

Abstract
Mild traumatic brain injury (mTBI) represents a serious public health concern. Although much is understood about long-term changes in cell signaling and anatomical pathologies associated with mTBI, little is known about acute changes in neuronal function. Using large scale Ca2+ imaging in vivo, we characterized the intracellular Ca2+ dynamics in thousands of individual hippocampal neurons using a repetitive mild blast injury model in which blasts were directed onto the cranium of unanesthetized mice on two consecutive days. Immediately following each blast event, neurons exhibited two types of changes in Ca2+ dynamics at different time scales. One was a reduction in slow Ca2+ dynamics that corresponded to shifts in basal intracellular Ca2+ levels at a time scale of minutes, suggesting a disruption of biochemical signaling. The second was a reduction in the rates of fast transient Ca2+ fluctuations at the sub-second time scale, which are known to be closely linked to neural activity. Interestingly, the blast-induced changes in basal Ca2+ levels were independent of the changes in the rates of fast Ca2+ transients, suggesting that blasts had heterogeneous effects on different cell populations. Both types of changes recovered after ∼1 h. Together, our results demonstrate that mTBI induced acute, heterogeneous changes in neuronal function, altering intracellular Ca2+ dynamics across different time scales, which may contribute to the initiation of longer-term pathologies.
Keywords: : brain injury, GCaMP, head trauma, in vivo imaging
Introduction
Traumatic brain injury (TBI) caused by blast represents a serious public health concern, particularly in military settings.1,2 Four categories of blast injury have been established. Primary injury results directly from exposure to the blast wave, whereas secondary, tertiary, and quaternary blast injuries are associated with collisions with projectiles set in motion by the blast, hitting other nearby objects, or being exposed to the heat and noxious chemicals accompanying the blast, respectively.3–7 Of the different types of blast-related injuries, primary injury is the most complex and remains the least understood, as there is often clear functional deficit in the absence of anatomical pathology.8,9
The pathophysiology of TBI can vary depending on the nature and severity of the injury. Although moderate and severe TBI receive considerable attention, mild TBI (mTBI) accounts for ∼75% of injuries.8,10 Damages from mild injury can result in a broad spectrum of neurological deficits. For example, repeated mTBI has been associated with memory impairment, increased susceptibility to temporal lobe epilepsy, post-traumatic stress disorder (PTSD), and neurodegeneration, as observed in chronic traumatic encephalopathy.1,5,11–16 Blast-related impairments in learning and memory,12 as well as increased susceptibility to seizures,13 have motivated studies focused on the hippocampus, because of its integral role in learning and memory and its contribution to temporal lobe seizures in humans.14,15 Several studies have reported that blast reduces hippocampal long-term potentiation, and leads to the loss of GABAergic interneurons and other anatomical structural changes.8,17,18 These pathologies have been linked to a number of biochemical and synaptic changes, involving various neurotransmitters and intracellular signaling pathways across all hippocampal sub-regions, including the dentate gyrus, Cornu Ammonis (CA)1, and CA3.
Over the years, several animal models of blast injury have been developed using devices such as shock tubes, blast tubes, and detonation of explosives in open fields, to study blast injury.11,19,20 To understand the acute effects of mTBI on neuronal function, we here used a previously published cranium-only blast injury model,20 to determine the feasibility of using wide-field imaging to examine Ca2+ changes in large numbers of individual CA1 neurons. This model has the advantage that the blast is confined to the cranium, and because no surgery is required, mice can be blasted in the absence of any anesthetics, allowing us to perform real time analysis in mice with full head mobility.
Recent developments in scientific complementary metal-oxide semiconductor (CMOS) cameras and high-performance genetically encoded Ca2+ sensors such as GCaMP6, have enabled ultra-large-scale Ca2+ imaging of thousands of individual neurons in the brain using wide-field microscopy.21 Studies using GCaMP6 and earlier generations of genetically encoded Ca2+ sensors have consistently demonstrated that transient, sub-second changes of intracellular Ca2+ levels are closely correlated with neuronal spiking in many brain regions.21–26 In the hippocampal CA1 region, patterns of sub-second Ca2+ transients recorded in individual neurons closely paralleled the spiking patterns reported in electrophysiological studies, demonstrating that sub-second Ca2+ transients can be used to estimate neural activities.27–30 Although a single spike can produce detectable changes in GCaMP6 fluorescence, most of the sub-second time scale GCaMP6 fluorescence transients observed in living brains are likely associated with bursts of spikes.21 We previously demonstrated that wide-field Ca2+ imaging with GCaMP6 allows the ability to simultaneously image the activity of hundreds to thousands of individual hippocampal neurons in awake behaving mice.30 Because of the simple instrumentation of wide-field Ca2+ imaging, this imaging modality can be easily adopted by many researchers to study acute changes in neuronal function following exposure to mild blasts. This study highlights the utility of this technique for studying mTBI, and reports some initial results using this technique in animals that experience a blast injury over two consecutive days.
Methods
Blast model
The blast injury model used was adapted from the previously developed Cranium Only Blast Injury Apparatus (COBIA).20 The central component of the COBIA was a Mastershot (a 22 caliber, single-shot, powder-actuated tool, Ramset RS22; ITW Ramset, Glendale Heights, IL) mounted vertically on a custom-fabricated stand. The Mastershot was modified by removing the piston that normally drove the fastener, making it function like a firearm allowing the blast wave to propagate undampened through the barrel (Fig. 1B). The muzzle of the Mastershot was snugly fit into a custom blast director, constructed from polyvinyl chloride pipe (10 cm long with an inner diameter of 2 cm), (Fig. 1B). The blast wave was generated by firing a 22 caliber crimped brass blank cartridge (power hammer loads power level 4, yellow color coding, with 179 ± 5 mg of smokeless powder, Ramset 42CW, Ramset, Glendale Heights, IL).
FIG. 1.

Experimental design for wide-field Ca2+ imaging of hippocampal neurons in mice exposed to a cranial blast (A) A wide-field fluorescence microscope coupled to a scientific complementary metal-oxide semiconductor (sCMOS) camera was used to image neurons expressing a genetically encoded Ca2+ sensor (GCaMP6f) in vivo. (B) The Cranium Only Blast Injury Apparatus (COBIA) consisted of a modified nail gun coupled to a blast director to direct the blast wave vertically onto the freely moving head of unanesthetized mice. The distance from the animal's head to the opening of the blast director was 2 cm. (C) Waveform of average overpressures (n = 5 tests) generated from the COBIA. Inset shows the zoom in of the waveform over 10 ms. (D) Experimental timeline. (E) Ca2+ imaging protocol during each blast session.
Blast wave measurement and calibration
A precision dynamic high frequency piezoelectric pressure transducer (Model: 113B21 High Frequency ICP® pressure sensor, PCB Piezotronics, Inc., Depew, NY), powered by a power supply (Model 5421, Columbia Research Laboratories, Inc., Woodlyn, PA), was used to measure the pressure of the blast waves produced by the COBIA (Fig. 1C). The sensor was positioned ∼2 cm from the dissipation chamber, where the head of the mouse was positioned. Transducer outputs were digitized using a NADAQ data acquisition system (National Instruments, NI-USB-6259) at 350 kHz, and analyzed offline.
We calibrated our COBIA device using this sensor, which had a 500 kHz resonant frequency (100 kHz maximum sampling frequency). To compare to the pressure measurements reported in the study by Kuehn and coworkers, we filtered the pressure trace at 12 kHz to measure a comparable transient overpressure.20 To compare to the shock tube device reported by Goldstein and coworkers, we filtered the blast pressure trace at 2 KHz.11 Although the severity of the blast injury is typically attributed to the magnitude of the pressure produced (peak blast overpressure), and the duration of peak overpressure,5,19,20,31 none of the animals exposed to the blast lost consciousness or exhibited noticeable abnormal behavior either immediately following the blast or the week after. In addition, upon dissection, the brains were unremarkable with no contusions. Because of the similarities of the blast overpressure in this study to those in prior studies, we consider results of this study most relevant to mild blast injury.11,20
Wide-field imaging and blast procedure
All animal procedures were approved by the Boston University Institutional Animal Care and Use Committee. Nine adult female C57BL/6 mice (Taconic; Hudson, NY) 2–12 months old at the start of the experiments, were imaged as described previously.30 Briefly, under isoflurane anesthesia, mice were stereotaxically injected in CA1 (anteroposterior [AP], −2 mm; mediolateral [ML], 1.4 mm; dorsoventral [DV], −1.6 mm) with 0.25 μL of AAV9-Syn-GCaMP6f.WPRE.SV40 virus (titer ∼6e12 GC/mL, University of Pennsylvania Vector Core) using a microsyringe pump (UltraMicroPump3-4; World Precision Instruments, Sarasota, FL) at a speed of 40 nL/min. On complete recovery (at least 5 days), animals were then surgically implanted over the CA1 viral injection site with a custom imaging window constructed using a stainless steel cannula (outer diameter [OD], 0.317 in; inner diameter [ID], 0.236 in; height, 2 mm) adhered to a cover-slip (size 0; OD, 3mm), and a custom aluminum head-plate to allow awake head fixed imaging. On complete recovery from implant surgery (∼ 2 weeks), mice were accustomed to head fixation before imaging sessions began (Fig. 1D).
Awake, head-fixed animals were imaged using a custom-built fluorescence microscope as previously described (Fig. 1A).30 Briefly, the imaging setup consisted of standard optics for GCaMP6 imaging and a scientific CMOS (sCMOS) camera (ORCA-Flash4.0 LT Digital CMOS camera C11440-42U; Hamamatsu, Boston, MA). Imaging data (1024 x 1024 pixels, 16 bit) were collected at 20 Hz and analyzed offline. No anesthetics or analgesics were used for any of the blast or imaging protocols.
Mice were first imaged for 100 sec to obtain baseline activity prior to the blast, and then removed from the imaging setup and inserted into a mouse restrainer (Stoelting Co. Wood Dale, IL) that permitted full mobility of the head (Fig. 1B). For the blast group (n = 4 mice), restrained mice were placed 2 cm under the blast dissipation chamber and positioned so that the blast was directed dorsally between bregma and lambda. For the sham group (n = 5 mice), restrained mice were positioned adjacent to the COBIA, so that mice experienced the sound of the blast but not the blast pressure. Prior to blasts, tape was placed over the imaging cannula and a wetted custom paper cone was fitted to cover the head to allow reproducible positioning of the head, to allow free movement of the head, and to prevent potential quaternary damage from gun powder during blasts. Immediately following blasts, animals were removed from the restrainer and quickly repositioned and head-fixed in the imaging setup. This transfer process typically took <5 min. Calcium imaging was then performed for 100 sec for all mice at each of the following intervals: immediately following head-fixation after blast (0 min), and at ∼10 min (range, 6–15 min; mean, 10 min 45 sec), 20 min (range, 16–26 min; mean, 21 min 19 sec), 35 min (range, 29–46 min; mean, 35 min 32 sec), and 65 min (range, 57–76 min; mean, 65 min 28 sec) after blast/sham (Fig. 1E). Each animal underwent these procedures once a day for two consecutive days.
Calcium imaging data analysis
For each imaging session, the 100 sec videos for the five time points following the blast/sham-exposure (0, 10, 20, 35, and 65 min after blast/sham exposure and head re-fixation) were first concatenated, and then processed as one video (500 sec total duration) to provide a continuous registration of regions of interest (ROIs) across all time points. Each of the 100 sec baseline videos taken immediately prior to blast or sham exposure was processed separately, because it was difficult to accurately align the ROIs between the videos taken before and after the blast, when the mice were transferred from the imaging setup to the COBIA device. Ca2+ imaging videos were first filtered with a homomorphic filter to enhance contrast, and then motion corrected using a rigid correction as previously described.30 Circular ROIs with a radius of 6 pixels (corresponding to 7.8 μm) were manually selected as being centered on the cell body on a maximum projection image across all video frames using a code we developed (https://github.com/HanLabBU/mTBI_Ca_Hippocampus).
Raw calcium traces were extracted for each identified ROI as the mean intensity of all pixels within a given ROI, where the absolute value of each pixel intensity was determined from the motion-corrected videos without the homomorphic filter. The raw calcium intensity for each cell was used to determine whether a cell was elevated or suppressed. A probability density estimate (PDE) of raw pixel intensities was calculated using MATLAB 8.1 (MathWorks Inc., Natick, MA, 2013) for each cell for the imaging periods immediately after blast (Period 1) and 65 min (Period 5) after blast. The difference in mean pixel intensity between Period 5 and Period 1 for each cell was calculated. To establish a confidence interval, pixel intensity differences were pooled across all sham mice for all imaging sessions. A 95% confidence interval was determined from the 0.025 and 0.975 quantiles of these pooled differences. The width of this confidence interval was applied to the unity line, which would suggest no difference between Period 1 and Period 5. Differences in mean pixel intensity that fell outside of this confidence interval for both blasted and sham-exposed mice were classified as elevated (Period 1 > Period 5) or suppressed (Period 1 < Period 5).
For additional analyses, we also calculated normalized Ca2+ traces as 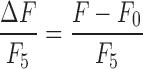 , where F is the linearly detrended instantaneous fluorescence intensity, F0 is the linearly detrended mean fluorescence for a given imaging period, and F5 is the mean fluorescence for the final imaging period (Period 5, ∼65 min after blast). Binarized traces were then created from the normalized traces by setting the rising phase of calcium activation events to 1s, with 0s elsewhere along the binary trace. Specifically, normalized Ca2+ traces (
, where F is the linearly detrended instantaneous fluorescence intensity, F0 is the linearly detrended mean fluorescence for a given imaging period, and F5 is the mean fluorescence for the final imaging period (Period 5, ∼65 min after blast). Binarized traces were then created from the normalized traces by setting the rising phase of calcium activation events to 1s, with 0s elsewhere along the binary trace. Specifically, normalized Ca2+ traces (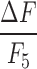 ) were low-pass filtered with a sixth order Butterworth filter at a cutoff of 2 Hz. A global standard deviation (SD) was calculated across all concatenated videos (SDall) for each trace. Calcium activation events were identified as peak values that were at least 3 SDall above the mean for each imaging period. Each of these peaks was counted to determine an integer value of the calcium event rate. From each peak point, the data points prior to the peak with a positive derivative were set to a binary 1, with all other data points were assigned as 0s. To determine if a cell showed a significant decrease or increase in dynamic activity after blast, a bootstrapping method was used. The percentage of time a cell was active was measured during a 10 sec window that was randomly permuted 1000 times from the trace over the course of each period to determine how active the cell was for that time period. These bootstrapped samples for Period 1 and Period 5 were compared for each cell using a two tailed Wilcoxon rank sum test. Cells that were statistically different (p < 0.05) between the two time periods were classified as showing increased activity or decreased activity, where decreased cells were those with less activity in Period 1 than in Period 5, and increased cells were those with more activity in Period 1 than in Period 5.
) were low-pass filtered with a sixth order Butterworth filter at a cutoff of 2 Hz. A global standard deviation (SD) was calculated across all concatenated videos (SDall) for each trace. Calcium activation events were identified as peak values that were at least 3 SDall above the mean for each imaging period. Each of these peaks was counted to determine an integer value of the calcium event rate. From each peak point, the data points prior to the peak with a positive derivative were set to a binary 1, with all other data points were assigned as 0s. To determine if a cell showed a significant decrease or increase in dynamic activity after blast, a bootstrapping method was used. The percentage of time a cell was active was measured during a 10 sec window that was randomly permuted 1000 times from the trace over the course of each period to determine how active the cell was for that time period. These bootstrapped samples for Period 1 and Period 5 were compared for each cell using a two tailed Wilcoxon rank sum test. Cells that were statistically different (p < 0.05) between the two time periods were classified as showing increased activity or decreased activity, where decreased cells were those with less activity in Period 1 than in Period 5, and increased cells were those with more activity in Period 1 than in Period 5.
Each cell was determined to be either suppressed, elevated, or consistent in its basal levels, as well as determining whether the calcium event rate was increased, decreased, or unmodulated by comparison of time Period 1 to time Period 5 for each cell. Each of these classifications was quantified as the fraction of total neurons within each mouse, and those values were used for population analyses between sham and blasted mice.
Immunocytochemistry
One week following the second blast, mice were perfused with 0.9% NaCl heparinized saline followed with 4% paraformaldehyde in 0.1M phosphate buffer (PB) pH 7.4 (containing sodium phosphate monobasic [Sigma, Cat# S0751], and sodium phosphate dibasic [Sigma, Cat# S3264]). Brains were post-fixed overnight at 4°C, and then cryoprotected in 30% sucrose in PB overnight at 4°C. Brains were then blocked from bregma −0.94 mm to −2.92 mm using an adult mouse brain slicer matrix (BSMAS001-1, Zivic Instruments, Pittsburgh, PA), and sectioned at 40 μm using a freezing sliding microtome (Reichert Jung Inc. Depew, NY). Brain slices were stored at −20°C in cryoprotectant (containing 30% sucrose [Sigma, Cat#S0389], 30% ethylene glycol [Fisher Scientific, Cat#BP230-1], and 1% polyvinylpyrrolidone [Fisher Scientific, Cat#BP431-500]) in Tris-buffered saline pH 7.6 (0.4 M Trizma HCl [Sigma, Cat# T-3253], 0.01 M Trizma base [Sigma T-1503], and 0.15 M sodium chloride (Fisher Scientific, Cat# BP358-212]) for subsequent immunostaining.
Free-floating sections were first mounted and dried onto Colorfrost Plus slides (Fisher Scientific, Waltham, MA). Mounted slices were rehydrated in PB prior to incubation with 5% normal donkey serum (Jackson ImmunoResearch Laboratory, Inc. West Grove, PA) in PB containing 0.3% TritonX100 (PBTx). Slices from each animal were immunostained overnight at 4°C using either mouse monoclonal anti-glial fibrillary acidic protein (GFAP 1:250, Clone No. N206 A/8, UC Davis/NIH NeuroMab Facility Cat# 75-240, RRID:AB_10672299), or rabbit anti-Iba1 (1:1000, Wako, Catalog No. 019-19741), followed by the appropriate fluorescently conjugated secondary antibodies, donkey anti-mouse Cy3 (1:500, Jackson Immuno Research, Catalog No. 715-165-150) or donkey anti-rabbit 647 (1:500, Jackson Immuno Research, catalog No. 711-605-152), for 2 h at room temperature. These primary antibodies have been previously characterized in the mouse brain.32,33 Slices were then cover-slipped in Gelvatol (10% polyvinyl alcohol [Sigma, Cat#P8136], 20% glycerol, Sigma G-9012, 0.02% sodium azide [Fisher Scientific, Cat#-227-100], and 0.2 M Tris [Amresco, Cat#0497], pH 8.5]) or Vectashield with 4′6-diamindino-2-phenylindole (DAPI) (Vector Labs, Burlingame, CA) to visualize nuclei.
Sections were imaged using a Nikon Eclipse motorized microscope (Nikon Instruments, Inc.) at 10x. ImageJ software (National Institutes of Health, Bethesda, Maryland, USA) was used to analyze the stained slices.
Immunocytochemistry quantification
The percent area occupied by GFAP was analyzed using ImageJ software. Immunostained images were first inverted so that signal appeared black and then binarized using the Iterative Self-Organizing Data Analysis Technique (ISODATA) algorithm in ImageJ. Five randomly selected cells per slice, and three slices per hemisphere with visible somata were analyzed for each mouse. An area large enough to enclose a single cell was used for all analyses. The area fraction, defined as the percent coverage (percent area) of immunoreactivity within each area, was obtained for each binary image using the area fraction selection from the ImageJ measure plugin. It was expected that evidence of glial activation would be reflected by a greater percent area because the cells would be larger in size and therefore occupy a greater percentage of the area. Values for percent area were obtained for individual cells (n = 5 cells per hemisphere). To account for any differences caused by the presence of the imaging window (ipsilateral), the hemisphere opposite to the window (contralateral) was analyzed separately as an internal control.
Two broad classifications of Iba1 phenotypes were chosen for evaluation: ramified and unramified. Manual counts of Iba1 positive cells identified as ramified or unramified were performed within the same ROI applied to the hemisphere ipsilateral to the imaging window as well as to the contralateral hemisphere.
GFAP and Iba1 immunoreactivity were first evaluated using a paired t test (p < 0.05) for the ipsilateral and contralateral hemispheres for sham (n = 5) and blast exposed (n = 6) animals. A two-tailed unpaired t test (p < 0.05) was then performed to compare the results from sham and blasted animals. Data are presented as mean ± SD. Statistical analysis of immunocytochemistry was performed using SPSS (IBM Corp. Released 2016. IBM SPSS Statistics for Windows, Version 24.0, Armonk, NY)
Results
Wide field imaging of hippocampal CA1 neurons in awake mice upon mild traumatic brain injury (mTBI)
To examine the acute effects of mTBI on individual hippocampal neurons, we performed Ca2+ imaging using wide-field fluorescence microscopy (Fig. 1A) in awake, head-fixed mice,30 before and immediately after a mild cranial blast injury. The mTBI was produced with a COBIA, which directs blast waves onto the freely moving head of a mouse (Fig. 1B), adapted from that of Kuehn and coworkers.20 We note that although the mouse bodies were restrained in the COBIA, their heads moved freely to ensure the success of the blast injury model.
We calibrated our COBIA device using a sensor that had a 500 kHz resonant frequency (100 kHz maximum sampling frequency). With our calibration setup, the blast peak overpressure reached 1399 ± 252 kPa (mean ± SD, n = 5 tests) at a single time point (∼2 μs) (Fig. 1C). To compare to the measurements reported in the study by Kuehn and coworkers, we filtered the pressure trace at 12 kHz and obtained a transient overpressure of 322 ± 92 kPa (mean ± SD), which was slightly below the maximum overpressure that they reported.20 To compare with the shock tube device reported by Goldstein and coworkers, we filtered the blast pressure trace at 2 kHz, and detected a peak pressure of 178 ± 26 kPa, which is above the 77 kPa that they reported.11 The severity of blast injury is typically attributed to the magnitude of the pressure produced (peak blast overpressure), and the duration of peak overpressure.5,19,20,31 Because none of our blasted animals lost consciousness or exhibited noticeable abnormal behavior immediately following blast or the week after, and the general agreement of the blast overpressure of our experiment with prior studies, we consider our COBIA model to be mTBI.11,20
Several weeks prior to testing, mice were surgically injected with the AAV9-syn-GCaMP6f virus encoding the genetically encoded Ca2+ sensor GCaMP6f into the CA1, and implanted with a custom imaging window over the injection site. Upon complete recovery from the surgeries, mice were blasted and imaged each day on two consecutive days (Fig. 1D). Prior to each blast, we first imaged spontaneous Ca2+ dynamics for 100 sec in mice that were awake and head-fixed under the imaging setup. We then transferred the mice to the COBIA, where they received a single cranial blast with free head movement. Immediately after the blast, we transferred the animals back to the imaging setup, and imaged them awake and head-fixed for 100 sec every 10–20 min until ∼1 h after the blast (Fig. 1E). Because the COBIA also produced a transient loud noise, we performed a sham study in mice that were prepared in an identical fashion, but positioned next to the COBIA during the blast portion of the protocol. Sham-exposed mice were also allowed free head movement, and thus they experienced the blast related noise without being exposed to any blast pressure waves. Each mouse was blasted and imaged each day on two consecutive days (n = 4 blasted mice) or sham-exposed on each of the two consecutive days (n = 5 sham-exposed mice).
Blast exposure induced both sustained and transient intracellular Ca2+ changes in a large number of individual neurons
The wide-field imaging system, equipped with a sCMOS camera and a 10x objective lens, allowed us to image over a 1.343 x 1.343 mm2 brain area containing hundreds of individual neurons in each mouse (Fig. 2A). In one representative session, we recorded 554 neurons before blast, and 467 neurons after blast (Fig. 2B). The difference in the number of neurons identified between the pre- and post- blast sessions in the same mice was because the blast procedure was conducted in mice whose heads were not restrained. Thus, the same neurons often could not be easily registered before and after the blast. However, because we imaged the same brain area from the same imaging window, a majority of cells recorded before and after the blast were expected to come from the same neuronal population.
FIG. 2.

Blast altered intracellular Ca2+ dynamics in individual hippocampal neurons. (A) Maximum projection of GCaMP6 fluorescence intensity across all imaging frames during a 100 sec long recording period for a representative blasted mouse (left) and a representative sham noise-exposed mouse (right). Post-blast/sham Period 1 was immediately after the blast, and post-blast/sham Period 5 was ∼65 min after the blast. Images were contrast enhanced using the same contrast scaling factor for both Periods 1 and 5. (B, C) Ca2+ traces (normalized by the mean fluorescence in Period 5) of individual neurons identified in a representative blasted mouse (B) and in a representative sham noise-exposed mouse (C), before and after blast. (i) Traces were sorted by the mean fluorescence in the pre-blast or post-blast Period 1 respectively. Sorting order of a given neuron from Period 1 was maintained across the remaining blast periods. (ii) Representative Ca2+ traces of specific representative neurons with different levels of intracellular Ca2+, as indicated by the colored asterisks in (i).
Before the blast, most neurons exhibited transient and dynamic GCaMP6f signals on a sub-second time scale, consistent with Ca2+ changes associated with neural activities.21–26,34–36 These Ca2+ transients, on the sub-second time scale, were sparse over time, and were present in cells across the entire imaging field (Supplementary Video 1). The overall dynamics of these patterns were consistent with those reported previously for spontaneous CA1 Ca2+ transients associated with spiking.27–30 Immediately after the blast, we observed a drastic change in the pattern of GCaMP6f signals (Fig. 2A). A substantial fraction of cells exhibited a sustained reduction in their sustained basal intracellular Ca2+ levels, as well as a reduction in the frequency of sub-second Ca2+ transients. In addition, we found a very small fraction of cells that exhibited sustained elevation of basal intracellular Ca2+ levels after blast, which appeared constantly bright in the imaging field (Supplementary Video 1). Changes in both the sub-second Ca2+ transients and the slow sustained basal intracellular Ca2+ levels slowly recovered over time. In most blast sessions, patterns of Ca2+ dynamics observed at the end of the recording session, ∼1 h after the blast, were visually indistinguishable from that observed during the pre-blast baseline period. However, there was one blast session on the 2nd day (out of a total of eight sessions in four animals), where Ca2+ dynamics remained attenuated and sparse 1 h after blast.
In sham mice, we did not detect noticeable changes in either the sub-second Ca2+ transients or the slow sustained basal intracellular Ca2+ levels (Fig. 2C and Supplementary Video 2). Because it took ∼5 min to transfer animals from the COBIA blast device back to the imaging setup, we could not determine the precise time when the changes in Ca2+ dynamics were first initiated by the blast.
Mild blast injury reduced basal intracellular Ca2+ levels in most neurons
Because we could not easily register a one-to-one match among imaged neurons before and after the blast, we first examined population neural activity observed ∼65 min after blast (Period 5) versus that observed during the pre-blast period, as those seemed qualitatively similar. We compared the histogram distributions of mean basal Ca2+ levels across all neurons for each mouse between the pre-blast period and Period 5, and found no statistically significant difference (Wilcoxon rank sum test, p > 0.05), consistent with our visual evaluation (Supplementary Video 1). Then, to examine the immediate effects of the blast at the individual neuron level, we compared the first 100 sec long imaging session immediately after the blast (Period 1) and the last 100 sec imaging session ∼65 min after the blast (Period 5) when activity largely recovered to that of pre-blast period in most cases.
To estimate the slow, sustained, basal intracellular Ca2+ changes induced by blast, we constructed a PDE for each neuron's fluorescence intensity in Period 1 and Period 5 (Fig. 3). The fluorescence intensity of a given neuron sampled at each image frame was used to calculate the PDE. The mean of the PDE represents the overall basal intracellular Ca2+ levels averaged over the given 100 sec long imaging period, and the distribution of the PDE estimates the temporal variation within the imaging period.
FIG. 3.

Blast reduced intracellular basal Ca2+ levels. (A) Left: spatial distribution of cells whose intracellular Ca2+ levels were elevated (red), suppressed (blue), or unmodulated (gray) in a representative blasted mouse. Middle: Ca2+ traces for the three representative neurons indicated in the spatial map. Right: the probability density estimates (PDE) of the neurons (shown in the middle) for imaging Period 1 and Period 5. The short vertical lines above the density distributions correspond to the mean of each respective density and highlight the shift in intracellular calcium levels between the imaging sessions. A.U., arbitrary units, as measured by 16 bit pixel intensities. (B) Spatial distribution map, Ca2+ traces, and probability density estimates for a representative sham noise-exposed mouse. (C) The mean fluorescence intensity of all cells during Period 1 plotted against the mean fluorescence intensity during Period 5 for the representative blasted mouse (red) and the representative sham-blasted mouse (blue) shown in A and B respectively. Dotted black line is the unity line. (D) All neurons recorded in all blast sessions (red, 2781 neurons) and all sham sessions (blue, 3259 neurons) during Period 1 plotted against Period 5. Dotted black line is the unity line, and dotted gray lines indicate 95% confidence interval used to classify elevated and suppressed cells. (E) The fraction of cells that were suppressed, elevated, or unmodulated, averaged across mice, for the first blast (day 1) and the second blast (day 2). More neurons were suppressed after the second blast (*p < 0.05, Wilcoxon rank sum). Error bars represent the quartile range.
In the blast group, the PDEs of most neurons during Period 1 were drastically shifted toward smaller intensity values when compared with those during Period-5 (Fig. 3Aii), suggesting that blast reduced basal intracellular Ca2+ levels in most neurons. Interestingly, the PDEs of a small portion of neurons (a representative one is shown in Fig. 3Ai) were shifted to higher intensity values, suggesting that blast increased the intracellular Ca2+ levels of these neurons. For the few cells that showed little change in the overall mean intensity, they exhibited a small shift in the distribution, suggesting that these neurons exhibit more variable changes in intracellular Ca2+ levels than sham-blasted mice (Fig. 3Aiii compared with Fig. 3B).
We then calculated the mean of the PDEs of all neurons recorded in this blast session, and compared between Period 1 and Period 5 (Fig. 3C). As a population, basal intracellular Ca2+ levels of most neurons in blasted mice were above the unity line, demonstrating that blast reduced the intracellular Ca2+ levels in most neurons. A few neurons were below the unity line, representing the neurons whose intracellular Ca2+ levels were elevated by the blast. This phenomenon was consistent across most blast sessions (Fig. 3D). Our estimation of blast-induced effects represent a conservative measure, as it is possible that blast-induced changes may remain at some level during Period 5 that is still different from the pre-blast period, though not statistically significant. In addition, the comparison between Period 1 and Period 5 allows us to assess how individual neurons recover over time after each blast. Across sham exposure sessions, the mean basal Ca2+ levels remained on the unity line, which confirmed that sham exposure did not alter intracellular basal Ca2+ levels (n = 10 sessions in five mice).
To further quantify the changes in individual neurons, we estimated the variation of the PDEs across all neurons recorded in the sham group where blast did not alter the mean basal calcium levels. We then used the 95% confidence interval of the width of this distribution as a threshold to determine significance along the unity line. If a neuron's mean fluorescence intensity during Period 1 was significantly smaller than its intensity during Period 5, we classified this neuron as significantly suppressed by the blast. If a neuron's mean fluorescent intensity during Period 1 was significantly larger during Period 5, we classified this neuron as significantly elevated by the blast. If a neuron's mean fluorescence intensity was within a 95% confidence interval between Period 1 and Period 5, it was considered to have a consistent basal calcium level. For the blasted mice, 55.5 ± 24.8% of cells were suppressed, whereas for sham mice, only 5.83 ± 9.26% of cells were suppressed for the second blast day (Wilcoxon rank sum W = 15, nb = 4 ns = 5 mice, p = 0.016, two tailed). It is interesting to note that the percentage of cells in this population that were suppressed doubled between the blasts, going from 20% of the cells on day 1 to 55% of the cells on day 2 of blasting (Fig. 3E).
Together, these results suggest that mild blast caused widespread, sustained reductions in basal intracellular Ca2+ levels in a large fraction of individual hippocampal neurons. A larger fraction of hippocampal neurons was impacted during the second blast compared with the first blast. Although the intracellular Ca2+ levels in most neurons were reduced following blast, a small fraction of cells exhibited long-lasting increase in intracellular Ca2+. These changes in intracellular Ca2+ recovered on a timescale of minutes, and returned to similar patterns as that observed before blast. Considering the importance of intracellular Ca2+ in cell signaling pathways and gene expression, such a shift in basal intracellular Ca2+ levels over a time course of minutes could lead to longer-term changes in membrane excitability, synaptic remodeling, and structural plasticity.
Mild blast injury reduces individual neuronal activity measured as Ca2+ transients
In addition to altering slow basal intracellular Ca2+ levels, blast also impacted the faster, sub-second time scale Ca2+ transients, known to be correlated with neural activity.21–26,34–36 Immediately after blast, many neurons were largely inactive and exhibited few Ca2+ transients for an extended period of time (Fig. 2B and 4A post-blast 1–2 and Supplemental Video 1). Ca2+ transients recovered gradually, and in most blast sessions by the last recording session (∼1 h after blast), they were similar to the pre-blast state. In the sham group, Ca2+ transients showed little change following exposure to the sound of the blast, thus confirming that noise alone did not cause the changes in Ca2+ transient events recorded in the blast group.
FIG. 4.

Blast decreased calcium transient event rates in individual neurons. (A, B) Sub-second Ca2+ transients, events closely related to neural activity, plotted for individual neurons for different time periods after blast in a blasted mouse (A), and a sham noise-exposed mouse (B). Cells were sorted using the same order as shown in Figure 2. Transient Ca2+ events were calculated using ΔF/F5 rather than F/F5 to highlight transient events occurring on top of the slower basal changes in intracellular Ca2+ levels. (C) The average number of calcium events before and after the first blast (day 1) and the second blast (day 2), plotted for the six recording periods each day. (*p < 0.05, Wilcoxon rank sum). Error bars represent the quartile range. (D) The fraction of neurons exhibiting decreased, increased, and unmodulated Ca2+ event rates. (*p < 0.05, Wilcoxon rank sum). Error bars represent the quartile range. (n = 4 blast mice, and 5 sham mice).
To quantify the effect of blast on Ca2+ transients for individual neurons, we calculated the frequency of the sub-second long Ca2+ transients. To dissociate the sub-second time scale of Ca2+ transients from the sustained long-lasting basal Ca2+ level shifts, we first calculated the changes in fluorescence from the mean fluorescence (ΔF) for each neuron within a given 100 sec long imaging period. We then normalized ΔF to the mean of the last imaging period (F5) to highlight any change in the frequency of Ca2+ transients (Fig. 4A, B). This normalization allowed optimal detection of changes in transient Ca2+ events, without any bias from effects of the basal Ca2+ level changes. To calculate the rate of Ca2+ transient events, we first converted normalized Ca2+ traces to binary traces to identify the rising phase of GCaMP6 signals (1s), and counted the number of Ca2+ events occurred for each 100 sec recording time period, before and after each blast.
We found that mean Ca2+ event rates of all neurons simultaneously recorded in each imaging session before the blast were comparable between the blasted and the sham groups (blast: 2.20 ± 0.19 events, mean ± SD, n = 4 mice; sham: 2.28 ± 0.42 events, mean ± SD, n = 5 mice). Ca2+ event rate dropped significantly to 0.47 ± 0.3 events per 100 sec for the blast group, compared wity 1.45 ± 0.47 events for the sham group (Wilcoxon rank sum W = 35, nb = 4 ns = 5 mice, p = 0.016, two tailed). Ca2+ events rates then quickly recovered over the subsequent tens of minutes following blast (Fig. 4C, p > 0.05). On the 2nd day, the average calcium event rate returned to approximately two events per 100 sec for both sham and blast groups before the blast. Following the second blast, the event rate again dropped sharply and recovered more slowly on the 2nd day compared with the 1st day, although this pronounced decline was not statistically significant.
To determine the fraction of cells in each mouse that exhibited a change in Ca2+ event rates, we compared the event rate in Period 1 with that in Period 5 for each cell. A bootstrapping method was used to determine whether the frequency of Ca2+ transients for each neuron was significantly different between Period 1 and Period 5, to classify neurons as significantly increased, decreased, or unmodulated. We found that following the first blast, 58.5 ± 20.3% (mean ± SD) of cells showed a decrease in response to blast, significantly different from the 26.5 ± 6.3% of cells upon sham exposure (Wilcoxon rank sum W = 16, nb = 4 ns = 5 mice, p = 0.032, two tailed). In addition, we found that 44.6 ± 11.3% of neurons in sham mice showed an increase in the frequency of Ca2+ transients over time, but only 13.6 ± 11.8% of neurons in the blasted mice exhibited such increase (Wilcoxon rank sum W = 35, nb = 4 ns = 5 mice, p = 0.0159, two tailed). These proportions were different for the 2nd day of blasting, with the number of calcium events being more comparable to that for the sham mice (Fig. 4D).
Heterogeneous effects of blast on intracellular Ca2+ dynamics at different time scales
Interestingly, when comparing changes between basal intracellular Ca2+ levels and the frequency of Ca2+ transients, we failed to find a clear relationship (Fig. 5). When plotting the full population of neurons comparing the difference in the average number of Ca2+ events with the difference in the basal Ca2+ levels, we found no linear relationship between the two for either blasting days (blast, R2 = 0.1141 and 0.0031 for the first and second blast respectively; sham, R2 = 0.0371 and 0.0206 for the first and second sham exposure respectively). Sham mice had a tighter distribution of the average basal Ca2+ levels, whereas blast mice showed a broader distribution of differences in basal Ca2+ levels. Interestingly, a few neurons in the sham group exhibited changes throughout the full range of basal Ca2+ level changes and the frequency of Ca2+ transient changes, suggesting that blast-induced changes are physiologically achievable. These results demonstrate heterogeneous effects of blast on both basal and transient Ca2+ changes, which could involve different cellular mechanisms, and also confirmed that our imaging technique and analysis methods can independently detect changes in transient sub-second Ca2+ events and sustained basal shift in intracellular Ca2+ levels.
FIG. 5.

Blast induced changes in baseline calcium levels are independent of the changes in Ca2+ event rates. Each dot representing a single cell is plotted comparing the change in the mean baseline Ca2+ level with the change in the number of Ca2+ events over the 100 sec recording period immediately after blast. Day 2 showed a greater divergence in the baseline mean, although both days showed similar cellular responses in the change in Ca2+ events. Although the range of values was observed in both blast and sham conditions was similar, the fraction of cells exhibiting stronger deviations from the center was much larger in the blast condition than the sham condition. Large colored dots represent the mean of each value across mice for either the sham or the blast group, and error bars represent the quantile range spanning 95% of the data.
No significant differences in glia immunoreactivity 1 week following repetitive blasts
Immunocytochemistry was performed 1 week after the second blast to identify morphological changes in cytoarchitecture and to evaluate the status of glia (Fig. 6). We found no significant differences in the percent area occupied by GFAP-labeled astrocytes between sham and blasted groups, either in the ipsilateral area directly underneath the imaging window (t = −0.404, df = 10, p = 0.695), or in the contralateral intact hemisphere (t = −0.876, df = 6.698, p = 0.411). We observed a slight increase in the area occupied by GFAP in the ipsilateral side underneath the imaging window, compared with the contralateral intact hemisphere in both blast and sham groups (sham: n = 5 t = 2.167, df = 4, p = 0.096; blast: n = 6, t = 1.580, df = 6, p = 0.165), consistent with local immune responses to imaging implants. In addition, no significant difference was observed in Iba1 phenotypes between sham and blasted groups under the imaging window (t = −0.329, df = 9, p = 0.749), as well as in the contralateral hemisphere (t = −1.299, df = 6.475, p = 0.238). Similar to GFAP-labeled astrocytes, we observed a greater percentage of unramified Iba1-positive microglia in the ipsilateral area directly underneath the imaging window compared with the contralateral hemisphere, in both sham and blasted animals, although the difference was not significant (sham: n = 5 t = 1.693, df = 4, p = 0.166; blast: n = 6, t = 1.829, df = 5, p = 0.127).
FIG. 6.

No significant differences in glial fibrillary acidic protein (GFAP) and ionized Ca2+ binding adaptor molecule 1 (Iba1) immunoreactivity following blasts. (A, B) 4′6-diamindino-2-phenylindole (DAPI) labeled coronal sections from a representative sham-exposed mouse (Ai.-iii.) and a representative blasted mouse (Bi.-iii.). DAPI = 4′,6-Diamidino-2-Phenylindole. GFAP = glial fibrillary acidic protein. CA1, Cornu Ammonis 1; SM, stratum moleculare; SP, stratum pyramidale. SO, stratum oriens. (C,D) GFAP immunofluorescence (C) and Iba1 immunofluorescence (D) from a sham mouse (top) and a blast exposed mouse (bottom), ipsilateral (i., iii.) and contralateral to the imaging window (ii., iv.). (E) No significant difference in the percent area of GFAP immunofluorescence was observed between sham and blast exposed animals, either ipsilateral or contralateral to the imaging window. (F) No significant difference in the percentage of unramified cells positive for Iba1 was observed between sham and blast exposed animals ipsilateral or contralateral to the imaging window.
Discussion
To examine the immediate functional effect of mild blast, we integrated a recently developed in vivo wide-field imaging technique to monitor intracellular Ca2+ changes in large numbers of individual hippocampal neurons in unanesthetized mice, with an mTBI COBIA model that produced no drastic anatomical pathology. We found a significant effect on neural function after exposure to a single blast or repeated blasts. Blast produced heterogeneous effects on different neuron populations, across different time scales. Basal Ca2+ levels, on the time scale of minutes, were reduced in most neurons, suggesting that biochemical signaling may be affected. Transient Ca2+ events, on the sub-second time scale, were reduced in a large fraction of neurons, suggesting an impact on neural activity, likely caused by changes in membrane excitability and/or synaptic signaling. Blast-induced transient Ca2+ activity changes showed little correlation with the shift of basal intracellular Ca2+ levels, suggesting different modes of impact on membrane biophysics and synaptic processing versus biochemical pathophysiology of the blast. The observed functional changes over the time course of tens of minutes in the absence of anatomical pathology may be directly relevant to the immediate cognitive impact of blast and may contribute to the long-lasting cellular changes. In addition, this study also highlights a novel technology platform that enables simultaneous analysis of a large number of individual neurons immediately after blast injury.
Blast model
The severity of primary blast injury is typically attributed to the peak blast overpressure, duration of peak overpressure, extent of head movement, distance from the blast, and mortality rate.5,19,20,31,37,38 However, major differences in the construction of the devices used to generate the blasts,39–41 administration of anesthetics,42–44 types of animals used,45 their orientation with respect to the blast,46,47 and their recovery post-blast can significantly affect injury outcome. A consensus to calibrate blast intensity based on the sampling frequency analyzed has not been reported to our knowledge, and the reported pressure measurements vary widely depending on the experimental setups. Kuehn and coworkers20 used a sensor with a 60 kHz resonant frequency and collected data at 333 kHz to obtain the peak pressure of 517 kPa using the same experimental setup that we adapted for the present study, whereas Goldstein and coworkers11 reported a peak pressure of only 77 kPa using a shock tube. Although our calibration results are in general agreement with these studies reporting mild blast injuries, future studies focused on standardizing calibration pressures would be beneficial for cross-study comparisons of blast-related injuries. Although we had a very high blast overpressure (1399 ± 252 kPa), its very short duration (∼2 μs) probably contributed to the mild nature of the blast.
Evaluation of astrocytes and microglia immunoreactivity
Although morphological changes used to infer the status of glial activation resulting from blasts have been reported, particularly following moderate and severe injuries, it is known that functional impairments can exist in the absence of morphological changes.8 For example, Pun and coworkers48 reported acute microglial activation 24 h following exposure to a mild blast, which was then absent 4 and 7 days post-blast, likely because of compensatory upregulation of reparative genes following blast exposure. Consistent with Pun and coworkers, we did not observe significant differences in astrocytes or microglia immunoreactivity 1 week following the final blast exposure. However, we cannot rule out the possibility that transient changes in glial morphology occurred prior to the subacute time point examined. Future studies are warranted to assess the temporal characteristics of glia morphologies and how the dynamic nature of glia may contribute to the functional responses following blast. Using the same experimental technique reported here, the acute changes of intracellular calcium in glia could also be studied in response to blast, as in vivo calcium imaging in glia has been performed previously.49
Neuronal functional changes
To our knowledge, we report the first in vivo evidence of how neurons in unanesthetized brains responded to blasts within minutes (∼ 5 min) after blast exposure. Although in vivo imaging has been utilized to examine a variety of neural networks involved in various behaviors,22,49–55 recent improvements in wide-field optical imaging allow simultaneous recording of cellular responses from hundreds to thousands of individual neurons, over relatively large brain areas, providing the opportunity to obtain finer details regarding the dynamic features of large neural networks.30,56 Using in vivo imaging in a blast injury rodent model, we identified acute changes in a large fraction of neurons within 10s of seconds, and the extended effects over the time course of minutes, which recovered after ∼1 h. Even though the activity of most neurons was drastically reduced, a small number of neurons exhibited striking elevations in intracellular calcium. Although the functional relationship between the intracellular Ca2+ changes at both the modulatory and synaptic time scales is unclear, future studies using Ca2+ imaging will likely establish their role in the manifestation of blast pathology.
At a population level, we found an overall loss of Ca2+ activation events in the hippocampus after blast, suggesting that blast led to a network state where the neurons are less active. This reduced activity may be related to the temporary loss of memory or confusion often observed in patients experiencing head trauma.57 Future Ca2+ imaging studies may provide important links to the mechanisms underlying memory deficits related to brain injuries.
Potential causes of the mTBI-induced changes in intracellular Ca2+ levels
Blast-induced changes in sustained basal intracellular Ca2+ levels on the order of minutes to 10s of minutes are indicative of modulation of biochemical, cellular signaling cascades, whereas the change in the frequency of sub-second Ca2+ transients is suggestive of altered spiking patterns that are likely associated with changes in membrane biophysics or synaptic mechanisms. Although the sub-second transient Ca2+ changes can be explained by changes in neuronal activity patterns, it is still unclear how blast induces sustained shifts in basal intracellular Ca2+ level. It is possible that the reduction in basal intracellular Ca2+ levels could arise from spreading depression.58 However, this does not seem likely, in that a small number of cells with increased Ca2+ levels were interspersed with those with decreased Ca2+ levels.
Although many studies indicated that blast induces a long-term increase in intracellular Ca2+ levels over the course of days,59–62 a recent study also showed immediate suppression of neuronal activity lasting for 5–20 min followed by increased cortical activity by 2 h post-injury upon sustained cortical compression in the barrel cortex.63 Our results showed an overall suppression in a large fraction of the neuronal population immediately after the blast within 1 h, indicating that blast injuries could cause a dramatic shift in the Ca2+ level on various time scales. The suppression and the increase in Ca2+ levels are likely caused by distinct cellular mechanisms. For example, it has been suggested that there can be a switch in AMPA receptor composition from Ca2+ impermeable glutamate receptors (GluRs) to Ca2+ permeable GluR receptors lacking GluR2 following mechanical injury in cortical neurons in vitro,64,65 which could explain the long-term increase in basal levels of intracellular Ca2+. In contrast, it has also been suggested that there is a decrease in GluR1 expression in the cortex 15 min after closed head injury,66 that could lead to the acute suppression of intracellular Ca2+ observed here. Further, long-lasting loss of Ca2+ permeable glutamate N-methyl-d-aspartate (NMDA) receptor function, following short-lived (< 1 h) hyperactivation may also contribute to the reduction in intracellular Ca2+.67 In a lateral fluid percussion model in the hippocampus there was decreased net synaptic efficacy and reduced excitatory postsynaptic potentials in CA1, whereas both evoked and spontaneous miniature inhibitory potentials were larger in injured brains.68 These results suggest that increased activation of NMDA receptors and Ca2+ permeable AMPA receptors may be responsible for the long-term increased basal Ca2+ levels that have been reported previously, whereas enhanced spontaneous inhibitory activity in area CA1 following blast may cause the immediate reduced levels of Ca2+ as we observed. It is also possible that direct alterations in GABAA receptors may contribute to inhibitory dysfunction after trauma.69
Interestingly, a recent in vitro study has shown that simulated blast primarily affects calcium signaling in human astrocytes, in contrast to neurons, producing calcium waves that propagate through astrocytic networks via purinergic signaling.70 This suggests that astrocytes may also play a significant role in modulating Ca2+ levels in response to blast. It also raises the possibility that astrocytes may be involved in our present results even though we found no pathological changes in astrocytes. The imaging methods and reagents employed in the present study could easily be adapted to study blast induced changes in Ca2+ in astrocytes.
In addition to these potential synaptically based mechanisms, it is also possible that changes in the homeostatic regulation of Ca2+ may be involved in the changes in basal Ca2+ levels. Homeostatic regulation of Ca2+ in response to TBI has been shown to occur by a wide variety of mechanisms including: voltage and receptor activated Ca2+ channels; Ca2+ transporters; release and sequestration of Ca2+ from intracellular stores in the nucleus, mitochondria, and endoplasmic reticulum; intracellular Ca2+ binding proteins; and changes in the permeability of neuronal membranes to Ca2+.71 Changes in any of these homeostatic mechanisms could influence both the basal and sub-second transient Ca2+ changes in response to blast.
Quantitative analysis indicated that there was a complex relationship between the slower modulatory changes in basal Ca2+ levels and the sub-second scale Ca2+ dynamics. For example, cells with increased modulatory cytoplasmic levels of Ca2+ could have either increased, decreased, or unaltered levels of presumptive synaptic Ca2+ activity at the millisecond time scale. These different responses indicate there are multiple neuronal types and synaptic circuits being influenced by blast, and that several different signal transduction pathways are involved. Given that only a small population of hippocampal neurons had increased cytoplasmic levels of Ca2+ in response to blast, it will be important for future studies to determine why these neurons are unique. Further characterization of the specific cell types involved, their synaptic properties, and the signal transduction pathways being affected could form the basis for future treatments to prevent or treat neuronal blast injury. Given that synaptic processing requires a precise functional balance of inhibition and excitation, the increased and decreased levels of basal Ca2+ seen in response to blast may produce overall changes in network functionality. These changes in intracellular Ca2+ and their related signaling pathways may underlie the immediate cognitive effects of blast exposure, and contribute to longer-term anatomical pathologies.
Supplementary Material
Acknowledgments
X.H. acknowledges funding from the National Institutes of Health (NIH) director's new innovator award (1DP2NS082126), the National Institute of Neurological Disorders and Stroke (NINDS) (1R01NS087950-01), the Pew Foundation, the Alfred P. Sloan Foundation, Defense Advanced Research Projects Agency (DARPA) Young Faculty Award, Boston University Biomedical Engineering Department, and Boston University Photonic Center. K.R.H. is supported by a National Science Foundation Graduate Research Fellowship under Grant No. DGE-1247312. We thank members of the Han and Eldred Labs for technical support of the study and suggestions on the manuscript. K.R.H. and H.T. performed all data analysis. A.I.M. and S.B. performed surgical window procedures. A.I.M. and M.E.A., performed in vivo imaging. G.J.D. performed immunocytochemistry and aided in animal handling during blast procedures. B.N. and V.S. assisted with analysis methods. W.D.E, and X.H. supervised the study. All authors contributed to manuscript preparation.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Walker K.R., and Tesco G. (2013). Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front. Aging Neurosci. 5, 1–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maas A.I. (2007). Traumatic brain injury: simple data collection will improve the outcome. Wien. Klin. Wochenschr. 119, 20–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morganti-Kossmann M.C., Yan E., and Bye N. (2010). Animal models of traumatic brain injury: is there an optimal model to reproduce human brain injury in the laboratory? Injury 41, Suppl. 1, S10–13 [DOI] [PubMed] [Google Scholar]
- 4.Burgess P., Sullivent E., Sasser M., Wald M., Ossmann E., and Kapil V. (2010). Managing traumatic brain injury secondary to explosions. J. Emerg. Trauma Shock 3, 164–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenfeld J.V., McFarlane A.C., Bragge P., Armonda R.A., Grimes J.B., and Ling G.S. (2013). Blast-related traumatic brain injury. Lancet Neurol. 12, 882–893 [DOI] [PubMed] [Google Scholar]
- 6.Stuhmiller J.H. (1997). Biological response to blast overpressure: a summary of modeling. Toxicology 121, 91–103 [DOI] [PubMed] [Google Scholar]
- 7.Mayorga M.A. (1997). The pathology of primary blast overpressure injury. Toxicology 121, 17–28 [DOI] [PubMed] [Google Scholar]
- 8.Girgis F., Pace J., Sweet J., and Miller J.P. (2016). Hippocampal neurophysiologic changes after mild traumatic brain injury and potential neuromodulation treatment approaches. Front. Syst. Neurosci. 10, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lyeth B.G., Jenkins L.W., Hamm R.J., Dixon C.E., Phillips L.L., Clifton G.L., Young H.F., and Hayes R.L. (1990). Prolonged memory impairment in the absence of hippocampal cell death following traumatic brain injury in the rat. Brain Res. 526, 249–258 [DOI] [PubMed] [Google Scholar]
- 10.Gerberding J.L., and Binder S. (2003). Steps to Prevent a Serious Public Health Problem. Atlanta, GA: Centers for Disease Control and Prevention [Google Scholar]
- 11.Goldstein L.E., Fisher A.M., Tagge C.A., Zhang X.L., Velisek L., Sullivan J.A., Upreti C., Kracht J.M., Ericsson M., Wojnarowicz M.W., Goletiani C.J., Maglakelidze G.M., Casey N., Moncaster J.A., Minaeva O., Moir R.D., Nowinski C.J., Stern R.A., Cantu R.C., Geiling J., Blusztajn J.K., Wolozin B.L., Ikezu T., Stein T.D., Budson A.E., Kowall N.W., Chargin D., Sharon A., Saman S., Hall G.F., Moss W.C., Cleveland R.O., Tanzi R.E., Stanton P.K., and McKee A.C. (2012). Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 4, 134ra160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sajja V.S., Hubbard W.B., Hall C.S., Ghoddoussi F., Galloway M.P. and VandeVord P.J. (2015). Enduring deficits in memory and neuronal pathology after blast-induced traumatic brain injury. Sci. Rep. 5, 15075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kovacs S.K., Leonessa F., and Ling G.S. (2014). Blast TBI models, neuropathology, and implications for seizure risk. Front. Neurol. 5, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geddes D.M., LaPlaca M.C., and Cargill R.S., 2nd (2003). Susceptibility of hippocampal neurons to mechanically induced injury. Exp. Neurol. 184, 420–427 [DOI] [PubMed] [Google Scholar]
- 15.Atkins C.M. (2011). Decoding hippocampal signaling deficits after traumatic brain injury. Transl. Stroke Res. 2, 546–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hall R.C., Hall R.C., and Chapman M.J. (2005). Definition, diagnosis, and forensic implications of postconcussional syndrome. Psychosomatics 46, 195–202 [DOI] [PubMed] [Google Scholar]
- 17.Almeida–Suhett C.P., Prager E.M., Pidoplichko V., Figueiredo T.H., Marini A.M., Li Z., Eiden L.E., and Braga M.F. (2015). GABAergic interneuronal loss and reduced inhibitory synaptic transmission in the hippocampal CA1 region after mild traumatic brain injury. Exp. Neurol. 273, 11–23 [DOI] [PubMed] [Google Scholar]
- 18.Schwarzbach E., Bonislawski D.P., Xiong G., and Cohen A.S. (2006). Mechanisms underlying the inability to induce area CA1 LTP in the mouse after traumatic brain injury. Hippocampus 16, 541–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Svetlov S.I., Larner S.F., Kirk D.R., Atkinson J., Hayes R.L., and Wang K.K. (2009). Biomarkers of blast-induced neurotrauma: profiling molecular and cellular mechanisms of blast brain injury. J. Neurotrauma 26, 913–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuehn R., Simard P.F., Driscoll I., Keledjian K., Ivanova S., Tosun C., Williams A., Bochicchio G., Gerzanich V., and Simard J.M. (2011). Rodent model of direct cranial blast injury. J. Neurotrauma 28, 2155–2169 [DOI] [PubMed] [Google Scholar]
- 21.Chen T.W., Wardill T.J., Sun Y., Pulver S.R., Renninger S.L., Baohan A., Schreiter E.R., Kerr R.A., Orger M.B., Jayaraman V., Looger L.L., Svoboda K., and Kim D.S. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wachowiak M., Economo M.N., Diaz-Quesada M., Brunert D., Wesson D.W., White J.A. and Rothermel M. (2013). Optical dissection of odor information processing in vivo using GCaMPs expressed in specified cell types of the olfactory bulb. J. Neurosci. 33, 5285–5300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akerboom J., Chen T.W., Wardill T.J., Tian L., Marvin J.S., Mutlu S., Calderon N.C., Esposti F., Borghuis B.G., Sun X.R., Gordus A., Orger M.B., Portugues R., Engert F., Macklin J.J., Filosa A., Aggarwal A., Kerr R.A., Takagi R., Kracun S., Shigetomi E., Khakh B.S., Baier H., Lagnado L., Wang S.S., Bargmann C.I., Kimmel B.E., Jayaraman V., Svoboda K., Kim D.S., Schreiter E.R., and Looger L.L. (2012). Optimization of a GCaMP calcium indicator for neural activity imaging. J. Neurosci. 32, 13819–13840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deneux T., Kaszas A., Szalay G., Katona G., Lakner T., Grinvald A., Rozsa B., and Vanzetta I. (2016). Accurate spike estimation from noisy calcium signals for ultrafast three-dimensional imaging of large neuronal populations in vivo. Nat. Commun. 7, 12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Badura A., Sun X.R., Giovannucci A., Lynch L.A., and Wang S.S. (2014). Fast calcium sensor proteins for monitoring neural activity. Neurophotonics 1, 025008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berlin S., Carroll E.C., Newman Z.L., Okada H.O., Quinn C.M., Kallman B., Rockwell N.C., Martin S.S., Lagarias J.C., and Isacoff E.Y. (2015). Photoactivatable genetically encoded calcium indicators for targeted neuronal imaging. Nat Methods 12, 852–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dombeck D.A., Harvey C.D., Tian L., Looger L.L. and Tank D.W. (2010). Functional imaging of hippocampal place cells at cellular resolution during virtual navigation. Nat. Neurosci. 13, 1433–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ziv Y., Burns L.D., Cocker E.D., Hamel E.O., Ghosh K.K., Kitch L.J., El Gamal A., and Schnitzer M.J. (2013). Long-term dynamics of CA1 hippocampal place codes. Nat. Neurosci. 16, 264–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zaremba J.D., Diamantopoulou A., Danielson N.B., Grosmark A.D., Kaifosh P.W., Bowler J.C., Liao Z., Sparks F.T., Gogos J.A., and Losonczy A. (2017). Impaired hippocampal place cell dynamics in a mouse model of the 22q11.2 deletion. Nat Neurosci. 20, 1612–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohammed A.I., Gritton H.J., Tseng H.A., Bucklin M.E., Yao Z., and Han X. (2016). An integrative approach for analyzing hundreds of neurons in task performing mice using wide-field calcium imaging. Sci. Rep. 6, 20986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mishra V., Skotak M., Schuetz H., Heller A., Haorah J., and Chandra N. (2016). Primary blast causes mild, moderate, severe and lethal TBI with increasing blast overpressures: experimental rat injury model. Sci Rep 6, 26992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L., Hagemann T.L., Kalwa H., Michel T., Messing A., and Feany M.B. (2015). Nitric oxide mediates glial-induced neurodegeneration in Alexander disease. Nat. Commun. 6, 8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huuskonen M.T., Tuo Q.Z., Loppi S., Dhungana H., Korhonen P., McInnes L.E., Donnelly P.S., Grubman A., Wojciechowski S., Lejavova K., Pomeshchik Y., Periviita L., Kosonen L., Giordano M., Walker F.R., Liu R., Bush A.I., Koistinaho J., Malm T., White A.R., Lei P., and Kanninen K.M. (2017). The Copper bis(thiosemicarbazone) Complex cuii(atsm) is protective against cerebral ischemia through modulation of the inflammatory milieu. Neurotherapeutics 14, 519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grewe B.F., Langer D., Kasper H., Kampa B.M., and Helmchen F. (2010). High-speed in vivo calcium imaging reveals neuronal network activity with near-millisecond precision. Nat. Methods 7, 399–405 [DOI] [PubMed] [Google Scholar]
- 35.Svoboda K., Denk W., Kleinfeld D., and Tank D.W. (1997). In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature 385, 161–165 [DOI] [PubMed] [Google Scholar]
- 36.Vogelstein J.T., Packer A.M., Machado T.A., Sippy T., Babadi B., Yuste R., and Paninski L. (2010). Fast nonnegative deconvolution for spike train inference from population calcium imaging. J. Neurophysiol. 104, 3691–3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meaney D.F., and Smith D.H. (2011). Biomechanics of concussion. Clin. Sports Med. 30, 19–31, vii [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Browne K.D., Chen X.H., Meaney D.F., and Smith D.H. (2011). Mild traumatic brain injury and diffuse axonal injury in swine. J. Neurotrauma 28, 1747–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hines-Beard J., Marchetta J., Gordon S., Chaum E., Geisert E.E., and Rex T.S. (2012). A mouse model of ocular blast injury that induces closed globe anterior and posterior pole damage. Exp. Eye Res. 99, 63–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heldt S.A., Elberger A.J., Deng Y., Guley N.H., Del Mar N., Rogers J., Choi G.W., Ferrell J., Rex T.S., Honig M.G., and Reiner A. (2014). A novel closed-head model of mild traumatic brain injury caused by primary overpressure blast to the cranium produces sustained emotional deficits in mice. Front. Neurol. 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koliatsos V.E., Cernak I., Xu L., Song Y., Savonenko A., Crain B.J., Eberhart C.G., Frangakis C.E., Melnikova T., Kim H., and Lee D. (2011). A mouse model of blast injury to brain: initial pathological, neuropathological, and behavioral characterization. J. Neuropathol. Exp. Neurol. 70, 399–416 [DOI] [PubMed] [Google Scholar]
- 42.Kawaguchi M., Furuya H., and Patel P.M. (2005). Neuroprotective effects of anesthetic agents. J. Anesth. 19, 150–156 [DOI] [PubMed] [Google Scholar]
- 43.Rowe R.K., Harrison J.L., Thomas T.C., Pauly J.R., Adelson P.D., and Lifshitz J. (2013). Using anesthetics and analgesics in experimental traumatic brain injury. Lab. Anim. 42, 286–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Statler K.D., Alexander H., Vagni V., Dixon C.E., Clark R.S., Jenkins L., and Kochanek P.M. (2006). Comparison of seven anesthetic agents on outcome after experimental traumatic brain injury in adult, male rats. J. Neurotrauma 23, 97–108 [DOI] [PubMed] [Google Scholar]
- 45.Xiong Y., Mahmood A., and Chopp M. (2013). Animal models of traumatic brain injury. Nat. Rev. Neurosci. 14, 128–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guley N.H., Rogers J.T., Del Mar N.A., Deng Y., Islam R.M., D'Surney L., Ferrell J., Deng B., Hines-Beard J., Bu W., Ren H., Elberger A.J., Marchetta J.G., Rex T.S., Honig M.G., and Reiner A. (2016). A novel closed-head model of mild traumatic brain injury using focal primary overpressure blast to the cranium in mice. J. Neurotrauma 33, 403–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeMar J., Sharrow K., Hill M., Berman J., Oliver T., and Long J. (2016). Effects of primary blast overpressure on retina and optic tract in rats. Front. Neurol. 7, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pun P.B., Kan E.M., Salim A., Li Z., Ng K.C., Moochhala S.M., Ling E.A., Tan M.H., and Lu J. (2011). Low level primary blast injury in rodent brain. Front Neurol. 2, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gee J.M., Smith N.A., Fernandez F.R., Economo M.N., Brunert D., Rothermel M., Morris S.C., Talbot A., Palumbos S., Ichida J.M., Shepherd J.D., West P.J., Wachowiak M., Capecchi M.R., Wilcox K.S., White J.A., and Tvrdik P. (2014). Imaging activity in neurons and glia with a Polr2a-based and cre-dependent GCaMP5G-IRES-tdTomato reporter mouse. Neuron 83, 1058–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Allen W.E., Kauvar I.V., Chen M.Z., Richman E.B., Yang S.J., Chan K., Gradinaru V., Deverman B.E., Luo L., and Deisseroth K. (2017). Global representations of goal-directed behavior in distinct cell types of mouse neocortex. Neuron 94, 891–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cui G., Jun S.B., Jin X., Pham M.D., Vogel S.S., Lovinger D.M., and Costa R.M. (2013). Concurrent activation of striatal direct and indirect pathways during action initiation. Nature 494, 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hofer S.B., Ko H., Pichler B., Vogelstein J., Ros H., Zeng H., Lein E., Lesica N.A., and Mrsic-Flogel T.D. (2011). Differential connectivity and response dynamics of excitatory and inhibitory neurons in visual cortex. Nat. Neurosci. 14, 1045–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Issa J.B., Haeffele B.D., Agarwal A., Bergles D.E., Young E.D., and Yue D.T. (2014). Multiscale optical Ca2+ imaging of tonal organization in mouse auditory cortex. Neuron 83, 944–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poort J., Khan A.G., Pachitariu M., Nemri A., Orsolic I., Krupic J., Bauza M., Sahani M., Keller G.B., Mrsic-Flogel T.D., and Hofer S.B. (2015). Learning enhances sensory and multiple non-sensory representations in primary visual cortex. Neuron 86, 1478–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scott B.B., Brody C.D., and Tank D.W. (2013). Cellular resolution functional imaging in behaving rats using voluntary head restraint. Neuron 80, 371–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Homma R., Baker B.J., Jin L., Garaschuk O., Konnerth A., Cohen L.B., Bleau C.X., Canepari M., Djurisic M., and Zecevic D. (2009). Wide-field and two-photon imaging of brain activity with voltage- and calcium-sensitive dyes. Methods Mol. Biol. 489, 43–79 [DOI] [PubMed] [Google Scholar]
- 57.Yan H., Feng Y., and Wang Q. (2016). Altered effective connectivity of hippocampus-dependent episodic memory network in mTBI survivors. Neural Plast. 2016, 6353845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hartings J.A., Strong A.J., Fabricius M., Manning A., Bhatia R., Dreier J.P., Mazzeo A.T., Tortella F.C., Bullock M.R., and Co-Operative Study of Brain Injury. (2009). Spreading depolarizations and late secondary insults after traumatic brain injury. J. Neurotrauma 26, 1857–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deshpande L.S., Sun D.A., Sombati S., Baranova A., Wilson M.S., Attkisson E., Hamm R.J., and DeLorenzo R.J. (2008). Alterations in neuronal calcium levels are associated with cognitive deficits after traumatic brain injury. Neurosci. Lett. 441, 115–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun D.A., Deshpande L.S., Sombati S., Baranova A., Wilson M.S., Hamm R.J., and DeLorenzo R.J. (2008). Traumatic brain injury causes a long-lasting calcium (Ca2+)-plateau of elevated intracellular Ca levels and altered Ca2+ homeostatic mechanisms in hippocampal neurons surviving brain injury. Eur. J. Neurosci. 27, 1659–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hemphill M., Tummala S. and Meaney D. (2017). In vivo calcium imaging of hippocampal ca1 neurons reveals a functional injury signature of primary blast neurotrauma. J. Neurotrauma 34, A3 [Google Scholar]
- 62.Sam K., Srnak T., and Sengupta P. (2017). Low-force mechanical perturbations altered calcium dynamics in networks of GCaMP6s expressing cortical neurons. Biophys. J. 112, 159a [Google Scholar]
- 63.Ding M.C., Wang Q., Lo E.H., and Stanley G.B. (2011). Cortical excitation and inhibition following focal traumatic brain injury. J. Neurosci. 31, 14,085–14,094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bell J.D., Park E., Ai J., and Baker A.J. (2009). PICK1-mediated GluR2 endocytosis contributes to cellular injury after neuronal trauma. Cell Death Differ. 16, 1665–1680 [DOI] [PubMed] [Google Scholar]
- 65.Spaethling J.M., Klein D.M., Singh P., and Meaney D.F. (2008). Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J. Neurotrauma 25, 1207–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schumann J., Alexandrovich G.A., Biegon A., and Yaka R. (2008). Inhibition of NR2B phosphorylation restores alterations in NMDA receptor expression and improves functional recovery following traumatic brain injury in mice. J. Neurotrauma 25, 945–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Biegon A., Fry P.A., Paden C.M., Alexandrovich A., Tsenter J., and Shohami E. (2004). Dynamic changes in N-methyl-D-aspartate receptors after closed head injury in mice: Implications for treatment of neurological and cognitive deficits. Proc. Natl. Acad. Sci. U. S. A. 101, 5117–5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Witgen B.M., Lifshitz J., Smith M.L., Schwarzbach E., Liang S.L., Grady M.S., and Cohen A.S. (2005). Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: a systems, network and cellular evaluation. Neuroscience 133, 1–15 [DOI] [PubMed] [Google Scholar]
- 69.Kao C.Q., Goforth P.B., Ellis E.F., and Satin L.S. (2004). Potentiation of GABA(A) currents after mechanical injury of cortical neurons. J. Neurotrauma 21, 259–270 [DOI] [PubMed] [Google Scholar]
- 70.Ravin R., Blank P.S., Busse B., Ravin N., Vira S., Bezrukov L., Waters H., Guerrero-Cazares H., Quinones-Hinojosa A., and Lee P.R. (2016). Blast shockwaves propagate Ca2+ activity via purinergic astrocyte networks in human central nervous system cells. Sci. Rep. 6, 25713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weber J.T. (2012). Altered calcium signaling following traumatic brain injury. Front. Pharmacol. 3, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.