

Abstract
The clinical successes in immunotherapy have been both astounding and at the same time unsatisfactory. Countless patients with varied tumor types have seen pronounced clinical response with immunotherapeutic intervention; however, many more patients have experienced minimal or no clinical benefit when provided the same treatment. As technology has advanced, so has the understanding of the complexity and diversity of the immune context of the tumor microenvironment and its influence on response to therapy. It has been possible to identify different subclasses of immune environment that have an influence on tumor initiation and response and therapy; by parsing the unique classes and subclasses of tumor immune microenvironment (TIME) that exist within a patient’s tumor, the ability to predict and guide immunotherapeutic responsiveness will improve, and new therapeutic targets will be revealed.
The past decade has seen a revolution in cancer treatments by moving away from drugs that target tumors broadly (for example, chemotherapy and radiation) and toward the use of antibody-based immunotherapies that modulate immune responses against tumors. The first generation of antibody-based immunotherapy, so-called immune-checkpoint blockade (ICB), works by blocking the receptor and/or ligand interactions of molecules, such as CTLA-4 and PD-1, that are involved in dulling T cell activation or function1. ICB therapies have shown significant clinical benefit for a minority of patients, who demonstrate durable responses. Unfortunately, there is still an unmet clinical need for the majority of patients, who do not respond to ICB. Retrospective analyses of patient populations treated with ICB have revealed that there are classes of TIME that are associated with those tumors more prone to ICB responsiveness.
Deeper analysis of complexity within the TIME is likely to reveal advanced biomarkers that will prove fruitful in identifying patient populations responsive to current ICB therapy and will benefit the search for novel targets for therapeutic modulation. Past efforts to characterize the TIME have provided a foundation for future efforts in which recent technological advances in techniques such as high-resolution single-cell RNA sequencing, flow cytometry and imaging are expected to provide an unprecedented view of the composition, function and location of immune cells within the TIME. In this Review, we provide a summary of the current knowledge centered around classes of TIME, focusing on the use of new technologies to study the TIME with increased granularity and the roles of systemic immune-and nonimmune-related factors in influencing TIME character and quality, and hence how tumors respond to immunotherapy.
Classification of the TIME
Predicting responsiveness to ICB on the basis of high-resolution data on the character and quality of tumor immune infiltrates is a critical next step in improving the success of current ICB and developing next-generation immunotherapies. To date, large bodies of work have established moderate-resolution TIME data from low-resolution sources, such as bulk tissue microarrays and immunohistochemistry2,3. Techniques such as CIBERSOrT3 and XCell4 can estimate the abundance of immune infiltrate into the tumor by using gene expression data from bulk tissues. Immunoscore5 uses a combination of immunohistochemistry and bulk tissue gene expression data to stratify patients according to immune-related criteria and subsequently predict disease outcome.
Unfortunately, because of the nature of the datasets being used, most of these studies can estimate the immunological frequency and cellular status in the tumor microenvironment, but they lack information related to actual cellular proportions, cellular heterogeneity and deeper spatial distribution. Nonetheless, these techniques have gleaned substantial information that has provided a basis for classifying TIME according to broad criteria—the composition of the immune infiltrate and the character of the inflammatory response. Using next-generation technologies to improve TIME classifications should expand understanding of how the immunological composition and quality vary in tumor types (such as breast) and subtypes (such as luminal B), inform the success or failure of current ICB, and encourage the discovery of new immunotherapeutics. Currently, three broad classes of moderate-resolution TIME can be described according to recent human and mouse data. These three classes almost certainly miss key subclasses that should be revealed by ongoing studies using higher-resolution techniques to uncover heterogeneity in immunological composition, spatial distribution and function.
TIMEs that are broadly populated with immune cells but are relatively void of cytotoxic lymphocytes (CTLs) in the tumor core are termed infiltrated-excluded (I-E) TIMEs herein. I-E TIMEs have CTLs localized along the border of the tumor mass in the invasive margin or ‘caught’ in fibrotic nests (Fig. 1a). I-E TIMEs are associated with various epithelial cancers such as colorectal carcinoma (CRC)6, melanoma7 and pancreatic ductal adenocarcinoma (PDAC)8, in which Ly6Clo F4/80hi tumor-associated macrophages (TAMs) along the tumor margins have been hypothesized to prevent CTL infiltration into the tumor core9. Tumors classified as I-E TIMEs have been hypothesized to be poorly immunogenic or ‘cold’, although this hypothesis remains to be clearly verified10. I-E TIMEs, compared with more inflamed TIMEs, contain CTLs with low expression of the activation markers GZMB (GRZB) and IFNG and poor infiltration of CTLs into the tumor core. A lack of activation-marker expression and exclusion from the tumor core are characteristics indicative of immunological ignorance, an immunological state in which adaptive immunity is unable to recognize or respond to a pathogen or malignancy11.
Fig. 1. General classes of TIME.
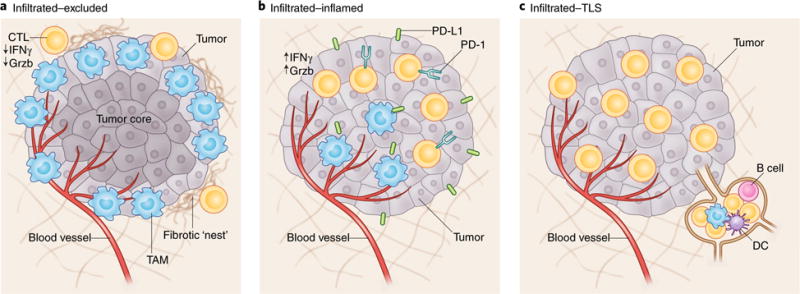
Three classes of TIME are displayed. a, I-E TIMEs are characterized by the exclusion of CTLs from the tumor core. CTLs in I-E TIMEs are instead present along the tumor periphery, where they can be found in contact with Ly6Clo F4/80+ tumor-associated macrophages or ‘stuck’ in fibrotic nests. b, In comparison, I-I TIMEs are defined by an abundance of PD-L1 expression on tumor and myeloid cells and highly activated CTLs characterized by expression of Grzb, IFNγ and PD-1. In some subsets of I-I TIME, tumor cells will have defects in DNA mismatch repair (MSI-H), thus resulting in an increased number of neoepitopes. c, TLS-TIMEs have histological evidence of containing TLSs, aggregates of immune cells with a composition similar to that in lymph nodes, including B cells, dendritic cells and Treg cells.
Infiltrated-inflamed (I-I) TIMEs (Fig. 1b) are considered to be immunologically ‘hot’ tumors and are characterized by high infiltration of CTLs expressing PD-1 and leukocytes and tumor cells expressing the immune-dampening PD-1 ligand PD-L1. A subset of CRC, known as microsatellite instability high (MSI-H), bears a higher rate of nonsynonymous single-nucleotide polymorphisms, thus leading to increased numbers of neoepitopes and of tumor-infiltrating PD-1+ CTLs, which have significantly higher responses to ICBs than do microsatellite instability low (MSI-L) or microsatellite stable (MSS) CRCs.
A subclass of I-I TIMEs, here termed TLS-TIMEs (Fig. 1c), display histological evidence of tertiary lymphoid structures (TLSs), lymphoid aggregates whose cellular composition is similar to that in lymph nodes. TLSs are often12,13 but not always correlated with a positive prognosis14. Similarly to lymph nodes, TLSs can contain a substantial diversity of lymphocytes, including naive and activated conventional T cells, regulatory T (Treg) cells, B cells and dendritic cells (DCs)15. TLSs are generally present at the invasive tumor margin and in the stroma, and are thought to act as sites of lymphoid recruitment and immune activation that are typically formed in settings of enhanced inflammation, such as after administration of an autologous tumor vaccine16. The ability to characterize a TLS thoroughly (for example, spatially, compositionally and functionally) is an important step in describing the TIME at a high resolution. For example, the TIME can be characterized in terms of not only the total number and type of cells present within a tissue but also the unique spatial collection of cells that may share a common program—in this case, a geographical feature established to recruit and activate adaptive immune cells. Spatial information paired with immunological composition and cellular status can help identify the presence of micro-niches within the TIME.
Broad classifications of immune context within a tumor microenvironment represent the first level of addressing how immunological composition and status (i.e., activated or suppressed) affect overall survival and dictate responsiveness to therapy. Beyond parsing the TIME with higher-resolution techniques, these classifications improve understanding of how mutational burden, oncogenes and distinct tumor types affect the establishment and maintenance of specific immunological compositions.
Interconnectivity of tumor genotypes and phenotypes and the TIME
It remains to be understood how tumor-produced cytokines and chemokines, tumor oncogenes and mutation landscapes determine the composition of the TIME. There are several examples strong enough to indicate relationships between both tumor genotype/phenotype and immunological composition, but these examples are not sufficiently strong for this understanding to be immediately applied toward therapeutic intervention (Fig. 2a)
Fig. 2. How tumor genotypes and phenotypes shape the TIME.
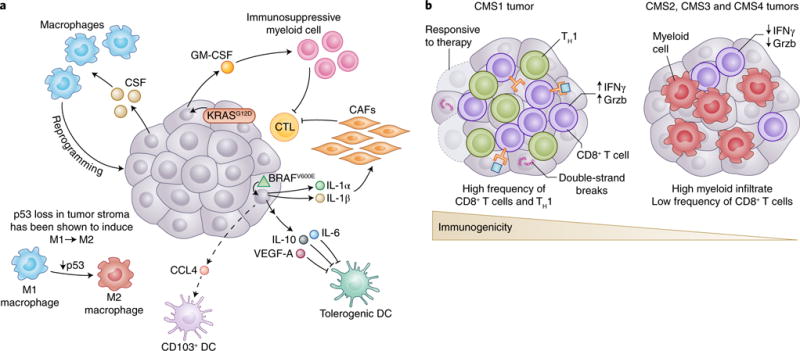
a, Tumors are known to establish protumoral and immunosuppressive environments to support their growth and promote immune evasion. Central to building an immunosuppressive TIME are oncogenes and aberrant signaling pathways that lead to the production of cytokines and chemokines with potent effects. The tumor shown is representative of a spectrum of cancer types. In melanoma, BRAFV600E (green triangle) has been shown to induce constitutive WNT/β-catenin signaling, which in turn decreases production of CCL4, a chemokine important for the recruitment of CD103+ DCs. Additionally, BRAFV600E has been shown to induce expression of factors such as IL-10 and IL-1α, which can induce tolerogenic forms of DC and cancer-associated fibroblasts (CAFs), respectively. Oncogenic KRASG12D in PDAC leads to the secretion of GM-CSF, corresponding to increased development of CD11b+ myeloid cells with reported immunosuppressive function. Deficiency in p53 in hepatic stellate cells, a stromal population, leads to production of factors that polarize TAMs from the immunoactivating M1 phenotype to the immunosuppressive M2 phenotype. Interestingly, many tumors have been shown to secrete high levels of the monocyte/macrophage-promoting cytokine CSF-1. b, The mutational landscape of tumors can profoundly affect the quality and character of the TIME. In CRC, there are four consensus molecular subtypes (CMS1-4). CMS1 is defined by defects in DNA mismatch repair leading to microsatellite instability or hypermutation rates. Because of the abundance of possible neoepitopes, CTL infiltration is generally high, and CTLs display gene expression patterns indicative of an ongoing immune response. Patients with CMS1 tumors have generally more favorable outcomes with checkpoint-blockade treatment than do patients with CMS2-4. Although there are differences in the histological and immunological character of CMS2, 3 and 4 CRC subtypes, they are generally less immune infiltrated, as is suggestive of antigenically cold tumors.
Tumor genotype contribution to cytokine production
Oncogene-driven expression of cytokines critical for the recruitment and phenotype of immune cells, particularly cells of the myeloid lineage, has been reported. In human melanoma, BRAFV600E, a mutated and highly oncogenic form of the MAPK family member BRAF, and STAT3, a potent transcriptional regulator often linked to oncogenic signaling, have been shown to drive expression of IL-6, IL-10 and VEGF, cytokines that promote a tolerogenic monocyte-derived DC phenotype in vitro, a process that would theoretically affect antitumor T cell function in vivo17.
Multiple reports have demonstrated that KRASG12D-driven PDAC secretes high levels of the growth factor GM-CSF, which is associated with an increase in tumor-associated Gr-1+CD11b+ myeloid cells of reported immunosuppressive function18,19. Interestingly, genetic ablation or neutralization of GM-CSF in mice leads to decreased myeloid infiltration, improved CD8+ T cell infiltration into tumors and markedly smaller lesion size. These data demonstrate that an oncogene promotes the establishment of an immunosuppressive TIME that supports malignant development. Missing from these studies, however, is an assessment of the character of DC infiltration, because GM-CSF has been shown to induce the generation of CD11b+ DCs, a DC population ill defined in the tumor20.
Tumor-derived chemokines
Secretion of tumor-derived chemokines, driven by specific oncogenes, is another critical point of interaction between tumor genotype and recruited immune cells. Recent data in a BRAFV600E and Pten-deficient mouse model of melanoma suggest that constitutive tumor-intrinsic WNT/β-catenin signaling is associated with poor immune infiltration and ineffective antitumor T cells, largely because of a decrease in the recruitment and frequency of CD103+ DCs21,22. Transcriptional analysis of tumor cells and in vitro DC migration assays have revealed that constitutive WNT/β-catenin signaling leads to decreased production of Ccl4, a potent chemoattractant for a variety of myeloid cells including CD103+ DCs, thus potentially explaining the decreased recruitment of CD103+ DCs and the corresponding poor infiltration of CD8+ T cells into the tumor microenvironment. Although the direct oncogenic determinant of expression is unclear, several studies in mice have reported that tumor-secreted CCL2 causes the recruitment of CCR2+ classical monocytes to the tumor, where they differentiate into TAMs, a protumoral myeloid population23.
Humoral factors
There is also evidence for a role for humoral factors in regulating the TIME. Recent data from mice suggest that TIME-derived PTX3, a critical component regulating complement activation through interaction with factor H, plays an essential role in suppressing tumor growth by indirectly controlling monocyte recruitment and TAM phenotype24. Epigenetic profiling of human tumors has revealed hypermethylation of the PTX3 promoter, thus suggesting that human PTX3 may similarly affect the architecture of the TIME.
Paracrine feedback loops
Paracrine feedback loops of cytokines between specific immune infiltrates and tumor cells play critical roles in influencing tumor phenotype and ultimately metastasis. TAMs are prominent components of the TIME and are involved in cross-talk with tumor cells, thus resulting in tumorigenic reprogramming25. Tumors in both mice26 and humans27 have been found to secrete high levels of colony stimulating factor 1 (CSF-1), a potent chemoattractant, survival and differentiation factor for monocytes and macrophages26, in addition to CCL2. T helper 2-polarized CD4+ T cells, through secretion of IL-4 and IL-13, have been shown to potentiate the ability of TAMs to secrete angiogenic growth factors, proteases and protumoral survival factors28, including VEGF-A, MMP-9, EGF and uPA26,29.
Modulating the stroma
In addition to tumor-intrinsic factors directly affecting immune cells within the TIME, tumor cells can elicit profound phenotypic changes in nonimmune stromal components that reside within the local tumor microenvironment and affect the immune component of the TIME. Indeed, oncogenic BRAFV600E signaling in human melanoma cells has been shown to perturb T cell-mediated antitumor responses by modulating the phenotype of cancer-associated fibroblasts. BRAFV600E in melanoma drives production of IL-1α and IL-1β, thereby enhancing the ability of cancer-associated fibroblasts to suppress melanoma-specific CTLs, in part through COX-2 secretion and upregulation of the PD-1 ligands PD-L1 and PD-L2 (ref. 30). Interestingly the loss of specific tumor suppressors in stromal cell types, has also been shown to influence the type and character of immune cells present within the TIME. For example, in a mouse model of chronic liver damage, p53-deficient hepatic stellate cells, a stromal cell type, secrete factors that polarize TAMs toward a more protumorigenic M2-like phenotype often associated with immunosuppression31. Interestingly, that study has also revealed that natural killer cells, TAMs and resident Kupffer cells are less able to eliminate p53-deficient proliferating hepatic stellate cells in vitro, although the mechanism of dampened elimination of hepatic stellate cells is unclear.
The mutational landscape of the tumor and the TIME
Beyond the effects of tumor-derived cytokines, chemokines and nonimmune cells on the character of the TIME, the overall mutational landscape of tumor cells, a direct reflection of tumor immunogenicity, can dictate the extent and phenotype of immune infiltrate. A particularly strong example of this influence is in CRC (Fig. 2b). As mentioned briefly in the previous section, CRC can be stratified through gene-expression-based subtyping into four consensus molecular subtypes (CMS1-4) (ref. 32). For example, in CMS1 CRC, there are DNA mismatch-repair defects, as indicated by microsatellite instability or hypermutation rates. CMS1 tumors have been found to be deeply infiltrated with CD8+ T cells and to display global gene expression patterns consistent with a high number of T helper 1 (TH1) cells6, as is indicative of an antitumor immune response. However, the antitumor response is likely to be moderated by the presence of immunosuppressive cell types, a protumor cytokine milieu and/or the expression of immune-checkpoint proteins including CTLA-4, PD-1, PD-L1 and IDO-1 (refs 33–35). The expression of immune-checkpoint proteins by CMS1 CRC is notable, because those tumors show substantial responses to anti-PD-1 ICB, thus suggesting that the large mutational burden and high frequencies of tumor-infiltrating CD8+ CTLs and TH1 cells has opened up the potential for many T cell clones to become potently antitumor after tolerance is broken36.
CMS4 CRC, characterized by tumor cells with a mesenchymal-like phenotype, is associated with poor prognosis and high expression of protumoral genes, including those associated with T helper 17 cells, the TGF-β pathway and the monocyte/macrophage lineage34, on the basis of bulk tissue RNA expression. Hence, CMS4 CRC antitumor responses might be easily overwhelmed by a TIME skewed toward immunosuppression. CMS2 and CMS3 CRCs, tumors that are microsatellite stable, nonhypermutated and epithelial according to their gene expression, exhibit low-immune and low-inflammatory signatures and are typically PD-L1 negative. CMS2 and CMS3 CRCs have phenotypes suggestive of antigenically cold tumors, and in both cases the tumors have lower lymphocyte infiltration into the tumor than that observed in CMS1. CMS2-4 are thought to respond poorly to ICBs, partly as a result of low antigenic diversity and generally low tumor-infiltrating lymphocyte (TIL) numbers34,36.
Analyzing progressive development of the TIME at primary and metastatic sites
In progressing cancers, neither the tumor nor the TIME is static. Reciprocal interactions between tumor and associated immune and stromal cell types evolve as the tumor grows, thus allowing for modulation of both tumor cell intrinsic and extrinsic processes37–39. The evolution of the TIME during tumor growth and dissemination is surmised to occur broadly and not just at the level of specific T cell clones recognizing variations in antigenic identities.
The role of the TIME in establishing primary tumors
A major factor determining tumor progression over time is the overall proportion and character of T cells within the TIME (Fig. 3). Several studies in mouse models have revealed that during de novo carcinogenesis, antitumor T cells cannot control tumor growth, owing to tumor-induced tolerance mechanisms40–42. Interestingly, T cell dysfunction in cancer shares many features with the T cell exhaustion (Fig. 3a) observed in chronic viral infections43 and is generally characterized by high surface expression of inhibitory receptors (CTLA-4, PD-1, TIM-3, LAG-3 and 2B4) on T cells; loss of effector functions, such as the production of cytokines IFNγ, IL-2 and TNFα ; and loss of proliferative capacity43,44. The plasticity and reversibility of T cell exhaustion is an important and open question in studies of tumor immunology. Reversible and irreversible states of T cell exhaustion have been identified45, and the irreversible exhausted cells are unresponsive to ICB-like anti-PD-1/anti-PD-L1 therapy46. Preventing or reversing T cell exhaustion for long-term tumor control will be challenging, and perhaps simultaneous targeting of other tolerance pathways, such as the immunosuppressive TIME, or encouraging the priming of new T cell clones, might be required to obtain durable antitumor T cell responses. T cell exhaustion and establishment of an immunosuppressive TIME are likely to be linked events, such that exhaustion occurs as a result of the combination of chronic exposure to tumor antigen47, unproductive interactions from DCs present in the TME48 and exposure to immunosuppressive cytokines and cell types.
Fig. 3. Character of the TIME during progressive tumor development.
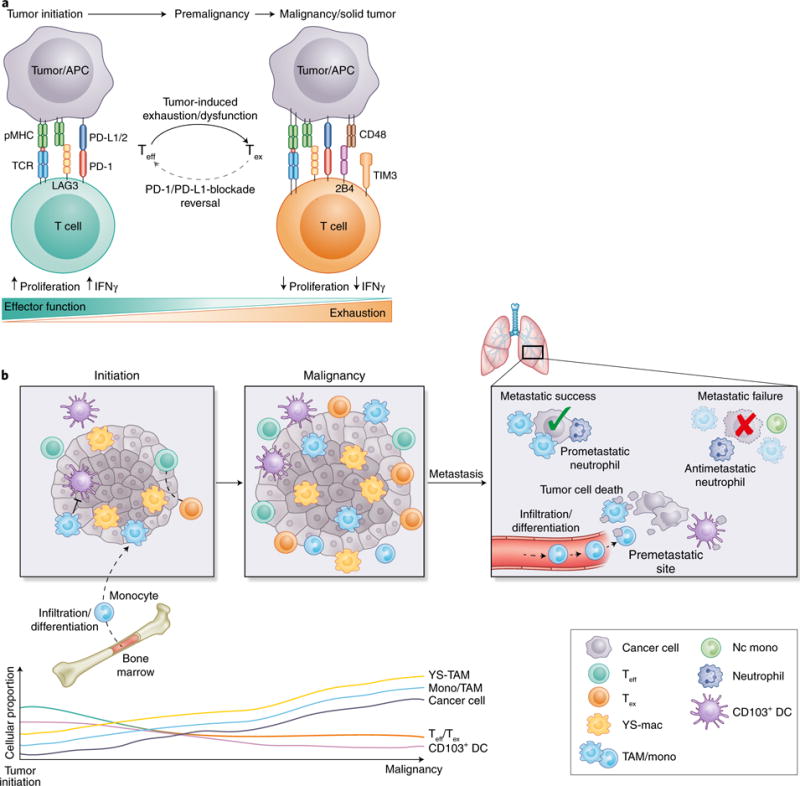
a, A major contributing factor in the failure to immunologically reject tumors stems from induction of T cell exhaustion, a state in which T cells become less responsive to antigens and are ineffective at providing T cell help or eliminating appropriate targets. Recent evidence suggests that T cell exhaustion occurs rapidly after oncogenic initiation, possibly because of chronic antigen exposure on tumor cells. As a T cell transitions from effector (Teff) to exhausted (Tex), there is increased expression of exhaustion-associated molecules such as LAG3, 2B4 and TIM3, and downregulation of effector cytokines such as IFNγ. From a therapeutic standpoint, epigenetic evidence suggests that there are types of T cell exhaustion that are irreversible, thus potentially explaining why some patients are completely unresponsive to some forms of ICB. APC, antigen-presenting cell; pMHC, peptide-bound major histocompatibility complex; TCR, T cell receptor. b, During tumor development, progressive changes in the character and quality of the TIME occur that coincide with disease severity and influence responsiveness to therapeutics. Recent evidence in mice suggests that YS-TAMs are more potently immunosuppressive than their monocyte-derived counterparts (TAMs). Whereas TAMs have been shown to suppress CD103+ DCs through secretion of IL-10, only depletion of YS-TAMs in a model of PDAC has been found to lessen tumor burden. Because YS-TAMs are theoretically present in the tumor site during oncogenic initiation, they may make the first contact with antitumor T cells and may be responsible for early T cell exhaustion. As tumors progress, TAMs of both types expand, whereas Teff cells slowly become Tex cells, and stimulatory CD103+ DCs become a more marginal and rare population. During metastatic seeding, pioneering metastatic tumor cells arrive and die. Their debris is taken up by distinct waves of myeloid cells, with monocytes and macrophages being the dominant phagocytes in the premetastatic site. CD103+ DCs have been shown to uptake minimal tumor debris/antigen, and this process is thought to partially contribute to reduced antitumor T cell priming. Whereas populations such as neutrophils have been associated with both metastatic success and failure, macrophages are often ascribed a prometastatic function, because their absence leads to pronounced metastatic failure. Mac, macrophage; mono, monocyte; nc mono, nonclassical monocyte.
Numerous populations of immune cells have been reported to have suppressive functions in the TIME (for example, neutrophils49 and Treg cells50); however, TAMs are the most extensively studied and well characterized. Recent data in mice suggest that the immunological origin of TAMs (yolk sac or monocyte derived) can substantially affect their overall suppressiveness. In studies of spontaneous mouse PDAC, yolk-sac-derived tumor-associated macrophages (YS-TAMs), which are seeded into tissues in early development and thus before malignant transformation, have been shown to be more tumor supporting than monocyte-derived TAMs51 (Fig. 3b). Moreover, that study has found comparable expression of immunoinhibitory and immunostimulatory receptors on both YS-TAMs and monocyte-derived TAMs, but has demonstrated that tumor burden is significantly decreased only by loss of YS-TAMs but not monocyte-derived TAMs, thus suggesting a more immunologically suppressive role for YS-TAMs. Because YS-TAMs are theoretically more dominant during the early stages of malignant development and consequently during the initial adaptive antitumor response, elimination of nascent tumors may be fundamentally different from the targeting of more advanced tumors. Although the results of that paper are intriguing, the origins of TAMs in different tumor types might be ontologically and functionally distinct. In the MMTV-PyMT breast cancer model, the proportion of exhausted PD-1+ CD8+ T cells has been found to increase in parallel with monocyte-derived TAMs52. In that study, depletion of monocyte-derived TAMs and not tissue-resident macrophage equivalents relieved suppression of cytotoxic T cells.
The inconsistencies regarding which ontologically distinct macrophage subset is the dominantly immunosuppressive and tumor-promoting type might have more to do with macrophage phenotype than origin. Although the specific macrophage phenotype or subset most involved in T cell dysfunction in tumor progression is unclear, and the diversity of macrophage states in vivo53 remains an open question, in vitro macrophages can be generated with two extremes of phenotypes suggested to be tumoricidal (M1) and pro-tumorigenic (M2)54,55. The plasticity of these cells makes therapeutic targeting challenging, but recent studies have shed more light on the molecular switches that control macrophage phenotype. One source of phenotypic switching from immunostimulatory to immunoinhibitory transcriptional macrophage programming may be controlled by either BTK56, a signal transducer downstream of the bacterial lipopolysaccharide receptor TLR4, or PI3Kγ57, a complex signaling molecule linked to the regulation of central myeloid transcriptional regulators NF-κB and C/EBPβ. Inhibition of either BTK or PI3Kγ has been found to restore antitumor cytotoxic T cell responses, thus demonstrating the involvement of these pathways in promoting immune tolerance. In pancreatic cancer, signaling of the innate immune receptor Dectin-1 promotes a tolerogenic macrophage phenotype and T cell suppression, thereby facilitating tumor progression58. Together, these data indicate that therapeutic targeting of macrophages to alter their phenotype may alleviate immunosuppression and improve antitumor immunity.
Although TAM numbers increase in tumors over time in mice and humans, mouse studies have demonstrated a progressive slow loss of CD103+ DCs, which are potent activators of antitumor CD8+ T cells20,48, 59–61. Migratory DCs, such as CD103+ DCs, use expression of the chemokine receptor CCR7 to traffic antigen from the periphery to the source of CCR7 ligand, CCL19 and CCL21, in draining lymph nodes. CD103+ DCs are critical for directing CD8+ T cell immunity, because depletion of CD103+ DCs abrogates CD8+ T cell priming and decreases the response to anti-PD-L1 ICB. Importantly, expansion and activation of CD103+ DCs has been found to synergize with ICB in multiple experimental models60,62. Although these findings demonstrate that enhancing the functionality of these cells can improve the efficacy of immunotherapy, their loss in mouse models over time suggests that the ability to prime T cells will slowly wane. Gene expression analysis of human cancer biopsies has revealed a correlations among a ‘CD103-associated gene signature’, T cell infiltration into tumors and improved prognosis20,59; however, longitudinal studies have not yet confirmed that these cells are also progressively lost in human tumors. As studies advance understanding of how the human TIME correlates with therapeutic response, it will also be interesting to determine whether patients with elevated frequencies of BDCA3+ DCs, the human equivalent of mouse CD103+ DCs, have a superior response to ICB. Beyond this possibility, other less well-defined DC populations exist within the TIME in mice and humans, and their function and importance has yet to be fully determined.
The immune environment in metastasis
Tumor-induced immunological changes affect the progression to metastatic disease, even before disseminated cancer cells have reached a secondary organ. Systemic immune tolerance and changes in the character of circulating myeloid cells can predispose a tumor for success in seeding a metastatic site. As tumor cells metastasize to distant tissue sites, they are immediately swarmed by distinct sets of immune populations that can both aid in and inhibit metastasis formation (Fig. 1b).
An overwhelming amount of data support the prometastatic function of classical inflammatory monocytes as well as macropha ges23,26,28,63,64. A seminal study using the MMTV-PyMT breast cancer mouse model has found that mice lacking Csj-1, which is required for the development of CSF-1-dependent cells, including monocytes and macrophages, exhibit delayed progression of mammary tumors to metastasis26. Recent data suggest that, beyond being promoted by TAMs in the primary tumor, metastasis is potently promoted by macrophages and their precursor populations present in premetastatic sites64,65.
Mouse studies have indicated that CD4+ T cell-derived IL-4 indirectly promotes breast cancer metastasis by regulating macrophage phenotype, thus demonstrating a role of both the innate and the adaptive immune system in suppressing productive antitumor effects28. A recent study using multiphoton intravital imaging of the lung premetastatic site in mice has revealed that as pioneering metastatic tumor cells arrive and die, distinct waves of myeloid cells ingest tumor material, thereby supplying antigen to both pro- and antitumor immune compartments65. However, monocytes engulf most of the tumor material and consequently may sequester valuable tumor antigen from stimulatory DC populations; moreover, a reduction in monocytes result in higher antigen loads in those DCs. Although classical inflammatory monocytes have a known metastatic-promoting function, nonclassical or ‘patrolling’ monocytes have been shown to have antimetastatic properties66.
Like monocytes and macrophages, neutrophils play critical roles in tumor development. Several preclinical mouse cancer models have revealed that, similarly to observations in patients, neutrophil proportions are elevated in the circulation and accumulate in peripheral organs during tumor progression67–70. However, the roles of neutrophils in metastasis remain controversial. Whereas some studies have reported antimetastatic functions of neutrophils71,72, others have demonstrated prometastatic properties67,69,73–76. In the 4T1 mouse breast tumor model, tumor-entrained neutrophils have been found to inhibit metastatic seeding in the lung via direct cytotoxicity toward disseminated cancer cells71. Moreover, a recent study has reported that a subpopulation of neutrophils expressing the MET proto-oncogene protects against the formation of metastasis72. In contrast, in the MMTV-PyMT mammary tumor model, neutrophils have been found to facilitate metastasis to the lung by propagating the number of metastasis-initiating cancer cells via the secretion of leukotrienes67. Furthermore, in a model of lobular breast cancer, neutrophils have been found to promote metastasis by dampening antitumor immunity69. Systemic expansion and polarization of prometastatic neutrophils is driven by tumor-induced IL17-producing γδ T cells69, thus demonstrating the tight interplay between the innate and the adaptive immune system during metastasis. Although much of what is known about immunological composition at the metastatic site relates to cells with immunosuppressive functions, emerging evidence suggests that stimulatory myeloid cells can also potentiate antitumor T cell responses.
Although macrophages take up most tumor antigen, they often fail to successfully activate T cells in vitro20, in agreement with their previously described protumoral role. However, although their presence in tumors and metastatic lesions is scarce, CD103+ DCs are far better T cell activators20,59, and their loss results in a significant increase in pulmonary metastasis65, thus suggesting that even in the metastatic site, CD103+ DCs are important for eliciting potent antitumor CD8+ T cell responses. Together, these data support that therapeutic strategies that target myeloid cells to alleviate immunosuppression and reinvigorate T cell responses may be a feasible immunotherapeutic approach to treat patients with metastatic cancer.
The contribution of systemic factors to the TIME
Understanding the spatiotemporal dynamics of the TIME necessitates dissecting the potential roles that systemic factors may have in predisposing certain TIMEs to be fostered. As the broad effects of factors such as exercise77, age78, diet79, adiposity80, the microbiome81 and sex82 on the immune system have become clearer, an understanding of how these factors directly affect the quality of the antitumor immune response has also emerged. Both these patient-intrinsic and tumor-dependent effects intersect on many levels and will be important considerations in improving the efficacy of existing therapies or developing orthogonal immunotherapeutic approaches.
The systemic inflammatory state of an individual can affect the character of the TIME in premalignancies, thus leading to an occult tumor’s eventual elimination or supporting progression to advanced disease. A recent study has found that patients with atherosclerosis treated with anti-IL-1β had lower incidence of lung cancer than did patients who had received placebo83. IL-1β has been shown to induce synthesis of COX-2, which in turn leads to high-level production of PGE2, a potent immunosuppressive molecule, in a subset of cancers. Aspirin, a COX inhibitor, has only very modest protective benefits when viewed across all cancers84, although its use is associated with lower disease incidence in patient populations predisposed to develop specific types of cancer85. These findings may indicate key differences in the TIMEs of patient populations. Interestingly, COX inhibition may also have utility in cancer treatments, because it has been shown to synergize with anti-PD-1 therapy in established tumors86.
As discussed above, tumors can make numerous cytokines and chemokines that attract and inform specific components of the immune system. Although these factors affect the local TIME itself, they can also become systemic, inducing broader changes in the tumor macroenvironment. Tumor cell production of the growth factors G-CSF70 and GM-CSF, as well as of IL-6 (ref. 87), can affect bone marrow myeloid progenitor expansion, thus leading to enhanced release of myeloid cells into circulation, and ultimately affect the number of circulating and tumor-infiltrating immunosuppressive myeloid cells and contribute to more severe disease and greater metastatic burden74,88. Tumor-induced systemic factors can affect the bone marrow and in turn promote tumor infiltration of cancer-promoting immune components, including neutrophils89, monocytes90 and platelets91.
Certain aspects that affect the tumor microenvironment predate tumor establishment. Both aging92,93 and obesity94 have been reported to produce a proinflammatory state and to lead to an increase in the number of suppressive immature myeloid cells in circulation. Moreover, sex hormones may lead to altered TIME responses in male as compared with female patients, because estrogen has been shown to activate the STAT3 pathway in human and mouse bone marrow myeloid progenitor cells, thereby leading to an increased presence of potentially suppressive myeloid cells in circulation95. In contrast, estrogen may also induce a more tolerogenic phenotype or subset in tumor DC populations96, thus partially explaining the difference in tumor growth between male and female mice. There is an added uncertainty, at present, of the heterogeneity of the myeloid lineage as it exists in circulation and whether each of these features of patients may influence the exact same or different subpopulations of cells.
More clearly, the microbiome has been found to have an important role in determining DC functionality. Two recent studies have reported that responses to checkpoint blockade are dependent on the microbiomes of the mice studied97,98. Moreover, patients can be stratified according to their microbiomes, and this stratification is predictive of the response to anti-CTLA-4 therapy98. Both of these studies have hypothesized that this effect may be at least partly due to improvements in DC functionality either through improved maturation and cross-presentation leading to improved CD8+ T cell priming97 or through improved CD11b+ DC migration from the tumor and improved TH1 responses98. DC phenotype is also affected by the temperature of the animal being studied: placing mice under mild cold stress in laboratory conditions leads to increased tumor growth and reduced immune control99, effects at least partly due to decreased DC functionality100. As such, the immune macroenvironment of a patient can dramatically affect the tumor microenvironment.
Tumor-derived factors, as well as those affecting myeloid cell production from the bone marrow, can also alter patient metabolic status, which in turn can influence antitumor immunity. Recent research has revealed that in the CT26 and KPC tumor models, tumor-derived IL-6 alters liver metabolism and consequently, in the context of caloric restriction, leads to increased corticosterone and suppressed antitumor immunity101. In this setting, tumor-derived factors alter systemic metabolic tone and consequently lead to alterations in the tumor microenvironment. Interestingly in other models, caloric restriction101 or the administration of a fasting mimetic102, both of which trigger autophagy, have led to improved antitumor immunity in mice. These fasting-related effects have been linked to potentiated responses to chemotherapy, partially as a result of increased TIL infiltration103 and loss of tumoral Treg cells102.
These findings indicate that there may be more complexity to uncover regarding the effects of nutrition on tumor immunity and that the effects may be model dependent, in a manner analogous to the opposing effects of fasting on responses to bacterial and viral disease104. These factors should thus be taken into account when considering potential orthogonal immunotherapeutic approaches as well as when deciding upon appropriate animal models for preclinical evaluation. The use of sex-matched, young and lean mice in most animal studies may explain some of the failures of mouse studies to predict therapeutic responses in the more diverse human population. Indeed, given that population obesity rates are increasing and that most tumors develop in elderly patients, understanding these factors is likely to prove critical for understanding of the tumor microenvironment. Moreover, other factors such as housing temperature (although this factor may not affect patient treatment, because hospitals are kept relatively thermoneutral) may greatly affect the findings from experimental systems.
Future directions
Further characterization of the tumor immune microenvironment
Major successes with ICB and the potential for substantial clinical effects are driving thousands of clinical trials. These successes include alternative ICB-like targets and drugs that modulate myeloid biology105, which may be paired with nonimmunological drug approaches. Pharmaceutical companies and clinical investigators alike are well aware of the value of tracking biomarkers associated with tumor growth, but more attention must be paid to how the TIME of a specific patient is altered before, during and after a trial. Using high-dimensional techniques to characterize patients with improved granularity should reveal as much about human immunology in an in vivo setting as any experiment in a mouse could. Similarly to the cases of anti-CTLA-4 and anti-PD-1/PD-L1, breakthroughs will occur when basic-science discoveries are translated into actionable improvements in human disease. Therefore, the fastest route to demonstrable successes will depend on asking useful questions and using applicable animal models and valuable, innovative tools.
Because immunotherapeutic intervention is attempted in disparate tumor types, there is a growing need to identify the unifying features and critical differences that define distinct classes and subclasses of TIME, which relate to the likelihood of response to immunotherapeutics. For substantial progress to occur in this area, use of the highest-resolution methods will be critical to assess total cellular composition (for example, flow cytometry versus mass cytometry), functional status (for example, bulk RNA sequencing versus single-cell RNA sequencing) and cellular localization (for example, immunohistochemistry versus multidimensional immunohistochemistry) in parallel to define highly granular classes and subclasses of TIME. Major advances have already been made in stratifying patients according to tumor type. We believe that further stratification of patients on the basis of not only their tumor type but also their TIME type will yield better insight into overall survival and the likelihood of response to immunotherapeutics, and will provide vast datasets to help identify new druggable targets. This progress will be garnered by using the most cutting-edge techniques in multiparametric imaging106, mass cytometry107,108 and single-cell RNA sequencing109. Critical to this goal is that improved resolution of cellular composition and analysis of functional status and spatial distribution must be paired with relevant patient outcomes (Fig. 1b). In particular, by casting a wider unbiased net, it will be possible to detect subtle changes in rare populations while also appreciating prominent effector activation states in situ.
A major goal in moving into truly orthogonal pathways to treat cancers is to understand the fundamental conditions in which TIMEs are arrayed; these conditions almost certainly reflect genetic programs engaged by the tumors themselves, if not also by the tissue in which the tumor is located. Distinct collections of stroma, epithelium and immune cell types present nearly countless ways to parse a TIME, but it is still unknown how many of these cellular combinations help permit the rapid cellular proliferation and disorganization associated with a growing tumor. To that end, parallel studies to characterize tissue-specific responses to pathogens, healing wounds, chronic viral infections and tolerance in the gut may provide powerful datasets for comparison with the classes of TIME, because they are all analyzed at this detail.
Using preexisting drugs to modulate immune-associated targets
As immune-immune and immune-tumor interaction networks are better characterized, it will become possible to define classes of TIME and determine which cells, molecules and pathways are essential for suppressing antitumor immunity, and in what tumor contexts. In some cases, such definition may already be possible, because of the existence of failed, orphan, poorly efficacious drugs or drugs without an obvious direct application as immunotherapeutic agents.
To advance immunotherapy, the state of thinking must be revamped in terms of the treatment goals (i.e., decreasing disease incidence versus combating advanced disease), and drugs that have had previous marginal success should be revisited. After paltry early clinical success, recent preclinical data in a mouse model of PDAC suggest that a combination of chemotherapy and anti-CD40 agonistic antibody unleashes a potent antitumor immune response; moreover, early data in humans show enhanced lymphocyte infiltration. Although investigations are still in their early days, drugs to normalize vasculature, alter metabolism and suppress individual components of the immune system may find new life in the clinic, either as single agents or in combination with ICB, for long-term use as prophylactic measures.
Mouse to human and back again
Translating clinical insights into improvements in mouse models is necessary to ensure that discoveries made at the bench can derive applicable and high-quality therapeutics. As classes of human TIME are elaborated (as described above), it is critical that parallel efforts take place to ascertain the best ways to generate reflective TIME in mouse models. Solid human tumors develop in situ and over long periods of time, characteristics not reflected in ectopic mouse tumor models, which very often grow in the subcutaneous space and are formed through bolus injection of thousands of highly malignant tumor cells. Ectopic mouse tumor models have been invaluable for preclinical validation of countless therapeutics but have fallen short of being good indicators of therapeutic efficacy in humans. Although genetically engineered mouse models of cancer have brought immuno-oncology research a step closer toward recapitulating the stepwise progression of human disease, the resultant spontaneous tumors still leave something to be desired. The discovery of CRISPR-Cas9 now allows for rapid and parallel introduction of numerous mutations or engineered constructs into a single mouse110,111, thus changing how genetically engineered mouse models can be created, with less of an emphasis on severe oncogenic drivers and more of an emphasis on tunable oncogenic induction and mutational landscapes more similar to those in human disease.
Furthermore, major advances in the development of humanized mouse models have made xenografts with matching patient tumor and immune compartments possible, thereby enabling studies in which a patient’s own adoptively transferred TILs can be used to recapitulate the exhaustion or the introduction of targeted gene reporter loci to visualize intravital tumor immune interactions. Although these models have downsides, being able to implant human tumor tissue with a native mutational landscape into a partially reconstituted human immune repertoire represents major progress. Even if mouse models fail to ever fully recapitulate human disease, it is important to understand the minutiae that make the most difference in dictating therapeutic response versus nonresponse. Distilling a disease into a few critical parameters is challenging, but understanding what cell types can be modulated and when may enable the next biggest improvements in immunotherapy.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ali HR, Chlon L, Pharoah PD, Markowetz F, Caldas C. Patterns of immune infiltration in breast cancer and their clinical implications: a gene-expression-based retrospective study. PLoS Med. 2016;13:e1002194. doi: 10.1371/journal.pmed.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newman AM, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bindea G, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 6.Mlecnik B, et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 2016;44:698–711. doi: 10.1016/j.immuni.2016.02.025. [DOI] [PubMed] [Google Scholar]
- 7.Herbst RS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ademmer K, et al. Effector T lymphocyte subsets in human pancreatic cancer: detection of CD8+CD18+ cells and CD8+CD103+ cells by multi-epitope imaging. Clin Exp Immunol. 1998;112:21–26. doi: 10.1046/j.1365-2249.1998.00546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beatty GL, et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6Clow F4/80+ extratumoral macrophages. Gastroenterology. 2015;149:201–210. doi: 10.1053/j.gastro.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spranger S. Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int Immunol. 2016;28:383–391. doi: 10.1093/intimm/dxw014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans RA, et al. Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight. 2016;1:e88328. doi: 10.1172/jci.insight.88328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee HJ, et al. Tertiary lymphoid structures: prognostic significance and relationship with tumour-infiltrating lymphocytes in triple-negative breast cancer. J Clin Pathol. 2016;69:422–430. doi: 10.1136/jclinpath-2015-203089. [DOI] [PubMed] [Google Scholar]
- 13.Sautes-Fridman C, et al. Tertiary lymphoid structures in cancers: prognostic value, regulation, and manipulation for therapeutic intervention. Front Immunol. 2016;7:407. doi: 10.3389/fimmu.2016.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finkin S, et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat Immunol. 2015;16:1235–1244. doi: 10.1038/ni.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, Lambrecht BN. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol. 2012;33:297–305. doi: 10.1016/j.it.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lutz ER, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res. 2014;2:616–631. doi: 10.1158/2326-6066.CIR-14-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203:1651–1656. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–847. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bayne LJ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Broz ML, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014;26:638–652. doi: 10.1016/j.ccell.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31:711–723.e714. doi: 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 23.Qian BZ, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonavita E, et al. PTX3 is an extrinsic oncosuppressor regulating complement-dependent inflammation in cancer. Cell. 2015;160:700–714. doi: 10.1016/j.cell.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 25.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scholl SM, et al. Anti-colony-stimulating factor-1 antibody staining in primary breast adenocarcinomas correlates with marked inflammatory cell infiltrates and prognosis. J Natl Cancer Inst. 1994;86:120–126. doi: 10.1093/jnci/86.2.120. [DOI] [PubMed] [Google Scholar]
- 28.DeNardo DG, et al. CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16:91–102. doi: 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 30.Khalili JS, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012;18:5329–5340. doi: 10.1158/1078-0432.CCR-12-1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lujambio A, et al. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153:449–460. doi: 10.1016/j.cell.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guinney J, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Angelova M, et al. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015;16:64. doi: 10.1186/s13059-015-0620-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Becht E, et al. Immune and stromal classification of colorectal cancer is associated with molecular subtypes and relevant for precision immunotherapy. Clin Cancer Res. 2016;22:4057–4066. doi: 10.1158/1078-0432.CCR-15-2879. [DOI] [PubMed] [Google Scholar]
- 35.Gatalica Z, et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol Biomarkers Prev. 2014;23:2965–2970. doi: 10.1158/1055-9965.EPI-14-0654. [DOI] [PubMed] [Google Scholar]
- 36.Boland PM, Ma WW. Immunotherapy for colorectal cancer. Cancers (Basel) 2017;9:E50. doi: 10.3390/cancers9050050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Egeblad M, Nakasone ES, Werb Z. Tumors as organs: complex tissues that interface with the entire organism. Dev Cell. 2010;18:884–901. doi: 10.1016/j.devcel.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15:73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Willimsky G, Blankenstein T. Sporadic immunogenic tumours avoid destruction by inducing T-cell tolerance. Nature. 2005;437:141–146. doi: 10.1038/nature03954. [DOI] [PubMed] [Google Scholar]
- 41.Garbe AI, et al. Genetically induced pancreatic adenocarcinoma is highly immunogenic and causes spontaneous tumor-specific immune responses. Cancer Res. 2006;66:508–516. doi: 10.1158/0008-5472.CAN-05-2383. [DOI] [PubMed] [Google Scholar]
- 42.DuPage M, et al. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell. 2011;19:72–85. doi: 10.1016/j.ccr.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36:265–276. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 45.Philip M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017;545:452–456. doi: 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pauken KE, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354:1160–1165. doi: 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schietinger A, et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity. 2016;45:389–401. doi: 10.1016/j.immuni.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruffell B, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26:623–637. doi: 10.1016/j.ccell.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar V, et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell. 2017;32:654–668.e5. doi: 10.1016/j.ccell.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med. 2013;210:2435–2466. doi: 10.1084/jem.20130762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu Y, et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity. 2017;47:323–338.e6. doi: 10.1016/j.immuni.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franklin RA, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921–925. doi: 10.1126/science.1252510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murray PJ, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mantovani A, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 55.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164:6166–6173. [Google Scholar]
- 56.Gunderson AJ, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov. 2016;6:270–285. doi: 10.1158/2159-8290.CD-15-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaneda MM, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature. 2016;539:437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Daley D, et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med. 2017;23:556–567. doi: 10.1038/nm.4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roberts EW, et al. Critical role for CD103+/CD141+ dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. 2016;30:324–336. doi: 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salmon H, et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. 2016;44:924–938. doi: 10.1016/j.immuni.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hildner K, et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sánchez-Paulete AR, et al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov. 2016;6:71–79. doi: 10.1158/2159-8290.CD-15-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qian BZ, et al. FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J Exp Med. 2015;212:1433–1448. doi: 10.1084/jem.20141555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kitamura T, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. 2015;212:1043–1059. doi: 10.1084/jem.20141836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Headley MB, et al. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature. 2016;531:513–517. doi: 10.1038/nature16985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hanna RN, et al. Patrolling monocytes control tumor metastasis to the lung. Science. 2015;350:985–990. doi: 10.1126/science.aac9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. 2015;528:413–417. doi: 10.1038/nature16140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nywening TM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a singlecentre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17:651–662. doi: 10.1016/S1470-2045(16)00078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Coffelt SB, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522:345–348. doi: 10.1038/nature14282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Casbon AJ, et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci USA. 2015;112:E566–E575. doi: 10.1073/pnas.1424927112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Granot Z, et al. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. 2011;20:300–314. doi: 10.1016/j.ccr.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Finisguerra V, et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature. 2015;522:349–353. doi: 10.1038/nature14407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Steele CW, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. 2016;29:832–845. doi: 10.1016/j.ccell.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kowanetz M, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci USA. 2010;107:21248–21255. doi: 10.1073/pnas.1015855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jamieson T, et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J Clin Invest. 2012;122:3127–3144. doi: 10.1172/JCI61067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bald T, et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature. 2014;507:109–113. doi: 10.1038/nature13111. [DOI] [PubMed] [Google Scholar]
- 77.Koelwyn GJ, Quail DF, Zhang X, White RM, Jones LW. Exercise-dependent regulation of the tumour microenvironment. Nat Rev Cancer. 2017;17:620–632. doi: 10.1038/nrc.2017.78. [DOI] [PubMed] [Google Scholar]
- 78.Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol. 2013;13:875–887. doi: 10.1038/nri3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Julia V, Macia L, Dombrowicz D. The impact of diet on asthma and allergic diseases. Nat Rev Immunol. 2015;15:308–322. doi: 10.1038/nri3830. [DOI] [PubMed] [Google Scholar]
- 80.Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15:104–116. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robinson DP, Lorenzo ME, Jian W, Klein SL. Elevated 17β-estradiol protects females from influenza A virus pathogenesis by suppressing inflammatory responses. PLoS Pathog. 2011;7:e1002149. doi: 10.1371/journal.ppat.1002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ridker PM, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1833–1842. doi: 10.1016/S0140-6736(17)32247-X. [DOI] [PubMed] [Google Scholar]
- 84.Cook NR, et al. Low-dose aspirin in the primary prevention of cancer: the Women’s Health Study: a randomized controlled trial. J Am Med Assoc. 2005;294:47–55. doi: 10.1001/jama.294.1.47. [DOI] [PubMed] [Google Scholar]
- 85.Burn J, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet. 2011;378:2081–2087. doi: 10.1016/S0140-6736(11)61049-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zelenay S, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell. 2015;162:1257–1270. doi: 10.1016/j.cell.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marigo I, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPβ transcription factor. Immunity. 2010;32:790–802. doi: 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 88.Waight JD, Hu Q, Miller A, Liu S, Abrams SI. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS One. 2011;6:e27690. doi: 10.1371/journal.pone.0027690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Engblom C, et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecFhigh neutrophils. Science. 2017;358:eaal5081. doi: 10.1126/science.aal5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cortez-Retamozo V, et al. Angiotensin II drives the production of tumor-promoting macrophages. Immunity. 2013;38:296–308. doi: 10.1016/j.immuni.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pucci F, et al. PF4 promotes platelet production and lung cancer growth. Cell Rep. 2016;17:1764–1772. doi: 10.1016/j.celrep.2016.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Verschoor CP, et al. Blood CD33+HLA-DR− myeloid-derived suppressor cells are increased with age and a history of cancer. J Leukoc Biol. 2013;93:633–637. doi: 10.1189/jlb.0912461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ruhland MK, et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun. 2016;7:11762. doi: 10.1038/ncomms11762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hale M, et al. Obesity triggers enhanced MDSC accumulation in murine renal tumors via elevated local production of CCL2. PLoS One. 2015;10:e0118784. doi: 10.1371/journal.pone.0118784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Svoronos N, et al. Tumor cell-independent estrogen signaling drives disease progression through mobilization of myeloid-derived suppressor cells. Cancer Discov. 2017;7:72–85. doi: 10.1158/2159-8290.CD-16-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Thompson MG, Peiffer DS, Larson M, Navarro F, Watkins SK. FOXO3, estrogen receptor alpha, and androgen receptor impact tumor growth rate and infiltration of dendritic cell subsets differentially between male and female mice. Cancer Immunol Immunother. 2017;66:615–625. doi: 10.1007/s00262-017-1972-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sivan A, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084–1089. doi: 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vétizou M, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079–1084. doi: 10.1126/science.aad1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kokolus KM, et al. Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proc Natl Acad Sci USA. 2013;110:20176–20181. doi: 10.1073/pnas.1304291110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kokolus KM, et al. Stressful presentations: mild cold stress in laboratory mice influences phenotype of dendritic cells in naïve and tumor-bearing mice. Front Immunol. 2014;5:23. doi: 10.3389/fimmu.2014.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Flint TR, et al. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab. 2016;24:672–684. doi: 10.1016/j.cmet.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pietrocola F, et al. Caloric restriction mimetics enhance anticancer immunosurveillance. Cancer Cell. 2016;30:147–160. doi: 10.1016/j.ccell.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Di Biase S, et al. Fasting-mimicking diet reduces HO-1 to promote T cell-mediated tumor cytotoxicity. Cancer Cell. 2016;30:136–146. doi: 10.1016/j.ccell.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang A, et al. Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell. 2016;166:1512–1525. doi: 10.1016/j.cell.2016.07.026. e1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol. 2016;13:143–158. doi: 10.1038/nrclinonc.2015.209. [DOI] [PubMed] [Google Scholar]
- 106.Tsujikawa T, et al. Quantitative multiplex immunohistochemistry reveals myeloid-inflamed tumor-immune complexity associated with poor prognosis. Cell Rep. 2017;19:203–217. doi: 10.1016/j.celrep.2017.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Spitzer MH, et al. Systemic immunity is required for effective cancer immunotherapy. Cell. 2017;168:487–502.e15. doi: 10.1016/j.cell.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lavin Y, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. 2017;169:750–765.e17. doi: 10.1016/j.cell.2017.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tirosh I, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang H, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]