

Key Points
A tetravalent BsAb against CD33 treated AML human xenografts despite CD33 internalization.
BsAb with bivalent versus monolavent binding to CD33 and CD3 had more than 10-fold greater potency against leukemia.
Abstract
Acute myeloid leukemia (AML), the most common acute leukemia in adults and the second most common cancer in children, is still a lethal disease in the majority of patients, but immunologic approaches have improved outcome. Bispecific antibodies (BsAbs) are novel immunotherapeutics that can redirect immune cells against AML. We now report a tetravalent (2+2) humanized BsAb in the immunoglobulin G light chain single chain fragment variable [IgG(L)-scFv] format to engage polyclonal T cells to kill CD33+ AML targets. In vitro, this BsAb demonstrated strong antigen-specific T-cell–dependent cell-mediated cytotoxicity (TDCC) with an 50% effective concentration (EC50) in the femtomolar range that translated into treatment of established human AML IV xenografts in vivo. Importantly, it could redirect intraperitoneally injected T cells to ablate established and rapidly growing extramedullary subcutaneous AML xenografts in vivo. Furthermore, internalization of CD33 upon BsAb binding was identical to that of a bivalent (1+1) heterodimer, both being substantially less than anti-CD33 IgG. In contrast to the heterodimer, the tetravalent IgG-scFv BsAb was >10-fold more efficient in TDCC of AML cells in vitro and in vivo. This BsAb did not react with and did not kill CD38–CD34+ hematopoietic stem cells from cord blood. We conclude that the novel anti-CD33 IgG(L)-scFv BsAb construct reported here is a potential candidate for clinical development.
Visual Abstract
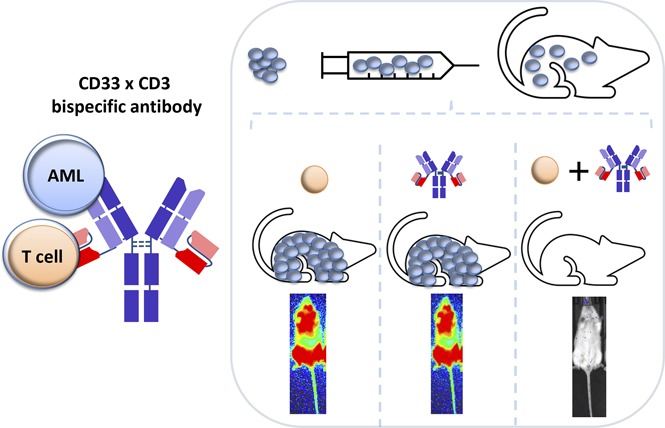
Introduction
Acute myeloid leukemia (AML) is the most common acute leukemia in adults with more than 20 000 new cases diagnosed and more than 10 000 deaths each year, in the United States alone.1 Among children, it is the second most common cancer. Contrary to cures in acute lymphoblastic leukemia in children, 5-year overall survival for all AML patients is only 15% to 27%.2,3 Antibody-based therapeutics have been developed against AML cell surface antigens. One such antigen, CD33, is a member of the sialic acid–binding immunoglobulin-like (Ig-like) lectin family expressed on the majority of AML cells.4 Importantly, CD33 is expressed in more than 87% of AML cases.5 Several anti-CD33 immunotherapeutic antibodies, including antibody-drug conjugates (ADCs), have been tested against AML. However, their toxicities and modest efficacy need to be improved. Lintuzumab, a naked IgG antibody directed at CD33, has failed in randomized clinical trials.6 Among ADCs,7,8 gemtuzumab ozogamicin (Mylotarg) has shown efficacy, although toxicity remains dose limiting.9
Bispecific antibodies (BsAbs) offer new opportunities to engage T cells in the treatment of AML.4 However, small platforms with monovalency toward the leukemia target (eg, bispecific T-cell engaging [BiTE]) suffer from fast clearance, as well as low avidity and low potency in vitro and in vivo. For antigens that endocytose (eg, CD33), multivalency could accelerate antigen loss from the cell surface. To overcome these obstacles, we generated a potent tetravalent (2+2 format) immunoglobulin G light chain single chain fragment variable [IgG(L)-scFv] humanized BsAb specific for human CD33 on AML cells and CD3 on human T cells. This BsAb (named BC133) could redirect the cytotoxic T cells against CD33+ AML cells without prior T-cell priming or HLA restriction. We tested our BsAb against AML cells in vitro and in vivo for the treatment of medullary and extramedullary AML using human AML xenografted mouse models. The potency of the BsAb was directly compared with that of an IgG heterodimeric BsAb, and the safety of our BsAb against hematopoietic stem cells (HSCs) was evaluated.
Methods
BsAbs
The murine M195 anti-CD33 antibody was humanized by grafting the heavy chain complementarity determining region sequences onto the human framework IGHV1-3*01 and IGHJ4*01 and the light chain complementarity determining region sequences onto the human framework IGKV3D11*02 and IGKJ4*01.
The anti-CD33 BsAb (BC133) was designed using heavy chain variable region fragment/light chain variable region fragment (VH/VL) domains from huM195 antibody and huOKT3 scFv fused to the C terminus of the light chain of a human IgG1 as previously described.10-12 The N297A and K322A mutations in the Fc region were made to remove glycosylation and complement binding, respectively. An IgG-based huM195×huOKT3 BsAb named heterodimer was generated using the controlled fragment antigen binding arm exchange.13 Briefly, the K409R and F405L mutations were made in the Fc domain of huM195 and huOKT3 IgG antibodies, respectively. The N297A and K322A mutations were also made in the Fc domain of these antibodies. Equimolar amounts of each IgG were mixed and reduced with 2-mercaptoethylamine at 31°C for 5 hours and then dialyzed to allow re-oxidization of the antibody into a heterodimer BsAb format. The antibody purity was determined using high performance liquid chromatography and isoelectric focusing gel.
Cytotoxicity assay (51Cr release assay)
CD33+ and CD33– leukemia cells were cultured in RPMI 1640 medium (Cellgro) supplemented with 10% fetal bovine serum (Life Technologies) at 37°C in a 5% CO2 humidified incubator. T cells were purified from human peripheral blood mononuclear cells (PBMCs) using a Pan T Cell Isolation Kit (Miltenyi Biotec). Target tumor cells were labeled with sodium 51Cr chromate (Amersham, Arlington Heights, IL) at 100 μCi/106 cells at 37°C for 1 hour. After the cells were washed twice, 5000 target cells per well were mixed with 50 000 effector cells at an effector to target (E:T) ratio of 10:1 in the presence of serial dilutions of BsAbs and incubated in 96-well polystyrene round-bottom plates (BD Biosciences) in a final volume of 250 μL per well at 37°C for 4 hours. After centrifugation at 400g for 10 minutes, the released 51Cr in the supernatant was counted in a γ-counter (Packard Bioscience Company, Downers Grove, IL). Percentage of specific lysis was calculated using the formula 100% (experimental cpm − background cpm)/(total cpm − background cpm), where cpm represented counts per minute of 51Cr released. Total release was assessed by lysis with 10% sodium dodecyl sulfate (Sigma, St. Louis, MO), and background release was measured in the absence of effector cells.
Cell lines and analysis by fluorescence-activated cell sorting
SET2-luciferase cells and MOLM13-luciferase cells were provided by David Scheinberg and Dmitry Pankov of Memorial Sloan Kettering Cancer Center, respectively. M-07e, U937, THP1, MOLT4, and HL60 cells were purchased from American Type Culture Collection. Cord and peripheral blood units were purchased from the New York Blood Center.
Antihuman antibodies against CD34, CD3, CD4, CD8, CD38, CD33, CCR7, CD45RO, and CD127 (BioLegend) were used to stain cells. Stained cells were processed with a FACSCalibur flow cytometer (BD Biosciences) and analyzed with FlowJo software (FlowJo, LLC, Ashland, OR).
In vivo studies
All mouse experiments were performed in compliance with the Institutional Animal Care and Use Committee guidelines. The immunodeficient BALB-Rag2−/−IL-2R-γc-KO (DKO) and NOD.Cg-Prkdcscid IL-2R-γtm1Wjl/SzJ (NSG) mice were purchased from Taconic Inc. (Hudson, NY) or maintained at Memorial Sloan Kettering Cancer Center and fed with Sulfatrim. For tumor challenge experiments, 6- to 10-week-old mice were inoculated with MOLM13-luciferase cells (subcutaneously with 3 × 106 cells in Matrigel [BD Biosciences] or IV with 1 × 106 cells). For the IV leukemia model, activated T cells that were in culture for 7 to 9 days were IV injected once per week for 3 weeks starting 7 days after tumor inoculation. In addition, 1000 IU of interleukin-2 (IL-2) was subcutaneously injected 2 to 3 times per week. For the subcutaneous model, PBMCs were given intraperitoneally once per week for 4 weeks. Tumor growth was measured once per week by using a Peira TM900 imaging device (Peira, Turnhout, Belgium) or an IVIS bioluminescence imaging system.
Results
Anti-CD33 BsAb (BC133) mediated strong T-cell–dependent cell-mediated cytotoxicity (TDCC) against CD33+ human leukemic cell lines in vitro
To build the tetravalent BC133, mouse M195 was re-humanized by genetic engineering and inserted into the IgG(L)-scFv cassette in which the anti-CD3 scFv was derived from huOKT3 (Figure 1A). To eliminate Fc receptor binding and complement activation, N297A and K322A mutations were introduced into the heavy chain of the BsAb Fc region. huOKT3 scFv was fused to the C terminus of the huM195 light chain using a (G4S)3 linker. The BsAb BC133 was stable after multiple freeze-thaw cycles (data not shown) and after prolonged incubation at 37°C.
Figure 1.

Anti-CD33 BsAb BC133 lysed AML cells in vitro at femtomolar EC50. (A) A schematic diagram of anti CD33×CD3 huM195 BsAb named BC133 in IgG(L)-scFv format. Heavy chains and light chains are shown in dark and light colors, respectively. (B) T-cell–mediated cytotoxicity against various CD33+ human AML cell lines in the presence of BC133 was assessed by a 4-hour chromium release assay. CD33– MOLT4 cells were used as negative control.
The specificity and potency of BC133 were tested in TDCC assays. In the presence of activated T cells in a 4-hour 51Cr release assay, all CD33+ human AML cell lines were killed (50% effective concentration [EC50] in the low picomolar to high femtomolar range); in contrast, no cytotoxicity was found against CD33– leukemic target MOLT4 (Figure 1B).
BC133 eradicated established human AML xenografts in vivo at very low BsAb doses
To test the in vivo potency of BC133, xenografts in NSG mice were established by IV injection of luciferase+ MOLM13 AML cells bearing fms-like tyrosine kinase internal tandem duplication (FLT3-ITD) mutations. Human peripheral blood T cells were activated with anti-CD3/CD28–coated microbeads for 7 days (supplemental Figure 1) and injected once per week for 3 weeks starting 7 days after leukemia implantation. T cells injected either IV or intraperitoneally were equally effective against AML. Thus, effector cells were injected intraperitoneally for the remainder of the experiments unless otherwise specified. The dose of BC133 injected 1 day before and 1 day after each T-cell administration was titrated down from 100 μg per dose to 0.01 μg per dose. Although injection of either BC133 or T cells alone did not elicit any antitumor effect, BC133 as low as 0.1 μg per dose inhibited leukemia growth, whereas 1 μg per dose was curative in the presence of human T cells (Figure 2A-B). To test whether BC133 was effective against AML with rapidly growing gross tumor burden, mice were treated with T cells and BC133 (100 μg per mouse per dose) starting ∼4 weeks after MOLM13 injection. As early as 1 week after BC133 treatment, tumor signal rapidly decreased (Figure 2C-D) and reached baseline by 4 weeks, which confirms the potency of BC133 in this in vivo AML xenograft model.
Figure 2.

High potency of BC133 in T-cell–mediated eradication of established human AML xenografts in vivo. (A-B) Female NSG mice were implanted IV with 1 million MOLM13 AML cells. Tumor growth was monitored by bioluminescence imaging (A) and expressed as total flux in photons/second (p/s) (B). Starting 7 days after leukemia implantation, activated T cells (ATCs; 5 million-10 million per dose) were injected once per week for 3 weeks. The dose of BC133 was titrated down (from 100 μg to 0.1 μg) and administered 1 day before and 1 day after each T-cell administration. To support T-cell survival in vivo, 1000 IU IL-2 was injected subcutaneously 2 to 3 times per week. Data from 2 independent experiments were pooled. (C-D) When tumor signal in mice treated with T cells only reached 1010 p/s, 100 μg per dose BC133 treatment was started (twice per week) without further T-cell injection. Tumor growth was monitored by bioluminescence imaging (C) and expressed as total flux in p/s (D).
Various BsAb platforms with different biologic properties have been designed, and some are already in the clinic.14 Unlike most BsAbs, this tetravalent (2+2) IgG(L)-scFv BsAb BC133 has 2 binding arms to CD33 on target cells and 2 to CD3 on T cells. To investigate the relevance of valency, we generated the bivalent (1+1) IgG-based BsAb named heterodimer using the controlled fragment antigen binding arm exchange method.13 This BsAb was composed of 2 half-IgG molecules against CD3 (huOKT3) and CD33 (huM195) that were preferentially paired to make an IgG heterodimer BsAb. The purity of the heterodimer was comparable to the purity of BC133 (both ∼85% by high performance liquid chromatography). To compare the potency of BC133 and the heterodimer, THP1 and MOLM13 cells were used in a killing assay. BC133 mediated TDCC of THP1 and MOLM13 AML cells 12-fold more potently than that mediated by the heterodimer (Figure 3). Next, BC133 and the heterodimer BsAb were compared in vivo. MOLM13 cells were implanted IV in NSG mice and treated on day 6 with very low doses of BsAb (0.1 μg per mouse per dose) plus activated T cells. Tumor growth was monitored by bioluminescence. Although the combination of T cells and the heterodimer was relatively ineffective in tumor suppression, being nearly identical to the T-cells-alone group, BC133 plus T cells markedly slowed down leukemia growth (Figure 4A-B). Activated T cells were cultured in the presence of IL-2, so we injected mice with IL-2 after administering T cells. The results showed that T cells redirected via BC133 had better antileukemic effect in the presence of IL-2, suggesting a beneficial role of this cytokine for in vivo T-cell function (Figure 5).
Figure 3.

The tetravalent BsAb (BC133) was more potent than the bivalent heterodimeric IgG platform (heterodimer) against human AML cells in vitro. TDCC in the presence of tetravalent BC133 vs bivalent heterodimer against CD33+ human AML cell lines was assessed by a 4-hour chromium release assay.
Figure 4.

BC133 outperformed heterodimer BsAb. Female NSG mice were implanted IV with 1 million MOLM13 AML cells. Tumor growth was monitored by bioluminescence imaging (A) and expressed as total flux in p/s (B). ATCs (5 million-10 million/dose) were injected once weekly for 3 weeks starting 7 days after leukemia implantation. BC133 or the heterodimeric BsAb (0.1 μg) was injected 1 day before and 1 day after each T cell administration. To support T-cell persistence in vivo, 1000 IU IL-2 was administered subcutaneously 2 times per week.
Figure 5.

IL-2 supports in vivo function of BC133-redirected T cells. Female NSG mice were implanted IV with 1 million MOLM13 AML cells. Tumor growth was monitored by bioluminescence imaging (A) and expressed as total flux in p/s (B). Starting 7 days after leukemia implantation, ATCs (5 million-10 million/dose) were injected once per week for 3 weeks. BC133 (10 μg) was administered 1 day before and 1 day after each T-cell administration. One group of ATC/BC133 recipients received IL-2 (1000 IU subcutaneously) 2 to 3 times per week and the other group did not receive any IL-2.
Extramedullary leukemia is a treatment challenge. We modeled this by implanting MOLM13 cells subcutaneously. Fresh human PBMCs (supplemental Figure 2) were injected intraperitoneally once per week starting 7 days after leukemia implantation, and BC133 was injected 1 day before and 1 day after each T-cell administration. In this model, PBMCs had to travel from the peritoneal cavity through the blood stream before infiltrating the subcutaneous tumor site. In the untreated or PBMC-alone control groups, tumors grew rapidly surpassing the 2000 mm3 volume that required the animals to be euthanized within the first 4 weeks after leukemia implantation. Among the treatment groups, after the initial growth spurt, there was a rapid regression in the PBMC/BC133 group (Figure 6A). To test the efficacy of BC133 against other AML xenografts, THP1 and HL60 cell lines were injected subcutaneously, and after 1 week, treatment with PBMCs with or without the BsAb was performed. BC133-redirected PBMCs significantly slowed down leukemia growth whereas PBMCs alone had minimal or no antitumor effect (Figure 6B-C). Collectively, these data supported the potency of tetravalent IgG(L)-scFv vs bivalent IgG heterodimer.
Figure 6.

BC133 is effective against CD33+AML in lymphoma models. (A) Female NSG mice were implanted subcutaneously with 3 million MOLM13 AML cells. PBMCs (10 million-30 million per dose) were injected once per week for 4 weeks starting at 7 days after leukemia injection. BC133 (50 μg per dose for the first 3 weeks and 150 μg per dose for the remaining time) were injected 1 day before and 1 day after each PBMC administration. No IL-2 was given to the mice. (B-C) Female NSG mice were implanted subcutaneously with 2 million THP1 cells (B) or 1 million HL60 AML cells (C). PBMCs (10 million per dose) were injected once per week for 4 weeks starting at 7 days after leukemia injection. BC133 (100 μg per dose) was injected 1 day before and 1 day after each PBMC administration. No IL-2 was given to the mice.
CD33 internalization did not compromise BC133 function
CD33 is internalized and will compromise engagement of effector cells after antibody binding. Although this phenomenon is beneficial for antibody-drug conjugates, it will render BsAbs ineffective. It is also assumed that monovalency has to be maintained toward CD33 (eg, BiTE or heterodimer) because crosslinking of CD33 will accelerate its endocytosis. MOLM13 AML cells were stained with huM195 (bivalent toward CD33), BC133 (bivalent toward CD33), or heterodimer at 37°C or 4°C (retarding internalization). Binding of BC133 and huM195 to CD33 was comparable at 4°C, but heterodimer binding was almost twofold less avid to CD33 (Figure 7A). At 37°C, both huM195 and heterodimer led to internalization of CD33 within the first 4 hours after staining, although internalization was stronger with huM195 than with the heterodimer (Figure 7B). Under the same conditions, BC133 behaved identically to the heterodimer, results that were entirely unexpected. Interestingly, BC133 binding to CD3 was 27-fold and 2.5-fold weaker than the binding of huOKT3 and heterodimer to CD3 (Figure 7C). Although this lower binding of BC133 to CD3 should reduce the likelihood of cytokine release syndrome, BC133 was more potent than the heterodimer in TDCC (Figure 3). In all of our in vivo experiments, BC133 was injected 1 day before T-cell administration. Under these conditions, internalization was expected, but the in vivo anti-AML effect was still substantial, suggesting that residual CD33 surface expression (20% for the MOLM13 cell line) was enough for the antileukemia function of BsAb.
Figure 7.

BC133 has distinct binding properties to CD33 and CD3. (A) MOLM13 cells were reacted for 30 minutes at 4°C using decreasing doses of hM195 (anti-CD33 IgG), the heterodimeric BsAb, and BC133. Cells were washed and immunostained with a fluorochrome-conjugated secondary antibody and mean fluorescence intensity (MFI) was assayed by flow cytometry. (B) MOLM13 cells were reacted for 30 minutes, 4 hours, or 24 hours at 37°C or 4°C with 1 μg/mL of huM195 (anti-CD33 IgG), the heterodimeric IgG BsAb, and BC133. Cells were washed and immunostained with a fluorochrome-conjugated secondary antibody at 4°C. After washing the unbound antibody, MFI was analyzed by flow cytometry. (C) Activated T cells were reacted for 30 minutes at 4°C with decreasing doses of huOKT3 (anti-CD3 IgG), the heterodimeric BsAb, and BC133. Cells were washed and immunostained with a fluorochrome-conjugated secondary antibody, and MFI was measured by flow cytometry.
CD33 was not expressed on cord blood HSCs
CD33 is considered to be a myeloid lineage–specific marker, and treatment with anti-CD33 antibodies may compromise long-term hematopoiesis.15,16 When cord blood CD34+ cells were stained with anti-CD33 BsAb (Figure 8A), almost all of the CD34+CD38– HSCs were negative, whereas all CD34+CD38+ hematopoietic progenitor cells were positive. Even for hematopoietic progenitor cells, the expression of CD33 was more than 20-fold lower than that on myeloid cells. CD34+ cells isolated from cord blood were tested for sensitivity to TDCC in a standard chromium release assay. BC133 lysed MOLM13 cells, but the CD34+ population was relatively insensitive to TDCC (Figure 8B). The low-level killing (<5%) seen at high concentrations (1 μg/mL) of BC133 could be accounted for by the residual CD34–CD33+ population (CD33 expression on this population was higher than that on CD34+CD38+ hematopoietic progenitor cells) (Figure 8C).
Figure 8.

BC133 did not cross react with CD34+CD38–HSCs. (A) Cord blood mononuclear cells were purified using Ficoll-Paque density gradient centrifugation and were immunostained with anti-human CD3, CD19, CD38, CD34, and BC133 antibodies. To exclude T cells and B cells from analysis, cells were gated on CD3– and CD19– populations. Different populations of cells (labeled 1 to 6) were assessed for their binding to BC133. (B) HSCs and progenitor cells were isolated from cord blood mononuclear cells using Miltenyi CD34 Microbeads. (C) TDCC by ATC (E:T ratio, 10) in the presence of BC133 against the purified CD34+ cells and MOLM13 AML cells was tested by using chromium release assay.
Discussion
In the last decade, there has been a surge in the development of BsAbs against human cancers, demonstrated in >60 different formats for a variety of cancer diagnoses.14,17 The key advantage of T-cell–based BsAbs over naked IgG or ADCs is the engagement of these professional killers against tumors with the potential to combine with immune checkpoint modulators. For AML, an effective T-cell–based curative strategy remains an unmet need. Our preclinical in vitro and in vivo data demonstrated the potency of the anti CD3×CD33 IgG(L)-scFv humanized BsAb BC133 in redirecting T cells administered IV or intraperitoneally against AML without clinical adverse effects.
For AML, several such BsAbs have been reported, although only a few are in phase 1 clinical trials.4 AMG330 is a CD3×CD33 BiTE.18 As a class of BsAbs, BiTE size is below the renal threshold, resulting in short in vivo half-lives necessitating continuous infusion over several weeks. Similarly, MGD006, a CD123×CD3 dual‐affinity retargeting BsAb, has a short in vivo half-life because of its small molecular size. A CD123×CD3 IgG heterodimer has the size of an IgG and is expected to have a longer half-life in vivo. However, all 3 BsAbs bind to their target antigen (CD33 or CD123) with monovalent affinity. In this study, we performed a side-by-side comparison of our IgG(L)-scFv (BC133) with the IgG heterodimer. In vitro, BC133 was greater than 10-fold more potent than the heterodimer, which translates to greater in vivo efficacy against AML. This superior functionality was not the result of stronger binding to effector cells because the reactivity of T cells with BC133 was weaker than with the heterodimer. In contrast, bivalent binding of BC133 to CD33+ AML targets was much stronger than the monovalent binding of heterodimer. These results were unexpected, given the fact that CD33 is an internalizing antigen upon binding to antibodies. In vitro, we showed that both BC133 and the heterodimer triggered similar levels of antigen endocytosis, although substantially less than anti-CD33 IgG. The 20% antigen retention on the cell surface was enough to allow highly effective TDCC in vitro. In the murine xenograft model, BsAb was administered 24 hours before T-cell injection. CD33 internalization was expected to be complete by 4 hours after antibody injection. The remaining surface CD33 density was adequate for the highly effective anti-AML effect. This bivalent IgG(L)-scFv platform could provide more potent anti-AML functionality than the univalent heterodimer format, an observation that could be highly relevant for designing other antileukemia BsAbs if it is generalizable.
To achieve a cure against cancer, targeting cancer stem cells is crucial. Depending on the type of leukemia, transformation can be traced back to either pluripotent or more mature progenitor stages.19 AML derived from mature progenitor cells has higher and more homogenous expression of CD33.19 Successful clinical trials that use gemtuzumab ozogamicin to treat acute promyelocytic and core binding factor leukemia suggest that these AML subtypes should be candidates for anti-CD33 therapies, including BC133.20,21 Among AMLs, higher expression of CD33 on those carrying the nucleophosmin (NPM1) and FLT3-ITD mutations may also render them more susceptible to anti-CD33 agents.5,22,23 However, despite the wide expression of CD33 among AMLs, the small percentage that is antigen negative or epitope negative will likely resist single antibody–based immunotherapy. Here, the combination with other immunotherapeutics with additional specificities, other biologics, or pathway-specific small molecules could be beneficial.
The expression of CD33 on normal HSCs has been extensively examined.16,24 In this study, we tested the activity of BC133 on phenotypically defined HSCs. Although there was a low expression of CD33 on CD34+CD38+ hematopoietic progenitors, none was found on CD34+CD38– HSCs and multipotent progenitors. Both HSCs and multipotent progenitors were spared in cytotoxicity assays in the presence of BC133-mediated TDCC. Although some hematopoietic progenitor cells that express CD33 could be targeted by BC133, our results suggest that the normal HSCs can be spared while AML cells are killed. Although we did not see clinical toxicity with BC133, our mouse model does not express the CD33 that reacts with BC133, nor can we evaluate nonhematopoietic tissues such as Kupffer cells that express CD33. Given the known clinical toxicities of CD33-based therapies, on-target/off-tumor effects are expected.25 The true toxicity of BC133 will need to be determined in a human phase 1 trial.
To conclude, the IgG(L)-scFv BsAb BC133 is a promising candidate for treating medullary and subcutaneous or extramedullary AML on the basis of preclinical models. With a reassuring safety profile against HSCs in vitro, BC133 deserves to be further explored.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Hoa Tran, Dmitry Pankov, Brian Santich, Hong Xu, Jeong Park, and Linlin Wang for their technical support and Irene Cheung for reviewing and editing the manuscript.
This work was supported in part by funds from Enid A. Haupt Endowed Chair, Kids Walk for Kids with Cancer NYC, Isabella Santos Foundation, Katie Find a Cure Foundation, the Robert Steel Foundation, YmAbs Therapeutics, Inc., and National Institutes of Health, National Cancer Institute Cancer Center Support Grant P30 CA008748.
Authorship
Contribution: S.S.H. conceived the study, performed the experiments, analyzed the data, designed the figures, and wrote the manuscript; H.G. helped with antibody generation; M.N.H. and Z.W. helped with the in vivo experiments; and N.-K.V.C. conceived the study, supervised the project, analyzed the data, and wrote and edited the manuscript.
Conflict-of-interest disclosure: S.S.H. and N.-K.V.C. were named as co-inventors in a patent on CD33 bispecific antibody filed by Memorial Sloan Kettering Cancer Center (MSKCC). BC133 was licensed to YmAbs Therapeutics by MSKCC. Both MSKCC and N.-K.V.C. have financial interest in YmAbs. The remaining authors declare no competing financial interests.
Correspondence: Nai-Kong V. Cheung, Department of Pediatrics, Memorial Sloan Kettering Cancer Center, 1275 York Ave, Box 170, New York, NY 10065; e-mail: cheungn@mskcc.org.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277-300. [DOI] [PubMed] [Google Scholar]
- 2.Smith A, Howell D, Patmore R, Jack A, Roman E. Incidence of haematological malignancy by sub-type: a report from the Haematological Malignancy Research Network. Br J Cancer. 2011;105(11):1684-1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.National Cancer Institute, Surveillance, Epidemiology, and End Results Program. Cancer Stat Facts: Acute Myeloid Leukemia (AML). 2017. https://seer.cancer.gov/statfacts/html/amyl.html.
- 4.Hoseini SS, Cheung NK. Acute myeloid leukemia targets for bispecific antibodies. Blood Cancer J. 2017;7(2):e522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ehninger A, Kramer M, Röllig C, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4(6):e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sekeres MA, Lancet JE, Wood BL, et al. Randomized phase IIb study of low-dose cytarabine and lintuzumab versus low-dose cytarabine and placebo in older adults with untreated acute myeloid leukemia. Haematologica. 2013;98(1):119-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laszlo GS, Estey EH, Walter RB. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014;28(4):143-153. [DOI] [PubMed] [Google Scholar]
- 8.Borthakur G, Rosenblum MG, Talpaz M, et al. Phase 1 study of an anti-CD33 immunotoxin, humanized monoclonal antibody M195 conjugated to recombinant gelonin (HUM-195/rGEL), in patients with advanced myeloid malignancies. Haematologica. 2013;98(2):217-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersdorf SH, Kopecky KJ, Slovak M, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121(24):4854-4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu H, Cheng M, Guo H, Chen Y, Huse M, Cheung NK. Retargeting T cells to GD2 pentasaccharide on human tumors using Bispecific humanized antibody. Cancer Immunol Res. 2015;3(3):266-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng M, Ahmed M, Xu H, Cheung NK. Structural design of disialoganglioside GD2 and CD3-bispecific antibodies to redirect T cells for tumor therapy. Int J Cancer. 2015;136(2):476-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiao P, Otto M, Geng Q, et al. Enhancing both CT imaging and natural killer cell-mediated cancer cell killing by a GD2-targeting nanoconstruct. J Mater Chem B Mater Biol Med. 2016;4(3):513-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Labrijn AF, Meesters JI, Priem P, et al. Controlled Fab-arm exchange for the generation of stable bispecific IgG1. Nat Protoc. 2014;9(10):2450-2463. [DOI] [PubMed] [Google Scholar]
- 14.Wu Z, Cheung NV. T cell engaging bispecific antibody (T-BsAb): from technology to therapeutics. Pharmacol Ther. 2018;182:161-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hauswirth AW, Florian S, Printz D, et al. Expression of the target receptor CD33 in CD34+/CD38-/CD123+ AML stem cells. Eur J Clin Invest. 2007;37(1):73-82. [DOI] [PubMed] [Google Scholar]
- 16.Taussig DC, Pearce DJ, Simpson C, et al. Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia. Blood. 2005;106(13):4086-4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017;9(2):182-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laszlo GS, Gudgeon CJ, Harrington KH, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood. 2014;123(4):554-561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walter RB, Appelbaum FR, Estey EH, Bernstein ID. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119(26):6198-6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lo-Coco F, Cimino G, Breccia M, et al. Gemtuzumab ozogamicin (Mylotarg) as a single agent for molecularly relapsed acute promyelocytic leukemia. Blood. 2004;104(7):1995-1999. [DOI] [PubMed] [Google Scholar]
- 21.Borthakur G, Faderl S, Verstovsek S, et al. Molecular response in core binding factor acute myelogenous leukemia with fludarbine, cytarabine, G-CSF and gemtuzumab ozogamicin [abstract]. Blood. 2008;112(11). Abstract 1937. [Google Scholar]
- 22.De Propris MS, Raponi S, Diverio D, et al. High CD33 expression levels in acute myeloid leukemia cells carrying the nucleophosmin (NPM1) mutation. Haematologica. 2011;96(10):1548-1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khan N, Hills RK, Virgo P, et al. Expression of CD33 is a predictive factor for effect of gemtuzumab ozogamicin at different doses in adult acute myeloid leukaemia. Leukemia. 2017;31(5):1059-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrews RG, Singer JW, Bernstein ID. Precursors of colony-forming cells in humans can be distinguished from colony-forming cells by expression of the CD33 and CD34 antigens and light scatter properties. J Exp Med. 1989;169(5):1721-1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ricart AD. Antibody-drug conjugates of calicheamicin derivative: gemtuzumab ozogamicin and inotuzumab ozogamicin. Clin Cancer Res. 2011;17(20):6417-6427. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.