

Abstract
Background
Promoter is an important sequence regulation element, which is in charge of gene transcription initiation. In prokaryotes, σ70 promoters regulate the transcription of most genes. The promoter recognition has been a crucial part of gene structure recognition. It’s also the core issue of constructing gene transcriptional regulation network. With the successfully completion of genome sequencing from an increasing number of microbe species, the accurate identification of σ70 promoter regions in DNA sequence is not easy.
Results
In order to improve the prediction accuracy of sigma70 promoters in prokaryote, a promoter recognition model 70ProPred was established. In this work, two sequence-based features, including position-specific trinucleotide propensity based on single-stranded characteristic (PSTNPss) and electron-ion potential values for trinucleotides (PseEIIP), were assessed to build the best prediction model. It was found that 79 features of PSTNPSS combined with 64 features of PseEIIP obtained the best performance for sigma70 promoter identification, with a promising accuracy and the Matthews correlation coefficient (MCC) at 95.56% and 0.90, respectively.
Conclusion
The jackknife tests showed that 70ProPred outperforms the existing sigma70 promoter prediction approaches in terms of accuracy and stability. Additionally, this approach can also be extended to predict promoters of other species. In order to facilitate experimental biologists, an online web server for the proposed method was established, which is freely available at http://server.malab.cn/70ProPred/.
Electronic supplementary material
The online version of this article (10.1186/s12918-018-0570-1) contains supplementary material, which is available to authorized users.
Keywords: sigma70 promoter, PSTNPSS, PseEIIP, SVM
Background
Transcription is strictly regulated and controlled by cis-regulatory DNA elements, which were known as promoters and enhancers. These elements control the level of gene expression and cell fate. Promoters are cis-acting DNA sequences that switch on or off the gene expression. They are generally located upstream of the transcription start sites of genes. In prokaryotes, promoters are identified by RNA polymerase and a related sigma factor [1]. Different σ factors interact with well-defined consensus promoter sequences. Each σ factor is marked according to its molecular weight. σ70 is a well-known factor that regulates the transcription of most housekeeping genes in normal circumstances [1]. For σ70 promoter, two short sequence elements approximately located at around -10 bp and -35 bp nucleotides upstream from the transcription start site (TSS) with consensus TATAAT and TTGACA respectively [2]. It is important to identify the promoters in a genome, because it can help clarify the regulatory mechanism in the genome and explain disease-causing variants within cis-regulatory elements [3, 4]. Meanwhile, it’s a crucial part of gene structure recognition and the core issue of building gene transcriptional regulation network. Man’s understanding of promoter is developing all the time. It’s an area of great concern as people place increasing attention on their importance not only in developmental gene expression but also in environmental response [5, 6].
Due to the rapid development of genome sequencing technology, large-scale data has been generated [7–9], the stable and accurate identification of promoter is an important problem. Because standard laboratory methods are time-consuming and performance overhead costing, bioinformatics technologies with perfect precision represent the ideal alternative for massive fast recognition of promoter.
The σ70 promoter recognition task is a binary classification task. Feature extraction and classifier design are the key problems in promoter identification technology. In the past 20 years, based on the feature of promoter sequences, a serious of approaches have been developed for detecting promoter region in prokaryotes [10–23]. In 2007, Zhang proposed an algorithm using increment of diversity with quadratic discriminant (IDQD) analysis [21]. Position weight matrix (PWM) is always regarded as a description of the sequence information, but sometimes it gives poor results [22]. After that, Wu proposed an improved Position Weight Matrix (IPWM) [23] in 2011. In the same year, Lin proposed a hybrid method (IPMD), which combines location-related scoring function and diversity increment with improved Mahalanobis Discriminant to predict promoters [16]. The next year, variable-window Z-curve was used for extracting basic features of prokaryotic promoter [14]. Recently, Lin developed an improved Z-curve called ‘multi-window Z-curve’ (PseZNC), which can express the frequency characteristics and three dimensionality characteristics of different length sequences [15], etc. They were mainly focused on the σ70 promoter recognition. Among these approaches, some typical machine learning algorithms have been used to develop prokaryotic promoter region prediction. Such as, SVM (Support Vector Machine), RF (Random Forest), NB (Naïve Bayes), PLS (Partial Least Square), etc. Although these approaches have contributed to the advancement of promoter recognition, their performance demonstrates that there is a long way to go to predict promoter accurately for the following reasons. (i) Most existing approaches overlooked the correlation of neighboring nucleotides in each position, especially their difference in positive and negative samples. (ii) The local biological and physical properties of DNA may have a certain relationship with the promoters, which plays an important role in identifying them but were utterly ignored. (iii) Few web-servers were provided as the predictors, and hence their usage is quite limited [24].
In current research, we exploited a new bioinformatics tool called 70ProPred, to predict σ70 promoter through a combination of position-specific trinucleotide propensity (PSTNP) and electron-ion interaction pseudopotentials (EIIPs) of nucleotides. Finally, based on the results analysis of jackknife test, 70ProPred significantly outperforms existing prediction models, and should be useful for identifying σ70promoter.
Methods
Briefly, 70ProPred is a prediction model based on support vector machine (SVM), which was built by PSTNPSS and PseEIIP sequence coding strategies. An outline of the computational framework of 70ProPred predictor is shown in Fig. 1.
Fig. 1.
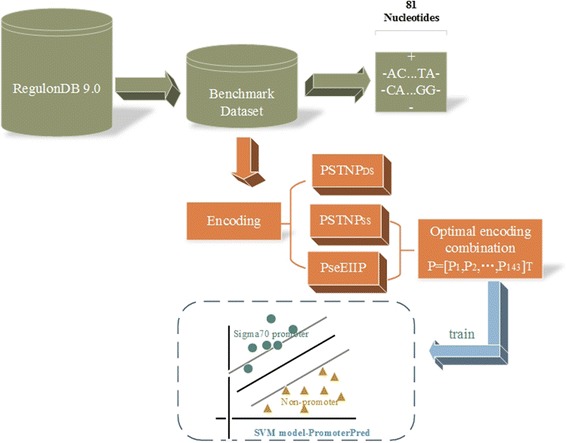
Overview of the proposed 70ProPred predictor. The diagram mainly contains datasets, sequence descriptors and 70ProPred prediction system. The optimal encoding combination PSTNPSS and PseEIIP are used as the input to train a SVM classifier. After optimization of the SVM parameters, the best SVM model is constructed based on the jackknife performance
Datasets
In the present study, we used the latest datasets in both [15] and [16]. A total of 741 σ70 promoter samples were selected from the E.coli K-12 genome, which have been verified by experiments and downloaded from the RegulonDB9.0 (http://regulondb.ccg.unam.mx/) [25]. The promoter region [TSS-60…TSS + 20] (the locus of TSS is 0) were prepared as positive samples with a length of 81 bp.
As there are not enough experimental confirmed negative sequences, negative samples are collected from both coding and non-coding regions. In simple terms, the benchmark dataset S used in this study can be expressed as:
| 1 |
Where S+ contains 741 σ70 promoter samples, S− contains 1400 non-promoter samples, contains 700 coding sequences, contains 700 convergent intergenic sequences. Each sample contains 81 nucleotides, which is selected by a sliding window. Additionally, symbol ∪ means union.
Analysis of σ70 promoter samples for conserved motif composition
The MEME Suite is designed to screen common sequence motifs from a set of sequences (amino acid or nucleotide). A motif can be assumed to be a conservative sequence pattern that repeats itself over a set of related sequences [26]. MEME is a useful sequence analysis tool that can rapidly detect new, non-gapped motifs for biological sequence data (protein, DNA and RNA) [27]. Then, we applied this tool to analyze the main motifs of σ70 promoter samples and found that only a small part of these samples which have corresponding motifs in Fig. 2. The maximum number of motif was set to 3 and the remaining arguments were set to default.
Fig. 2.
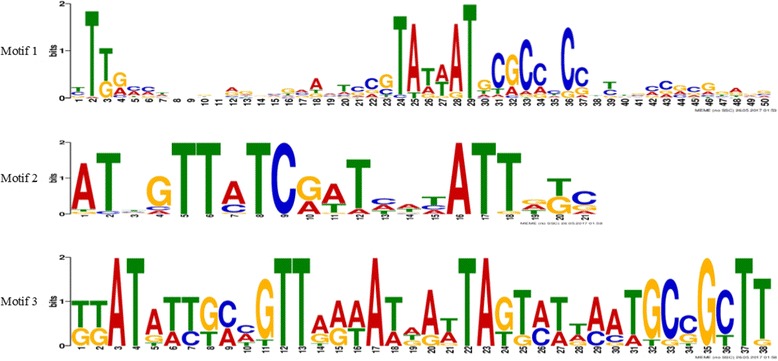
Motif of σ70 promoter samples as found by the MEME system. The corresponding three-motif logos as visualized for σ70 promoter samples (details in Table 1)
Although phylogenetic foot printing takes the advantage of relatively conservative of motifs between species [28], these motifs are short and not complete species [29, 30]. For example, in Table 1, the number of sites contributing to the construction of motif 1 only 47, which may result in a great deal of false positive results. Therefore, it would be practicable to turn to the machine learning-based methods and has been proved to be effective in many fields [26, 31–46].
Table 1.
Conserved motifs of σ70 promoter samples identified by the MEME system
| Motif | Width | Best possible match | Sites count |
|---|---|---|---|
| 1 | 50 | YTKRMMWNNBNRGNVGVAMTSCGTATWATGCGCCYCCNYBVMCVCGKRVV | 47 |
| 2 | 21 | ATBGTTATCRATHWHATTDKC | 20 |
| 3 | 38 | KKATATTGMHGTTRRWATDAWTAGTMTWAATGCSGCTT | 10 |
Feature vector construction
In order to get more useful information from the sequence, we extracted two kinds of features. Position-specific tendencies of trinucleotide (PSTNPss or PSTNPDS) was adopted to reveal the differences in the distribution of all positive and negative samples between trinucleotide. While the electron-ion interaction pseudopotentials (PseEIIP) were adopted to represent the interaction of trinucleotides.
Position-specific trinucleotide propensity based on single-stranded or double-stranded characteristic of DNA (PSTNPSS/PSTNPDS)
The recently proposed position-specific theory has been successfully applied to numerous fields of bioinformatics, such as identifying protein S-nitrosylation sites [47], hydroxyproline and hydroxylysine sites [48], DNA replication origin sites [49], Enhancer [41], etc. Besides, a series of studies have shown that the trinucleotides composition was effective in recognition of promoter [28, 50–52]. Inspired by the above studies, we presented a novel feature extraction strategy, which combined position-specific trinucleotide property (PSTNP) with the K-mer nucleotide composition information to predict σ70 promoter. A brief account of feature extraction is as follows.
Let S represents a sample which was consisted of A, G, C, and T, i.e.
| 2 |
In which L means the length of the sample and L = 81, and
| 3 |
represents the i-th position of corresponding nucleotide in the sequence.
PSTNPSS
Feature PSTNPSS using a statistical strategy based on single-stranded characteristics of DNA. There are 43 = 64trinucleotides: AAA, AAC, AAG, ..., TTT. So, for an 81 bp sample, its details of the trinucleotides position specificity can be expressed by the following 64 × 79 matrix [41]:
| 4 |
where the variable
| 5 |
F+(3meri| j) and F−(3meri| j) denote the frequency of the i-th trinucleotide (3meri) at the j-th position appear in the positive (S+) and negative (S−) data sets, respectively. In the formula, 3mer1 equals AAA,3mer2 equals AAC, …, 3mer64 equals TTT.
Therefore, the sample of Eq.2 can be expressed as:
| 6 |
where T is the operator of transpose and ϕu was defined as follows:
| 7 |
PSTNPDS
Feature PSTNPDS using a statistical strategy based on double-stranded characteristics of DNA according to complementary base pairing, so they have more evident statistical features. At this point, we deem A and T as identical, the same to C and G. Thus, for every sample, it can be converted into a sequence contained A and T only. As shown in Fig. 3, promoter-1 converted into promoter-1 AC.
Fig. 3.

promoter-1 converted into promoter-1 AC
So, there are 23 = 8 trinucleotides: AAA, AAC, ..., CCC. Therefore, for an 81 bp sample, its details of the trinucleotides position specificity can be expressed by the following 8 × 79 matrix:
| 8 |
where the variable
| 9 |
F+(3meri| j) and F−(3meri| j) denote the frequency of the i-th trinucleotide (3meri) at the j-th position appear in the positive (S+) and negative (S−) data sets, respectively. In the formula, 3mer1 equals AAA,3mer2 equals AAC, …, 3mer8 equals CCC.
Therefore, the sample of Eq.2 can be expressed as:
| 10 |
where T is the operator of transpose and ϕ'u was defined as follows:
| 11 |
Electron-ion interaction pseudopotentials of trinucleotide (PseEIIP)
Nair [53] came up with electron-ion interaction pseudopotentials (EIIP) value of nucleotides A, G, C, T. The EIIP value based methods have been shown effective through previous studies, such as the recognition of gene F56F11.4, prediction of the cystic-fibrosis gene [54], recognition of enhancer [41], and so on [55, 56].
The electron-ion interaction pseudopotentials value for the nucleotides [53] are shown in Table 2. We let EIIPA, EIIPT, EIIPG, and EIIPC denote the EIIP values of nucleotides A, T, G and C, respectively. Then, we employed the mean EIIP value of trinucleotides in each sample to construct feature vector, which can be formulated as:
| 12 |
where fxyz equal to the normalized frequency of the i-th trinucleotide (3meri), EIIPxyz = EIIPx + EIIPy + EIIPz expresses the EIIP value of one trinucleotide and X, Y, Z∈ [A, C, G, T]. Obviously, the dimension of vector V is 64.
Table 2.
EIIP values of nucleotides
| Nucleotide | EIIP(Ry) |
|---|---|
| A | 0.1260 |
| T | 0.1335 |
| G | 0.0806 |
| C | 0.1340 |
Model building and parameter selection
SVM classification algorithm plays a significant role in some areas of bioinformatics [18, 36, 40, 57]. In this work, SVM was implemented using the LIBSVM packet [58] to build models and execute predictions. The radial basis function (RBF) was selected as the kernel function. At the same time, penalty parameter C and kernel parameter γ were optimized using SVMcg in the LIBSVM package. The final parameters C = 22.6274 and γ = 2.8284 were selected for the prediction of σ70 promoters and non-promoters.
The jackknife test is regarded as a unique random test that can produce the unique result for a given dataset [59]. Therefore, all these parameters were optimized through jackknife test.
In order to evaluate the predictive performance of the model, four metrics are calculated: sensitivity (Sn), specificity (Sp), accuracy (Acc) and MCC:
| 13 |
| 14 |
| 15 |
| 16 |
In the formula, TP, TN, FP and FN represent the numbers of true positives (accurately predicted σ70 promoters), true negatives (accurately predicted non-promoters), false positives (falsely predicted σ70 promoters) and false negatives (falsely predicted non-promoters).
Results and discussion
Prediction of σ70 promoter using only PSTNP
PSTNP was first proposed for predicting enhancer [41]. The obvious advantage of this approach is that the feature vectors are encoded in a way that contains information from all training samples. In this work, the ability of PSTNPSS and PSTNPDS to discriminate σ70 promoter and non-promoter were first declared by jackknife test (Table 3). For σ70 promoter, the PSTNPDS model obtained a good performance, reaching at 75.98% sensitivity, 88.57% specificity, 84.21% accuracy and the 0.6493 of MCC value, while the PSTNPSS model obtained a Sn of 90.82%, a Sp of 96.57%, an Acc of 94.58% and a MCC of 0.8797.
Table 3.
Jackknife test performance of PSTNPSS and PSTNPDS
| Features | Sn (%) | Sp (%) | Acc (%) | MCC | SVM |
|---|---|---|---|---|---|
| PSTNPSS (79) | 90.82 | 96.57 | 94.58 | 0.8797 | -c 22.6274 -g 1.4142 |
| PSTNPDS (79) | 75.98 | 88.57 | 84.21 | 0.6493 | -c 1.4142 -g 2.8284 -w1 1.2 -w-1 1 |
A comparative figure (Fig. 4) with F-value of trinucleotides in different position also declared the difference in forecast results.
Fig. 4.
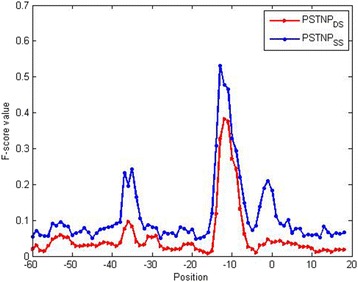
F-score value of trinucleotides in both PSTNPSS and PSTNPDS
Furthermore, we used the Entropy (Additional file 1) [23, 60] to demonstrate the conservative sites of trinucleotides property in σ70 promoter. In order to comparing, the calculated entropy values for the trinucleotide of the σ70 promoter and non-promoter are shown in Fig. 5. Obviously, the lower entropy, the more conservative the position is. From this figure, we can see that in σ70 promoter most sites are obviously of lower entropy values compared with non-promoter. It may prove that PSTNPSS feature extraction agreed well with the previous prediction that in different positions the trinucleotide is conservative in σ70promoter.
Fig. 5.
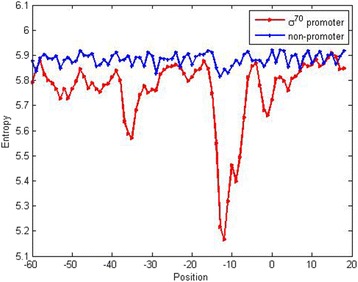
Entropy of trinucleotide in the σ70 promoter and non-promoter
From all above, the composition of trinucleotide which based on single-stranded characteristics of DNA contributes to the detection of σ70 promoter. This result indicated that the PSTNPSS model performs better than the PSTNPDS model in σ70 promoter prediction. Consequently, the training model optimized on the basis of the feature PSTNPSS.
Improving performance by incorporating PseEIIP
Because the physicochemical property indexes of nucleotides affect the recognition of promoter, incorporating the sample’s average energy of delocalized electrons (EIIP), especially, the EIIP value of trinucleotides with PSTNPSS might boost the performance of the training model, the prediction results are listed in Table 4.
Table 4.
Performances of our model on the jackknife test
| Features | Sn (%) | Sp (%) | Acc (%) | MCC |
|---|---|---|---|---|
| PSTNPSS (79) | 90.82 | 96.57 | 94.58 | 0.8797 |
| PSTNPSS (79) + PseEIIP (64) | 93.12 | 96.86 | 95.56 | 0.9018 |
Eventually, the prediction model was established using the PSTNPSS + PseEIIP feature extraction methods combined with the SVM classifier (cost parameter –c 22.6274, −g 2.8284) to predict σ70promoter.
In order to gauge the predictive performance of training model, the ROC curve and the area under the ROC curve (AUC) were adopted. The AUC value the 70ProPred model was 0.990 (Fig. 6).
Fig. 6.
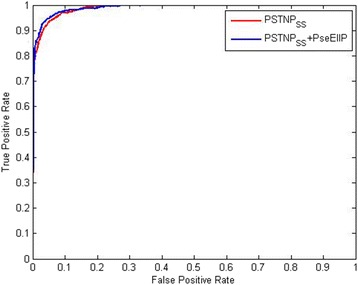
The ROC curves to assess the predictive performance based on different sequences encoding schemes for σ70promoter
Furthermore, we constructed a heat map to visually show the distribution of feature PseEIIP in positive dataset, as shown in Fig. 7. Each hotspot in the heat map corresponds to a unique trinucleotide; for instance, hotspot (1, 1) corresponds to triplet AAA. For more detailed information on the heat map, please see Additional file 2: Table S1. Red squares are positively associated with recognition ability.
Fig. 7.
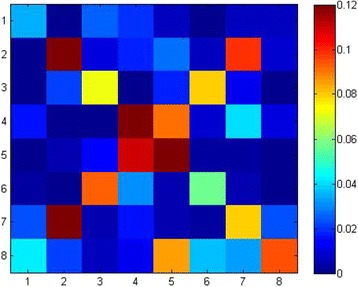
A heat map for the F-score values of the 64 trinucleotides with different EIIP values. The blue boxes indicate the features with a lower effect for recognition of the σ70 promoter, while the red boxes indicate the features that are useful for the recognition of the σ70promoter
Comparison of the performance between SVM and other classifiers
In order to select a better classifier to identify σ70 promoter, we analyzed the performances of KNN [61], NB [62], RF [63], EB [64], LibD3C [65], GBDT [66] and SVM, which are the most widely used classification algorithms in bioinformatics. Since it is known that the number of neighbors has an impact on the performance of KNN algorithm and the tree number has an impact on the performance of RF algorithm, the optimal parameters of KNN and RF are searched in the study, as shown in Additional file 3: Table S2 and Additional file 4: Table S3.
The performances for the above classifiers in the jackknife test are shown in Table 5. The results indicate that SVM performs better than the other classifiers with the highest MCC value 0.9018.
Table 5.
Comparison of different classifiers for identifying σ70promoter
| Classifier | Sn (%) | Sp (%) | Acc (%) | MCC |
|---|---|---|---|---|
| KNN (8) | 87.04 | 96.21 | 93.04 | 0.8450 |
| Naïve Bayes | 91.90 | 89.00 | 90.00. | 0.7891 |
| Random Forest (200) | 85.29 | 97.79 | 93.46 | 0.8548 |
| Ensembles for Boosting (200) | 89.88 | 95.29 | 93.41 | 0.8541 |
| LibD3C | 77.33 | 87.57 | 84.03 | 0.6478 |
| GBDT | 86.50 | 96.14 | 92.81 | 0.8397 |
| SVM | 93.12 | 96.86 | 95.56 | 0.9018 |
Comparison of 70ProPred with other σ70 promoter prediction methods
The 5-fold cross-validation results achieved by Z-curve [14], PseZNC [15], IPMD [16], PSTNPDS and 70ProPred on the benchmark dataset are listed in Table 6. Their marked difference is the feature extraction strategy. Therefore, the advantage of 70ProPred superior to other methods is mainly due to the combination of PSTNPSS and PseEIIP coding strategy. PSTNPSS employs primary sequence information of trinucleotides, and PseEIIP is closely related to the physicalchemical property of DNA sample. As shown in Table 3, based on the PSTNPSS feature only, the prediction result of 70ProPred was significantly higher than the other methods. The performance has been further improved by adding the feature PseEIIP, as shown in Table 4, especially in sensitivity (Sn). Taken together, the application of feature PSTNPSS and feature PseEIIP achieved a good performance in the prediction of σ70promoter.
Table 6.
performances of our model, Z-curve, PSTNPDS, PseZNC and IPMD on 5-fold cross-validation
| Methods | Sn (%) | Sp (%) | Acc (%) | MCC | AUC |
|---|---|---|---|---|---|
| Z-curve | 74.6 | 79.5 | 77.8 | 0.527 | 0.848 |
| PSTNPDS | 75.9 | 88.0 | 83.8 | 0.641 | 0.911 |
| PseZNC | 80.3 | 86.8 | 84.5 | 0.663 | 0.909 |
| IPMD | 82.4 | 90.7 | 87.9 | 0.731 | – |
| 70ProPred | 92.4 | 96.9 | 95.3 | 0.897 | 0.990 |
The results in Table 6 also show that the PSTNPDS-based model performs better than the multi-window Z-curve-based method. It can also be a supplement to the present methods for predicting other DNA related predictions.
Conclusions
The 70ProPred is a new bioinformatics tool for predicting σ70 promoter. This tool uses the feature extraction methods of PSTNPSS and PseEIIP. The combination of features and SVM could achieve an overall MCC value of 0.90. Compared to other σ70 promoter prediction models, 70ProPred produced better results. Although this method shows good performance in σ70 promoter prediction, there is still room to improve prediction performance due to the following reasons. (i) Since structural information is a supplementary to sequence information, the future work may build a model combine with the two aspects. (ii) The feature selection algorithms can be used to delete the redundant features to improve the prediction model. (iii) More species of promoters should be adopted to estimate the performance of 70ProPred method. In conclusion, our future work is to extend this method to other species promoter region prediction. We suspect that our feature extraction methods is not only suitable for identifying promoter, but also for other bioinformatics sequence classification tasks.
Availability
The web-server for 70ProPred has been established. It is now freely available to all interested users at http://server.malab.cn/70ProPred/. All the data sets used in this study can also be download on the website.
Additional files
Entropy. (DOC 94 kb)
Table S1. Rules of composition of heat maps. (DOC 41 kb)
Table S2. Comparison prediction results of different k neighbors. (DOC 47 kb)
Table S3. Comparison prediction results of different nTrees. (DOC 42 kb)
Acknowledgements
The work was supported by the Natural Science Foundation of China (No. 61771331).
Funding
Publication costs were funded by the Natural Science Foundation of China (No. 61771331).
Availability of data and materials
All data analyzed during this study are included in this published article.
About this supplement
This article has been published as part of BMC Systems Biology Volume 12 Supplement 4, 2018: Selected papers from the 11th International Conference on Systems Biology (ISB 2017). The full contents of the supplement are available online at https://bmcsystbiol.biomedcentral.com/articles/supplements/volume-12-supplement-4.
Abbreviations
- AUC
Area Under the ROC Curve
- EB
Ensembles for Boosting
- GBDT
Gradient Boosting Decision Tree
- KNN
k-Nearest Neighbor
- MCC
Matthew’s Correlation Coefficient
- NB
Naïve Bayes
- PseEIIP
Electron-ion interaction pseudopotentials of nucleotides
- PSTNPDS
Position-specific trinuclotide propensity based on double-stranded characteristic of DNA
- PSTNPss
Position-specific trinuclotide propensity based on single-stranded characteristic of DNA
- RF
Random Forest
Authors’ contributions
QZ conceived the research. WH performed the study. CJ, QZ and YD provided valuable suggestions and improvements. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12918-018-0570-1) contains supplementary material, which is available to authorized users.
Contributor Information
Wenying He, Email: hwying1234@sina.com.
Cangzhi Jia, Email: cangzhijia@dlmu.edu.cn.
Yucong Duan, Email: yucongduan@hotmail.com.
Quan Zou, Email: zouquan@nclab.net.
References
- 1.Barrios H, Valderrama B, Morett E. Compilation and analysis of sigma(54)-dependent promoter sequences. Nucleic Acids Res. 1999;27(22):4305–4313. doi: 10.1093/nar/27.22.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hawley DK, McClure WR. Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res. 1983;11(8):2237–2255. doi: 10.1093/nar/11.8.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y, Chen CY, Wasserman WW. Deep feature selection: theory and application to identify enhancers and promoters. J Comput Biol. 2016;23(5):322–336. doi: 10.1089/cmb.2015.0189. [DOI] [PubMed] [Google Scholar]
- 4.Geng Y, Zhao ZM, Zhang XP, Wang WK, Cui XJ, Ye K, et al. An improved burden-test pipeline for identifying associations from rare germline and somatic variants. BMC Genomics. 2017;18(7):55–62. [DOI] [PMC free article] [PubMed]
- 5.Bernardo LM, Johansson LU, Skarfstad E, Shingler V. sigma54-promoter discrimination and regulation by ppGpp and DksA. J Biol Chem. 2009;284(2):828–838. doi: 10.1074/jbc.M807707200. [DOI] [PubMed] [Google Scholar]
- 6.Lu C, Xie MC, Wendl MC, Wang JY, McLellan MD, Leiserson MDM, et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat Commun. 2015;6 [DOI] [PMC free article] [PubMed]
- 7.Mrozek D, Malysiak-Mrozek B, Klapcinski A. Cloud4Psi: cloud computing for 3D protein structure similarity searching. Bioinformatics. 2014;30(19):2822–2825. doi: 10.1093/bioinformatics/btu389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mrozek D, Danilowicz P, Malysiak-Mrozek B. HDInsight4PSi: boosting performance of 3D protein structure similarity searching with HDInsight clusters in Microsoft azure cloud. Inform Sciences. 2016;349:77–101. doi: 10.1016/j.ins.2016.02.029. [DOI] [Google Scholar]
- 9.Mrozek D, Gosk P, Malysiak-Mrozek B. Scaling ab initio predictions of 3D protein structures in Microsoft azure cloud. J Grid Comput. 2015;13(4):561–585. doi: 10.1007/s10723-015-9353-8. [DOI] [Google Scholar]
- 10.Zhou X, Li Z, Dai Z, Zou X. Predicting promoters by pseudo-trinucleotide compositions based on discrete wavelets transform. J Theor Biol. 2013;319:1–7. doi: 10.1016/j.jtbi.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 11.Wang H, Benham CJ. Promoter prediction and annotation of microbial genomes based on DNA sequence and structural responses to superhelical stress. BMC Bioinformatics. 2006;7:248. doi: 10.1186/1471-2105-7-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Towsey MW, Gordon JJ, Hogan JM. The prediction of bacterial transcription start sites using SVMs. Int J Neural Syst. 2006;16(5):363–370. doi: 10.1142/S0129065706000767. [DOI] [PubMed] [Google Scholar]
- 13.Towsey M, Hogan JM, Mathews S, Timms P. The in silico prediction of promoters in bacterial genomes. Genome Inform. 2007;19:178–189. [PubMed] [Google Scholar]
- 14.Song K. Recognition of prokaryotic promoters based on a novel variable-window Z-curve method. Nucleic Acids Res. 2012;40(3):963–971. doi: 10.1093/nar/gkr795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin H, Liang ZY, Tang H, Chen W. Identifying sigma70 promoters with novel pseudo nucleotide composition. IEEE/ACM Trans Comput Biol Bioinform. 2017;PP(99):1–1. [DOI] [PubMed]
- 16.Lin H, Li QZ. Eukaryotic and prokaryotic promoter prediction using hybrid approach. Theory Biosci. 2011;130(2):91–100. doi: 10.1007/s12064-010-0114-8. [DOI] [PubMed] [Google Scholar]
- 17.Kanhere A, Bansal M. A novel method for prokaryotic promoter prediction based on DNA stability. BMC Bioinformatics. 2005;6:1. doi: 10.1186/1471-2105-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia CZ, He WY, Yao YH. OH-PRED: prediction of protein hydroxylation sites by incorporating adapted normal distribution bi-profile Bayes feature extraction and physicochemical properties of amino acids. J Biomol Struct Dyn. 2017;35(4):829–835. doi: 10.1080/07391102.2016.1163294. [DOI] [PubMed] [Google Scholar]
- 19.Huerta AM, Collado-Vides J. Sigma70 promoters in Escherichia coli: specific transcription in dense regions of overlapping promoter-like signals. J Mol Biol. 2003;333(2):261–278. doi: 10.1016/j.jmb.2003.07.017. [DOI] [PubMed] [Google Scholar]
- 20.de Avila ESS, Gerhardt GJ, Echeverrigaray S. Rules extraction from neural networks applied to the prediction and recognition of prokaryotic promoters. Genet Mol Biol. 2011;34(2):353–360. doi: 10.1590/S1415-47572011000200031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ZHANG Y, JIA Y, Lü J. THE RECOGNITION OF σ~(70) PROMOTERS IN Escherichia coli K-12. Acta Biophysica Sinica. 2007;6:011. [Google Scholar]
- 22.Barrick D, Villanueba K, Childs J, Kalil R, Schneider TD, Lawrence CE, et al. Quantitative analysis of ribosome binding sites in E.Coli. Nucleic Acids Res. 1994;22(7):1287–1295. doi: 10.1093/nar/22.7.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu Q, Wang J, Yan H. An improved position weight matrix method based on an entropy measure for the recognition of prokaryotic promoters. Int J Data Min Bioinform. 2011;5(1):22–37. doi: 10.1504/IJDMB.2011.038575. [DOI] [PubMed] [Google Scholar]
- 24.Mrozek D, Malysiak-Mrozek B, Siaznik A. Search GenBank: interactive orchestration and ad-hoc choreography of web services in the exploration of the biomedical resources of the National Center for Biotechnology Information. Bmc Bioinformatics. 2013;14(1):1–19. [DOI] [PMC free article] [PubMed]
- 25.Gama-Castro S, Salgado H, Santos-Zavaleta A, Ledezma-Tejeida D, Muniz-Rascado L, Garcia-Sotelo JS, et al. RegulonDB version 9.0: high-level integration of gene regulation, coexpression, motif clustering and beyond. Nucleic Acids Res. 2016;44(D1):D133–D143. doi: 10.1093/nar/gkv1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao Z, Wang X, Zeng Y, Zou Q. Identification of DEP domain-containing proteins by a machine learning method and experimental analysis of their expression in human HCC tissues. Sci Rep. 2016;6:39655. doi: 10.1038/srep39655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bailey TL, Johnson J, Grant CE, Noble WS. The MEME suite. Nucleic Acids Res. 2015;43(W1):W39–W49. doi: 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin H, Deng EZ, Ding H, Chen W, Chou KC. iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Res. 2014;42(21):12961–12972. doi: 10.1093/nar/gku1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abeel T, Saeys Y, Bonnet E, Rouze P, Van de Peer Y. Generic eukaryotic core promoter prediction using structural features of DNA. Genome Res. 2008;18(2):310–323. doi: 10.1101/gr.6991408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abeel T, Saeys Y, Rouze P, Van de Peer Y. ProSOM: core promoter prediction based on unsupervised clustering of DNA physical profiles. Bioinformatics. 2008;24(13):i24–i31. doi: 10.1093/bioinformatics/btn172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liao Z, Wang X. Chen X. Comb Chem High Throughput Screen: Zou Q. Prediction and Identification of Kruppel-like transcription factors by machine learning method; 2017. [DOI] [PubMed] [Google Scholar]
- 32.Xiong H, Liu D, Li Q, Lei M, Xu L, Wu L, et al. RED-ML: a novel, effective RNA editing detection method based on machine learning. Gigascience. 2017;6(5):1–8. doi: 10.1093/gigascience/gix012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen W, Xing P, Zou Q. Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble support vector machines. Sci Rep. 2017;7:40242. doi: 10.1038/srep40242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen W, Feng P, Yang H, Ding H, Lin H, Chou KC. iRNA-AI: identifying the adenosine to inosine editing sites in RNA sequences. Oncotarget. 2017;8(3):4208–4217. doi: 10.18632/oncotarget.13758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meher PK, Sahu TK, Saini V, Rao AR. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou's general PseAAC. Sci Rep. 2017;7:42362. doi: 10.1038/srep42362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jia CZ, Liu T, Wang ZP. O-GlcNAcPRED: a sensitive predictor to capture protein O-GlcNAcylation sites. Mol BioSyst. 2013;9(11):2909–2913. doi: 10.1039/c3mb70326f. [DOI] [PubMed] [Google Scholar]
- 37.Jia C, Lin X, Wang Z. Prediction of protein S-nitrosylation sites based on adapted normal distribution bi-profile Bayes and Chou's pseudo amino acid composition. Int J Mol Sci. 2014;15(6):10410–10423. doi: 10.3390/ijms150610410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen W, Feng PM, Lin H, Chou KC. iRSpot-PseDNC: identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res. 2013;41(6):e68. doi: 10.1093/nar/gks1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kabir M, Hayat M. iRSpot-GAEnsC: identifing recombination spots via ensemble classifier and extending the concept of Chou's PseAAC to formulate DNA samples. Mol Gen Genomics. 2016;291(1):285–296. doi: 10.1007/s00438-015-1108-5. [DOI] [PubMed] [Google Scholar]
- 40.Jia C, He W. EnhancerPred: a predictor for discovering enhancers based on the combination and selection of multiple features. Sci Rep. 2016;6:38741. doi: 10.1038/srep38741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He W, Jia C. EnhancerPred2.0: predicting enhancers and their strength based on position-specific trinucleotide propensity and electron-ion interaction potential feature selection. Mol BioSyst. 2017;13(4):767–774. doi: 10.1039/C7MB00054E. [DOI] [PubMed] [Google Scholar]
- 42.Liu B, Fang L, Wang S, Wang X, Li H, Chou KC. Identification of microRNA precursor with the degenerate K-tuple or Kmer strategy. J Theor Biol. 2015;385:153–159. doi: 10.1016/j.jtbi.2015.08.025. [DOI] [PubMed] [Google Scholar]
- 43.Liu B, Fang L, Long R, Lan X, Chou KC. iEnhancer-2L: a two-layer predictor for identifying enhancers and their strength by pseudo k-tuple nucleotide composition. Bioinformatics. 2016;32(3):362–369. doi: 10.1093/bioinformatics/btv604. [DOI] [PubMed] [Google Scholar]
- 44.Chen W, Feng PM, Deng EZ, Lin H, Chou KC. iTIS-PseTNC: a sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Anal Biochem. 2014;462:76–83. doi: 10.1016/j.ab.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 45.Liu Z, Xiao X, Qiu WR, Chou KC. iDNA-methyl: identifying DNA methylation sites via pseudo trinucleotide composition. Anal Biochem. 2015;474:69–77. doi: 10.1016/j.ab.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 46.Jia J, Zhang L, Liu Z, Xiao X, Chou KC. pSumo-CD: predicting sumoylation sites in proteins with covariance discriminant algorithm by incorporating sequence-coupled effects into general PseAAC. Bioinformatics. 2016;32(20):3133–3141. doi: 10.1093/bioinformatics/btw387. [DOI] [PubMed] [Google Scholar]
- 47.Xu Y, Shao XJ, Wu LY, Deng NY, Chou KC. iSNO-AAPair: incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins. PeerJ. 2013;1:e171. doi: 10.7717/peerj.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu Y, Wen X, Shao XJ, Deng NY, Chou KC. iHyd-PseAAC: predicting hydroxyproline and hydroxylysine in proteins by incorporating dipeptide position-specific propensity into pseudo amino acid composition. Int J Mol Sci. 2014;15(5):7594–7610. doi: 10.3390/ijms15057594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiao X, Ye HX, Liu Z, Jia JH, Chou KC. iROS-gPseKNC: predicting replication origin sites in DNA by incorporating dinucleotide position-specific propensity into general pseudo nucleotide composition. Oncotarget. 2016;7(23):34180–34189. doi: 10.18632/oncotarget.9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chou K-C. Some remarks on protein attribute prediction and pseudo amino acid composition. J Theor Biol. 2011;273(1):236–247. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen W, Lin H, Chou K-C. Pseudo nucleotide composition or PseKNC: an effective formulation for analyzing genomic sequences. Mol BioSyst. 2015;11(10):2620–2634. doi: 10.1039/C5MB00155B. [DOI] [PubMed] [Google Scholar]
- 52.Pei Li MG. Chunyu Wang, Xiaoyan Liu, Quan Zou. An overview of SNP interactions in genome-wide association studies. Briefings in Functional Genomics. 2015;14(2):143–155. doi: 10.1093/bfgp/elu036. [DOI] [PubMed] [Google Scholar]
- 53.Nair AS, Sreenadhan SP. A coding measure scheme employing electron-ion interaction pseudopotential (EIIP) Bioinformation. 2006;1(6):197–202. [PMC free article] [PubMed] [Google Scholar]
- 54.Rao KD, Swamy M. Analysis of genomics and proteomics using DSP techniques. IEEE Transactions on Circuits and Systems I: Regular Papers. 2008;55(1):370–378. doi: 10.1109/TCSI.2007.910541. [DOI] [Google Scholar]
- 55.Sahu SS, Panda G. Efficient localization of hot spots in proteins using a novel S-transform based filtering approach. IEEE/ACM Transactions on Computational Biology and Bioinformatics (TCBB) 2011;8(5):1235–1246. doi: 10.1109/TCBB.2010.109. [DOI] [PubMed] [Google Scholar]
- 56.Khan A. G-protein-coupled receptor prediction using pseudo-amino-acid composition and multiscale energy representation of different physiochemical properties. Anal Biochem. 2011;412(2):173–182. doi: 10.1016/j.ab.2011.01.040. [DOI] [PubMed] [Google Scholar]
- 57.Wei L, Liao M, Gao X, Zou Q. Enhanced protein fold prediction method through a novel feature extraction technique. IEEE Transactions on Nanobioscience. 2015;14(6):649–659. doi: 10.1109/TNB.2015.2450233. [DOI] [PubMed] [Google Scholar]
- 58.Chang C-C, Lin C-J. LIBSVM: a library for support vector machines. ACM Transactions on Intelligent Systems and Technology (TIST) 2011;2(3):27. [Google Scholar]
- 59.Chou K-C, Shen H-B. Cell-PLoc 2.0: An improved package of web-servers for predicting subcellular localization of proteins in various organisms. Natural Science. 2010;2(10):1090. doi: 10.4236/ns.2010.210136. [DOI] [PubMed] [Google Scholar]
- 60.Fickett JW. Quantitative discrimination of MEF2 sites. Mol Cell Biol. 1996;16(1):437–441. doi: 10.1128/MCB.16.1.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cover T, Hart P. Nearest neighbor pattern classification. IEEE Trans Inf Theory. 1967;13(1):21–27. doi: 10.1109/TIT.1967.1053964. [DOI] [Google Scholar]
- 62.Rish I. An empirical study of the naive Bayes classifier. Journal of Universal Computer Science. 2001;1(2):127.
- 63.Ho TK. Random decision forests. International Conference on Document Analysis and Recognition. IEEE Computer Society. 1995;278.
- 64.Opitz D, Maclin R. Popular ensemble methods: an empirical study. J Artif Intell Res. 1999;11:169–198. [Google Scholar]
- 65.Lin C, Chen W, Qiu C, Wu Y, Krishnan S, Zou Q. LibD3C: ensemble classifiers with a clustering and dynamic selection strategy. Neurocomputing. 2014;123:424–435. doi: 10.1016/j.neucom.2013.08.004. [DOI] [Google Scholar]
- 66.Friedman JH. Greedy function approximation: a gradient boosting machine. Ann Stat. 2001;29(5):1189–232.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Entropy. (DOC 94 kb)
Table S1. Rules of composition of heat maps. (DOC 41 kb)
Table S2. Comparison prediction results of different k neighbors. (DOC 47 kb)
Table S3. Comparison prediction results of different nTrees. (DOC 42 kb)
Data Availability Statement
All data analyzed during this study are included in this published article.