

Keywords: nerve regeneration, Alzheimer's disease, APP/PS1 mice, Morris water maze test, corpus callosum, demyelination, oligodendrocytes, myelin basic protein, monocarboxylic acid transporter 1, neural regeneration
Abstract
The APPSwe/PSEN1dE9 (APP/PS1) transgenic mouse model is an Alzheimer’s disease mouse model exhibiting symptoms of dementia, and is commonly used to explore pathological changes in the development of Alzheimer’s disease. Previous clinical autopsy and imaging studies suggest that Alzheimer’s disease patients have white matter and oligodendrocyte damage, but the underlying mechanisms of these have not been revealed. Therefore, the present study used APP/PS1 mice to assess cognitive change, myelin loss, and corresponding changes in oligodendrocytes, and to explore the underlying mechanisms. Morris water maze tests were performed to evaluate cognitive change in APP/PS1 mice and normal C57BL/6 mice aged 3 and 6 months. Luxol fast blue staining of the corpus callosum and quantitative reverse transcription-polymerase chain reaction (qRT-PCR) for myelin basic protein (MBP) mRNA were carried out to quantify myelin damage. Immunohistochemistry staining for NG2 and qRT-PCR for monocarboxylic acid transporter 1 (MCT1) mRNA were conducted to assess corresponding changes in oligodendrocytes. Our results demonstrate that compared with C57BL/6 mice, there was a downregulation of MBP mRNA in APP/PS1 mice aged 3 months. This became more obvious in APP/PS1 mice aged 6 months accompanied by other abnormalities such as prolonged escape latency in the Morris water maze test, shrinkage of the corpus callosum, upregulation of NG2-immunoreactive cells, and downregulation of MCT1 mRNA. These findings indicate that the involvement of early demyelination at 3 months and the oligodendrocyte dysfunction at 6 months in APP/PS1 mice are in association with Alzheimer’s disease pathogenesis.
Introduction
Alzheimer’s disease (AD) is characterized by progressive cognitive decline and is the leading cause of dementia (Akram et al., 2017; Hane et al., 2017). Early impairments in memory and learning are the main clinical features of AD. To date, amyloid β plaques and neurofibrillary tangles in the AD brain gray matter have been considered the primary pathogenic triggers (Hyman et al., 2012). However, anti-amyloid therapies have failed to show benefits in cognitive or functional outcomes (Castello et al., 2014; Akram et al., 2017), leaving the pathogenesis yet to be elucidated (Castello et al., 2014).
In recent years, increasing evidence has suggested that demyelination is one of important components of AD (Sachdev et al., 2013; Caso et al., 2016). Magnetic resonance imaging studies have found that white matter lesions increase with age (Ihara et al., 2010; Amlien et al., 2014) and these lesions show in the early stages of AD prior to the presence of clinical symptoms (Selnes et al., 2012). Postmortem studies have discovered myelin degeneration in brain tissue staining from frontal and temporal lobes (Ihara et al., 2010) and widespread white matter damage in both the preclinical and end stages of AD (de la Monte, 1989). These findings indicate that white matter lesions might be crucial in AD pathogenesis (Pietroboni et al., 2017). However, there have been few animal model studies on demyelination. Therefore, the present study aimed to assess myelin damage, and quantify its severity, in 3- and 6-month-old APP/PS1 mice—an AD mouse model.
It is known that oligodendrocytes play an important role in myelin formation. The myelin damage detected in AD raises the possibility that oligodendrocytes experience a pathophysiological assault during the course of AD (Desai et al., 2010). A postmortem study reported that oligodendrocyte loss, together with reduced myelin density, axonal loss and astrogliosis, are the main structural white matter changes in the AD brain (Zhan et al., 2014). It remains unclear whether this oligodendrocyte loss is secondary to white matter change or is a primary neuropathological event independent of myelin change. In the central nervous system, chondroitin sulphate proteoglycan NG2 is mainly expressed by the oligodendrocyte precursor cells. These NG2-labeled cells proliferate and are believed to give rise to remyelinating oligodendrocytes in pathological conditions with myelin damage (Kang et al., 2010). Recently, emerging evidence showed that oligodendrocytes metabolically support neurons through monocarboxylic acid transporter 1 (MCT1) (Kang et al., 2013). Therefore, the present study aimed to explore the underlying etiology of oligodendrocytes with NG2 and MCT1 assessment in 3- and 6-month-old APP/PS1 mice.
Materials and Methods
Animals
A total of 20 male APP/PS1 mice and 12 male C57BL/6 mice were acquired from the Experimental Animal Center of China Medical University, China [license No. SCXK (Liao) 2013–0001]. Mice were housed in cages and given a 12-hour light/dark cycle with free access to water and food. All experiments were approved by the Animal Care and Use Committee of the China Medical University (IACUC-2010888). All the procedures were done under anesthesia, and all possible efforts were made to minimize suffering.
The mice were divided into four subgroups: The APP/PS1 3 group (APP/PS1 mice aged 3 months, n = 10), the APP/PS1 6 group (APP/PS1 mice aged 6 months, n = 10), the control 3 group (C57BL/6 mice aged 3 months, n = 6), and the control 6 group (C57BL/6 mice aged 6 months, n = 6).
Morris water maze test
Working memory is a well modeled aspect of the memory deficits in AD. The spatial working memory task of the Morris water maze test is the most widely employed one (Webster et al., 2013). After one week of adaption, the Morris water maze was performed to assess spatial learning and memory. The Morris water maze was a circular tank (50 cm diameter and 150 cm deep) filled with 20 cm of water made opaque with the addition of 200 g powdered milk. The tank was divided into four quadrants: northeast, southeast, southwest, and northwest; and a circular platform 9 cm in diameter was hidden 1 cm below the water level in the northeast quadrant. On day 1, for adaption, mice swam freely in the pool without the hidden platform in it. On days 2–4, mice underwent four trials a day. With each trial mice were placed in the tank facing the wall of each quadrant and allowed 60 seconds to find the hidden platform. Those who failed to locate the platform within 60 seconds were gently pushed towards it and allowed to remain there for 10 seconds. On day 5, after training, the platform was removed, and the mice were placed in the water in a random quadrant and allowed 60 seconds to explore freely. Escape latencies (the swimming time spent before finding the submerged platform) and swimming route and time spent in the northeast quadrant where the hidden platform was located were monitored by a video tracking system (Chengdu Taimeng Tech. Chengdu, China). In the retention phase of the Morris water maze test, the ratio of swimming distance (swimming distance in the target quadrant where the hidden platform had been placed) against the total swimming distance of different groups was calculated.
Tissue processing
Twenty-four hours after the Morris water maze test, mice were deeply anesthetized with 1% sodium pentobarbital (1 mL/100 g body weight). For Luxol fast blue and immunohistochemistry staining, mice (APP/PS1 groups, n = 5 each; control groups; n = 3 each) were perfused transcardially with 4% paraformaldehyde. Brains were post-fixed in 4% paraformaldehyde overnight at 4°C, imbedded in paraffin, and sectioned 5 μm thick with a microtome. Brain sections were deparaffinied with xylene and rehydrated in a graded alcohol series, prior to Luxol fast blue and immunohistochemistry staining. For quantitative reverse transcription-polymerase chain reaction (qRT-PCR) of myelin basic protein (MBP) and MCT1 mRNA expression, mouse brains (APP/PS1 groups, n = 5 each; control groups; n = 3 each) were removed by cervical dislocation and the temporal lobes dissected out on ice.
Luxol fast blue staining and immunohistochemistry staining
The corpus callosum, the largest white matter structure in the brain, consists of millions of axons that provide a large connection between homologous cerebral cortical areas.
Therefore, Luxol fast blue staining of the corpus callosum was performed to determine the degree of myelination after tissue processing. Immunohistochemistry staining for NG2 was performed to assess oligodendrocyte change.
For corpus callosum Luxol fast blue staining, sections were stained with Luxol fast blue solution (YuanMu, Shanghai, China) at room temperature for 12–20 hours, rinsed with 95% ethanol and water, reacted with 1% lithium carbonate solution for 15 seconds and 70% ethanol for 30 seconds, and then rinsed in water. Counter staining with eosin was performed as necessary.
For immunohistochemistry staining, sections were treated with antigen retrieval buffer 0.4% Triton-X100 in phosphate buffer for 20 minutes at 95°C. Sections were then treated with 3% H2O2 in phosphate buffered saline for 10 minutes. After blocking with 10% normal goat serum in phosphate buffered saline for 30 minutes, sections were incubated with primary NG2 antibodies 55027-1-AP (1:200; Proteintech, Rosemont, IL, USA) overnight at 4°C. Following subsequently rinsed in phosphate buffer, sections were incubated with secondary antibodies SPN - 9001 (1:1000; ZSbio, Beijing, China). Finally, a peroxidase ABC system (ZSbio) with diaminobenzidine as the substrate was used to detect the antibody complex according to the manufacturer’s instructions.
All samples were examined using a phase contrast microscope, and images were obtained with a digital camera and image-capturing software (Diagnostic, Frankfurt, Germany). Images of sections through each anatomic region of interest (corpus callosum or temporal lobe) were captured. The corpus callosum intensity signals were divided by the corresponding intensity signals of the background and presented as the % optical density ratio. The number of NG2-immunoreactive cells was calculated in randomly sampled regions and was presented as the ratio of the total number of cells in the same region. Data were analyzed using an image analysis system (Image-Pro Plus v 6.0 image analysis software, Media Cybernetics, Silver Spring, MD, USA).
qRT-PCR analysis
After tissue processing the MiniBEST Universal RNA Extraction Kit (TaKaRa, Dalian, China) was used to extract total RNA according to the manufacturer’s instructions. The RNA concentration was determined by 260/280 nm absorbance using a Nanodrop Spectrophotometer (ND-1000, Thermo Fisher Scientific, Waltham, MA, USA).
For reverse transcription, PrimeScript™ RT Master Mix (TaKaRa) was used according to the manufacturer’s recommendation. Primers were designed using the Primer Express software (TaKaRa) as shown in Table 1.
Table 1.
Primers used for quantitative reverse transcription-polymerase chain reaction

qRT-PCR was performed on an ABI 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using the one-step SYBR Premix Ex Taq™ II kit (TaKaRa) in a total volume of 20 µL: 1 cycle of 95°C for 30 seconds, 40 cycles of 95°C for 3 seconds, 60°C for 30 seconds, and finally 1 cycle of 95°C for 15 seconds, 60°C for 60 seconds, and 95°C for 15 seconds. β-actin was used as the reference gene (Desai et al., 2009). Experiments were repeated in triplicates. The average threshold cycle (Ct) and the comparative ΔΔCt were automatically calculated. Relative quantification of the genes was performed by determination of the n-fold differential expression with the 2-??Ct method (Livak et al., 2001) and was expressed as relative fold change compared to β-actin.
Statistical analysis
Results were expressed as the mean ± standard deviations (SD). Statistical analysis was performed by one-way analysis of variance followed by the least significant difference post-hoc test using SPSS 13.0 software (SPSS, Chicago, IL, USA). A P value ≤ 0.05 was considered to be statistically significant, and P < 0.01 was deemed highly significant.
Results
Morris water maze test
The swimming route and the time spent on northeast quadrant, where the hidden platform was located, were digitally monitored and are shown in Figure 1A.
Figure 1.

Morris water maze test of APP/PS1 mice and C57BL/6 mice aged 3 and 6 months.
(A) A typical swimming routine of APP/PS1 mouse in the retention phase of the Morris water maze test. (B) In the acquisition phase of the Morris water maze test, the escape latency shortened in all groups. (C) In the retention phase of the Morris water maze test, the ratio of swimming distance [swimming distance in the target quadrant (the former location of the hidden platform) against the total swimming distance of different groups] was calculated. The ratio of the swimming distance of the 6-month APP/PS1 mice is much lower than that of 6-month C57BL/6 mice and 3-month APP/PS1 mice. **P < 0.01. Results are expressed as the mean ± SD (APP/PS1 groups, n = 5 each; control groups, n = 3 each; one-way analysis of variance followed by least significant difference post-hoc test).
In the acquisition phase (days 2–4) of the Morris water maze test, the escape latency in all groups showed downward trends with the training time (Figure 1B). Mice in the APP/PS1 groups, particularly the APP/PS1 6 group, spent longer reaching the hidden platform compared with their age-matched controls.
In the retention phase (day 5) of the Morris water maze test, the frequency that the mice crossed the former position of the hidden platform was lower in the APP/PS1 groups, especially in the APP/PS1 6 group, when compared with that of their age-matched control groups. However, one-way analysis of variance did not find differences between the groups (data not shown).
We further calculated the ratio of the swimming distance in the northeast quadrant, the former location of the hidden platform, with the total swimming distance. We found that the ratios of the swimming distance in the northeast quadrant in the APP/PS1 groups were lower than that of the control groups (Figure 1C). Comparison between groups revealed a significant deterioration between the APP/PS1 6 group and the control 6 group (P = 0.000), and an age-related deterioration between the APP/PS1 6 group and the APP/PS1 3 group (P = 0.000), suggesting memory retention disabilities. No significant differences were found between the APP/PS1 3 group and the control 3 group.
Luxol fast blue staining
Myelin damage in the white matter was assessed using Luxol fast blue staining to visualize the corpus callosum as shown in Figure 2A and B. The intensity signals in the corpus callosum were divided by the corresponding intensity signals of the background and presented as the % optical density ratio (Zhou et al., 2013) (Figure 2C). Comparison between groups revealed a significant myelin damage in the APP/PS1 6 group compared with the control 6 group (P = 0.000) and the APP/PS1 3 group (P = 0.003), as well as an age-related myelin formation in the control 6 group compared with the control 3 group (P = 0.000). No significant differences were found between the APP/PS1 3 group and the control 3 group (P = 0.871).
Figure 2.

Luxol fast blue staining of the corpus callosum of APP/PS1 mice and C57BL/6 mice aged 3 and 6 months.
(A) Corpus callosum of a C57BL/6 mouse aged 3 months visualized by Luxol fast blue staining. Scale bars: 200 μm, 100 μm. (B) Examples of Luxol fast blue staining of the corpus callosum of the different groups. Scale bar: 100 μm. (C) Intensity signals of CC of different groups divided by the corresponding intensity signals of the background and presented as the % optical density ratio. The optical density ratio of CC (%) of 6-month APP/PS1 mice is much lower than that of 6-month C57BL/6 mice and 3-month APP/PS1 mice. The optical density ratio of CC (%) of 3-month C57BL/6 mice is lower than that of 6-month C57BL/6 mice. *P < 0.05, **P < 0.01. Results are expressed as the mean ± SD (APP/PS1 groups, n = 5 each; control groups, n = 3 each; one-way analysis of variance followed by the least significant difference post-hoc test). CC: Corpus callosum; Cg: cingulate gyrus; dHC: dorsal hippocampal commissural.
Immunohistochemistry staining for NG2 and qRT-PCR for MBP and MCT1
In the central nervous system, chondroitin sulphate proteoglycan NG2 is mainly expressed by the oligodendrocyte precursor cells. These NG2-labeled cells proliferate and are believed to give rise to remyelinating oligodendrocytes in pathological conditions with myelin damage (Kang et al., 2010).
We performed immunohistochemistry staining for NG2 on the temporal lobe (Figure 3A). The number of the NG2-immunoreactive cells was calculated in randomly sampled regions and was presented as ratio of the total number of cells in the same region (Figure 3B).
Figure 3.
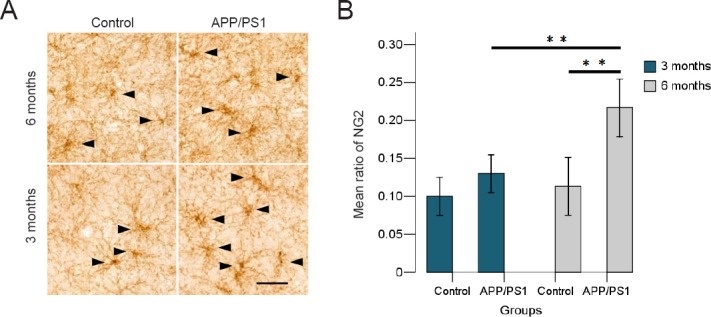
NG2 cells in the temporal lobe of APP/PS1 mice and C57BL/6 mice aged 3 and 6 months.
(A) NG2-labeled cells shown by immunohistochemistry staining in the different groups. Black arrows show NG2-labeled cells. Scale bar: 50 μm. (B) Number of NG2-immunoreactive cells are present as a ratio of the total numbers of cells in the temporal lobe among the different groups. The ratio of NG2-immunoreactive cells of 6-month APP/PS1 mice is much higher than that of 6-month C57BL/6 mice and 3-month APP/PS1 mice. **P < 0.01. Results are expressed as the mean ± SD (APP/PS1 groups, n = 5 each; control groups, n = 3 each; one-way analysis of variance followed by the least significant difference post-hoc test).
Immunohistochemistry staining showed a proliferation of NG2 cells in the temporal cortices of APP/PS1 mice. Comparisons between groups revealed a significant proliferation of NG2 cells between the APP/PS1 6 group and the control 6 group (P = 0.007), and an age-related proliferation between the APP/PS1 3 group and the APP/PS1 6 group (P = 0.013). No differences were found between the APP/PS1 3 group and the control 3 group (P = 0.866).
The qRT-PCR results showed that MBP mRNA expression in the temporal lobe reduced by approximately 27 % in the APP/PS1 3 group compared with the control 3 group (P = 0.000), and reduced by approximately 38 % in the APP/PS1 6 group compared with the control 6 group (P = 0.000) (Figure 4A).
Figure 4.
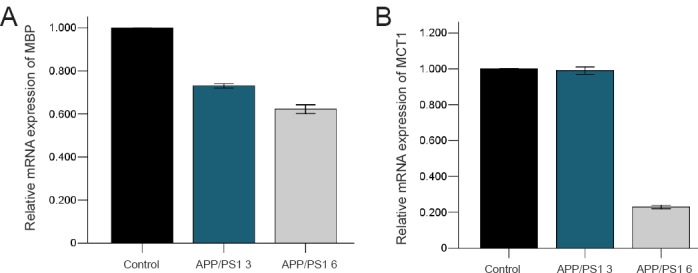
Quantitative reverse transcription-polymerase chain reaction of MBP and MCT1 expression at the mRNA level in the temporal lobe of APP/PS1 mice and C57BL/6 mice aged 3 and 6 months.
(A) Relative expression of MBP mRNA in 3- and 6-month APP/PS1 mice (%). Control: β-Actin; APP/PS1 3: the relative expression of MBP mRNA in 3-month APP/PS1 mice; APP/PS1 6: the relative expression of MBP mRNA in 6-month APP/PS1 mice. (B) The relative expression of MCT1 mRNA in 3- and 6-month APP/PS1 mice (%). Control: β-Actin; APP/PS1 3: the relative expression of MCT1 mRNA in 3-month APP/PS1 mice; APP/PS1 6: the relative expression of MCT1 mRNA in 6-month APP/PS1 mice.
MCT1 qRT-PCR was performed to further assess metabolic changes in oligodendrocytes and revealed that MCT1 mRNA expression reduced by approximately 1% in the APP/PS1 3 group compared with the control 3 group (P = 0.428), and reduced by approximately 77% in the APP/PS1 6 group compared with the control 6 group (P = 0.000) (Figure 4B).
Discussion
Our study revealed cognitive deficits, corpus callosum shrinkage, and oligodendrocyte disorder in APP/PS1 mice at the age of 6 months. However, MBP mRNA downregulation in temporal lobe tissue was observed as early as 3 months of age.
The typical progression of AD starts with memory and learning decline, followed by executive dysfunction, language barriers, and finally cognitive deficits (Carmeli et al., 2013). Similarly, in AD mouse models, cognitive deficits in APP/PS1 mice in the Morris water maze spatial memory test have been reported by 6 months of age, as well as impairments in reference memory and associative learning (Webster et al., 2014). In this study, impaired spatial memory in the Morris water maze test appeared at 6 months of APP/PS1 mice. Although cognitive deficits in APP/PS1 mice have also been seen the spatial working memory in radial arm water maze task as early as 3 months (Webster et al., 2014), the present study failed to find spatial memory and learning impairments in 3-month-old APP/PS1 mice.
According to convergent findings, myelin degradation is an important component of AD (Zhan et al., 2014; Caso et al., 2016; Hoy et al., 2017). Loss of myelin integrity in AD patients has been found in both autopsy and imaging studies (Carmeli et al., 2013; Sachdev et al., 2013; Agosta et al., 2015; Zhan et al., 2015; Hoy et al., 2017; Sachdev et al., 2017; Tascone et al., 2017). In this study, we chose to assess myelin damage in the corpus collosum and temporal lobe in AD mice model because these regions are vulnerable and commonly involved in AD (Ting et al., 2015). Shrinkage of the corpus collosum in 6-month-old APP/PS1 mice was observed and is supported by another animal study of APPswe mice at the ages of 12 and 15 months using diffusion tensor imaging (Desai et al., 2009) and by longitudinal MRI studies in AD patients (Weiler et al., 2015). Such morphological changes in myelin tissue in other brain regions such as the hippocampus have also been reported in APP/PS1 mice (Wu et al., 2017), indicating persistent and widespread myelin damage in AD progression. In the present study, a reduction of MBP mRNA expression was discovered in APP/PS1 mice aged 3 months when no spatial memory decline was found in the Morris water maze test, and before morphological changes in the corpus collosum, indicating that the loss of myelin integrity precedes the onset of cognitive impairment (Zhan et al., 2015).
The white matter damage observed both in AD patients and in AD mouse models inevitably leads to speculation that alterations in oligodendrocytes may in part underlie the white matter damage found in AD (Kang et al., 2010; Nielsen et al., 2013), particularly because oligodendrogenesis continues throughout life, contributes to myelin formation and remodeling, and therefore plays an important role in learning and memory (El Waly et al., 2014). As oligodendrocytes arise from a large population of precursor cells defined by expression of chondroitin sulfate proteoglycan NG2 (Kang et al., 2010), the present study performed immunohistochemistry staining for NG2.
In accordance with previous findings, the current study observed a proliferation of NG2 cells in the temporal lobe of APP/PS1 mice at the age of 6 months, indicating that NG2 cells are mitotically active and exhibit enhanced proliferation in response to acute demyelination in the central nervous system (Reynolds et al., 2001). The upregulation of NG2 cells has been reported with western blots analysis in an even earlier phase of AD-like pathogenesis in APP/PS1 mice aged 2 months (Wu et al., 2017). The difference in the timing of NG2 upregulation in this study may result from the different methods used and different regions of interest investigated. This enhanced proliferation of NG2 cells in the present study appeared after myelin lesion onset, which suggests that NG2 proliferation might be a secondary neuropathological event to white matter change. Also, there might exist other possible mechanisms, such as amyloid deposition, ischemic vascular changes, oxidative stress, and immune-mediated damage, that underlie white matter damage, and/or trigger aberrant oligodendrocyte responses (Sachdev et al., 2013). For example, in cerebral amyloid angiopathy, the amyloid may induce ischemic injury to the white matter, causing luminal stenosis and thrombosis, finally reducing blood supply to the white matter (Weller et al., 1998), or by poor clearance of soluble amyloid β peptides from the brain parenchyma (Chalmers et al., 2005).
The upregulation of NG2 cells observed in the present study may represent an increase in oligodendrogenesis to compensate for oligodendrocyte loss found in AD patients (Nielsen et al., 2013). A recent study found that the lactate transporter-MCT1 in the central nervous system was highly enriched within oligodendrocytes and disruption of MCT1 caused axon damage and neuron loss in cell culture and animal models (Lee et al., 2012). The present study is the first to assess MCT1 mRNA expression in AD mouse models and found a decrease in MCT1 mRNA expression at the age of 6 months, coinciding with an enhanced proliferation of NG2 cells.
Taken together, despite the continuous upregulation of oligodendrocyte progenitors, there were reductions in myelin levels and failure of MCT1 expression by oligodendrocytes in the current study, indicating impaired maturation of oligodendrocytes. The dysfunction of oligodendrocytes as well as the early demyelination in APP/PS1 mice may contribute to AD disease progression. The assessment of the proliferation, apoptosis, and metabolic changes of oligodendrocytes is required in the future study to get better understanding of the pathogenesis of AD.
Additional file (78.5KB, pdf) : Open peer review report 1.
Open peer review report 1
Footnotes
Conflicts of interest: We declare that we have no conflict of interest.
Financial support: This work was supported by the National Natural Science Foundation of China, No. 81371395; the Liaoning Scientific and Technological Preferential Finance for Returned Overseas 2015 of China, No. 2015]125; the Natural Science Foundation of Liaoning Province of China, No. 20170541021, No. 2015020547; a grant from the Shenyang Science Technology Project, No. F16-206-9-12; and the China Post-doctoral Science Foundation, No. 2015M581375. Funders had no involvement in the study design; data collection, analysis, and interpretation; paper writing; or decision to submit the paper for publication.
Institutional review board statement: The procedures were in accordance with ethical standards of the University Animal Welfare and Ethics Review Committees (No. 2010888). The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1985).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Ivan Fernandez-Vega, Hospital Universitario Central de Asturias, Asturias, Spain.
Funding: This work was supported by the National Natural Science Foundation of China, No. 81371395; the Liaoning Scientific and Technological Preferential Finance for Returned Overseas 2015 of China, No. [2015]125; the Natural Science Foundation of Liaoning Province of China, No. 20170541021, 2015020547; a grant from the Shenyang Science Technology Project, No. F16-206-9-12; and the China Post-doctoral Science Foundation, No. 2015M581375.
(Copyedited by Wang J, Li CH, Qiu Y, Song LP, Zhao M)
References
- 1.Agosta F, Galantucci S, Magnani G, Marcone A, Martinelli D, Antonietta Volontè M, Riva N, Iannaccone S, Ferraro PM, Caso F, Chiò A, Comi G, Falini A, Filippi M. MRI signatures of the frontotemporal lobar degeneration continuum. Hum Brain Mapp. 2015;36:2602–2614. doi: 10.1002/hbm.22794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akram M, Nawaz A. Effects of medicinal plants on Alzheimer’s disease and memory deficits. Neural Regen Res. 2017;12:660–670. doi: 10.4103/1673-5374.205108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amlien IK, Fjell AM. Diffusion tensor imaging of white matter degeneration in Alzheimer’s disease and mild cognitive impairment. Neuroscience. 2014;276:206–215. doi: 10.1016/j.neuroscience.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 4.Carmeli C, Donati A, Antille V, Viceic D, Ghika J, von Gunten A, Clarke S, Meuli R, Frackowiak RS, Knyazeva MG. Demyelination in mild cognitive impairment suggests progression path to Alzheimer’s Disease. PLoS One. 2013;8:e72759. doi: 10.1371/journal.pone.0072759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caso F, Agosta F, Filippi M. Insights into white matter damage in alzheimer’s disease: from postmortem to in vivo diffusion tensor MRI studies. Neurodegener Dis. 2016;16:26–33. doi: 10.1159/000441422. [DOI] [PubMed] [Google Scholar]
- 6.Castello MA, Jeppson JD, Soriano S. Moving beyond anti-amyloid therapy for the prevention and treatment of Alzheimer’s disease. BMC Neurol. 2014;14:169. doi: 10.1186/s12883-014-0169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chalmers K, Wilcock G, Love S. Contributors to white matter damage in the frontal lobe in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2005;31:623–631. doi: 10.1111/j.1365-2990.2005.00678.x. [DOI] [PubMed] [Google Scholar]
- 8.de la Monte SM. Quantitation of cerebral atrophy in preclinical and end-stage Alzheimer’s disease. Ann Neurol. 1989;25:450–459. doi: 10.1002/ana.410250506. [DOI] [PubMed] [Google Scholar]
- 9.Desai MK, Sudol KL, Janelsins MC, Mastrangelo MA, Frazer ME, Bowers WJ. Triple-transgenic Alzheimer’s disease mice exhibit region specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia. 2009;57:54–65. doi: 10.1002/glia.20734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desai MK, Mastrangelo MA, Ryan DA, Sudol KL, Narrow WC, Bowers WJ. Early oligodendrocyte/myelin pathology in Alzheimer’s disease mice constitutes a novel therapeutic target. Am J Pathol. 2010;177:1422–1435. doi: 10.2353/ajpath.2010.100087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desai MK, Sudol KL, Janelsins MC, Mastrangelo MA, Frazer ME, Bowers WJ. Triple-transgenic Alzheimer’s disease mice exhibit region specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia. 2009;57:54–65. doi: 10.1002/glia.20734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Waly B, Macchi M, Cayre M, Durbec P. Oligodendrogenesis in the normal and pathological central nervous system. Front Neurosci. 2014;8:145. doi: 10.3389/fnins.2014.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hane FT, Robinson M, Lee BY, Bai O, Leonenko Z, Albert MS. Recent progress in Alzheimer’s disease research, part 3: diagnosis and treatment. J Alzheimers Dis. 2017;57:645–665. doi: 10.3233/JAD-160907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoy AR, Ly M, Carlsson CM, Okonkwo OC, Zetterberg H, Blennow K, Sager MA, Asthana S, Johnson SC, Alexander AL, Bendlin BB. Microstructural white matter alterations in preclinical Alzheimer’s disease detected using free water elimination diffusion tensor imaging. PLoS One. 2017;12:e0173982. doi: 10.1371/journal.pone.0173982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ. National institute on aging–Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ihara M, Polvikoski TM, Hall R, Slade JY, Perry RH, Oakley AE, Englund E, O’Brien JT, Ince PG, Kalaria RN. Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer’s disease, and dementia with Lewy bodies. Acta Neuropathol. 2010;119:579–589. doi: 10.1007/s00401-009-0635-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68:668–681. doi: 10.1016/j.neuron.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16:571–579. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.Reynolds R, Cenci di Bello I, Dawson M, Levine J. The response of adult oligodendrocyte progenitors to demyelination in EAE. Prog Brain Res. 2001;132:165–174. doi: 10.1016/s0079-6123(01)32073-3. [DOI] [PubMed] [Google Scholar]
- 22.Pietroboni AM, Scarioni M, Carandini T, Basilico P, Cadioli M, Giulietti G, Arighi A, Caprioli M, Serra L, Sina C, Fenoglio C, Ghezzi L, Fumagalli GG, De Riz MA, Calvi A, Triulzi F, Bozzali M, Scarpini E, Galimberti D. CSF β-amyloid and white matter damage: a new perspective on Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2018;89:352–357. doi: 10.1136/jnnp-2017-316603. [DOI] [PubMed] [Google Scholar]
- 23.Nielsen HM, Ek D, Avdic U, Orbjörn C, Hansson O; Netherlands Brain Bank, Veerhuis R, Rozemuller AJ, Brun A, Minthon L, Wennström M. NG2 cells, a new trial for Alzheimer’s disease mechanisms. Acta Neuropathol Commun. 2013;1:7. doi: 10.1186/2051-5960-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sachdev PS, Zhuang L, Braidy N, Wen W. Is Alzheimer’s a disease of the white matter. Curr Opin Psychiatry. 2013;26:244–251. doi: 10.1097/YCO.0b013e32835ed6e8. [DOI] [PubMed] [Google Scholar]
- 25.Selnes P, Fjell AM, Gjerstad L, Bjørnerud A, Wallin A, Due-Tønnessen P, Grambaite R, Stenset V, Fladby T. White matter imaging changes in subjective and mild cognitive impairment. Alzheimers Dement. 2012;8:S112–S121. doi: 10.1016/j.jalz.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 26.Tascone LDS, Payne ME, MacFall J, Azevedo D, de Castro CC, Steffens DC, Busatto GF, Bottino CMC. Cortical brain volume abnormalities associated with few or multiple neuropsychiatric symptoms in Alzheimer’s disease. PLoS One. 2017;12:e0177169. doi: 10.1371/journal.pone.0177169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ting WK, Fischer CE, Millikin CP, Ismail Z, Chow TW, Schweizer TA. Grey matter atrophy in mild cognitive impairment / early Alzheimer disease associated with delusions: a voxel-based morphometry study. Curr Alzheimer Res. 2015;12:165–172. doi: 10.2174/1567205012666150204130456. [DOI] [PubMed] [Google Scholar]
- 28.Webster SJ, Bachstetter AD, Nelson PT, Schmitt FA, Van Eldik LJ. Using mice to model Alzheimer’s dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front Genet. 2014;5:88. doi: 10.3389/fgene.2014.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Webster SJ, Bachstetter AD, Van Eldik LJ. Comprehensive behavioral characterization of an APP/PS-1 double knock-in mouse model of Alzheimer’s disease. Alzheimers Res. 5:28. doi: 10.1186/alzrt182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weiler M, Agosta F, Canu E, Copetti M, Magnani G, Marcone A, Pagani E, Balthazar ML, Comi G, Falini A, Filippi M. Following the spreading of brain structural changes in alzheimer’s disease: a longitudinal, multimodal MRI study. J Alzheimers Dis. 2015;47:995–1007. doi: 10.3233/JAD-150196. [DOI] [PubMed] [Google Scholar]
- 31.Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE. Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol. 1998;153:725–733. doi: 10.1016/s0002-9440(10)65616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y, Ma Y, Liu Z, Geng Q, Chen Z, Zhang Y. Alterations of myelin morphology and oligodendrocyte development in early stage of Alzheimer’s disease mouse model. Neurosci Lett. 2017;642:102–106. doi: 10.1016/j.neulet.2017.02.007. [DOI] [PubMed] [Google Scholar]
- 33.Zhan X, Jickling GC, Ander BP, Liu D, Stamova B, Cox C, Jin LW, DeCarli C, Sharp FR. Myelin injury and degraded myelin vesicles in Alzheimer’s disease. Curr Alzheimer Res. 2014;11:232–238. doi: 10.2174/1567205011666140131120922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhan X, Jickling GC, Ander BP, Stamova B, Liu D, Kao PF, Zelin MA, Jin LW, DeCarli C, Sharp FR. Myelin basic protein associates with AβPP, Aβ1-42, and amyloid plaques in cortex of Alzheimer’s disease brain. J Alzheimers Dis. 2015;44:1213–1229. doi: 10.3233/JAD-142013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–438. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou J, Zhuang J, Li J, Ooi E, Bloom J, Poon C, Lax D, Rosenbaum DM, Barone FC. Long-term post-stroke changes include myelin loss, specific deficits in sensory and motor behaviors and complex cognitive impairment detected using active place avoidance. PLoS One. 2013;8:e57503. doi: 10.1371/journal.pone.0057503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Open peer review report 1