

Key Points
The algorithms have high sensitivity and specificity to identify patients with hemoglobin SS/Sβ0 thalassemia and acute care pain encounters.
Codes conforming to common data model are provided to facilitate adoption of algorithms and standardize definitions for EHR-based research.
Abstract
Electronic health records (EHRs) are a source of big data that provide opportunities for conducting population-based studies and creating learning health systems, especially for rare conditions such as sickle cell disease (SCD). The objective of our study is to validate algorithms for accurate identification of patients with hemoglobin (Hb) SS/Sβ0 thalassemia and acute care encounters for pain among SCD patients within EHR warehouse. We used data for children receiving care at Children’s Hospital of Wisconsin from 2013 to 2016 to test the accuracy of the 2 algorithms. The algorithm for genotype identification used composite information (blood test results, transcranial Doppler) along with diagnoses codes. Acute pain encounters were identified using diagnoses codes and further refined by using prescription of IV pain medications. Sensitivities and specificities were calculated for the algorithms. Predictive values for the algorithm to identify SCD genotype were calculated. For all assessments, the local SCD registry and patients’ charts were considered gold standards. These included 360 children with SCD, of whom 51% were females. Our algorithm to identify patients with HbSS/Sβ0 thalassemia demonstrated sensitivity of 89.9% (confidence interval [CI], 85.1%-93.7%) and specificity of 97.1% (CI, 92.7%-99.2%). This algorithm had a positive and negative predictive value of 97.9% (CI, 94.8%-99.9%) and 88.7% (CI, 82.6%-93.3%), respectively. Acute pain crises encounters were identified with a sensitivity and specificity of 95.1% (CI, 86.3%-99.0%) and 96.1% (CI, 88.3%-99.6%). This study demonstrates the feasibility to accurately identify patients with specific types of SCD and pain crises within an EHR.
Visual Abstract
Introduction
Electronic health records (EHRs) are increasingly being used by institutions across the world to continually collect patient information every time a patient makes an encounter within a health care system.1 These data, although not primarily collected for research purposes, are housed within a data repository almost on a real-time basis and offer great potential to be used for a learning health system (LHS) and population-based studies. Harnessing the information that is continually stored in EHRs can facilitate research and LHSs not only within a site but also across multiple sites nationally.
An LHS uses a feedback loop model to draw knowledge from various data sources at the patient-level to provide near real time data that allow for continuous improvement and innovation. In addition, the LHS lends itself to comparative effectiveness research conducted within a real-world setting. The EHR data repository can be particularly valuable for creating an LHS, especially for children with rare and potentially life-threatening disorders like sickle cell disease (SCD). SCD is a chronic disease diagnosed at birth affecting ∼1 out of 400 African American births. This disease is characterized by recurrent painful crises, which is one of the most common manifestation of the disease among children. The initial steps to create an LHS using EHR data, however, require accurate identification of a patient cohort and outcomes within the EHR warehouse. In addition to accurate identification of the patient cohort in SCD, it is necessary for appropriate care of the patient to correctly ascertain an individual patient’s genotype. Patients with genotypes hemoglobin (Hb) SS/Sβ0 thalassemia will be defined as sickle cell anemia throughout the text. Children with sickle cell anemia are considered to have the more severe form of disease and require specific surveillance care and monitoring of the therapy provided. For example, the National Heart, Lung, and Blood Institute guidelines for prescribing hydroxyurea and conducting annual transcranial Doppler (TCD) screens are directed toward children with these severe genotypes.2 Thus, knowledge of a patient’s genotype is eminent when tracking health outcomes and/or quality improvement efforts.
Our prior work supports identifying the cohort of patients with SCD.3 However, within these EHR data warehouses, there are no standard definitions or a common data language to identify children with sickle cell anemia. In addition, one of the most common complications for children with SCD are acute pain crises. Similarly, there are no standard data definitions to capture pain crises information within the EHRs.
The objective of this project was to test the diagnostic accuracy of common data definitions that use multiple elements of EHR data to identify children with HbSS and HbSβ0 thalassemia disease and identify acute care encounters for vaso-occlussive pain among children with SCD. The assessment of the diagnostic accuracy of these algorithms forms a critical first step for demonstrating the feasibility of using these EHR data for SCD population health research in children and building a LHS to support quality improvement endeavors.
Methods
Study design and population
This study used retrospective EHR data collected at Medical College of Wisconsin/Children’s Hospital Wisconsin in the years 2013-2016 and stored in the i2b2 data warehouse. This data warehouse contains stored data from the Epic EHR of the Children’s Hospital Wisconsin, including information on patient demographics, visit encounters, laboratory tests, diagnosis, procedures, and medications ordered. The study was deemed exempt by our institution’s review board as it involves systematic investigation for research development, testing, and evaluation and is designed to develop generalizable knowledge.
We identified children with SCD (age ≤18 years) using a previously validated and published algorithm.3 This published algorithm was slightly modified to incorporate the International Classification of Diseases (ICD), version 10 codes and is detailed in supplemental Table 1. The modified algorithm includes both ICD-9 and ICD-10 codes to identify children with SCD with a sensitivity of 93.3% and a positive predictive value of 97.9%.
Table 1.
Metrics to test the diagnostic accuracy of algorithms
| Metric name | Numerator | Denominator | Interpretation |
|---|---|---|---|
| SCD genotype algorithm | |||
| Sensitivity | Number of patients correctly identified as sickle cell anemia by the algorithm | Number of patients with sickle cell anemia as determined by chart review/registry | Ability of algorithm to identify patients with sickle cell anemia among SCD patients |
| Specificity | Number of patients correctly identified as not having sickle cell anemia by the algorithm | Number of SCD patients who did not have sickle cell anemia as determined by the registry | Ability of algorithm to identify patients without sickle cell anemia among SCD patients |
| Positive predictive value | Number of patients correctly identified as having sickle cell anemia by the algorithm | Total number of patients identified as sickle cell anemia by the algorithm | Probability of the patient to truly have sickle cell anemia if identified by the algorithm |
| Negative predictive value | Number of patients correctly identified as not having sickle cell anemia by the algorithm | Total number of patients identified as not having sickle cell anemia by the algorithm | Probability of the patient to truly not have sickle cell anemia if identified as such by the algorithm |
| Pain encounters algorithm* | |||
| Sensitivity | Number of acute care encounters correctly identified as pain encounters | Number of acute care encounter for pain as determined by chart review of the sample | Ability of algorithm to identify acute care encounters for pain |
| Specificity | Number of acute care encounters correctly identified as encounter for reasons other than pain crises | Number of acute care encounter for reasons other than pain crises as determined by chart review of the sample | Ability of algorithm to identify acute care encounters for reasons other than pain |
The diagnostic accuracy is based on random samples selected for each year.
We developed an algorithm to identify children with sickle cell anemia and another to identify acute pain crises requiring an emergency department visit or hospitalization within the pediatric SCD cohort that uses data elements conforming to the Patient Centered Clinical Network (PCORnet) common data model format. The PCORnet common data model specifies standard organization and representation of data for the PCORnet Distributed Research Network,4 enabling consistent data definitions and formats across multiple sites. The PCORnet common data model ensures harmonized data definitions are independent of EHR type, thus overcoming the limitation of interoperability across EHR vendors. The SAS programs for the 2 algorithms are provided in supplemental Data (Programs 1 and 2).
Algorithm to identify children with HbSS and HbSβ0 thalassemia disease (SCD-genotype algorithm).
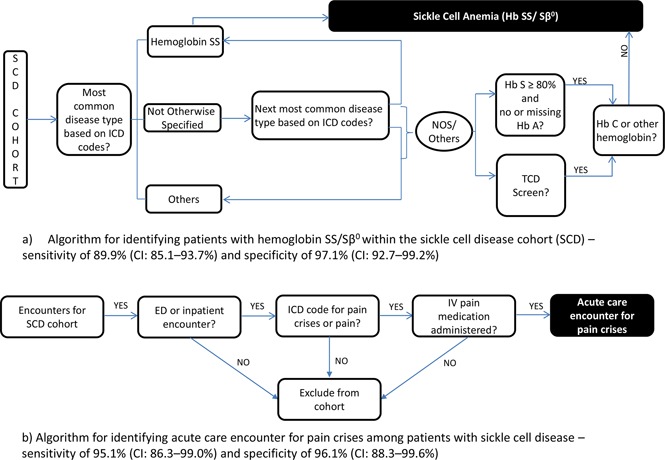
The algorithm (Figure 1A) to identify children with sickle cell anemia within the SCD cohort uses the union of the following criteria: (1) ICD-9 and ICD-10 diagnoses codes. The PCORnet table for diagnosis includes information on diagnoses codes. We specifically used the data elements of DX_TYPE, DX, DX_SOURCE for these criteria. (2) Hemoglobin identification. Results of patients – the PCORnet table for laboratory results (Lab_result_cm) has elements for identification of test using Logical Observation Identifiers Names and Codes for Hb tests (data variable LAB_LOINC) and the numerical results (variable RESULT_NUM). (3) TCD screening test. The data elements in PX, PX_TYPE in the table for procedures were used for this criteria. The specific codes for the PCORnet common data elements are listed in supplemental Table 2.
Figure 1.

Diagrammatic representation of the algorithms. (A) Algorithm for identifying patients with HbSS and HbSβ0 thalassemia disease within SCD cohort. (B) Algorithm for identifying acute care encounters for pain crises among patients with SCD. ED, emergency department; NOS, not otherwise specified.
ICD classification.
The first step in ICD classification determined patient’s genotype based on the most commonly occurring ICD code in the patient’s record. However, the ICD-10 code for Hemoglobin SS disease without crisis is the same code as Sickle Cell Disease Not Otherwise Specified (D57.1). Therefore, we used a second step to identify the patients’ genotype more specifically in this situation. The second most common code was identified and, if specific to the genotype (D57.00, Hb-SS Disease With Crisis, Unspecified; D57.01, Hb-SS Disease With Acute Chest Syndrome; D57.02 Hb-SS Disease With Splenic Sequestration; D57.20, Sickle-Cell/Hb-C Disease Without Crisis; D57.211, Sickle-Cell/Hb-C Disease With Acute Chest Syndrome; D57.212, Sickle-Cell/Hb-C Disease With Splenic Sequestration; D57.219, Sickle-Cell/Hb-C Disease With Crisis, Unspecified; D57.40 Sickle-Cell Thalassemia Without Crisis; D57.411, Sickle-Cell Thalassemia With Acute Chest Syndrome; D57.412, Sickle-Cell Thalassemia With Splenic Sequestration; D57.419, Sickle-Cell Thalassemia With Crisis; D57.80, Other Sickle-Cell Disorders Without Crisis; D57.811, Other Sickle-Cell Disorders With Acute Chest Syndrome; D57.819, Other Sickle-Cell Disorders With Crisis, Unspecified), was then used to classify the patient. If a child’s genotype still remained as SCD not otherwise specified using these steps, the laboratory and TCD criteria described below were used.
Laboratory criteria for Hb identification.
Because children with these genotypes have HbS levels higher than in other types of SCD, we used the laboratory criteria of a HbS level of ≥80% on Hb identification testing as the threshold to categorize patients as having sickle cell anemia. In addition, if a child’s laboratory test showed evidence of HbC, then the patient was classified as not having sickle cell anemia. The descriptive names for Logical Observation Identifiers Names and Codes for Hb test are listed in supplemental Table 3.
TCD criteria.
TCD screening is a test currently recommended only for those children with sickle cell anemia2; therefore, we used the criteria that having had a TCD exam classified the patients as having the more severe genotypes of SCD. The TCD exam was identified using the Current Procedural Terminology codes.
Testing of the SCD genotype algorithm
We used our locally developed registry for SCD to assess the diagnostic accuracy of the algorithm to identify children with sickle cell anemia. The local SCD registry, created by our SCD provider team, is housed within EPIC and managed by the clinical team at our institution. It includes children based on their encounter with the hematology specialty clinic and newborn screening results. This registry has been validated against the known clinic patient population and abstracted charts. In addition, the local team regularly provides oversight of the data registry to ensure quality data, including accurate specification of the genotype of patients in the registry. The provider team tracks updated information for patients who receive care at our institution; therefore, we used it as the gold standard for validating the algorithm to identify children with sickle cell anemia. The registry is designed to include patients who receive clinical care in our health system. Deceased patients are removed from the registry. In case of a mismatch between the i2b2 data warehouse and the registry data, we adjudicated patient’s genotype using the individual’s EHR. The chart abstraction was carried out in a structured format by experienced research personnel. The genotype was ascertained using the information on the newborn screening scanned document. If newborn screening was not available, then genotype ascertainment was done using complete Hb profile laboratory results and problem list diagnoses.
Algorithm to identify acute care encounters for vaso-occlusive pain crises (pain crises algorithm).
The algorithm to identify vaso-occlusive pain crises encounters within the SCD cohort used composite information based on ICD diagnoses codes and administration of IV pain medication (Figure 1B). We included generic pain ICD codes along with the ICD codes for SCD crisis (unspecified) to create a sensitive algorithm. In addition, to increase specificity we combined the ICD codes for pain with the prescription of an IV pain medication identified by RXCUI (a unique concept identifier for a normalized naming system for generic and branded drugs) or raw medication names. An encounter was identified as a pain encounter if it had an ICD code for diagnoses of SCD crisis (unspecified) or any pain, along with IV pain medication (morphine, hydromorphone or fentanyl). The PCORnet common data model tables of Diagnosis and “Prescribing” include the required information for the algorithm. The specific data elements and codes are detailed in supplemental Table 4. The ICD codes that used to identify pain diagnoses include those that have been used in prior administrative data research.5
Testing of the pain crises algorithm
The patients’ EHRs were reviewed to assess the accuracy of the algorithm used to identify acute care encounters for pain. To validate our algorithm for identification of vaso-occlusive pain episodes, we randomly selected 15 acute care encounters for pain and 15 for reasons other than pain (that is, 30 acute care encounters each year) among children with SCD. This resulted in a review of a total of 120 acute care encounters over the study period of 2013-2016 to determine the overall diagnostics of the algorithm. The random selection was done by simple random sampling such that each member had an equal chance of being included in the sample.
Statistical analyses
We determined the sensitivity and specificity of the algorithms to identify children with sickle cell anemia within the SCD cohort and acute care encounters for painful vaso-occlusive episodes. Table 1 provides the definitions and interpretations of sensitivity, specificity, positive predictive value, and negative predictive value as calculated for the respective algorithms. Exact binomial confidence intervals (CIs) (95%) were reported for all proportions. Two-by-2 contingency tables are presented to illustrate the true positive, true negative, false positive, and false negative values identified by the algorithms as compared with the chart abstractions. All analyses were carried out using SAS software version 9.4 (SAS, Inc., Cary, NC).
Results
There were 343 patients with SCD identified within the i2b2 data warehouse. The mean age of these patients by the end of study period was 8.6 years (standard deviation, 4.7 years), and 51% were females; the majority were African American (94.7%) and non-Hispanic (97.6%).
Diagnostics of SCD genotype algorithm
For identification of children with sickle cell anemia within the SCD cohort, only 75 of the 343 patients (22%) were classified as having a severe genotype using the most common ICD code for these genotypes (ICD-9: 282.61, 282.62; ICD-10: D57.00, D57.01, D57.02) in the patients’ medical records. Subsequent steps of the algorithm increased the number of children with sickle cell anemia to 192.
The local SCD registry, which was used to validate our algorithm, had 358 children with SCD. There were 2 children that were correctly identified as having SCD in the i2b2 warehouse but not included in the registry because they died during the study period and were no longer in the patient registry. Hence, the total number of SCD patients used in validation of the SCD-genotype algorithm were 360 who were ≤18 years of age, and 51% of these were females (Figure 2).
Figure 2.

Cohort of patients with SCD in the local registry and those identified in the i2b2 warehouse.
Table 2 shows the contingency table for the validation of the SCD-genotype algorithm. Of the 360 children with SCD, 209 had sickle cell anemia and 151 had other genotype SCD as per the local SCD registry/chart review. The algorithm correctly identified 188 of the 209 patients with sickle cell anemia, demonstrating a sensitivity of 89.9% (CI, 85.1%-93.7%). There were a total of 21 children who had sickle cell anemia as per the registry but were not identified by our algorithm (false negatives). Eleven out of these 21 were those who had just one visit with sickle cell diagnoses and hence were not identified in the i2b2 warehouse. The reasons for discrepancies of the remaining 10 false negatives are illustrated in Figure 3.
Table 2.
Two-by-two table for validating the SCD genotype algorithm
| Genotype based on the algorithm | Genotype based on registry/chart reviews | Do not have SCD | |
|---|---|---|---|
| HbSS/HbSβ0 thalassemia | Other sickle cell genotype | ||
| HbSS/HbSβ0 thalassemia | 188 | 4 | 0 |
| Other sickle cell genotype | 10 | 134 | 7 |
| Patients with SCD not identified by the algorithm | 11 | 13 | — |
| Total number of SCD patients | 209 | 151 | — |
Figure 3.

Classification of genotype of patients with SCD.
Among the 151 children who did not have sickle cell anemia, 138 were identified within the EHR warehouse. Most of these children (134 out of 138) were correctly classified as not having sickle cell anemia, demonstrating a specificity of 97.1% (CI, 92.7%-99.2%). The discrepancies for the 4 patients are described in Figure 3.
The positive and negative predictive values for the SCD genotype algorithm were 97.9% (CI, 94.8%-99.4%) and 88.7% (CI, 82.6%-93.3%), respectively, at our institution, wherein cell sickle cell anemia represents 58% of the population of SCD pediatric patients.
Diagnostics of pain crises encounter algorithm
The algorithm for identifying acute care encounters for pain also demonstrated a high sensitivity and specificity of 95.1% (CI, 86.3%-99.0%) and 96.6% (CI, 88.3%-99.6%), respectively. Table 3 shows the algorithm results vs the chart review as a 2-by-2 table. There were 2 encounters in the years 2013 and 2014 that were coded as SCD crises, and the patients received IV morphine. Upon review of individual patient charts, these were identified as splenic sequestration only and hence classified as false positive. Of the 3 false negatives, 1 was an encounter during which the patient had acute chest syndrome and pain crises but the associated pain crises codes were not present in the warehouse, and the other 2 were missed because only oral pain medications were used for pain management.
Table 3.
Two-by-two table for validating the pain crises algorithm
| Type of acute care encounters based on the algorithm | Type of acute care encounters based on chart review | ||
|---|---|---|---|
| For pain crises | Not for pain crises | Row total | |
| Pain crises | 58 | 2 | 60 |
| No pain crises | 3 | 57 | 60 |
| Column total | 61 | 59 | 120 |
Based on random samples of encounters selected for validation purposes.
Discussion
Our results support that the algorithms we created can identify children with sickle cell anemia within the SCD cohort and identify vaso-occlussive pain crises encounters with a high degree of accuracy. The strength of our algorithms lies in 2 areas. First, we use composite laboratory criteria such as laboratory values (HbS >80% for identification of patients with sickle cell anemia) and recommended clinical practices (TCD screens for identification of children with sickle cell anemia and IV opioid administration for identification of pain crises) along with standardized ICD codes to enhance our accuracy. Second, we base our algorithm on common data elements of the PCORnet common data model, which enables sites to adopt and implement the algorithm at their site using the SAS codes that we provide in the supplemental Data (Programs 1 and 2). In the past, the scientific community has been reluctant to use EHR and administrative data for research purposes given the limitations and inaccuracies of these data, which are primarily collected for billing purposes.6,7 Our results, however, provide the foundation needed to use the EHR data to develop an LHS for SCD.
The advancement of EHR platforms and the application of appropriate algorithms make the EHR an appealing data source for an LHS for quality improvement and research purposes. An LHS uses information from multiple sources of patient data to generate evidence in near real time and feeds it back to the clinical practice forming a continuous cycle of data to support new evidence generation and up-to-date clinical care.8-10 An exemplary prototype of such an LHS is ImproveCareNow, an inflammatory bowel disease–specific LHS.11,12 The network is a collaborative effort across 107 care centers that has resulted in quality improvement initiatives leading to better outcomes for patients with inflammatory bowel disease and has demonstrated continual improvement over time toward reaching the targeted and recommended population level outcomes. SCD, which is a rare disorder affecting an underserved population of the country, can also benefit from such a network by improving adherence to recommended care, reducing unnecessary variation in care, improving health outcomes, and communicating and sharing implementation strategies and outcomes across institutions, along with supporting research. For example, an LHS for SCD that includes accurate identification of genotype of children with SCD can help define a cohort of patients with sickle cell anemia and their adherence to hydroxyurea, annual TCD screening, and surveillance magnetic resonance imaging brain scans, which will ultimately aid in improving patient care and health outcomes. Likewise, knowledge of acute care encounters for painful vaso-occlusive crises among children with SCD is essential to understand the burden of the disease and long-term effectiveness of care. Work being done to advance the use of EHRs and incorporate additional data such as electronic patient-reported outcomes offer opportunities to include patient’s perspectives such as quality of life during a health encounter. This would help us achieve the patient-centered care goals and improve care as informed by patient-reported outcomes in an LHS.13
Although our work focuses on using EHR information for a rare disorder, it is extendible to other chronic diseases. Moreover, an LHS that incorporated multiple diseases would allow us to study and compare multiple chronic diseases and their impact on patient outcomes. The importance of computational algorithms is being increasingly recognized across disciplines in the medical field.14-18 A few examples in the pediatric field are computable phenotype to identify cohorts of patients with pulmonary hypertension (positive predictive value, 85%),14 autism spectrum disorder (positive predictive value, 86%),15 and also certain outcomes like neurological and critical care events in children with traumatic brain injury.16 These algorithms support the use of health information technology and big data to form an LHS, which many have advocated for recently.19,20 However, there are no algorithms to identify children with sickle cell anemia. Operationalization of an LHS using these algorithms provides a strong foundation for quality improvement and comparative effectiveness research.
Prior studies using large data have been done with administrative data sets21-23 and cannot identify patients' genotype. Moreover, it is well known that within administrative data sets and within the EHR, many patients with SCD have multiple genotypes coded across admissions and are often miscoded.22,24 This supports the need for the development of standard methods to accurately identify a patients’ genotype within existing data. There are ongoing efforts to create standard measures to collect data for SCD research.25 This project adds to the field by creating standard computational algorithms for using preexisting data to identify genotypes and acute pain encounters in patients with SCD. These algorithms make it possible to leverage the power of big data that stakeholders can use to understand the natural history and epidemiology of this rare disorder.
This study has a few limitations. Though the algorithms demonstrated high sensitivity and specificity at our site, these have not been tested at other institutions. However, we expect the algorithms to perform similarly given the composite criteria we incorporate to improve accuracy. These criteria include HbS/HbA levels and receipt of TCD procedures that help ensure a high sensitivity for identification of specific severe genotypes of SCD. Finally, we included administration of IV opioid along with pain and crises codes to capture vaso-occlusive pain encounters to improve errors from using ICD codes alone. We did not include codes for splenic sequestration to be specific to pain encounters which might be considered as misses by some experts in SCD. Future work involves the use natural language processing to extract information from physician’s notes to improve our algorithm and extend to other aspects of SCD such as results of imaging studies.
In conclusion, our study demonstrates accurate identification of patients with HbSS and HbSβ0 thalassemia and acute care encounters for pain using composite algorithms within an EHR warehouse. To facilitate dissemination of our work, we provide SAS codes that map our algorithms to the PCORnet common data model. These computational algorithms provide the necessary backbone to develop an LHS for SCD that incorporates EHR data from multiple institutions.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgment
The Midwest Athletes Against Childhood Cancer, Inc. Fund provided support to the study investigators (A.S. and J.A.P.).
Authorship
Contribution: A.S. designed and performed research, analyzed data, and wrote the first draft of the manuscript; J.M. investigated, researched, and performed data curation and reviewed and edited manuscript; and J.A.P. designed research, supervised research and methodology, and reviewed and edited manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ashima Singh, Department of Pediatrics, Medical College of Wisconsin, 8701 W Watertown Plank Rd, Suite 3050, Milwaukee, WI 53226; e-mail: ashimasingh@mcw.edu.
References
- 1.Henry J, Pylypchuk Y, Searcy T, Patel V. Adoption of Electronic Health Record Systems among U.S. Non-Federal Acute Care Hospitals: 2008-2015. ONC Data Brief. Washington, DC: Office of the National Coordinator for Health Information Technology; 2016. [Google Scholar]
- 2.Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(10):1033-1048. [DOI] [PubMed] [Google Scholar]
- 3.Michalik DE, Taylor BW, Panepinto JA. Identification and validation of a sickle cell disease cohort within electronic health records. Acad Pediatr. 2017;17(3):283-287. [DOI] [PubMed] [Google Scholar]
- 4.PCORnet. Common Data Model 3.1. Available at: http://www.pcornet.org/wp-content/uploads/2017/05/2017-05-01_Integrated-CDM-Specification-Implemeantion-Guidance-CDM-v....pdf. Accessed 31 January 2018.
- 5.Brousseau DC, Owens PL, Mosso AL, Panepinto JA, Steiner CA. Acute care utilization and rehospitalizations for sickle cell disease. JAMA. 2010;303(13):1288-1294. [DOI] [PubMed] [Google Scholar]
- 6.Weng CY. Data accuracy in electronic medical record documentation. JAMA Ophthalmol. 2017;135(3):232-233. [DOI] [PubMed] [Google Scholar]
- 7.Bayley KB, Belnap T, Savitz L, Masica AL, Shah N, Fleming NS. Challenges in using electronic health record data for CER: experience of 4 learning organizations and solutions applied. Med Care. 2013;51(8 suppl 3):S80-S86. [DOI] [PubMed] [Google Scholar]
- 8.Seid M, Margolis PA, Opipari-Arrigan L. Engagement, peer production, and the learning healthcare system. JAMA Pediatr. 2014;168(3):201-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greene SM, Reid RJ, Larson EB. Implementing the learning health system: from concept to action. Ann Intern Med. 2012;157(3):207-210. [DOI] [PubMed] [Google Scholar]
- 10.Olsen LA, Aisner D, and McGinnis JM IOM Roundtable on Evidence-Based Medicine. The Learning Healthcare System: Workshop Summary. Washington, DC: National Academies Press; 2007:374. [PubMed] [Google Scholar]
- 11.Improve Care Now. Available at: http://www.improvecarenow.org. Accessed 21 August 2017.
- 12.Crandall WV, Margolis PA, Kappelman MD, et al. ; ImproveCareNow Collaborative. Improved outcomes in a quality improvement collaborative for pediatric inflammatory bowel disease. Pediatrics. 2012;129(4):e1030-e1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harle CA, Lipori G, Hurley RW. Collecting, integrating, and disseminating patient-reported outcomes for research in a learning healthcare system. EGEMS (Wash DC). 2016;4(1):1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geva A, Gronsbell JL, Cai T, et al. ; Pediatric Pulmonary Hypertension Network and National Heart, Lung, and Blood Institute Pediatric Pulmonary Vascular Disease Outcomes Bioinformatics Clinical Coordinating Center Investigators. A computable phenotype improves cohort ascertainment in a pediatric pulmonary hypertension registry. J Pediatr. 2017;188:224-231.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lingren T, Chen P, Bochenek J, et al. Electronic health record based algorithm to identify patients with autism spectrum disorder. PLoS One. 2016;11(7):e0159621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennett TD, DeWitt PE, Dixon RR, et al. Development and prospective validation of tools to accurately identify neurosurgical and critical care events in children with traumatic brain injury. Pediatr Crit Care Med. 2017;18(5):442-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tasker RC. Why everyone should care about “computable phenotypes”. Pediatr Crit Care Med. 2017;18(5):489-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirby JC, Speltz P, Rasmussen LV, et al. PheKB: a catalog and workflow for creating electronic phenotype algorithms for transportability. J Am Med Inform Assoc. 2016;23(6):1046-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carroll AE. How health information technology is failing to achieve its full potential. JAMA Pediatr. 2015;169(3):201-202. [DOI] [PubMed] [Google Scholar]
- 20.Myers SR, Carr BG, Branas CC. Uniting big health data for a national learning health system in the united states. JAMA Pediatr. 2016;170(12):1133-1134. [DOI] [PubMed] [Google Scholar]
- 21.Paulukonis ST, Harris WT, Coates TD, et al. Population based surveillance in sickle cell disease: methods, findings and implications from the California registry and surveillance system in hemoglobinopathies project (RuSH). Pediatr Blood Cancer. 2014;61(12):2271-2276. [DOI] [PubMed] [Google Scholar]
- 22.Brunson A, Lei A, Rosenberg AS, White RH, Keegan T, Wun T. Increased incidence of VTE in sickle cell disease patients: risk factors, recurrence and impact on mortality. Br J Haematol. 2017;178(2):319-326. [DOI] [PubMed] [Google Scholar]
- 23.Reeves S, Garcia E, Kleyn M, et al. ; Identifying Sickle Cell Disease Cases Using Administrative Claims. Identifying sickle cell disease cases using administrative claims. Acad Pediatr. 2014;14(5 suppl):S61-S67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Snyder AB, Lane PA, Zhou M, Paulukonis ST, Hulihan MM. The accuracy of hospital ICD-9-CM codes for determining sickle cell disease genotype. J Rare Dis Res Treat. 2017. [PMC free article] [PubMed]
- 25.Eckman JR, Hassell KL, Huggins W, et al. Standard measures for sickle cell disease research: the PhenX Toolkit sickle cell disease collections. Blood Adv. 2017;1(27):2703-2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.