

Abstract
Background
Even though coexistence of multiple phenotypes sharing the same genomic background is interesting, it remains incompletely understood. Epigenomic profiles may represent key factors, with unknown contributions to the development of multiple phenotypes, and social-insect castes are a good model for elucidation of the underlying mechanisms. Nonetheless, previous studies have failed to identify genes associated with aberrant gene expression and methylation profiles because of the lack of suitable methodology that can address this problem properly.
Methods
A recently proposed principal component analysis (PCA)-based and tensor decomposition (TD)-based unsupervised feature extraction (FE) can solve this problem because these two approaches can deal with gene expression and methylation profiles even when a small number of samples is available.
Results
PCA-based and TD-based unsupervised FE methods were applied to the analysis of gene expression and methylation profiles in the brains of two social insects, Polistes canadensis and Dinoponera quadriceps. Genes associated with differential expression and methylation between castes were identified, and analysis of enrichment of Gene Ontology terms confirmed reliability of the obtained sets of genes from the biological standpoint.
Conclusions
Biologically relevant genes, shown to be associated with significant differential gene expression and methylation between castes, were identified here for the first time. The identification of these genes may help understand the mechanisms underlying epigenetic control of development of multiple phenotypes under the same genomic conditions.
Electronic supplementary material
The online version of this article (10.1186/s12859-018-2068-7) contains supplementary material, which is available to authorized users.
Keywords: Tensor decomposition, Principal component analysis, Feature extraction, Gene expression, Methylation, Social insect
Background
Organisms often exhibit different phenotypes despite a common genomic background. For example, juveniles and adults frequently have different body plans (e.g., tadpoles and frogs, caterpillars and butterflies, and megalopas and clubs). Nonetheless, juvenile and adult organisms have different sizes or must survive in distinct environments, and these conditions require different phenotypes. More striking examples are castes of social insects, such as ants and bees, which can form two distinct forms: queens and workers, both female [1]. They are usually closely related, but queens and workers have different sizes and lifespans. The mechanism that potentially allows social insects to form castes with distinct body plans is the epigenome [2, 3], which is flexible and can lead to the formation of different phenotypes without genomic alterations. Therefore, it is important to determine the correlation between an epigenome and phenotype by analyzing gene expression.
In actuality, there have been many discussions regarding how an epigenome can affect a phenotype [4]. Because a gene can affect the phenotype through the regulation of gene expression, it is natural to expect that the epigenome can also affect the phenotype through the regulation of gene expression [5]. The epigenome is even expected to be heritable and thus affect phenotypes through generations [6]. In this field, the relation between the epigenome and phenotype has been comprehensively investigated; through regulation of gene expression, epigenetic mechanisms have the potential to determine and alter cell phenotypes, and epigenetic mechanisms also mediate dosage compensation, chromosomal silencing, and imprinting [7]. In this regard, castes of social insects are expected to be affected by various epigenomic alterations.
Some pioneering studies in this field have been conducted [8, 9], but statistical analyses in these studies have not been satisfactory, for example,
Gene ontology (GO) enrichment analyses had an insufficiently small false discovery rate (FDR): <0.5 [8];
identification of differentially expressed genes (DEGs) was performed at insufficiently large q>0.6, corresponding to FDR <0.4 [9];
highly methylated regions significantly different between phenotypes (castes) were not identified [8].
Even though these issues do not always reduce quality of the studies, addressing them should increase the confidence in the conclusions.
Inadequate statistical analyses may be due to disregarding the multivariate nature of variables. All the performed statistical analyses have been single–gene–based, meaning that group behaviors were considered only after identification of genes. Recently, I proposed a principal component analysis (PCA)-based unsupervised feature extraction (FE) as a method that can perform multivariate analysis before gene selection and applied it to various bioinformatic problems [10–31]. Therefore, applying PCA-based unsupervised FE to the analysis of datasets may yield more reliable results. PCA-based unsupervised FE was also extended to tensor decomposition (TD) to integrate multiway [32–34] and multiview [35–37] datasets. TD-based unsupervised FE applied to the integrated analysis of gene expression and methylation profiles may allow for identification of relations between gene expression and methylation, essential for identification of the mechanisms behind epigenetic regulation of phenotype development.
Methods
A flow chart showing the experimental design is presented in Fig. 1.
Fig. 1.
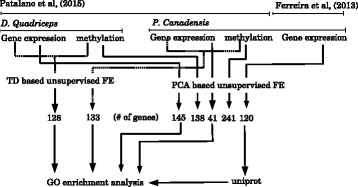
A flow chart showing the design of this study. Three gene expression profiles and two methylation profiles were retrieved from two studies on two social insect species, Polistes canadensis and Dinoponera quadriceps. All of them were processed by PCA-based unsupervised FE, whereas two pairs of gene expression and methylation profiles were analyzed by TD-based unsupervised FE. Differential expression between castes was analyzed in seven obtained gene sets. Analysis of GO term enrichment was performed on three sets of genes derived from gene expression analysis and on two sets of genes generated by TD-based unsupervised FE. The full list of the selected genes is presented in Additional file 1
Gene expression and methylation profiles
All gene expression and methylation profiles [8] were retrieved from Gene Expression Omnibus (GEO). Gene expression profiles of P. canadensis and D. quadriceps are available as Supplementary Files in GEO ID GSE59525: GSE59525_RPKM_Pcan.txt.gz and SE59525_RPKM_Dqua.txt.gz. Methylation profiles of P. canadensis and D. quadriceps are in GSM14388XX_PcanYYY_CX.txt.gz and GSM14388XX_DquaYYY_CX.txt.gz, which are also available as Supplementary Files in GEO ID GSE59525 (XX and YYY are presented in Table 1). An additional gene expression profile of P. canadensis [9], 13059_2012_3057_MOESM9_ESM.CSV, was retrieved from the supplementary file presented in the study (Additional file 9). Gene expression values were used as-is, but methylation profile values were integrated so that they represented the relative methylation within genes. Assuming ms1 and ms2 are methylation and nonmethylation values respectively at locus s, then the relative methylation within the ith gene can be defined as
where is taken over s bases within DNA sequences corresponding to the ith gene body.
Table 1.
The list of files containing methylation profiles retrieved from GEO
| XX | YYY | XX | YYY | Description |
|---|---|---|---|---|
| P. canadensis | D. quadriceps | |||
| 50 | _Synthetic | 57 | _Synthetic | Control |
| 51 | 21Q | 58 | 1AQ | Queen 1st replicate |
| 52 | 43Q | 59 | 2AQ | Queen 2nd replicate |
| 53 | 75Q | 60 | 3AQ | Queen 3rd replicate |
| 54 | 26W | 61 | 1CW | Worker 1st replicate |
| 55 | 42W | 62 | 3CW | Worker 2nd replicate |
| 56 | 76W | 63 | 3DW | Worker 3rd replicate |
PCA-based unsupervised FE
A flow chart showing PCA- and TD-based unsupervised FE is presented in Fig. 2.
Fig. 2.
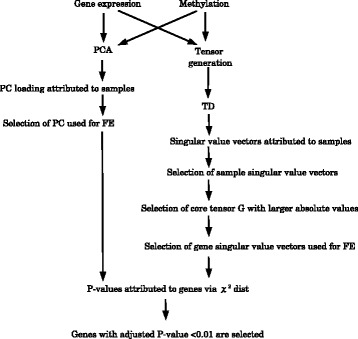
A flow chart of PCA- and TD-based unsupervised FE. Gene expression and methylation profiles were examined by PCA or TD. For PCA, gene expression and methylation profiles were processed separately, whereas TD was applied after generating a tensor from them. For PCA, principal component (PC) loading attributed to samples was studied and selected for FE. Because PC loadings and PC scores attributed to genes show one-to-one correspondence, PC scores corresponding to the selected PC loadings were subjected to FE. For TD, one-sample singular value vectors used for FE were selected. Afterwards, during analysis of core tensors, G, gene singular value vectors associated with Gs with larger absolute values were selected. By means of the identified PC scores or gene singular value vectors, P-values were determined for genes, assuming a χ2 distribution, and genes associated with adjusted P-values less than 0.01 were finally selected
PCA
Assume that N×M matrix X represents gene expression or methylation of the ith gene in the jth sample, , which can be standardized as . The kth PC score, , attributed to the ith gene can be obtained as the ith component of the kth eigenvector uk of XXT, where XT is the transposed matrix of X, so that
where λk is the kth eigenvalue. The kth PC loading, , attributed to the jth sample can be obtained as the jth component of the kth vector vk, which is defined as
This is also the kth eigenvector of matrix XTX because
PCA-based unsupervised FE
To carry out FE by PCA, PC loadings, vk, of interest that can be used for FE must be identified, and there are several approaches. Suppose that Ω represents a set of ks of the identified vks. Then, gene i, primarily contributing to the kth (k∈Ω) PC score, uk, should be identified, and this task can be accomplished by selecting the outliers within the space spanned by the PC scores:
uki(k∈Ω,1≤i≤N) was assumed to follow a multiple normal distribution, and P-values, Pis, were attributed to each gene i via the χ2 distribution,
| 1 |
where represents the cumulative probability of the χ2 distribution that the argument is greater than x, whereas σk is standard deviation. Afterwards, genes is, associated with the Benjamini–Hochberg criterion–adjusted Pis [38] lower than the threshold value, e.g., 0.01, were selected as outliers.
TD-based unsupervised FE
When two sets of experimental factors are affecting each sample, e.g., tissues and diseases, gene expression and methylation profiles should be presented as a tensor , where j and ℓ correspond to the tissue and disease, respectively.
Equivalence of PCA and singular value decomposition (SVD)
PCA can be extended to a tensor as follows. PCA is known to be equivalent to SVD,
| 2 |
where U and V are N×M and M×M orthogonal matrices, respectively. Λ represents a M×M diagonal matrix. Assume that U=(u1,…,uM) and V=(v1,…,vM). The diagonal component of Λ can be written as λk. Then, using vk=XTuk and XXTuk=λuk,
Therefore, U and V composed of PC scores and loadings satisfy Eq. (2). Subsequently,
or this relation can be written as
| 3 |
Extension to TD
Equation (3) can be easily generalized to TD [32] by extending the matrix to a tensor,
where , , and with (K1,K2,K3)=(N,M,L) were assumed to be orthogonal matrices. Hereafter, and are referred to as singular value matrix and singular value vector, respectively. Given that the core tensor, , is as large as xijk, this situation represents an overcomplete problem, i.e., there is no unique decomposition. In this study, higher-order SVD (HOSVD) [39], which is known to frequently give a global minimum [40], was employed to perform TD, assuming , as in the PCA cases.
TD-based unsupervised FE
First, (k2,k3) of interest, which were attributed to samples, were selected, and, as for PCA, there are different approaches. Next, G(k1,k2,k3)s associated with selected k2,k3 were ranked based on their absolute values, and by means of top-ranked k1s with the set defined as Ω, the same procedure applied to Eq. (1) was repeated by replacing PC score uki with a gene singular value vector, . Currently, we do not have any specific criterion specifying how many k1s should be considered.
TD-based unsupervised FE for integrated analysis of multi-omics data
For two distinct omics datasets, such as a gene expression profile, xij, and a methylation profile, xiℓ, TD-based unsupervised FE can be used for the integrated analysis by generating a tensor,
to which TD-based unsupervised FE can be applied [35]. The subsequent procedure was the same as the standard one described in the previous subsection.
GO enrichment analysis
To perform GO enrichment analysis of the previously described dataset [8], the list of genes associated with GO terms was downloaded [41]: PCAN.v01.GO.tsv for P. canadensis and DQUA.v01.GO.tsv for D. quadriceps.
Because genes presented in the second study we used [9] were not fully annotated but contained protein sequence gene IDs, a list of gene IDs was uploaded to UniProt [42] and GO term associations were downloaded. GO enrichment analyses were performed on the retrieved list.
Fisher’s exact test was selected for evaluation of the overlaps between the set of provided genes and genes associated with a specific GO term. The obtained P-values were adjusted in accordance with the Benjamini–Hochberg criterion [38]. GO terms associated with adjusted P-values less than 0.01 were selected.
Results
PCA-based unsupervised FE applied to the dataset provided by Patalano et al. [8]
PCA-based unsupervised FE was performed on the dataset presented in another study [8].
The methylation profile
PCA-based unsupervised FE was applied to the methylation profiles of P. canadensis and D. quadriceps. In Fig. 3(a) and (c), the first PC loadings, v1, are presented, attributed to seven samples, comprising one control, three queen samples, and three worker samples of P. canadensis and D. quadriceps each. For both, v1 mainly denotes the difference between the control and queen or worker samples. In Fig. 3(b) and (d), the histogram of the first PC score, u1, attributed to genes is presented, and red areas represent the selected genes, with adjusted P-values computed using Eq. (1) lower than 0.01 (241 and 138 selected genes for P. canadensis and D. quadriceps, respectively). Because in Fig. 3(a) hypermethylated genes are presented, and the results in Fig. 3(b) suggest that only the genes with positive PC scores are selected, all the selected genes were found to be hypermethylated. Similarly, because in Fig. 3(c), hypomethylation is presented, and the results in Fig. 3(d) indicate that only the genes with negative PC scores are selected, all the selected genes were found to be associated with hypermethylation as well. This finding is in agreement with the results of the other study [8].
Fig. 3.
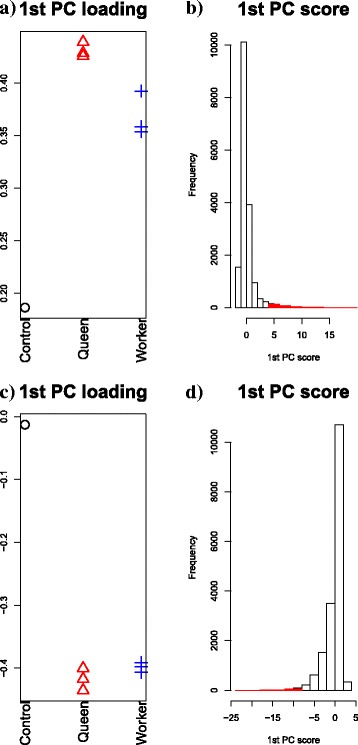
The results of PCA performed on methylation profiles. PCA-based unsupervised FE was tested on methylation profiles. a and b: P. canadensis. c and d: D. quadriceps. a and c: A boxplot of PC loadings v1, (b) and (d): A PC score histogram, u1. Red areas in (b) and (d) represent selected genes
Patalano et al. [8] did not find genes associated with the emergence of distinct methylation patterns between queens and workers, but the results presented in Fig. 3(a) and (c) show minor differences between queens and workers, suggesting that the selected genes may have different methylation profiles between queens and workers overall. Three statistical tests were carried out to analyze the differences between queens and workers (Table 2) and demonstrated that PCA-based unsupervised FE could identify gene-associated methylation patterns between queens and workers, unlike the analyses in the other study [8]. Given that in Fig. 3(a) and (c), the upregulation and downregulation of methylation in queens are presented, whereas in Fig. 3(b) and (d), genes associated with positive and negative PC scores are shown, the selected genes were found to be associated with hypermethylation in queens. This finding suggests that hypermethylated genes are associated with relative hypermethylation in queens.
Table 2.
Statistical tests for differences in the methylation rates of selected genes between queens and workers
| t | Wilcox | KS | |
|---|---|---|---|
| P. canadensis | 8.59×10−5 | 3.10×10−3 | 1.83×10−4 |
| D. quadriceps | 1.11×10−2 | 5.88×10−3 | 1.75×10−3 |
t: the t test, Wilcox: the Wilcoxon rank sum test, KS: the Kolmogorov–Sinai test, all two-sided
Gene expression
PCA-based unsupervised FE was performed on the gene expression profiles of P. canadensis and D. quadriceps. Because the gene expression profile of P. canadensis was log2-ratio converted, it was scaled back to the original one as before the application of PCA. In Fig. 4(a) and (c), v3 and v4 are presented, showing the most significant differences in gene expression levels between queens and workers of P. canadensis and D. quadriceps, respectively. In Fig. 4(b) and (d), the distributions of v3 and v4 for P. canadensis and D. quadriceps, respectively, are depicted. Red areas represent the selected genes, with adjusted P-values, determined using Eq. (1), lower than 0.01 (41 and 145 genes for P. canadensis and D. quadriceps, respectively).
Fig. 4.
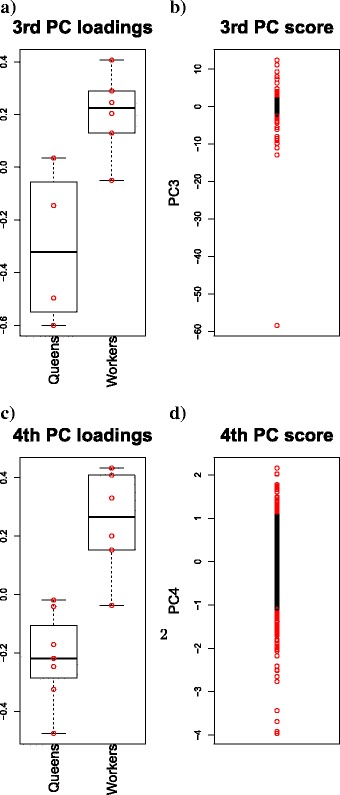
Results of gene expression PCA. PCA-based unsupervised FE was applied to the gene expression profiles. a and b: P. canadensis. c and d: D. quadriceps. a and c: A boxplot of PC loadings v1, (b) and (d): A distribution of PC scores u1. Red areas in (b) and (d) represent the selected genes
To determine whether the expression of selected genes differs between queens and workers, three statistical tests were carried out (Table 3). The majority (five of six) of the applied tests confirmed that expression of the selected genes differs between queens and workers of P. canadensis and D. quadriceps, in line with the findings reported elsewhere [8].
Table 3.
Statistical tests for differences in expression of the selected genes between queens and workers
| t | Wilcox | KS | |
|---|---|---|---|
| P. canadensis | 4.37×10−4 | 0.07 | 6.45×10−3 |
| D. quadriceps | 1.73×10−12 | 2.24×10−12 | 5.26×10−12 |
t: the t test, Wilcox: the Wilcoxon rank sum test, KS: the Kolmogorov–Sinai test; all two-sided
PCA-based unsupervised FE applied to the dataset provided by Ferreira et al. [9]
PCA-based unsupervised FE was performed on the expression profiles reported in another study [9]. In Fig. 5(a), the third PC loadings, v3, are presented, which demonstrate the most significant class dependence based on categorical regression (ANOVA). In Fig. 5(b), the distribution of the third PC score, u3, is shown, where the red areas represent the selected genes (120 genes associated with the adjusted P-values, as determined by means of Eq. (1), lower than 0.01). Because this situation corresponds to q>0.99 in the analysis provided by Ferreira et al., we demonstrated that we were able to identify a set of genes more significant than those identified in the other study, where q was > 0.6.
Fig. 5.
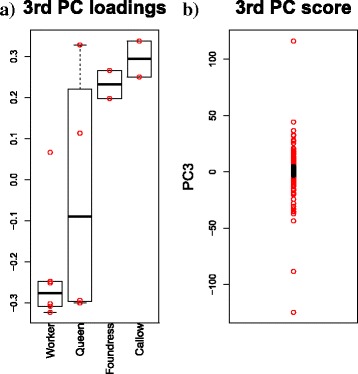
The results of PCA applied to gene expression profiles. PCA-based unsupervised FE was performed on the gene expression profiles obtained from the study by Ferreira et al. a: A boxplot of PC loadings v1. b: A distribution of PC scores u1. Red areas in panel (b) represent 120 selected genes. PC3 > 0: 77 genes, PC3 < 0: 43 genes
To test whether the selected genes reflect the class-specific up- and downregulation of expression, three statistical tests were conducted (Table 4). We separated genes into two classes based on the sign of u3i, to identify the upregulation or downregulation of these genes for further comparisons. As shown in Table 4, 120 selected genes were found to be significantly upregulated or downregulated, excluding the upregulation of genes in Foudress, with the smallest number of genes identified in the other study. Therefore, we successfully identified genes that manifest class-specific upregulation or downregulation. Furthermore, the genes identified here are common for all four classes, while those identified in the other study differed between the comparisons.
Table 4.
Statistical tests for upregulation and downregulation of gene expression in four categories vs. others
| PC3 | t | Wilcox | KS | ||
|---|---|---|---|---|---|
| Worker vs others | + | down | 3.96×10−5 | ∗ | ∗ |
| − | up | 0.22 | ∗ | ∗ | |
| Queen vs others | + | up | 0.36 | 0.10 | 8.87×10−4 |
| − | down | 0.32 | 0.69 | 4.69×10−3 | |
| Foundress vs others | + | up | 0.05 | 0.13 | 0.17 |
| − | down | 0.11 | 3.49×10−10 | 6.27×10−11 | |
| Callow vs others | + | up | 0.99 | ∗ | ∗ |
| − | down | 1.14×10−6 | 2.77×10−7 | 3.34×10−6 |
∗: < 2.2×10−16. t: the t test, Wilcox: the Wilcoxon rank sum test, KS: the Kolmogorov–Sinai test, all are two-sided
TD-based unsupervised FE applied to the integrated analysis of gene expression and methylation profiles
By PCA-based unsupervised FE, we successfully identified genes associated with gene expression and methylation profiles showing significant differences between castes. Genes found by means of gene expression profiles and methylation profiles did not overlap. Fisher’s test results yielded an odds ratio lower than 1.0 (data not shown because of negative results). This finding indicated that the genes identified using different datasets are quite distinct. Therefore, it is difficult to understand the mechanisms by which the epigenetic modifications affect gene expression and regulate phenotype development.
One may wonder if a gene expression alteration must not always be associated with altered methylation. Nevertheless, many authors have employed the strategy where genes associated with both altered gene expression and methylation are sought, to identify biologically important genes. Heng et al. [43, 44] have tried to find genes associated with both altered gene expression and methylation to discover genes crucial for breast cancers. Li et al. [45] have attempted to find genes associated with both altered gene expression and methylation to identify key genes in severe oligozoospermia. Mallik et al. [46] have looked for genes associated with both altered gene expression and methylation for tumor prediction. These are only a few examples of studies that involve the association of altered gene expression and methylation. Thus, altered gene expression and promoter methylation may have to be considered together to identify genes specific for castes of social insects, too.
To explore the possibility of correlating gene expression and methylation profiles via our strategy, we applied TD-based unsupervised FE to tensor xijℓ=xijxiℓ, where i represents a gene, xij is the gene expression of the jth sample, and xiℓ denotes methylation of the ℓth sample. In Fig. 6(a), the first sample singular value vector for gene expression, (at k2=1) for P. canadensis is shown, which has the most significant dependence upon class labels based upon categorical regression. Because the presented results are similar to those shown in Fig. 3(a), TD-based unsupervised FE was demonstrated to successfully generate biologically relevant singular value vectors for gene expression, (at k2=1). Similarly, results depicted in Fig. 6(b) represent the third sample singular value vectors for the methylation profiles of P. canadensis, (with k3=3), which were also found to be similar to those presented in Fig. 4(a). This finding indicates that TD-based unsupervised FE can successfully generate biologically relevant sample singular value vectors.
Fig. 6.
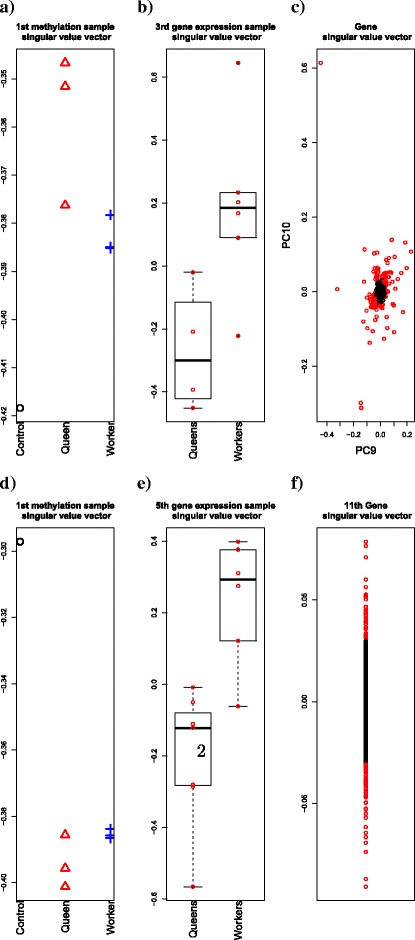
TD analysis of gene expression profiles. TD-based unsupervised FE was applied to gene expression and methylation profiles of P. canadensis (a, b, c)and D. quadriceps (d, e, f). a: The first sample singular value vector for methylation profiles (with k3=1), (b) A boxplot of the third sample singular value vectors for gene expression profiles, (with k2=3). c The ninth and 10th gene singular value vector, (with 9≤k1≤10). Red areas in (c) represent 133 selected genes. d: The first sample singular value vector for methylation profiles, (at k3=1). e A boxplot of the fifth sample singular value vectors for gene expression profiles, (at k2=5). f The 11th gene singular value vector, (at k1=11). Red areas in (f) represent 128 selected genes
Next, we aimed to identify core tensor G(k1,k2,k3) (at (k2,k3)=(1,3)) associated with the larger absolute values, to select k1s used for the gene selection based on P. canadensis profiles (Table 5). Given that G(k1,k2,k3) (with 9≤k1≤10,(k2,k3)=(1,3)) were shown to be top-ranked, the ninth and 10th singular value vectors, (with 9≤k1≤10) were used for the selection of 133 genes, associated with the adjusted P-values, determined via Eq. (1), lower than 0.01 (Fig. 6(c)).
Table 5.
The top 10 core tensors, G, with large absolute values
| P. canadensis | D. quadriceps | ||
|---|---|---|---|
| k 1 | G(k1,k2,k3) | k 1 | G(k1,k2,k3) |
| (k2,k3)=(1,3) | (k2,k3)=(1,5) | ||
| 9 | −79.8 | 11 | −54.8 |
| 10 | 75.4 | 12 | 4.1 |
| 7 | −61.4 | 25 | 3.4 |
| 11 | 38.4 | 2 | −2.9 |
| 5 | −23.4 | 23 | 2.8 |
| 4 | −16.0 | 9 | 2.4 |
| 12 | −11.9 | 20 | −2.2 |
| 1 | −5.4 | 8 | 2.2 |
| 13 | 5.4 | 10 | −1.7 |
| 6 | −4.5 | 22 | −1.4 |
To determine whether the selected genes show differential expression and methylation between workers and queens, three statistical tests were applied (Table 6). We observed that 133 selected genes have significant differences in expression and methylation between the samples under study. Therefore, using TD-based unsupervised FE, we successfully identified a set of genes that have both differential gene expression and methylation between queens and workers; this accomplishment was not possible with the PCA-based unsupervised FE performed individually on gene expression and methylation profiles.
Table 6.
Statistical tests of the differences (between queens and workers) in gene expression and methylation
| t | Wilcox | KS | ||
|---|---|---|---|---|
| P. canadensis | gene expression | 1.71×10−3 | 1.89×10−2 | 0.08 |
| methylation | 1.74×10−4 | 5.06×10−3 | 1.02×10−3 | |
| D. quadriceps | gene expression | 2.73×10−12 | 9.05×10−12 | 4.41×10−11 |
| methylation | 0.3757 | 0.7163 | 0.4413 |
The genes identified by TD-based unsupervised FE were analyzed by t (the t test), Wilcox (the Wilcoxon rank sum test), and KS (the Kolmogorov–Sinai test), all two-sided
In Fig. 6(d), the first sample singular value vector for gene expression, (with k2=1) for D. quadriceps is presented, which was shown to have the most significant dependence upon class labels based on categorical regression. Because these results were shown to be similar to those presented in Fig. 3(c), TD-based unsupervised FE was demonstrated to successfully generate biologically relevant sample singular value vectors of gene expression, (with k2=1). Similarly, in Fig. 6(e), the fifth sample singular value vectors for the methylation profile of D. quadriceps are depicted, (with k3=5), which were found to be similar to those in Fig. 4(c). This finding indicates that TD-based unsupervised FE can lead to successful generation of biologically relevant sample singular value vectors.
Furthermore, we aimed to identify the core tensor G(k1,k2,k3) (at (k2,k3)=(1,5)) associated with the increased absolute values, to identify the k1s used for the gene selection in D. quadriceps datasets (Table 5). Because G(k1,k2,k3) (with (k1,k2,k3)=(11,1,5)) is top-ranked, the 11th gene singular value vectors, , were employed for the selection of 128 genes associated with the adjusted P-values, determined using Eq. (1), lower than 0.01 (Fig. 6(f)).
To confirm that the selected genes are associated with differential gene expression and methylation between workers and queens, three statistical tests were applied (Table 6). We demonstrated that the 128 selected genes are associated with significant differences in gene expression but not methylation between queens and workers. Therefore, by TD-based unsupervised FE, we successfully found a set of genes associated with the differential gene expression but not methylation between queens and workers, suggesting that this analysis was not successful in the case of D. quadriceps.
GO enrichment analysis
We demonstrated that PCA- and TD-based unsupervised FE can be used for the successful identification of genes associated with differential gene expression and methylation between workers and queens, but these results may be improved by showing that these sets of genes are biologically relevant as well. GO enrichment analysis was performed on three sets of genes selected on the basis of the gene expression profiles obtained from other studies (two P. canadensis datasets and one D. quadriceps dataset [8, 9], Table 7).
Table 7.
GO enrichment analysis of the genes selected using gene expression profiles
| P. canadensis | |
| Dataset provided by Patalano et al. [8] | |
| Results obtained in this study (PCA) | |
| GO:0005319 | Lipid transporter activity |
| GO:0006869 | Lipid transport |
| Results obtained in this study (TD) | |
| GO:0005319 | Lipid transporter activity |
| GO:0005811 | Lipid particle |
| GO:0006869 | Lipid transport |
| Patalano et al. [8] | |
| No enrichments | |
| Dataset provided by Ferreira et al. [9] | |
| Results obtained in this study (PCA) | |
| GO:0004129 | Cytochrome-c oxidase activity |
| GO:0003735 | Structural constituent of ribosome |
| GO:0006412 | Translation |
| GO:0005743 | Mitochondrial inner membrane |
| GO:0008137 | NADH dehydrogenase (ubiquinone) activity |
| D. quadriceps | |
| Dataset provided by Patalano et al. [8] | |
| Results obtained in this study (PCA) | |
| GO:0005506 | Iron ion binding |
| GO:0009055 | Electron carrier activity |
| GO:0016705 | Oxidoreductase activity, acting on paired donors, |
| with incorporation or reduction of molecular oxygen | |
| GO:0020037 | heme binding |
| GO:0055114 | Oxidation-reduction process |
| Results obtained in this study (TD) | |
| No enrichments | |
| Patalano et al. [8] | |
| GO:0003735 | Structural constituent of ribosome |
| GO:0005622 | Intracellular |
| GO:0005840 | Ribosome |
| GO:0005842 | Cytosolic large ribosomal subunit |
| GO:0006412 | Translation |
In contrast to the results of the other study on P. canadensis gene expression profiles [8], which showed no enrichment data, by means of the same dataset, we identified two enriched GO terms using the results obtained by PCA-based unsupervised FE. In the TD analysis, the number of enriched GO terms increased to three. These results indicate that we successfully performed the integrated analysis via TD-based unsupervised FE on P. canadensis datasets.
For D. quadriceps profiles, both genes identified by Patalano et al. [8] and those selected by PCA-based unsupervised FE were found to be associated with the same number of enriched GO terms, five, although the identified terms were not identical. Nevertheless, in the TD analysis, we did not observe any enrichment, and this result coincides with the fact that TD-based unsupervised FE failed to identify genes associated with differences in methylation profiles between queens and workers (Table 6).
Finally, in the analysis of genes identified by PCA-based unsupervised FE in the dataset provided by Ferreira et al. [9], five enriched GO terms were identified as well. Therefore, PCA- and TD-based unsupervised FE methods were shown to successfully identify biologically relevant sets of genes associated with significant enrichment in GO terms.
Discussion
Biological importance of the obtained results
We observed some instances of enrichment of GO terms, but the biological importance of our results should be examined further. In P. canadensis analysis, GO terms related to lipid transport were found to be enriched. Recently, Ament et al. [47] reported that worker honey bees undergo a socially regulated, highly stable lipid loss as part of their behavioral maturation. Given that P. canadensis is a bee species as well, the observed GO term enrichment of genes with differential gene expression and methylation profiles may be promising. Altered methylation of these genes may induce changes in gene expression that result in a highly stable lipid loss. This arrangement may enable the coexistence of multiple phenotypes.
In contrast, oxidation-reduction processes, regulated by genes expressed differently between queens and workers of D. quadriceps, have been reported to be upregulated in the queens of multiple ant species [48]. Because D. quadriceps is an ant species, this means that we correctly identified DEGs between workers and queens, via the proposed strategy. In addition to the heme-binding–associated genes, shown to be differently expressed between queens and workers of D. quadriceps, they were associated with aberrant methylation in termites [49], another species of social insects.
Furthermore, oxidoreductase activity–related genes, found to be differently expressed between queens and workers of D. quadriceps, have been reported to be expressed in the queens of honey bees too [50].
Alaux et al. [51] determined genes associated with electron carrier activity—shown to be differentially expressed between the queens and workers of D. quadriceps in this study as well—in another study, which analyzed the relation between aggressiveness and behavioral evolution in honey bees.
When genes of D. quadriceps, identified by Patalano et al. were analyzed, more general GO terms were obtained, e.g., those related to translation and ribosomes, which are unlikely to be related to social-insect–specific features. In contrast, identification of more specific GO terms and the results of other studies point to biological importance of the analyses presented here.
Future directions
This paper shows the importance of integrated analyses of gene expression and promoter methylation for finding genes that might link castes of social insects to the epigenome. At the moment, only two species were investigated, but because castes of social insects have been examined in multiple species that do not belong to even the same family from the genetic point of view [52], this approach should be extended other species. Inclusion of more species may clarify how castes of social insects have evolved and have been maintained from the standpoint of epigenetics.
Conclusions
Here, we tested newly developed PCA- and TD-based unsupervised FE for the analysis of gene expression and methylation profiles of P. canadensis and D. quadriceps. The issues observed in other studies were solved as follows:
GO enrichment analysis was performed successfully on P. canadensis gene expression profiles [8] with a strict criterion of FDR less than 0.01 (Table 7);
Genes found to be differentially expressed among four castes [9] were analyzed by means of a strict criterion of FDR less than 0.01 (Table 6);
Genes associated with differential methylation between queens and workers of P. canadensis were analyzed successfully [8] (Table 2).
Therefore, PCA- and TD-based unsupervised FE methods were successfully performed on ’omics datasets comprising gene expression and methylation profiles. The obtained sets of genes will help us understand how development of caste phenotypes is regulated epigenetically.
Additional file
Genes selected by PCA- and TD-based unsupervised FE. (XLSX 16 kb)
Acknowledgements
Not applicable.
Funding
This study was supported by KAKENHI 17K00417 and a Chuo University Specific Topics grant. Funding for the publication of this article was provided by the above KAKENHI.
Availability of data and materials
All data used in this study was obtained from GEO.
About this supplement
This article has been published as part of BMC Bioinformatics Volume 19 Supplement 4, 2018: Selected articles from the 16th Asia Pacific Bioinformatics Conference (APBC 2018): bioinformatics. The full contents of the supplement are available online at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-19-supplement-4.
Abbreviations
- FE
Feature extraction
- PC
Principal component
- PCA
Principal component analysis
- TD
Tensor decomposition
Authors’ contributions
YHT planned the project, performed all the analyses, and wrote the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares that he has no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12859-018-2068-7) contains supplementary material, which is available to authorized users.
References
- 1.Corona M, Libbrecht R, Wheeler DE. Molecular mechanisms of phenotypic plasticity in social insects. Current Opinion Insect Sci. 2016;13:55–60. doi: 10.1016/j.cois.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 2.Lockett GA, Kucharski R, Maleszka R. DNA methylation changes elicited by social stimuli in the brains of worker honey bees. Genes Brain Behavior. 2011;11(2):235–242. doi: 10.1111/j.1601-183X.2011.00751.x. [DOI] [PubMed] [Google Scholar]
- 3.Becker N, Kucharski R, Rössler W, Maleszka R. Age-dependent transcriptional and epigenomic responses to light exposure in the honey bee brain. FEBS Open Bio. 2016;6(7):622–639. doi: 10.1002/2211-5463.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peaston AE, Whitelaw E. Epigenetics and phenotypic variation in mammals. Mammalian Genome. 2006;17(5):365–374. doi: 10.1007/s00335-005-0180-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohtat D, Susztak K. Fine tuning gene expression: The epigenome. Semin Nephrol. 2010;30(5):468–476. doi: 10.1016/j.semnephrol.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Triantaphyllopoulos KA, Ikonomopoulos I, Bannister AJ. Epigenetics and inheritance of phenotype variation in livestock. Epigenetics & Chromatin. 2016; 9(1). https://doi.org/10.1186/s13072-016-0081-5. [DOI] [PMC free article] [PubMed]
- 7.Duncan EJ, Gluckman PD, Dearden PK. J Exp Zool Part B: Mol Dev Evol. 2014; 322(4):208–220. https://doi.org/10.1002/jez.b.22571. [DOI] [PubMed]
- 8.Patalano S, Vlasova A, Wyatt C, Ewels P, Camara F, Ferreira PG, Asher CL, Jurkowski TP, Segonds-Pichon A, Bachman M, González-Navarrete I, Minoche AE, Krueger F, Lowy E, Marcet-Houben M, Rodriguez-Ales JL, Nascimento FS, Balasubramanian S, Gabaldon T, Tarver JE, Andrews S, Himmelbauer H, Hughes WOH, Guigó R, Reik W, Sumner S. Molecular signatures of plastic phenotypes in two eusocial insect species with simple societies. Proc Natl Acad Sci. 2015;112(45):13970–13975. doi: 10.1073/pnas.1515937112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferreira PG, Patalano S, Chauhan R, Ffrench-Constant R, Gabaldón T, Guigó R, Sumner S. Transcriptome analyses of primitively eusocial wasps reveal novel insights into the evolution of sociality and the origin of alternative phenotypes. Genome Biol. 2013;14(2):20. doi: 10.1186/gb-2013-14-2-r20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taguchi Y-h, Iwadate M, Umeyama H, Murakami Y. Principal component analysis based unsupervised feature extraction applied to bioinformatics analysis. In: Tsai JJP, Ng K-L, editors. Computational Methods with Applications in Bioinformatics Analysis. Singapore: World Scientific; 2017. [Google Scholar]
- 11.Taguchi Y-h. microrna-mrna interaction identification in wilms tumor using principal component analysis based unsupervised feature extraction. In: 2016 IEEE 16th International Conference on Bioinformatics and Bioengineering (BIBE): 2016. p. 71–78. https://doi.org/10.1109/BIBE.2016.14.
- 12.Taguchi Y-h. Principal Components Analysis Based Unsupervised Feature Extraction Applied to Gene Expression Analysis of Blood from Dengue Haemorrhagic Fever Patients. Sci Rep. 2017;7:44016. doi: 10.1038/srep44016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taguchi Y-h. Principal component analysis based unsupervised feature extraction applied to publicly available gene expression profiles provides new insights into the mechanisms of action of histone deacetylase inhibitors. Neuroepigenetics. 2016;8:1–18. doi: 10.1016/j.nepig.2016.10.001. [DOI] [Google Scholar]
- 14.Taguchi YH, Iwadate M, Umeyama H. Principal component analysis-based unsupervised feature extraction applied to in silico drug discovery for posttraumatic stress disorder-mediated heart disease. BMC Bioinforma. 2015;16:139. doi: 10.1186/s12859-015-0574-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taguchi Y-h, Okamoto A. Principal component analysis for bacterial proteomic analysis. In: Shibuya T, Kashima H, Sese J, Ahmad S, editors. Pattern Recognition in Bioinformatics. LNCS. Heidelberg: Springer; 2012. [Google Scholar]
- 16.Ishida S, Umeyama H, Iwadate M, Taguchi Y-h. Bioinformatic Screening of Autoimmune Disease Genes and Protein Structure Prediction with FAMS for Drug Discovery. Protein Pept Lett. 2014;21(8):828–39. doi: 10.2174/09298665113209990052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinoshita R, Iwadate M, Umeyama H, Taguchi Y-h. Genes associated with genotype-specific DNA methylation in squamous cell carcinoma as candidate drug targets. BMC Syst Biol. 2014;8 Suppl 1:4. doi: 10.1186/1752-0509-8-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taguchi Y-h, Murakami Y. Principal component analysis based feature extraction approach to identify circulating microRNA biomarkers. PLoS ONE. 2013;8(6):66714. doi: 10.1371/journal.pone.0066714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taguchi Y-h, Murakami Y. Universal disease biomarker: can a fixed set of blood microRNAs diagnose multiple diseases? BMC Res Notes. 2014;7:581. doi: 10.1186/1756-0500-7-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murakami Y, Toyoda H, Tanahashi T, Tanaka J, Kumada T, Yoshioka Y, Kosaka N, Ochiya T, Taguchi Y-h. Comprehensive miRNA expression analysis in peripheral blood can diagnose liver disease. PLoS ONE. 2012;7(10):48366. doi: 10.1371/journal.pone.0048366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murakami Y, Tanahashi T, Okada R, Toyoda H, Kumada T, Enomoto M, Tamori A, Kawada N, Taguchi Y-h, Azuma T. Comparison of Hepatocellular Carcinoma miRNA Expression Profiling as Evaluated by Next Generation Sequencing and Microarray. PLoS ONE. 2014;9(9):106314. doi: 10.1371/journal.pone.0106314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murakami Y, Kubo S, Tamori A, Itami S, Kawamura E, Iwaisako K, Ikeda K, Kawada N, Ochiya T, Taguchi Y-h. Comprehensive analysis of transcriptome and metabolome analysis in Intrahepatic Cholangiocarcinoma and Hepatocellular Carcinoma. Sci Rep. 2015;5:16294. doi: 10.1038/srep16294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Umeyama H, Iwadate M, Taguchi Y-h. TINAGL1 and B3GALNT1 are potential therapy target genes to suppress metastasis in non-small cell lung cancer. BMC Genomics. 2014;15 Suppl 9:2. doi: 10.1186/1471-2164-15-S9-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taguchi Y-h, Iwadate M, Umeyama H. Heuristic principal component analysis-based unsupervised feature extraction and its application to gene expression analysis of amyotrophic lateral sclerosis data sets. In: Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), 2015 IEEE Conference On: 2015. p. 1–10. https://doi.org/10.1109/CIBCB.2015.7300274.
- 25.Taguchi Y-h, Iwadate M, Umeyama H, Murakami Y, Okamoto A. Heuristic principal component analysis-aased unsupervised feature extraction and its application to bioinformatics In: Wang B, Li R, Perrizo W, editors. Big Data Analytics in Bioinformatics and Healthcare: 2015. p. 138–162.
- 26.Taguchi Y-h. Integrative analysis of gene expression and promoter methylation during reprogramming of a non-small-cell lung cancer cell line using principal component analysis-based unsupervised feature extraction. In: Huang D-S, Han K, Gromiha M, editors. Intelligent Computing in Bioinformatics. LNCS. Heidelberg: Springer; 2014. [Google Scholar]
- 27.Taguchi Y-h. Identification of aberrant gene expression associated with aberrant promoter methylation in primordial germ cells between E13 and E16 rat F3 generation vinclozolin lineage. BMC Bioinformatics. 2015;16 Suppl 18:16. doi: 10.1186/1471-2105-16-S18-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taguchi Y-h. Identification of More Feasible MicroRNA-mRNA Interactions within Multiple Cancers Using Principal Component Analysis Based Unsupervised Feature Extraction. Int J Mol Sci. 2016;17(5):696. doi: 10.3390/ijms17050696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taguchi Y-h. Principal component analysis based unsupervised feature extraction applied to budding yeast temporally periodic gene expression. BioData Min. 2016;9:22. doi: 10.1186/s13040-016-0101-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taguchi Y-h, et al. SFRP00001 is a possible candidate for epigenetic therapy in non-small cell lung cancer. BMC Med Genomics. 2016;9 Suppl 1:28. doi: 10.1186/s12920-016-0196-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taguchi Y-h, Wang H. Genetic association between amyotrophic lateral sclerosis and cancer. Genes. 2017;8(10):243. doi: 10.3390/genes8100243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taguchi Y-h. Tensor decomposition-based unsupervised feature extraction identifies candidate genes that induce post-traumatic stress disorder-mediated heart diseases. BMC Medical Genomics. in press. [DOI] [PMC free article] [PubMed]
- 33.Taguchi Y-h. Tensor decomposition based unsupervised feature extraction identified universal nature of sequence-non-specific off-target regulation of mrna mediated by microrna transfection. BMC Med Genomics. 2017. in press. [DOI] [PMC free article] [PubMed]
- 34.Tensor decomposition/principal component analysis based unsupervised feature extraction applied to brain gene expression and methylation profiles of social insects with multiple castes. BMC Bioinformtics. 2018. in press. [DOI] [PMC free article] [PubMed]
- 35.Taguchi Y-h. Intelligent Computing Theories and Application. Heidelberg: Springer; 2017. Identification of candidate drugs for heart failure using tensor decomposition-based unsupervised feature extraction applied to integrated analysis of gene expression between heart failure and DrugMatrix datasets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taguchi Y-h. Tensor decomposition-based unsupervised feature extraction applied to matrix products for multi-view data processing. Plos ONE. 2017;12(8):0183933. doi: 10.1371/journal.pone.0183933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taguchi Y-h. Identification of candidate drugs using tensor-decomposition-based unsupervised feature extraction in integrated analysis of gene expression between diseases and drugmatrix datasets. Scientific Report. 2017. in press. [DOI] [PMC free article] [PubMed]
- 38.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995;57(1):289–300. [Google Scholar]
- 39.Lathauwer LD, Moor BD, Vandewalle J. A multilinear singular value decomposition. SIAM Journal on Matrix Analysis and Applications. 2000;21(4):1253–1278. doi: 10.1137/S0895479896305696. [DOI] [Google Scholar]
- 40.Luo D, Ding C, Huang H. Are tensor decomposition solutions unique? on the global convergence HOSVD and ParaFac algorithms. In: Advances in Knowledge Discovery and Data Mining. Springer: 2011. p. 148–159. https://doi.org/10.1007/978-3-642-20841-6_13.
- 41.Paper Wasp and Denosaur Ant Project. Accessed 18 Nov. 2017. http://wasp.crg.eu/download.html.
- 42.Uniprot. Accessed 18 Nov. 2017. http://www.uniprot.org/uploadlists/.
- 43.Heng J, Guo X, Wu W, Wang Y, Li G, Chen M, Peng L, Wang S, Dai L, Tang L, Wang J. Integrated analysis of promoter mutation, methylation and expression of AKT1 gene in chinese breast cancer patients. PLOS ONE. 2017;12(3):0174022. doi: 10.1371/journal.pone.0174022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heng J, Zhang F, Guo X, Tang L, Peng L, Luo X, Xu X, Wang S, Dai L, Wang J. Integrated analysis of promoter methylation and expression of telomere related genes in breast cancer. Oncotarget. 2017. https://doi.org/10.18632/oncotarget.16036. [DOI] [PMC free article] [PubMed]
- 45.Li Z, Zhuang X, Zeng J, Tzeng C-M. Integrated analysis of DNA methylation and mRNA expression profiles to identify key genes in severe oligozoospermia. Frontiers in Physiology. 2017; 8. https://doi.org/10.3389/fphys.2017.00261. [DOI] [PMC free article] [PubMed]
- 46.Mallik S, Mukhopadhyay A, Maulik U, Bandyopadhyay S. Integrated analysis of gene expression and genome-wide DNA methylation for tumor prediction: An association rule mining-based approach. In: 2013 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB). IEEE: 2013. https://doi.org/10.1109/cibcb.2013.6595397.
- 47.Ament SA, Chan QW, Wheeler MM, Nixon SE, Johnson SP, Rodriguez-Zas SL, Foster LJ, Robinson GE. Mechanisms of stable lipid loss in a social insect. Journal of Experimental Biology. 2011;214(22):3808–3821. doi: 10.1242/jeb.060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Warner MR, Mikheyev AS, Linksvayer TA. Genomic signature of kin selection in an ant with obligately sterile workers. Molecular Biology and Evolution. 2017;34(7):1780–1787. doi: 10.1093/molbev/msx123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glastad KM, Gokhale K, Liebig J, Goodisman MAD. The caste- and sex-specific DNA methylome of the termite zootermopsis nevadensis. Scientific Reports. 2016; 6(1). https://doi.org/10.1038/srep37110. [DOI] [PMC free article] [PubMed]
- 50.Cristino AS, Nunes FMF, Lobo CH, Bitondi MMG, Simões ZLP, da Fontoura Costa L, Lattorff HMG, Moritz RFA, Evans JD, Hartfelder K. Caste development and reproduction: a genome-wide analysis of hallmarks of insect eusociality. Insect Molecular Biology. 2006;15(5):703–714. doi: 10.1111/j.1365-2583.2006.00696.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alaux C, Sinha S, Hasadsri L, Hunt GJ, Guzman-Novoa E, DeGrandi-Hoffman G, Uribe-Rubio JL, Southey BR, Rodriguez-Zas S, Robinson GE. Honey bee aggression supports a link between gene regulation and behavioral evolution. Proceedings of the National Academy of Sciences. 2009;106(36):15400–15405. doi: 10.1073/pnas.0907043106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fischman BJ, Woodard SH, Robinson GE. Molecular evolutionary analyses of insect societies. Proceedings of the National Academy of Sciences. 2011;108(Supplement_2):10847–10854. doi: 10.1073/pnas.1100301108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genes selected by PCA- and TD-based unsupervised FE. (XLSX 16 kb)
Data Availability Statement
All data used in this study was obtained from GEO.