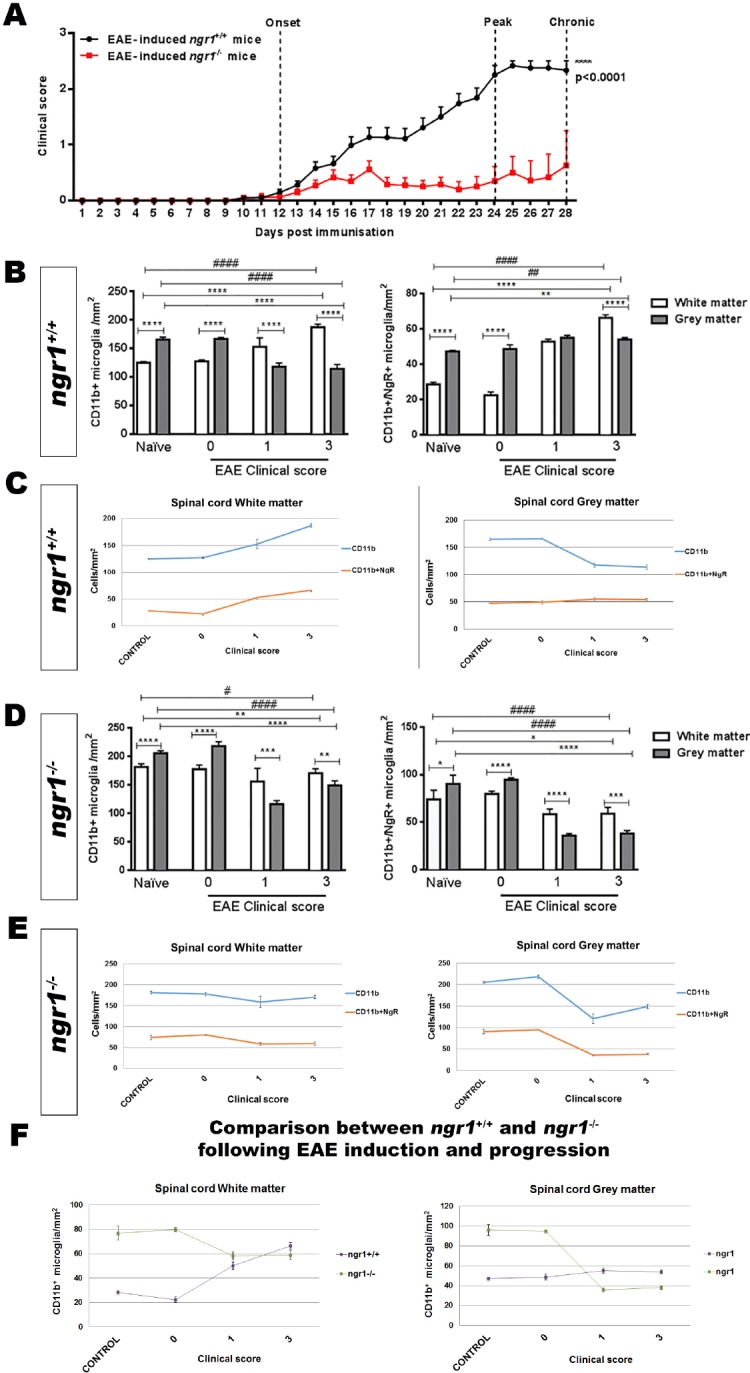
Substantial delay in the development of EAE in ngr1–/– mice after immunization with MOG35–55 associated with reduction in microglial/macrophage cell number that is unrelated to NgR1 expression within these cell types.
(A) Delay in the onset and severity of disease in ngr1–/– mice (n = 51, red line) post-immunization with MOG35–55 when compared to ngr1+/+ mice (n = 68, black line). EAE clinical scores were determined as following: Pre-onset stage (7–12 dpi), onset stage (12–23 dpi), peak stage (24–28 dpi) and chronic stage (28–30 dpi). Data are represented as the mean ± SEM (****P < 0.0001, two-way ANOVA and Friedman’s non-parametric repeated measures). (B) There were significant differences in the number of CD11b-positive microglial/macrophage cells or microglial cells that were positive for NgR between white and grey matter for naïve ngr1+/+ mice and during the disease progression of EAE-induced ngr1+/+ (one-way ANOVA with post-hoc Tukey’s multiple comparison test; ##P < 0.01, **P < 0.01, ####P < 0.0001, and ****P < 0.0001; n = 3 for naïve ngr1+/+ mice; n = 5 for clinical score 0; n = 12 for clinical score 1 and n = 7 for clinical score 3). (C) The linear elevation in numbers of microglial/macrophage cells along with those that were co-labeled with NgR during the progression of EAE in female ngr1+/+ mice occurs in the white matter but not the grey matter. (D) In EAE-induced ngr1–/– mice, NgR-labeling was detected in microglial/macrophage cell populations, with significant differences in the number of these cells between the white and grey matter areas of naïve ngr1–/– and EAE-induced ngr1–/– mice at clinical score 0 (one-way ANOVA with post-hoc Tukey’s multiple comparison test; #P < 0.05, *P < 0.05, **P < 0.01, ####P < 0.0001 and ****P < 0.0001; n = 5 for naïve ngr1–/– mice; and n = 5 for clinical score 0). In addition, significant reductions in the number of these NgR-positive microglial cells within white matter and grey matter areas could be demonstrated in ngr1–/– mice when EAE progression could be documented in these mice (***P < 0.001 and ****P < 0.0001; n = 5 for naïve ngr1–/– mice; and n = 5 for clinical score 0. Also, n = 6 for clinical score 1 and n = 5 for clinical score 3). (E) The linear graphical representation of these data illustrates the reduction in numbers of microglia/macrophages localized within the grey matter of the ngr1–/– mice when disease progresses. The numbers of single labeled CD11b+ macrophage/microglial cells with those cells co-labeled with CD11b+ and NgR+ following EAE-induction, are represented for both white and grey matter. This pattern cannot be replicated in the white matter. (F) Synthesis of the linear representation of CD11b-positive macrophage/microglial cells in the spinal cord grey and white matter areas of ngr1–/– compared with ngr1+/+ mice as EAE progresses. The numbers of CD11b-positive macrophage/microglial cells in the spinal cord white matter of naïve, ngr1–/– mice are very high when compared to ngr1+/+ mice. As EAE progresses independently in these two genotypes, the numbers of CD11b-positive macrophage/microglial cells reach similar levels, suggesting that numbers of these cells are increased substantially only in ngr1+/+ mice. However, in grey matter, the numbers of these cells were stable in EAE-induced ngr1+/+ mice but significantly dropped in ngr1–/– mice as EAE progressed. EAE: Experimental autoimmune encephalomyelitis; NgR: Nogo receptor; MOG35–55: myelin oligodendrocyte glycoprotein peptide 35–55 amino acids; dpi: days post-induction.
Table 1.
Clinical scores of female ngr1–/– and ngr1+/+ MOG35-55 EAE-induced mice
Significant differences were demonstrated in the number of CD11b-positive microglia/macrophages along with NgR-positive cells expressed on these populations within the white matter when compared to grey matter (Figure 1B, C). In addition, we noticed that the numbers of NgR-positive microglia/macrophages were significantly increased in white matter areas during disease progression in EAE-induced ngr1+/+ mice when compared with similar areas in the naïve ngr1+/+ mice (Figure 1B, C).
To examine how NgR1-positive microglia/macrophages affect CNS axon integrity, we analyzed the relationship of these populations with the progression of EAE over time in white matter versus grey matter areas. Microglia/macrophages increased in number during myelin and axonal damage as EAE progressed (Additional Figure 2 (975KB, tif) ). However, when we examined the spinal cord grey matter compartment, the data showed that microglia/macrophages were elevated in the white matter tracts where chronic inflammatory lesions exist (Figure 1B, C, and Additional Figure 2 (975KB, tif) ). Therefore, we observed a decrease in the number of microglia/macrophages in grey matter with increases in white matter lesion areas, irrespective of NgR1 expression within these cells.
Caspr is localized in degenerative axons in the optic nerves of EAE-induced ngr1+/+ but not in ngr1–/– mice.
(A) Paraffin-embedded optic nerves of EAE-induced (peak-chronic stage of disease) ngr1+/+mice were immunostained with MBP, (B) Caspr and counter-stained with (C) DAPI. (D) Merged images are shown (Scale bar = 50μm). (E-P) Co-localization of markers of degenerative axons with Caspr in EAE (peak-chronic stage of disease) in ngr1+/+ optic nerves. The serial sections were immunostained with (E) βAPP, (I) AT8, or (M) SMI-32 (red), which were co-immunostained with Caspr (F, J & N), counterstaining with DAPI was performed in all images (G, K & O); merged images (H, L & P) (Scale bar = 10 μm). (Q) Stereological quantification revealed a significant co-localization of Caspr with markers for degenerative axon βAPP, AT8 and SMI-32 (**P<0.01, ****P<0.0001, n =8).
Despite deletion of ngr1, we still found NgR immunoreactivity on microglia/macrophages. Our results demonstrated that significant differences existed in the numbers of microglia/macrophages expressing NgR between grey and white matter areas of ngr1–/– mice that developed EAE symptoms and pathology (Figure 1D–F). However, significant reductions in both of these cell populations were shown within grey matter (Figure 1D–F).
In spinal cord white matter, the numbers of these double-positive cells within naïve mice and also at the pre-onset stage of EAE, were two-fold greater in ngr1–/– mice compared to those in ngr1+/+ mice; estimated numbers were approximately 79 ± 4.3 cells/mm2 and 28.5 ± 1.2 cells/mm2, respectively. However, as EAE progressed, these numbers reached similar levels in both ngr1+/+ and ngr1–/– mice (Figure 1F). Within spinal cord grey matter of untreated mice or EAE-induced mice at a pre-clinical disease score of 0, higher numbers of microglial/macrophage cells expressing NgR were detected in ngr1–/– mice (i.e., 95 ± 0.9 cells/mm2) compared to 48.6 ± 2.4 cells/mm2, enumerated in ngr1+/+ mice (Figure 1F). Notably, these NgR-positive macrophage/microglial cell numbers were significantly reduced in EAE-induced ngr1–/– mice but remained stable within grey matter areas of EAE-induced ngr1+/+ mice as EAE developed (Figure 1F). Collectively, these data may suggest that NgR1 does not regulate the numbers of microglia/macrophages at neuroinflammatory sites.
Isolated spinal cord microglia/macrophages in ngr1+/+ and ngr1–/– mice show expression of NgR homologs
The results from the immunostaining experiments and our semi-quantitative analysis illustrated that microglial/macrophage cells express NgR. However, we wished to differentiate which NgR homologs, namely NgR2 and NgR3, were being expressed within these inflammatory cells specifically. To address this question, we isolated microglial/macrophage cells from the spinal cords of both ngr1+/+ and ngr1–/– mice and western immunoblot analyses were performed on cell lysates.
To confirm the expression of NgR2 and NgR3 proteins, specific antibodies directed against these specific receptors were used. The results established the presence of these NgR homologs present in microglial/macrophage cell lysates during EAE progression in ngr1+/+ and ngr1–/– mice, as well as in those lysates from naïve ngr1+/+ and ngr1–/– mice (Figure 2B). These results were validated using NgR(310)-Fc, NgR2 and NgR3 peptides (i.e., as positive controls) for this analysis (Figure 2B).
Figure 2.
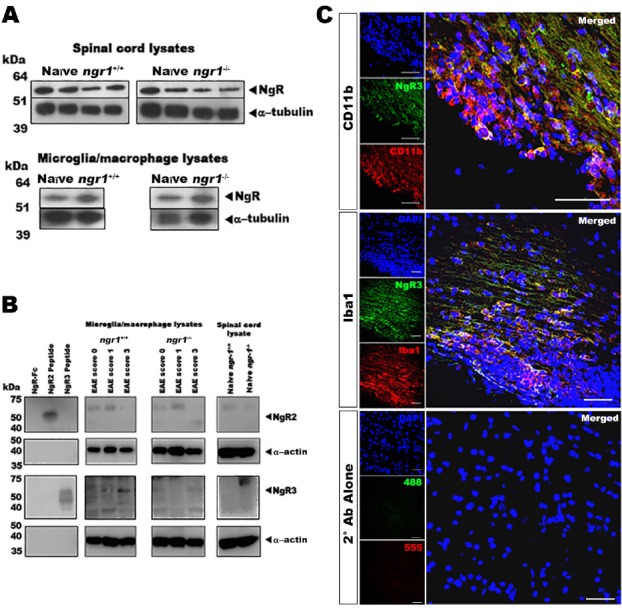
NgR homologs within spinal cord and microglial/macrophage cell lysates.
(A) NgR protein expression was detected in spinal cord and isolated microglial/macrophage cell lysates from ngr1+/+ and ngr1–/– mice. (B) Expression of NgR2 and NgR3 in microglial/macrophage cell lysates during EAE progression as well as spinal cord lysates from naïve ngr1+/+ and ngr1–/– mice. (C) Immunofluorescent co-labeling of either CD11b- or Iba1-positive microglial/macrophage cells, with the NgR3, present in spinal cord inflammatory lesions of ngr1+/+ mice at the chronic stage of EAE (i.e., clinical score 3). NgR2: Nogo66 receptor homolog 1; NgR3: Nogo66 receptor homolog 2; Iba1: ionizing binding adaptor molecule 1.
NgR3 expression in spinal cord macrophages/microglia following EAE induction
As stated above, our results showed the immunodetection of NgR in microglial/macrophage cells within demyelinating lesions from those ngr1–/– mice that actually progressed with EAE (n = 25 from 51 mice 49% of the knockout cohort; Table 1), suggesting that the antibody cross-reacts, potentially with the NgR homologs (i.e., NgR2 and/or NgR3). Expression of NgR3 was elevated at peak-chronic stage of EAE mainly in those macrophages/microglia isolated from ngr1+/+ mice. We identified extensive numbers of microglia/macrophages immunostained with either CD11b or Iba1 antibodies that were also co-labeled for NgR3 in severe inflammatory lesions of wild-type mice (clinical score 3; Figure 2C). These results may suggest that NgR3 is expressed in microglia/macrophages at the chronic stage of EAE, within demyelinated lesions that manifest at this stage.
NgR3-positive microglia/macrophages are increased in EAE-induced ngr-1+/+ mice as they progress in disability
To define the role of NgR in regulating microglial activity within neuroinflammatory lesions, microglia/macrophages were isolated from the freshly dissected spinal cord of ngr1+/+ and ngr1–/– mice both with and without EAE induction, and these endogenous populations were then examined by flow cytometry. Immunopanned Iba-1-positive microglia/macrophages that expressed the NgR homologs were identified. A significant increase in the percentage of isolated CD11b-positive microglia/macrophages were observed in the spinal cords of EAE-induced ngr1+/+ mice with a clinical score of 3 (made up 87.4 ± 1.8%) (Figure 3A). In terms of the population of microglia/macrophages expressing NgR, a significant increase (Δ5.4 ± 1.5%) was also observed in the population of CD11b-positive cells isolated from mice at the peak-chronic stage of EAE (clinical score 3) compared to either the percentage of these cells at the onset of disease (clinical score 1) or their isolation from naïve ngr1+/+ mice (Figure 3A).
Figure 3.
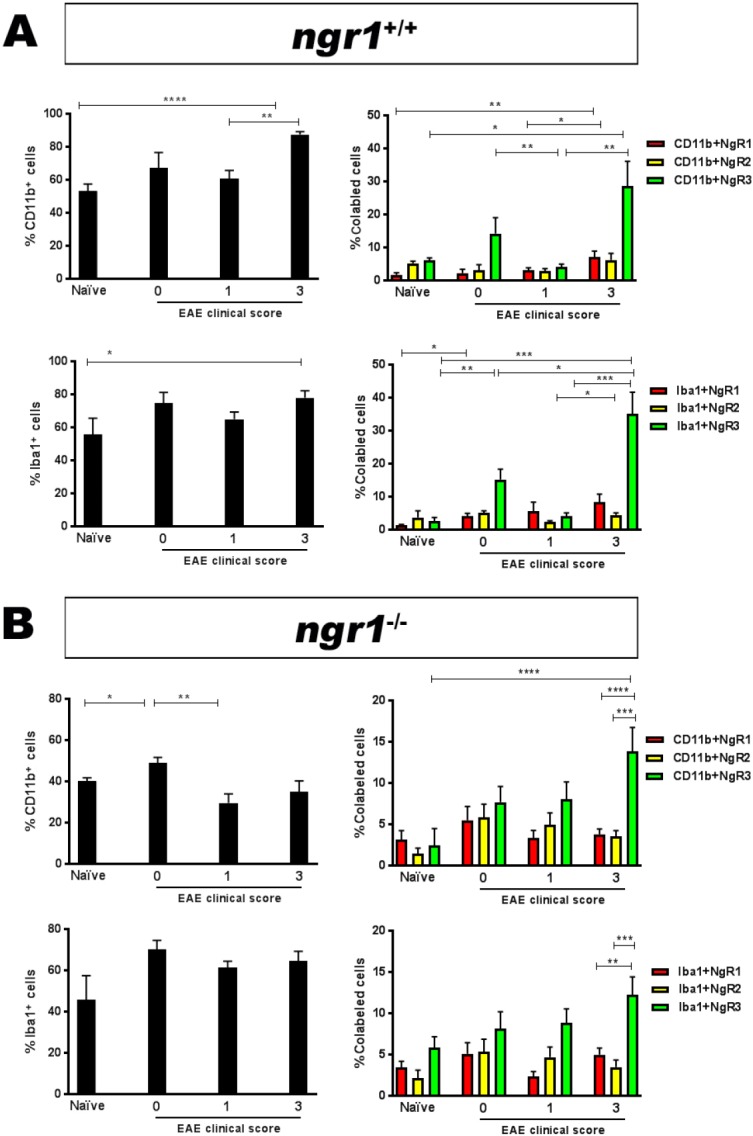
Flow cytometric analysis of microglial/macrophage cell populations isolated from both ngr1+/+ and ngr1–/– mice during the progression of EAE.
(A) A substantial elevation for NgR3-positive microglial/macrophage cells (either CD11b- or Iba1-positive) is demonstrated during the peak-chronic stage of EAE (clinical score 3). (B) No statistical differences exist in the numbers of NgR3-positive microglia/macrophages during the progression of EAE in ngr1–/– mice (from clinical scores 0 to 3), yet differences do exist between the expression of NgR3 on these cells compared to the expression of either NgR1 or NgR2 homologs on CD11b- or Iba1-positve cells at clinical score 3 (one-way ANOVA with post-hoc Tukey’s multiple comparison test; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). NgR2: Nogo66 receptor homolog 1; NgR3: Nogo66 receptor homolog 2; EAE: experimental autoimmune encephalomyelitis.
The percentage of the other NgR homologs (NgR2 and NgR3) was also studied on the isolated microglia/macrophages. However, despite being able to detect NgR2 expression on microglia isolated from ngr1+/+ mice with and without EAE induction, the percentages of these double-labeled cells were not statistically different during EAE progression. The percentage of the NgR3+/CD11b+ cells was considerably elevated at the peak-chronic stage of EAE when compared to the populations isolated at the naïve WT and onset of EAE (Figure 3A). Our data show that NgR3 expression can be induced during microglial activity as EAE progresses.
We corroborated these data by examining the percentage of microglia/macrophages utilizing the Iba1 antibody, either with and without NgR double labeling. Our results demonstrate that the population of Iba-1- positive microglia/macrophages within spinal cord of EAE-induced ngr1+/+ mice made up approximately 65 ± 4.4% and 78 ± 4.3% of the entire cell population at the induction and chronic stages of the disease, respectively (Figure 3A). Importantly, a substantial increase in NgR3- and Iba-1-positive cells was again observed at the peak-chronic stage of EAE (Figure 3A), 35 ± 7% of these cells being NgR3-positive at this stage of disease compared to ~2.6 ± 1.2% in naïve mice. Collectively, the induction of NgR3 expression in macrophages/microglia at the peak-chronic stage of EAE may indicate alternate signaling when there exist extensive demyelinating lesions and neurological progression. However, the mechanism by which NgR3 can influence microglial/macrophage activity during neuroinflammation needs to be specifically addressed.
NgR3-positive microglia/macrophages during the progression of EAE in ngr1–/– mice
As stated above, although the majority of ngr1–/– mice did not come down with severe clinical disease (Table 1 and Figure 1A), when we examined the isolated microglia/macrophages from the spinal cords of naïve ngr1–/– and those ngr1–/– mice where EAE was successfully induced, the number of either CD11b+ or Iba1+ cells that expressed both NgR2 and NgR3 homologs was not statistically different based on ANOVA comparisons assessed at different stages of EAE except at the clinical score of 3 (NgR3 vs. NgR1/NgR2, P < 0.0001; Figure 3B). The data may suggest that microglia/macrophages express NgR3 in the peak-chronic stage of EAE but are observed to be in greater numbers when NgR1 is present (i.e., in ngr1+/+ mice). This may imply that NgR1 can collaborate with NgR3 to signal through alternative ligands (Steinbach et al., 2011) during the chronic progressive phase of EAE; however, this hypothesis remains to be determined.
Early engulfment of myelin debris (including Nogo-A) by microglia isolated from ngr1–/– mice during the progression of EAE
To see the degree of NgR1-dependent macrophage/microglial activity, the amount of myelin engulfment was addressed in isolated spinal cord cells. We examined the levels of intracellular MOG, Nogo-A and Caspr from either naïve (ngr1+/+ and ngr1–/–) or EAE-induced mice. The results show that a significant increase in the level of engulfed MOG took place when we compared naïve and EAE-induced ngr1+/+ and ngr1–/– mice following their progression (Figure 4A). However, the MOG expression was enhanced in the ngr1–/– mice by clinical score of 3, in those mice that reached this disease score. Moreover, myelin debris engulfed by microglia/macrophages can be seen early during EAE induction in ngr1–/– mice, suggesting increased early phagocytic activity in these mutant mice (Figure 4B).
Figure 4.
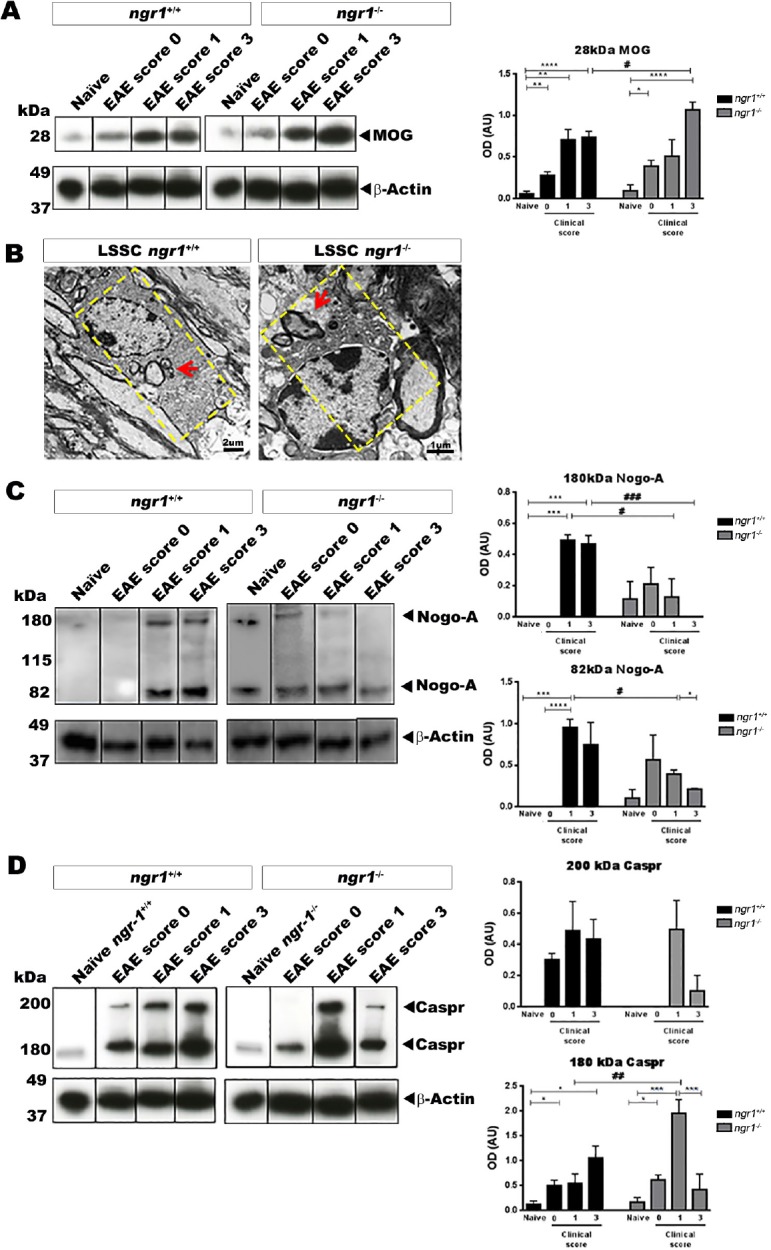
Detection of myelin/myelin degeneration products in microglia/macrophages during EAE progression in both ngr1+/+ and ngr1–/– mice.
(A) The levels of MOG within microglia/macrophages were significantly increased in both ngr1+/+ and ngr1–/– mice during EAE disease progression (one-way ANOVA with post-hoc Tukey’s multiple comparison test; #P < 0.05, *P < 0.05, ##P < 0.01, **P < 0.01, ####P < 0.0001, and ****P < 0.0001). Moreover, at the chronic stage of EAE, the levels of MOG within microglia/macrophages were significantly increased in EAE-induced ngr1–/– mice compared to EAE-induced ngr1+/+ mice, suggesting that an elevation in myelin engulfment occurred (n = 3 for clinical score 0, n = 4 for clinical score 1 and n = 5 for clinical score 3, one-way ANOVA with post-hoc Tukey’s multiple comparison test; #P < 0.05). (B) Investigation of the phagocytosis of myelin debris by microglia/macrophages in ngr1+/+ and ngr1–/– mouse spinal cord and optic nerve using transmission electron microscopy imaging. (C) The levels of Nogo-A increased in microglia/macrophages during EAE in ngr1+/+ mice. However, significant reductions were seen in EAE-induced ngr1–/– mice as disease progressed (clinical scores 1 and 3 respectively; n = 3/clinical score, one-way ANOVA with post-hoc Tukey’s multiple comparison test; *P < 0.05 and **P < 0.01). (D) The level of Caspr increased within microglia/macrophages during EAE progression in ngr1+/+ mice. However, Caspr levels decreased in EAE-induced ngr1–/– microglia/macrophages during peak-chronic stage of disease (n = 3/clinical score,*P < 0.05 and **P < 0.01). Data are represented as the mean ± SEM (one-way ANOVA with post-hoc Tukey’s multiple comparison test). EAE: Experimental autoimmune encephalomyelitis; MOG: myelin oligodendrocyte glycoprotein; Caspr: contactin associated protein 1.
We next assessed the level of Nogo-A (one of the myelin inhibitory molecules) in our isolated microglia/macrophages from the spinal cords of ngr1+/+ and ngr1–/– mice throughout the clinical course of EAE. The results suggest an early/fast elevation, followed by a significant reduction in the level of Nogo-A at the peak-chronic stage of EAE in microglia/macrophages isolated from the ngr1–/– mice (Figure 4C). On the other hand, the data generated from ngr1+/+ mice with EAE progression showed that continuous elevation of Nogo-A levels in microglia/macrophages along with the degradation of myelin that can potentiate EAE pathology seen in these mice (Figure 4C). Collectively, the results implicate increased myelin engulfment (including Nogo-A) when NgR1 and NgR3 are co-expressed on macrophage/microglial cell membranes.
The paranodal protein Caspr is elevated in microglia primarily at the induction of EAE in ngr1–/– mice
We also assessed the expression of Caspr, an integral paranodal protein that forms and preserves the paranodal junctions (Wolswijk and Balesar, 2003) and co-localizes with Nogo-A (Nie et al., 2003). Microglia/macrophages isolated from the spinal cords of the naïve (ngr1+/+ and ngr1–/–) mice as well as following EAE induction showed that the level of Caspr expression increased during EAE progression in ngr1+/+ mice but peaked in ngr1–/– mice at the stage of EAE induction followed with a sharp reduction at the chronic stage of disease (Figure 4D). These data may suggest early/fast clearance of paranodal Caspr by microglia/macrophages, in ngr1–/– mice as EAE progresses. However, whether this junctional protein clearance is directly related to the early Nogo-A phagocytosis at the paranode during EAE progression when NgR1 is not expressed remains to be elucidated.
Phenotyping of isolated microglia/macrophage populations during EAE progression in ngr1+/+ and ngr1–/– mice
In vivo studies have reported that microglia/macrophages can be quickly stimulated and activated based on stimuli present in the CNS milieu under the conditions of inflammation and degeneration (Fumagalli et al., 2013). Microglia/macrophages have two opposing phenotypes representative of their active states; namely M1, responsible for releasing pro-inflammatory cytokines such as TNF-α, IL-6 and IL1-β within an injured site (Fumagalli et al., 2011), thereby enhancing the severity of a disease; and M2, known as a neuroprotective phenotype, has the ability to produce anti-inflammatory cytokines such as IGF-1, bFGF and TGF-β that support CNS repair (Fumagalli et al., 2011). To define the properties of these cells analyzed during our study, we immunostained isolated spinal cord microglia/macrophages with specific antibodies against M1 and M2, which included CD38 and Erg-2 antibodies, believed to distinguish between these two cell phenotypes (Jablonski et al., 2015). At clinical score 0 of EAE-induced ngr1+/+ mice, the M1 population was significantly increased compared to the M2 population; semi-quantitatively measured to be 11.3 ± 2.8% and 23.6 ± 1.5%, respectively (Figure 5A). These data may suggest the ability of microglia/macrophages to start their activation of the M1-pathogenic phenotype as a first innate response to induce EAE pathology in these mice. During the chronic stage of EAE in these ngr1+/+ mice, a substantial increase in the percentage of CD38-positive M1 microglia/macrophages was detected more than Egr-2-positive M2 microglia/macrophages. This result was approximately 61.7 ± 3.9% for M1 and 33.3 ± 3.8% for M2 microglia/macrophages, suggesting the significant role played by pathogenic M1 microglia/macrophages during the chronic stage of EAE induced in ngr1+/+ mice (Figure 5A).
Figure 5.
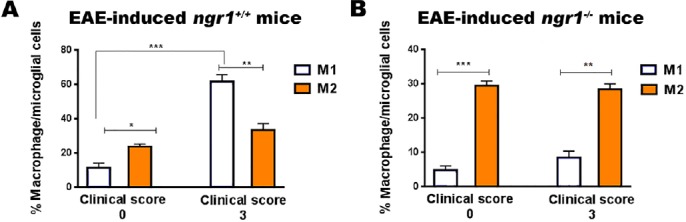
Phenotyping of microglia/macrophages in ngr1+/+ and ngr1–/– EAE-induced mice at clinical scores 0 and 3.
(A) There was a significant increase in the percentage of the M1 macrophage/microglial cell phenotype in the spinal cord of EAE-induced ngr1+/+ mice at the peak-chronic stage of disease when compared with the M2 phenotype. In addition, there was a substantial increase in these M1-pathogenic populations compared to cells within the pre-onset stage. Results are expressed as the mean ± SEM, Student’s t-test, n = 3/clinical score, *P < 0.05, **P < 0.01, and ****P < 0.0001. (B) In the spinal cord of EAE-induced ngr1–/– mice, there was a significant elevation in M2 macrophage/microglial cell percentages, more so than in the M1 phenotype at clinical score 3, even though more M2 than M1 cells were present at clinical score 0. Data are expressed as the mean ± SEM, Student’s t-test, n = 3/clinical score, **P < 0.01 and ****P < 0.0001. EAE: Experimental autoimmune encephalomyelitis; M1: classically activated macrophages; M2: alternatively activated macrophages.
The properties of microglia/macrophages isolated from ngr1–/– mice following EAE induction were also investigated during the pre-onset and chronic stages of the disease, respectively. The percentages of M2 microglia/macrophages during EAE progression were similar to the percentages detected at the pre-onset stage of EAE (Erg-2-positive M2 cells at the chronic stage of EAE were approximately 28.4 ± 1.6 %, compared with 8.4 ± 1.9 % for CD38-positive M1 cells; Figure 5B). These results implicate the predominance of M2 cells in ngr1–/– mice, suggesting that the deletion of NgR1 may play a role in limiting the macrophage/microglial cell pathogenic conversion in these mutant mice but does not play a role in MOG35-55 EAE pathogenesis (considering the macrophages/microglia were isolated from ngr1–/– mice with acute disease).
Discussion
Previous studies have shown that NgR and its co-receptor, Troy/Lingo-1, are expressed in microglia/macrophages in demyelinating MS lesions (Satoh et al., 2007; Yan et al., 2012). An interesting hypothesis was raised with respect to these data, suggesting microglial expression of NgR may be a key factor in regulating inflammation following CNS injury. In the current study, we investigated the importance of NgR in the activity of microglia/macrophages within inflammatory demyelinating lesions. The dynamics and phenotypes of these potentially pathogenic cells were investigated with minimal importance attributed to NgR1 expressed in microglia/macrophages to govern EAE pathogenesis. However, our study has concluded that the NgR3 may be an important component of a multimeric complex potentiating myelin phagocytosis. The limitation of this study was that microglial cell specific deletion of NgR and/or its homologs was not achieved during the induction and progression of EAE, to more effectively identify the physiological effects of these receptors upon these cells, during neuroinflammation.
It is well documented that integral myelin proteins, defined as the myelin associated inhibitory factors (MAIFs), can initiate failure in endogenous neural repair and possibly play a role in EAE progression (Petratos et al., 2012). The expression of Nogo-A, one of chief inhibitory proteins, has been reported to be upregulated in chronic-active MS lesions (Satoh et al., 2005). Furthermore, Nogo-A expression is increased in the spinal cord during EAE progression limiting regeneration of axons and associated with enhanced axonal damage (Theotokis et al., 2012). Moreover, inhibition of Nogo-A signaling in EAE can promote locomotor recovery (Yang et al., 2010) and enhance axonal regrowth (Yang et al., 2010). This raises the possibility that Nogo-A present in myelin debris can signal axonal degeneration in EAE and possibly MS lesions. Since myelin debris, glial scaring and axonal degeneration are major components of MS pathology, the binding of NgR with MAIFs (Schnell and Schwab, 1993; Wang et al., 2002; Cafferty and Strittmatter, 2006) and chondroitin sulphate proteoglycans (McKeon et al., 1999; Bradbury et al., 2002; Dickendesher et al., 2012) may be of central importance in MS-related pathological conditions. Targeting the Nogo-A signaling cascade could be considered an attractive method to limit MS progression.
How NgR1-dependent signaling potentiates neuroinflammatory axonopathy is not entirely clear, although the downstream signaling in axons following their interaction with myelin debris may be a mechanism (Petratos et al. 2012). Thus, faster clearance of myelin debris may be beneficial for an endogenous modulation of the diseased tissue environment, thereby promoting neurological improvement through neural regeneration (Neumann et al., 2009; Vargas et al., 2010; Lampron et al., 2015).
Evidence suggests that microglia/macrophages are the innate immune cells contributing to MS disease course and disability outcomes, with the extensive activation of those populations being a primary feature of the disease (Ferguson et al., 1997; Gao and Tsirka, 2011). The activity of these populations is reported as central to MS, participating, along with amoeboid microglia/macrophages, in the creation of its lesions histopathologically (Evangelou et al., 2000; van Horssen et al., 2012; Singh et al., 2013a), thus affecting these activated cells with substantial CNS degeneration and disease progression (Bjartmar et al., 2001; Deng and Sriram, 2005).
In line with our previously published work, we confirmed that a significant delay in EAE onset and pathological outcomes occurs in ngr1–/– mice as compared to wild type mice. In the current study, spinal cord tissues from both wild type and ngr1–/– mice post-EAE induction were subjected to an analysis of NgR expression, profiling of macrophages/microglia during active demyelinating disease and clinical progression. Moreover, the effect of this receptor on the migration of microglia/macrophages was determined, as well as its participation in demyelinated lesions, in the presence and absence of NgR1. The strong association between the presence of myelin debris in the white matter and increased microglial/macrophage cell numbers in these areas during disease progression was significant in wild type mice but not in ngr1–/– mice, suggesting that the expression of NgR did not inhibit microglial presence in lesion areas where myelin debris predominates following inflammatory demyelination. This finding supports the study by Clarneret al. (2012), in which it was demonstrated that myelin debris was important for microglial activation through the white and grey matter of the CNS (Clarner et al., 2012) but indicates microglia/macrophages are not limited by a NgR1-dependent mechanism.
Even though NgR1-dependent microglia/macrophages could not be attributed to the progression of EAE, in this study, we have demonstrated that NgR3 is expressed in microglia/macrophages at the peak stage of disease. NgR and its homologs are commonly localized in lipid rafts on the neuronal cell surface to facilitate signalling (Vinson et al., 2003; Yu et al., 2004). The inference is that NgR2 and NgR3 are able to elicit signal transduction events in a similar fashion to NgR, and to regulate similar cellular functions such as cytoskeletal reorganization and migratory inhibition (Pignot et al., 2003). NgR2 is a receptor, which can bind MAG with high affinity, and has been shown to inhibit neurite outgrowth (Domeniconi et al., 2002), whereas NgR3 was recently identified as a receptor that binds chondroitin sulphate proteoglycans (Dickendesher et al., 2012). Dickendesher et al. (2012) have also presented novel evidence showing chondroitin sulphate proteoglycans can bind both NgR1 and NgR3 as part of a multimeric complex to limit axonal growth following optic nerve crush injury. To our knowledge, the current study is the first to determine the presence of NgR3 in microglia/macrophages during EAE progression. Whether there exists a possible connection between two major families of inhibitory factors, specifically the MAIFs and proteoglycans, which have been correlated with CNS degeneration during the progressive phase of EAE, remains to be elucidated. It is plausible that NgR3 is part of the NgR1 signaling complex (NgR1/Lingo1/p75/TROY) in activated microglia, which can also mediate Nogo-66 inhibitory signaling in vivo, to increase neurodegeneration (Zhang et al., 2011). We postulated that the binding of NgR expressed on microglia/macrophages with myelin debris (consisting of Nogo-A), can initiate inhibitory signaling. However, we identified that NgR3 expression was increased in macrophage/microglial cells at the peak of neurological disability during EAE when profound axo-glial degeneration was observed and increased myelin debris within the predominantly M1 pathogenic microglia/macrophages could be demonstrated. These data may suggest that NgR3 expressed in these cells are important in demyelinating lesion pathology.
The collaborative interaction between NgR1/NgR3, although implied in this study, may argue that specifically targeting NgR3 expression during EAE progression may limit the pathogenic activity of M1 macrophages/microglia, with direct application for translation in human neuroinflammatory and neurodegenerative conditions. Reduced adhesion and migration to myelin in vitro and an association with microglial process retraction within the injured site has been suggested in studies on the effect of NgR on microglia/macrophages (Yan et al., 2012). However, these investigators utilized phosphatidylinositol-specific phospholipase C to remove GPI-anchored NgR from the microglia (Yan et al., 2012), leaving the possibility that other GPI-anchored proteins could be leached from cell membranes, thereby limiting downstream signaling.
The presence of myelin debris is a pathological marker of MS (Schnell and Schwab, 1993). Myelin debris can inhibit axonal regrowth and possibly affect the differentiation of oligodendrocyte precursor cells into their mature myelinating phenotype (Kotter et al., 2005). Therefore, the rapid clearance of myelin debris by phagocytic cells (i.e., microglia and macrophages) is essential during remyelination and axonal regeneration.
In this study, we identified a disparity in the microglia/macrophage numbers between the wild type (ngr1+/+) and ngr1–/– spinal cord sections. This may be attributed to the participation of macrophages/microglia in the enhanced phagocytic clearance of myelin when NgR1 and NgR3 are expressed in these cells, corroborated by the presence of myelin proteins (MOG and Nogo-A) in the predominant M1 pathogenic population of cells as EAE progressed. However, in the early onset stage of disease in ngr1–/– mice, the predominant M2 population was primarily isolated with both paranodal Nogo-A and Caspr engulfed but with higher levels of NgR3 expression. This finding raises the possibility that NgR3 is integral for myelin attachment and paranodal stripping of myelin and indicates that by targeting this receptor, paranodal myelin phagocytosis may be minimized. However, such a hypothesis remains to be tested.
A feature of ngr1–/– mice is that they do not develop severe symptoms following EAE induction (Petratos et al., 2012). However, EAE progression and pathology does not seem to depend on NgR1 expression in microglia/macrophages. The data presented here now implicate a role for NgR3 in inflammatory demyelinating lesion propagation and requires further investigation.
Additional file:
Additional Figure 1 (1.2MB, tif) : Microglial cells expressing NgR in the spinal cord of EAE-induced ngr1+/+ and ngr-1–/– mice during inflammatory demyelinating pathology.
Additional Figure 2 (975KB, tif) : Caspr is localized in degenerative axons in the optic nerves of EAE-induced ngr1+/+ but not in ngr1–/– mice.
Acknowledgments
The authors acknowledge the provision of ngr1–/– mice from Dr. Stephen Strittmatter (Yale University).
Footnotes
Conflicts of interest: We declare that no financial or non-financial conflicts of interest exist for any of the authors. Toolgen Inc. is the current address for JYL and have not contributed financially or intellectually to the current study.
Financial support: JYL was supported by Multiple Sclerosis Research Australia and Trish Multiple Sclerosis Research Foundation Postgraduate Scholarship. SP was supported by National Multiple Sclerosis Society Project Grant #RG4398A1/1, International Progressive Multiple Sclerosis Alliance Challenge Award #PA0065, Multiple Sclerosis Research Australia and Trish Multiple Sclerosis Research Foundation #15-022 and Bethlehem Griffiths Research Foundation #BGRF1706.
Institutional review board statement: The AMREP Animal Ethics Committee (AMREP AEC#E/1532/2015/M) approved the use of these animals for experimentation, in accordance with the National Health and Medical Research Council of Australia code for the care and use of animals for scientific purposes.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This study was supported by Multiple Sclerosis Research Australia and Trish Multiple Sclerosis Research Foundation Postgraduate Scholarship (to JYL); the National Multiple Sclerosis Society Project Grant #RG4398A1/1, International Progressive Multiple Sclerosis Alliance Challenge Award #PA0065, Multiple Sclerosis Research Australia and Trish Multiple Sclerosis Research Foundation #15-022 and Bethlehem Griffiths Research Foundation #BGRF1706 (to SP).
(Copyedited by Li CH, Song LP, Zhao M)
References
- 1.Bjartmar C, Kinkel RP, Kidd G, Rudick RA, Trapp BD. Axonal loss in normal-appearing white matter in a patient with acute MS. Neurology. 2001;57:1248–1252. doi: 10.1212/wnl.57.7.1248. [DOI] [PubMed] [Google Scholar]
- 2.Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- 3.Cafferty WB, Strittmatter SM. The Nogo-Nogo receptor pathway limits a spectrum of adult CNS axonal growth. J Neurosci. 2006;26:12242–12250. doi: 10.1523/JNEUROSCI.3827-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clarner T, Diederichs F, Berger K, Denecke B, Gan L, van der Valk P, Beyer C, Amor S, Kipp M. Myelin debris regulates inflammatory responses in an experimental demyelination animal model and multiple sclerosis lesions. Glia. 2012;60:1468–1480. doi: 10.1002/glia.22367. [DOI] [PubMed] [Google Scholar]
- 5.Cudrici C, Niculescu T, Niculescu F, Shin ML, Rus H. Oligodendrocyte cell death in pathogenesis of multiple sclerosis: Protection of oligodendrocytes from apoptosis by complement. J Rehabil Res Dev. 2006;43:123–132. doi: 10.1682/jrrd.2004.08.0111. [DOI] [PubMed] [Google Scholar]
- 6.David S, Lacroix S. Molecular approaches to spinal cord repair. Annu Rev Neurosci. 2003;26:411–440. doi: 10.1146/annurev.neuro.26.043002.094946. [DOI] [PubMed] [Google Scholar]
- 7.Deng X, Sriram S. Role of microglia in multiple sclerosis. Curr Neurol Neurosci Rep. 2005;5:239–244. doi: 10.1007/s11910-005-0052-x. [DOI] [PubMed] [Google Scholar]
- 8.Dickendesher TL, Baldwin KT, Mironova YA, Koriyama Y, Raiker SJ, Askew KL, Wood A, Geoffroy CG, Zheng B, Liepmann CD, Katagiri Y, Benowitz LI, Geller HM, Giger RJ. NgR1 and NgR3 are receptors for chondroitin sulfate proteoglycans. Nat Neurosci. 2012;15:703–712. doi: 10.1038/nn.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Domeniconi M, Cao Z, Spencer T, Sivasankaran R, Wang K, Nikulina E, Kimura N, Cai H, Deng K, Gao Y, He Z, Filbin M. Myelin-associated glycoprotein interacts with the Nogo66 receptor to inhibit neurite outgrowth. Neuron. 2002;35:283–290. doi: 10.1016/s0896-6273(02)00770-5. [DOI] [PubMed] [Google Scholar]
- 10.Evangelou N, Konz D, Esiri MM, Smith S, Palace J, Matthews PM. Regional axonal loss in the corpus callosum correlates with cerebral white matter lesion volume and distribution in multiple sclerosis. Brain. 2000;123(Pt 9):1845–1849. doi: 10.1093/brain/123.9.1845. [DOI] [PubMed] [Google Scholar]
- 11.Fang Y, Yan J, Li C, Zhou X, Yao L, Pang T, Yan M, Zhang L, Mao L, Liao H. The Nogo/Nogo Receptor (NgR) Signal Is Involved in Neuroinflammation through the Regulation of Microglial Inflammatory Activation. J Biol Chem. 2015;290:28901–28914. doi: 10.1074/jbc.M115.678326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120(Pt 3):393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- 13.Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- 14.Fumagalli M, Lecca D, Abbracchio MP. Role of purinergic signalling in neuro-immune cells and adult neural progenitors. Front Biosci. 2011;16:2326–2341. doi: 10.2741/3856. [DOI] [PubMed] [Google Scholar]
- 15.Fumagalli S, Perego C, Ortolano F, De Simoni MG. CX3CR1 deficiency induces an early protective inflammatory environment in ischemic mice. Glia. 2013;61:827–842. doi: 10.1002/glia.22474. [DOI] [PubMed] [Google Scholar]
- 16.Gao Z, Tsirka SE. Animal models of MS reveal multiple roles of microglia in disease pathogenesis. Neurol Res Int. 2011;2011:383087. doi: 10.1155/2011/383087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, Guerau-de-Arellano M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS One. 2015;10:e0145342. doi: 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim JE, Liu BP, Park JH, Strittmatter SM. Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron. 2004;44:439–451. doi: 10.1016/j.neuron.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 19.Kotter MR, Zhao C, van Rooijen N, Franklin RJ. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol Dis. 2005;18:166–175. doi: 10.1016/j.nbd.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 20.Lampron A, Larochelle A, Laflamme N, Prefontaine P, Plante MM, Sanchez MG, Yong VW, Stys PK, Tremblay ME, Rivest S. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med. 2015;212:481–495. doi: 10.1084/jem.20141656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee JY, Petratos S. Multiple sclerosis: does Nogo play a role. Neuroscientist. 2013;19:394–408. doi: 10.1177/1073858413477207. [DOI] [PubMed] [Google Scholar]
- 22.Lee JY, Taghian K, Petratos S. Axonal degeneration in multiple sclerosis: can we predict and prevent permanent disability. Acta Neuropathol Commun. 2014;2:97. doi: 10.1186/s40478-014-0097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu G, Ni J, Mao L, Yan M, Pang T, Liao H. Expression of Nogo receptor 1 in microglia during development and following traumatic brain injury. Brain Res. 2015;1627:41–51. doi: 10.1016/j.brainres.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 24.McKeon RJ, Jurynec MJ, Buck CR. The chondroitin sulfate proteoglycans neurocan and phosphacan are expressed by reactive astrocytes in the chronic CNS glial scar. J Neurosci. 1999;19:10778–10788. doi: 10.1523/JNEUROSCI.19-24-10778.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nie DY, Zhou ZH, Ang BT, Teng FY, Xu G, Xiang T, Wang CY, Zeng L, Takeda Y, Xu TL, Ng YK, Faivre-Sarrailh C, Popko B, Ling EA, Schachner M, Watanabe K, Pallen CJ, Tang BL, Xiao ZC. Nogo-A at CNS paranodes is a ligand of Caspr: possible regulation of K(+) channel localization. EMBO J. 2003;22:5666–5678. doi: 10.1093/emboj/cdg570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oudega M, Xu XM. Schwann cell transplantation for repair of the adult spinal cord. J Neurotrauma. 2006;23:453–467. doi: 10.1089/neu.2006.23.453. [DOI] [PubMed] [Google Scholar]
- 28.Petratos S, Ozturk E, Azari MF, Kenny R, Lee JY, Magee KA, Harvey AR, McDonald C, Taghian K, Moussa L, Mun Aui P, Siatskas C, Litwak S, Fehlings MG, Strittmatter SM, Bernard CC. Limiting multiple sclerosis related axonopathy by blocking Nogo receptor and CRMP-2 phosphorylation. Brain. 2012;135:1794–1818. doi: 10.1093/brain/aws100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pignot V, Hein AE, Barske C, Wiessner C, Walmsley AR, Kaupmann K, Mayeur H, Sommer B, Mir AK, Frentzel S. Characterization of two novel proteins, NgRH1 and NgRH2, structurally and biochemically homologous to the Nogo-66 receptor. J Neurochem. 2003;85:717–728. doi: 10.1046/j.1471-4159.2003.01710.x. [DOI] [PubMed] [Google Scholar]
- 30.Satoh J, Onoue H, Arima K, Yamamura T. Nogo-A and nogo receptor expression in demyelinating lesions of multiple sclerosis. J Neuropathol Exp Neurol. 2005;64:129–138. doi: 10.1093/jnen/64.2.129. [DOI] [PubMed] [Google Scholar]
- 31.Satoh J, Tabunoki H, Yamamura T, Arima K, Konno H. TROY and LINGO-1 expression in astrocytes and macrophages/microglia in multiple sclerosis lesions. Neuropathol Appl Neurobiol. 2007;33:99–107. doi: 10.1111/j.1365-2990.2006.00787.x. [DOI] [PubMed] [Google Scholar]
- 32.Schnell L, Schwab ME. Sprouting and regeneration of lesioned corticospinal tract fibres in the adult rat spinal cord. Eur J Neurosci. 1993;5:1156–1171. doi: 10.1111/j.1460-9568.1993.tb00970.x. [DOI] [PubMed] [Google Scholar]
- 33.Sicotte M, Tsatas O, Jeong SY, Cai CQ, He Z, David S. Immunization with myelin or recombinant Nogo-66/MAG in alum promotes axon regeneration and sprouting after corticospinal tract lesions in the spinal cord. Mol Cell Neurosci. 2003;23:251–263. doi: 10.1016/s1044-7431(03)00053-8. [DOI] [PubMed] [Google Scholar]
- 34.Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Bruck W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol. 2013a;125:595–608. doi: 10.1007/s00401-013-1082-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sozmen EG, Rosenzweig S, Llorente IL, DiTullio DJ, Machnicki M, Vinters HV, Havton LA, Giger RJ, Hinman JD, Carmichael ST. Nogo receptor blockade overcomes remyelination failure after white matter stroke and stimulates functional recovery in aged mice. Proc Natl Acad Sci U S A. 2016;113:E8453–8462. doi: 10.1073/pnas.1615322113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steinbach K, McDonald CL, Reindl M, Schweigreiter R, Bandtlow C, Martin R. Nogo-receptors NgR1 and NgR2 do not mediate regulation of CD4 T helper responses and CNS repair in experimental autoimmune encephalomyelitis. PLoS One. 2011;6:e26341. doi: 10.1371/journal.pone.0026341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation. 2004;1:14. doi: 10.1186/1742-2094-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Theotokis P, Lourbopoulos A, Touloumi O, Lagoudaki R, Kofidou E, Nousiopoulou E, Poulatsidou KN, Kesidou E, Tascos N, Spandou E, Grigoriadis N. Time course and spatial profile of Nogo-A expression in experimental autoimmune encephalomyelitis in C57BL/6 mice. J Neuropathol Exp Neurol. 2012;71:907–920. doi: 10.1097/NEN.0b013e31826caebe. [DOI] [PubMed] [Google Scholar]
- 39.van Horssen J, Singh S, van der Pol S, Kipp M, Lim JL, Peferoen L, Gerritsen W, Kooi EJ, Witte ME, Geurts JJ, de Vries HE, Peferoen-Baert R, van den Elsen PJ, van der Valk P, Amor S. Clusters of activated microglia in normal-appearing white matter show signs of innate immune activation. J Neuroinflammation. 2012;9:156. doi: 10.1186/1742-2094-9-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vargas ME, Watanabe J, Singh SJ, Robinson WH, Barres BA. Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc Natl Acad Sci U S A. 2010;107:11993–11998. doi: 10.1073/pnas.1001948107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venkatesh K, Chivatakarn O, Lee H, Joshi PS, Kantor DB, Newman BA, Mage R, Rader C, Giger RJ. The Nogo-66 receptor homolog NgR2 is a sialic acid-dependent receptor selective for myelin-associated glycoprotein. J Neurosci. 2005;25:808–822. doi: 10.1523/JNEUROSCI.4464-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vinson M, Rausch O, Maycox PR, Prinjha RK, Chapman D, Morrow R, Harper AJ, Dingwall C, Walsh FS, Burbidge SA, Riddell DR. Lipid rafts mediate the interaction between myelin-associated glycoprotein (MAG) on myelin and MAG-receptors on neurons. Mol Cell Neurosci. 2003;22:344–352. doi: 10.1016/s1044-7431(02)00031-3. [DOI] [PubMed] [Google Scholar]
- 43.Wang KC, Koprivica V, Kim JA, Sivasankaran R, Guo Y, Neve RL, He Z. Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature. 2002;417:941–944. doi: 10.1038/nature00867. [DOI] [PubMed] [Google Scholar]
- 44.Wolswijk G, Balesar R. Changes in the expression and localization of the paranodal protein Caspr on axons in chronic multiple sclerosis. Brain. 2003;126:1638–1649. doi: 10.1093/brain/awg151. [DOI] [PubMed] [Google Scholar]
- 45.Yan J, Zhou X, Guo JJ, Mao L, Wang YJ, Sun J, Sun LX, Zhang LY, Zhou XF, Liao H. Nogo-66 inhibits adhesion and migration of microglia via GTPase Rho pathway in vitro. J Neurochem. 2012;120:721–731. doi: 10.1111/j.1471-4159.2011.07619.x. [DOI] [PubMed] [Google Scholar]
- 46.Yang Y, Liu Y, Wei P, Peng H, Winger R, Hussain RZ, Ben LH, Cravens PD, Gocke AR, Puttaparthi K, Racke MK, McTigue DM, Lovett-Racke AE. Silencing Nogo-A promotes functional recovery in demyelinating disease. Ann Neurol. 2010;67:498–507. doi: 10.1002/ana.21935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu W, Guo W, Feng L. Segregation of Nogo66 receptors into lipid rafts in rat brain and inhibition of Nogo66 signaling by cholesterol depletion. FEBS Lett. 2004;577:87–92. doi: 10.1016/j.febslet.2004.09.068. [DOI] [PubMed] [Google Scholar]
- 48.Zhang L, Kuang X, Zhang J. Nogo receptor 3, a paralog of Nogo-66 receptor 1 (NgR1), may function as a NgR1 co-receptor for Nogo-66. J Genet Genomics. 2011;38:515–523. doi: 10.1016/j.jgg.2011.10.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Microglial cells expressing NgR in the spinal cord of EAE-induced ngr1+/+andngr1–/– mice during inflammatory demyelinating pathology.
(A) CD11b-positive microglial/macrophage cells were co-labeled with the anti-NgRantibody within the spinal cord, during the progression of EAE in ngr1+/+mice. (B)Microglial/macrophage cells were also immunostained with Iba-1 for further confirmation of their activation, along with anti-NgR, demonstrating the elevationin the numbers of these cells within spinal cord inflammatory lesions in ngr1+/+mice. (C) The ngr1–/–micethat developed EAE were also immunolabeled for both CD11b and NgR, utilizing the same antibodies as for ngr1+/+mouse tissue. NgR expression could still be seen in large numbers of these macrophage/microglial cells in the spinal cord sections of EAE-induced ngr1–/–mice. (D) The Iba-1-positive macrophage/microglial cells, immunopositive for NgR, were present in the spinal cords of ngr1–/–mice during the progression of their EAE. Scale bar: 50 μm.
Caspr is localized in degenerative axons in the optic nerves of EAE-induced ngr1+/+ but not in ngr1–/– mice.
(A) Paraffin-embedded optic nerves of EAE-induced (peak-chronic stage of disease) ngr1+/+mice were immunostained with MBP, (B) Caspr and counter-stained with (C) DAPI. (D) Merged images are shown (Scale bar = 50μm). (E-P) Co-localization of markers of degenerative axons with Caspr in EAE (peak-chronic stage of disease) in ngr1+/+ optic nerves. The serial sections were immunostained with (E) βAPP, (I) AT8, or (M) SMI-32 (red), which were co-immunostained with Caspr (F, J & N), counterstaining with DAPI was performed in all images (G, K & O); merged images (H, L & P) (Scale bar = 10 μm). (Q) Stereological quantification revealed a significant co-localization of Caspr with markers for degenerative axon βAPP, AT8 and SMI-32 (**P<0.01, ****P<0.0001, n =8).