

Key Points
A human monoclonal inhibitory PAR4 antibody is equally effective in platelets expressing the Ala120 or hyperreactive Thr120 PAR4 variant.
Antibody-mediated PAR4 inhibition is markedly anti-thrombotic in human blood, independent of PAR4 genotype.
Abstract
Thrombin activates human platelets via 2 protease-activated receptors (PARs), PAR1 and PAR4, both of which are antithrombotic drug targets: a PAR1 inhibitor is approved for clinical use, and a PAR4 inhibitor is in trial. However, a common sequence variant in human PAR4 (rs773902, encoding Thr120 in place of Ala120) renders the receptor more sensitive to agonists and less sensitive to antagonists. Here, we develop the first human monoclonal function-blocking antibody to human PAR4 and show it provides equivalent efficacy against the Ala120 and Thr120 PAR4 variants. This candidate was generated from a panel of anti-PAR4 antibodies, was found to bind PAR4 with affinity (KD ≈ 0.4 nM) and selectivity (no detectable binding to any of PAR1, PAR2, or PAR3), and is capable of near-complete inhibition of thrombin cleavage of either the Ala120 or Thr120 PAR4 variant. Platelets from individuals expressing the Thr120 PAR4 variant exhibit increased thrombin-induced aggregation and phosphatidylserine exposure vs those with the Ala120 PAR4 variant, yet the PAR4 antibody inhibited these responses equivalently (50% inhibitory concentration, 4.3 vs 3.2 µg/mL against Ala120 and Thr120, respectively). Further, the antibody significantly impairs platelet procoagulant activity in an ex vivo thrombosis assay, with equivalent inhibition of fibrin formation and overall thrombus size in blood from individuals expressing the Ala120 or Thr120 PAR4 variant. These findings reveal antibody-mediated inhibition of PAR4 cleavage and activation provides robust antithrombotic activity independent of the rs773902 PAR4 sequence variant and provides rationale for such an approach for antithrombotic therapy targeting this receptor.
Visual Abstract
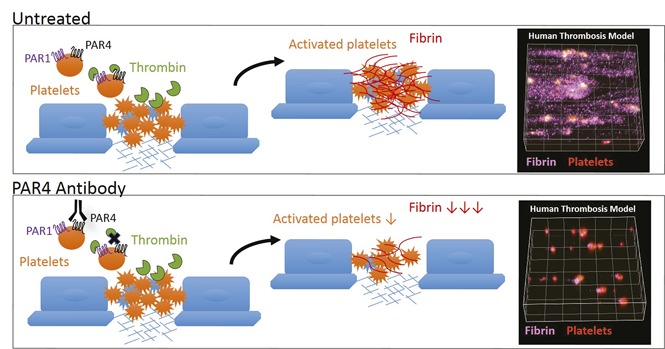
Introduction
Protease-activated receptors (PARs) are G protein-coupled receptors that are present on the surface of a range of cells and respond to a variety of proteases.1 Human platelets express 2 PARs, PAR1 and PAR4, and these receptors are primarily responsible for mediating the platelet-activating effects of the key coagulation protease, thrombin.2 Because of this central function in platelet biology, both platelet PARs have been the focus of antithrombotic drug development. PAR1 is the high-affinity thrombin receptor on human platelets, responding more sensitively and rapidly to thrombin than PAR4 as a result of a thrombin-binding domain in PAR1 that is absent in PAR4.3 On the basis of this difference, the initial clinical strategy was to block PAR1 function. This approach yielded vorapaxar, approved for the prevention of thrombotic events in patients with myocardial infarction or peripheral vascular disease when used in combination with standard-of-care therapy (aspirin and a thienopyridine such as clopidogrel).4,5 However, this triple therapy is contraindicated in patients with a history of stroke or transient ischemic attack resulting from an unacceptable increase in bleeding,6 limiting its clinical utility. We and others have recently shown that targeting PAR4 is less likely to invoke bleeding complications than targeting PAR1 because of its distinct mechanism of action and overall broader safety profile.7,8 As a result, there is now emerging interest in targeting PAR4 as a safer antithrombotic approach (for review, see French and Hamilton9 and Hamilton and Trejo10).
There is substantial rationale for developing PAR4 inhibitors as antithrombotics. One key point of distinction between PAR1 and PAR4 is the different signaling kinetics of the 2 receptors and the effect this has on the regulation of platelet function. Specifically, PAR4 contains an anionic sequence downstream of the thrombin cleavage site that serves to prolong the thrombin–receptor interaction.11 One effect of the lower-affinity but more prolonged interaction between thrombin and PAR4 vs PAR1 is that activation of PAR4 induces a more sustained, albeit weaker, intracellular signal than the robust and acute signal elicited downstream of PAR1.12 This has been most obviously observed with the kinetics of PAR-induced calcium signaling. In the setting of platelet function, prolonged calcium signaling drives the procoagulant response. Indeed, selective inhibition of PAR4, but not of PAR1, specifically impairs platelet procoagulant function, leading to marked reductions in thrombin generation and fibrin formation during human thrombus formation.8 This distinct antithrombotic mechanism of action suggests PAR4 inhibition is a viable alternative approach for novel therapy. Toward this goal, a series of small molecule PAR4 inhibitors has been developed, with at least 2 entering clinical trial. BMS-986120 afforded impressive antithrombotic activity in cynomolgous monkeys with a safety profile that exceeded that of the widely-used P2Y12 antagonist, clopidogrel,7 and was anti-thrombotic in an ex vivo human thrombosis model in healthy subjects in a recently completed phase 1 trial.13 Similarly, BMS-986141 has undergone a phase 2 trial for prevention of transient ischemic attack (NCT02671461). Together, these studies provide a strong rationale for pursuing PAR4 antagonists as novel antithrombotics.
However, recently described single nucleotide polymorphisms (SNPs) in PAR4 appear to affect the receptor’s pharmacology and may be clinically relevant. A SNP at rs773902 results in a sequence variant at amino acid position 120 (Thr120 or Ala120), whereas a SNP at rs2227346 causes a sequence variant at position 296 (Phe296 or Val296). Initial studies have focused on the SNP at rs773902 because the Thr120 variant is expressed in 20% to 80% of individuals (depending on the population)14,15 and renders the receptor more sensitive to agonists and less sensitive to antagonists.14,16 Specifically, platelets from individuals expressing the Thr120 variant of PAR4 exhibit increased responsiveness to a PAR4-selective activating peptide14,15 and resistance to the small molecule orthosteric PAR4 antagonist, YD-3, when compared with individuals expressing the Ala120 variant.14 In both cases, individuals heterozygous for the SNP displayed an intermediate phenotype. These surprising findings suggest the effectiveness of current strategies to inhibit PAR4 may vary significantly between individuals and indicate that a different approach to receptor inhibition may be required for indiscriminate PAR4 antagonism across the population.
To address this, we used VelocImmune HumAb mice to generate the first human monoclonal inhibitory antibodies against PAR4. We targeted the thrombin cleavage site of the receptor and examined the activity of antibodies against the Ala120 and Thr120 PAR4 variants in cultured cells, isolated platelets, and an ex vivo whole blood thrombosis assay. There have been a number of previous reports of function-blocking PAR4 antibodies,3,17 including our own,8 but none yet suitable for therapy. To date, the most effective inhibitory antibodies against human PAR4 are polyclonals,3,8,17 with limited inhibitory activity observed using monoclonal antibodies.18 However, our approach yielded a potent, specific, and highly effective inhibitory antibody that inhibits PAR4 and provides marked antithrombotic effects independent of the rs773902 PAR4 sequence variant. These findings reveal antibody-mediated inhibition of PAR4 cleavage and activation is sufficient to provide robust antithrombotic activity in a human thrombosis model and provide rationale for such an approach for improved antithrombotic therapy across the population when targeting PAR4.
Materials and methods
Materials
Antibodies used were Alexa Fluor 488-conjugated goat anti-mouse immunoglobulin G (IgG; BD Biosciences, San Jose, CA), mouse IgG1 isotype (Santa Cruz Biotechnology, Dallas, TX), anti-CD41a (BD Biosciences), Dylight650-conjugated anti-fibrin antibody (clone 59D819 gift from Vivien Chen, University of New South Wales, Sydney, Australia), anti-CD9-PE (BD Biosciences), and Alexa Fluor 488-conjugated annexin-V (Sigma-Aldrich, St. Louis, MO). Peptides used were hPAR4 immunizing peptide (GGDDSTPSILPAPRGYPGQVC-KLH), hPAR4 naked peptide (GGDDSTPSILPAPRGYPG QVC), hPAR4 biotinylated peptide (GDDSTPSILPAPRGYPGQVC-GGGGSKB), hPAR3 biotinylated peptide (AKPTLPIKTFRGAPPNSF-GGGGSKB), hPAR2 biotinylated peptide (SCSGTIQGTNRSSKGRSL-GGGGSKB), and hPAR1 biotinylated peptide (SKATNATLDPRS FLLRNP-GGGGSKB; all from Auspep, Melbourne, Australia). PAR4-activating peptide (PAR4-AP; AYPGKF) and PAR1-activating peptide (PAR1-AP; TFLLR) were synthesized by Philip Thompson (Monash University, Melbourne, Australia). Other reagents were vorapaxar (Axon Medchem, Reston, VA), human α-thrombin (Sigma-Aldrich), calcium ionophore A23187 (Sigma-Aldrich), bovine type 1 collagen (Sigma-Aldrich), and hirudin (lepirudin, Celgene, Summit, NJ). For thrombin measurements in whole blood, a fluorescence resonance energy transfer-based thrombin activity sensor (CPC Scientific, Sunnyvale, CA) was linked to the anti-CD41a antibody via CLICK chemistry, as previously described.20
Antibody production and purification
Immunizations were undertaken in VelocImmune HumAb mice (generously provided by Regeneron Pharmaceuticals), which have been genetically modified by replacing the variable regions of the mouse heavy and light chain Ig loci with the corresponding human sequence to produce antibodies that have human variable regions with the mouse constant regions.21,22 All mouse studies were approved by the Monash University Animal Ethics Committee. Fourteen-week-old HumAb mice were injected subcutaneously with a KLH-coupled peptide corresponding to a region spanning the thrombin cleavage site of PAR4 (GDDSTPSILPAPR/GYPGQVC-KLH, where / indicates the thrombin cleavage site; 25 µg in Freund’s complete adjuvant), with 3 boosts given every 2 weeks for 6 weeks. After the final boost, primary splenocytes were isolated and fused with myeloma Sp2/0 cells and plated onto 96-well plates to generate antibody-producing hybridomas. Supernatants from the resulting hybridomas were screened for high-affinity-specific hPAR4 antigen-positive lines using microarray (Arraviet Super Marathon, ArrayJet, Roslin, United Kingdom) and standard ELISA. Cross reactivity to hPAR1, hPAR2, and hPAR3 was also measured. Specific hPAR4 clones were subcloned to monoclonality by limiting dilution. Specific hybridoma lines were expanded and adapted to serum-free and suspension culture, and purified anti-hPAR4 antibody was isolated by standard-affinity chromatography procedures, using Protein A/G sepharose.
Surface plasmon resonance binding studies
The binding kinetics of purified antibody were determined via surface plasmon resonance, using a Bio-Rad ProteOn XPR36 array system. Analysis was performed using ProteOn NLC biosensor chips containing a NeutrAvidin surface bound to the alginate polymer for capture of biotinylated proteins and peptides. The NLC chip was conditioned with 50 mM NaOH, followed by 1 M NaCl, both at a flow rate of 30 µL/min. Biotinylated hPAR1, hPAR2, hPAR3, or hPAR4 peptides (25 µg/mL) were captured on the chip in the vertical channels at a flow rate of 30 µL/min. The chip was then rotated horizontally, and varying antibody concentrations (6.25-100 nM) were injected across channels at a flow rate of 100 µL/min. The data were analyzed using Proteon Manager software with a Langmuir interaction model that assumes a 1:1 binding interaction between the peptide and antibody.
Human blood samples
All human studies were approved by the Monash University Human Research Ethics Committee. Blood was collected after informed consent from healthy adults (21-50 years old, of both sexes) who had not taken antiplatelet medications in the last 10 days. Blood was drawn from the antecubital vein, using a 19-gauge butterfly needle, into syringes containing either one-seventh acid citrate dextrose (7:1 vol/vol, final concentration) for platelet isolation or one-tenth volume trisodium citrate (0.32% wt/vol, final concentration) for whole blood flow experiments and DNA extraction.
PAR4 genotyping
Genomic DNA was extracted from the buffy coat of blood samples collected in citrate, using the DNEasy Blood and Tissue Kit (Qiagen) as per manufacturer’s instructions. Samples were genotyped for the PAR4 SNP at rs773902 only, using a TaqMan Assay (Thermo Fisher Scientific).
Flow cytometry
Antibody binding to native PAR4 on the platelet surface was determined using flow cytometry. Platelets were isolated from human whole blood collected in acid citrate dextrose, as previously described.8 Isolated platelets (1 × 106 cells) were then incubated with anti-PAR4 antibody (10 µg/mL), anti-human CD41a (10 µg/mL), or isotype control (mouse IgG1, 10 µg/mL) for 30 minutes at 37°C. Cells were then fixed with paraformaldehyde (2% final concentration) for 30 minutes at room temperature. The suspension was centrifuged at 1000g for 2 minutes to obtain the platelet pellet, which was then resuspended in modified Tyrode’s buffer (12 mM NaHCO3, 10 mM HEPES at pH 7.4, 137 mM NaCl, 2.7 mM KCl, 5.5 mM D-glucose, 1.8 mM CaCl2, 5% bovine serum albumin) containing 10 µg/mL Alexa488-conjugated anti-mouse IgG. After 30 minutes at 37°C, the samples were centrifuged again and the platelet pellet was resuspended in modified Tyrode’s buffer and analyzed using a flow cytometer (FACSCalibur; BD Biosciences).
PAR4 cleavage assay
FLAG-tagged Ala120-PAR4 or Thr120-PAR4 expression vectors (PBJ-FLAG-PAR4-120A-296F and pBJ-FLAG-PAR4-120T-296F) were transiently transfected into HEK293T cells using Lipofectamine 2000 (Thermo Fisher Scientific), as per the manufacturer’s instructions. Note that both constructs contained the 296F sequence and varied only at position 120 (Ala120 vs Thr120). Cells were harvested 48 hours after transfection, washed twice with PBS, and resuspended to 1 × 106 cells/mL. Cells were then pretreated with either anti-PAR4 antibody (1-100 µg/mL) or matched isotype control (mouse IgG1; 100 µg/mL) for 15 minutes at 37°C, before being stimulated with thrombin (2 U/mL) for 10 minutes. The reaction was stopped with hirudin (8 U/mL). Samples were then incubated with FITC-conjugated anti-FLAG antibody for 1 hour at room temperature and analyzed by flow cytometry (FACSCalibur).
Platelet aggregation
Platelet aggregation was measured by light transmission aggregometry in a 96-well plate format, as previously described.23 Human isolated platelets (2 × 108/mL) were stimulated with varying concentrations of thrombin (0.0178-1 U/mL), PAR4-AP (10-100 μM), or PAR1-AP (3-30 μM). The plate was analyzed at 37°C in a FLUOstar OPTIMA plate reader (BMG Labtech, Ortenberg, Germany), using a 595-nm excitation filter, for a period of 50 min (10 read cycles with a 5-minute double orbital shake period between each read). Aggregation was calculated as [OD(No Agonist) − OD(Agonist)]/[OD(No Agonist) − OD(blank)] × 100 at the time point at which aggregation was at a maximum. Optical density was normalized against the blank (maximum) and unstimulated platelets (minimum) and expressed as percentage maximum. For antagonist studies, platelets were pretreated for 15 minutes at 37°C with anti-PAR4 antibody (1-100 μg/mL), vorapaxar (90 nM), a combination of both, or their vehicle controls (mouse IgG1 and 0.1% vol/vol dimethyl sulfoxide, respectively).
Phosphatidylserine exposure
Phosphatidylserine (PS) exposure on the platelet surface was used as a measure of platelet procoagulant activity. Here, isolated platelets (1 × 106 cells) were stimulated with increasing concentrations of PAR4-AP (0.1-3 mM), thrombin (0.1-3 U/mL), or calcium ionophore (20 µM) for 30 minutes at 37°C. Alexa Fluor 488-conjugated annexin V (1:100 dilution) was added after agonist stimulation for PS detection. After the 30-minute stimulation, samples were fixed with paraformaldehyde (2% final concentration) and resuspended in a modified Tyrode’s buffer for flow cytometry analysis, as earlier (FACSCalibur).
Whole-blood thrombosis assay
Human whole blood collected in citrate (3.2%) was preincubated for 15 minutes at 37°C with PE-conjuated anti-CD9 antibody (4 μg/mL), thrombin activity sensor (5 μM), and a Dylight650-conjugated anti-fibrin antibody (5 μg/mL), plus either the anti-PAR4 antibody (0.1 mg/mL) or hirudin (800 U/mL). Whole blood was recalcified with CaCl2 (10 mM final concentration) to initiate coagulation, and drawn over glass microslides (1 × 0.1 mm internal diameter; Vitrotubes, Vitrocom, Mountain Lakes, NJ) coated with bovine type 1 collagen (250 μg/mL), using a Harvard pump (Instech Laboratories, Plymouth Meeting, PA) at a fixed flow rate of 0.06 mL/min, resulting in a wall shear rate of 600 s−1. Tricolor confocal fluorescence images were recorded at 488, 561, and 647 nm excitation, collected through a 25× water immersion objective. Thrombi were formed over the course of 3 minutes of blood flow, after which calcium-free Tyrode’s buffer was flowed over the thrombi. Confocal z-stacks encompassing the entire height of the thrombus field were continuously recorded over a period of 10 minutes (Z step, 2 μm; Nikon A1r, with NIS software). Offline analysis of thrombi parameters was performed using NIS software. Image series were initially thresholded empirically, and then the same threshold applied to all subsequent experiments, as identical experimental and confocal settings were used throughout. Platelet thrombi were defined using anti-CD9-PE, and thrombin activity and fibrin volume were quantified using average fluorescence of the thrombus field. Data were normalized against the hirudin baseline and expressed as a percentage of the control.
Statistical analyses
Statistical analyses were performed using GraphPad Prism software (v6.0). Significance was defined at P < .05 as determined by either unpaired, 2-tailed, Student t test or 1-way ANOVA with Dunnett’s post hoc test for multiple comparisons, as indicated in the relevant figure legends.
Results
Development of a human monoclonal function-blocking antibody against PAR4
VelocImmune HumAb mice were immunized with a KLH-linked peptide corresponding to a region of hPAR4 spanning the thrombin cleavage and activation site (Figure 1A). Serum titers from routine bleeds were monitored, and spleens from the highest-producing mice were harvested. Splenocytes were then fused with Sp2/0 cells, and the resulting hybridomas (∼5000) were screened for binding to the naked version of the immunizing peptide by microarray and confirmed secondarily by ELISA. ELISA-based antigen screening showed that clone mAb-5A-RC3 (mAb-RC3) bound naked hPAR4 peptide with a 16-fold selectivity over any of hPAR1, hPAR2, or hPAR3 peptides (Figure 1B). Indeed, there was no detectable binding of mAb-5RC3 to any of hPAR1, hPAR2, or hPAR3 peptides above the media-only control (Figure 1B). Surface plasmon resonance confirmed binding of clone mAb-RC3 to the target antigen and estimated it bound the immunizing peptide with a KD of ∼0.4 nM (Figure 1C). We also confirmed mAb-RC3 bound native PAR4 on human platelets. Here, mAb-RC3 (10 μg/mL) bound to a purified human platelet preparation, with a rightward shift in fluorescence intensity over that produced by a mouse IgG1 isotype (Figure 1D).
Figure 1.

Development of a human monoclonal function-blocking antibody against human PAR4. (A) HumAb mice were immunized with a KLH-coupled peptide corresponding to a region in the N-terminus of hPAR4 spanning the thrombin cleavage site (GDDSTPSILPAPRGYPGQVC-KLH; bold font indicates thrombin cleavage site). (B) Initial ELISA-based antigen screening showed clone mAb-RC3-bound native human PAR4 peptide (hPAR4) with a 16-fold selectivity over human PAR1, PAR2, or PAR3 peptides; another clone (mAb-RB4) bound all 4 peptides similarly. Note the lack of binding of mAb-5RC3 to any of hPAR1, hPAR2, or hPAR3 peptides above control (media only). (C) Binding was confirmed by surface plasmon resonance analysis, which showed that mAb-RC3 bound native PAR4 peptide with a KD ∼0.4 nM. (D) mAb-RC3 (10 μg/mL) binding to human PAR4 on the platelet surface was confirmed by flow cytometry. Data are mean ± standard error of the mean in panel B and are representative traces (C-D) of n = 3 independent experiments.
mAb-RC3 inhibits thrombin cleavage of both Ala120 and Thr120 PAR4 variants
We next examined the function-blocking activity of the clone mAb-RC3 in a bioassay of thrombin-induced cleavage of PAR4. In HEK293Ts transfected with plasmids containing either Ala120-PAR4 or Thr120-PAR4 with an N-terminal FLAG-tag, thrombin caused a similar concentration-dependent loss of FLAG epitope, with a maximum cleavage of receptor of approximately 60% in both cases (Figure 2A). Under conditions of maximum PAR4 cleavage, mAb-RC3 inhibited receptor cleavage in a concentration-dependent manner (Figure 2B). This effect was not different between the Ala120 and Thr120 PAR4 variants (Figure 2B). Pretreatment with the highest concentration of mAb-RC3 examined (100 µg/mL) almost completely prevented thrombin cleavage of either PAR4 variant, with only ∼5% residual loss of the FLAG epitope detected (Figure 2B).
Figure 2.

mAb-RC3 inhibits thrombin cleavage of both Ala120 and Thr120 PAR4 variants. Thrombin cleavage of PAR4 was assessed in HEK293T cells transiently transfected with either Ala120-PAR4 or Thr120-PAR4 variants with an N-terminal FLAG tag. (A) Cells were stimulated with increasing concentrations of thrombin and receptor cleavage measured as a loss of FLAG-epitope by flow cytometry. (B) Preincubation of transfected cells with mAb-RC3 before thrombin stimulation provided equivalent concentration-dependent inhibition of thrombin cleavage of either PAR4 variant. Note near-complete inhibition of thrombin cleavage with higher concentrations of mAb-RC3. Data are mean ± standard error of the mean of n = 3 individual experiments each performed in duplicate.
mAb-RC3 inhibits the enhanced platelet aggregation elicited by the Thr120-PAR4 variant
To date, the heightened PAR4 response in platelets from individuals expressing the Thr120 PAR4 variant has only been demonstrated in response to the PAR4-specific agonist peptide, PAR4-AP.14 We first confirmed this phenotype in our local (Australian) cohort. In line with previous observations,14 we observed a significant increase in platelet aggregation induced by PAR4-AP in individuals expressing the Thr120 variant vs those expressing the Ala120 variant (Figure 3A). Platelets from heterozygous individuals displayed a similar increase in responsiveness to PAR4-AP (Figure 3A). We next determined whether this hyperreactivity in platelets from individuals with the Thr120 allele also occurred in response to the physiological PAR4 agonist, thrombin, as this has not been previously reported. Although thrombin-induced aggregation was increased in platelets from Thr120 individuals (homozygous or heterozygous) vs those from Ala120 individuals, this effect was less obvious than that observed in response to PAR4-AP (Figure 3B). To isolate the PAR4-dependant effects of thrombin in this assay, platelets were treated with the PAR1 antagonist vorapaxar at a concentration (90 nM) we have previously shown sufficiently blocks PAR1 activation under these conditions.23 Here, we observed a further dampening of the PAR4 genotype-dependent difference in platelet aggregation (Figure 3C). Regardless, mAb-RC3 caused a concentration-dependent inhibition of this thrombin-induced, PAR4-dependent, platelet aggregation. mAb-RC3 almost abolished aggregation at the highest concentrations examined (30 or 100 µg/mL), with indistinguishable effects across donors expressing the various PAR4 variants (Figure 3D; 50% inhibitory concentration [IC50] of 4.3 µg/mL for Ala120, 3.2 µg/mL for Thr120, and 6.8 µg/mL for heterozygotes) and with a combined IC50 of 5.1 µg/mL (Figure 3E). The inhibitory effect of mAb-RC3 in platelets was specific to PAR4, with no observable effects on PAR1 activation (supplemental Figure 1A-B) or cleavage (supplemental Figure 1C-D) in these cells.
Figure 3.

mAb-RC3 inhibits the enhanced platelet aggregation elicited by the Thr120-PAR4 variant. Aggregation of human isolated platelets from donors subsequently genotyped as homozygous at rs773902 for Thr120, Ala120, or as heterozygous. Shown are concentration-response curves to (A) PAR4-AP, (B) thrombin, and (C) thrombin in the presence of the PAR1 inhibitor vorapaxar (90 nM). (D) Also shown are concentration-inhibition curves to mAb-RC3 on responses to thrombin (0.1 U/mL) in the presence of vorapaxar (90 nM), indicating antibody-mediated PAR4 inhibition is equally effective across all genotypes (IC50s for Ala120, Thr120, and heterozygotes = 4.3, 3.1, and 6.8 μg/mL, respectively). (E) An IC50 for mAb-RC3 of 5.1 μg/mL was determined from the pooled data from all individuals shown in panel D. All data are mean ± standard error of the mean from n = 3 to 6 experiments per genotype. *P < .05 (1-way ANOVA with Dunnett’s test for multiple comparisons between each genotype).
mAb-RC3 inhibits the enhanced platelet procoagulant response elicited by the Thr120-PAR4 variant
The generation of procoagulant platelets is important for thrombus growth and stability,24,25 and thrombin-induced procoagulant platelet production is mediated predominantly by PAR4.8 Therefore, we next examined the sensitivity of thrombin-induced procoagulant platelet production (assessed by annexin V binding to surface-exposed PS) in donors expressing the Ala120 vs Thr120 PAR4 variant, and the ability of mAb-RC3 to inhibit this response. We observed a strikingly similar trend to the aggregation studies in Figure 3. First, PAR4-AP induced a greater increase in PS on platelets expressing the Thr120 PAR4 variant (heterozygous or homozygous) than on those expressing the Ala120 PAR4 variant (Figure 4A). Second, the same but less obvious trend was observed in response to thrombin (Figure 4B). Third, mAb-RC3 concentration-dependently inhibited thrombin-induced PS exposure independent of PAR4 genotype, again nearly abolishing the response at the highest concentrations examined (100 µg/mL) in platelets expressing either of the PAR4 rs773902 sequence variants (Figure 4C).
Figure 4.

mAb-RC3 inhibits the enhanced platelet procoagulant response elicited by the Thr120-PAR4 variant. Production of procoagulant (annexin V-binding) platelets in human isolated platelet preparations from donors subsequently genotyped as homozygous at rs773902 for Thr120 or Ala120, or as heterozygous. Shown are concentration-response curves to (A) PAR4-AP and (B) thrombin. (C) Also shown are concentration-inhibition curves to mAb-RC3 on responses to thrombin (1 U/mL), indicating antibody-mediated PAR4 inhibition is equally effective across all genotypes. All data are mean ± standard error of the mean from n = 3 to 6 experiments per genotype. *P < .05 (1-way ANOVA with Dunnett’s test for multiple comparisons between each genotype).
mAb-RC3 inhibits human thrombus formation independent of PAR4 variant expression
Finally, the antithrombotic activity of mAb-RC3 was assessed in a human whole blood thrombosis assay in which coagulation occurs.8 Specifically, we measured platelet deposition, thrombin generation, and fibrin formation in real time in blood from individuals expressing the Ala120 or Thr120 PAR4 variant (Figure 5A). Perhaps surprisingly, there was no significant difference in the extent of any of platelet deposition, thrombin generation, or fibrin deposition between donors expressing the various PAR4 variants (Figure 5B-C). Importantly, and in support of the procoagulant platelet response experiments of Figure 4, pretreatment of blood with mAb-RC3 alone was sufficient to significantly reduce thrombin generation (∼40%) and fibrin formation (∼60%) in formed thrombi (Figure 5D-E). As in the in vitro experiments, this reduction in thrombin and fibrin occurred independent of PAR4 genotype, as it was evident in blood taken from individuals expressing the Ala120 or Thr120 PAR4 sequence variant and in individuals who genotyped as heterozygous at the allele (Figure 5B-C). As predicted from the in vitro experiments in Figure 4, the effect of mAb-RC3 on fibrin formation (∼60% reduction; Figure 6C) was more pronounced than that on platelet deposition (∼30% reduction; Figure 6B). Together, the overall effect of mAb-RC3 pretreatment was a marked reduction in total thrombus volume (>50%; Figure 6D). Again, the antithrombotic effect of mAb-RC3 was independent of PAR4 genotype, occurring in 16 of 18 individuals examined (Figure 6E). Indeed, mAb-RC3 only failed to provide an antithrombotic effect in the 2 individuals with the lowest baseline thrombotic response (Figure 6E). These studies indicate that PAR4 inhibition with the function-blocking monoclonal antibody developed here affords equally effective antiplatelet and antithrombotic effects in individuals, regardless of their expression of the sequence variant of PAR4 at rs773902.
Figure 5.

mAb-RC3 inhibits platelet procoagulant activity during ex vivo human whole blood flow. (A) Representative images and (B-E) quantitation of human thrombi formed after 3 minutes of a whole blood thrombosis assay using donors subsequently genotyped as homozygous at rs773902 for Thr120 or Ala120, or as heterozygous. (A) Shown are thrombin (green, thrombin probe), fibrin (purple, anti-fibrin), and brightfield image of thrombi. Note that the direct thrombin inhibitor, hirudin (800 U/mL), abolished thrombin activity and fibrin accumulation despite continued platelet deposition. Scale bars, 20 μm. No differences in (B) thrombin activity or (C) fibrin volume were observed between thrombi formed in individuals expressing the indicated PAR4 genotypes. Pretreatment with mAb-RC3 (100 μg/mL) significantly inhibited both (D) thrombin activity and (E) fibrin formation. Note that all blood donors were pooled in panels D and E with the different genotypes highlighted by the colors as indicated. Data are mean ± standard error of the mean of N = 3 to 8 per genotype and was analyzed by 1-way ANOVA (B-C) or unpaired Student t test (D-E). *P < .05.
Figure 6.

mAb-RC3 inhibits human thrombus formation independent of PAR4 variant expression. (A) Representative 3-dimensional reconstructions and (B-E) quantitation of human thrombi formed after 3 minutes of a whole blood thrombosis assay, showing platelets (red, anti-CD9), thrombin (green, thrombin probe), and fibrin (purple, anti-fibrin). (B,C) Pretreatment of blood with mAb-RC3 (RC3, 100 μg/mL) significantly inhibited fibrin formation, but the effect on platelet deposition did not reach significance. (D) This translated to a significant reduction in total thrombus volume. Individual data points are shown. Bars are mean ± standard error of the mean of all N = 18 experiments. *P < .05; not significant (ns) (unpaired Student t test). (E) Pairwise analysis for all individuals, separated by PAR4 genotype, showing a reduction in total thrombus volume in all individuals except the 2 with the lowest baseline response.
Discussion
We have generated the first human monoclonal function-blocking antibody against the platelet thrombin receptor, PAR4. We used VelocImmune HumAb mice to generate human monoclonal antibodies directed against the thrombin cleavage site of PAR4, a strategy similar to one we have used previously to generate a specific antagonist of thrombin-induced PAR4 activation.8 One clone (mAb-RC3) demonstrated specific and high-affinity binding to PAR4, and effective inhibition of thrombin-induced PAR4 cleavage and activation that markedly improves on previous antibody-based approaches.3,17,18 Importantly, mAb-RC3 inhibits the Ala120 PAR4 variant and the hyperreactive Thr120 PAR4 variant equally well in isolated platelets and provides equivalent antithrombotic effects in blood from individuals regardless of their PAR4 genotype. These studies thereby provide a new agent for PAR4 inhibition that may have broad clinical utility.
Despite the development of a number of PAR4 inhibitors,7,13,26,27 including 2 small molecule agents that have been evaluated in early clinical trials (BMS-986120 and BMS-986141), specificity and efficacy have proven especially challenging. Binding analyses performed here indicate mAb-RC3 binds PAR4 with high affinity (KD ∼0.4 nM) and with no detectable cross reactivity to other PARs. Furthermore, mAb-RC3 appears significantly more effective than previous antibodies.3,8,17,18 In addition to the usual challenges of specificity and efficacy, the recent discovery of a clinically significant PAR4 variant (rs773902) has raised further issues for PAR4 antagonism, with the Thr120-PAR4 variant exhibiting increased sensitivity to agonists and an apparent insensitivity to at least 1 small molecule antagonist.14 The mAb-RC3 antibody developed here appears to completely overcome this. Given the prevalence of the Thr120-PAR4 variant (20%-80%, depending on the population),14,15 these studies suggest antibody-based inhibition of PAR4 may be required for effective and well-controlled antithrombotic activity across the population.
The Thr120-PAR4 variant has previously been associated with upregulated platelet responses to a PAR4 activating peptide.14 Our findings extend on this to indicate the heightened response also occurs in response to PAR4’s physiological agonist, thrombin, with platelets from individuals expressing the Thr120 PAR4 variant exhibiting increased thrombin-induced aggregation. The mechanism by which the amino acid change at position 120 (located in the second transmembrane domain) affects PAR4 function remains unknown. Our data suggest it is not a result of changes in receptor cleavage, as the cleavage-site spanning mAb-RC3 was equally effective at inhibiting PAR4 cleavage and activation in Thr120 and Ala120 PAR4 variants. Whether or not interactions at the ligand binding domain within the second extracellular loop are involved remain untested, but are consistent with the findings from us and others14 showing differences in responses to PAR4 activating peptides and small molecule antagonists that act at this site.
We have previously shown that PAR4 activation via thrombin drives procoagulant platelet activity.8 Here, we extended on these studies to provide the first evidence for a heightened production of procoagulant platelets in individuals expressing the Thr120-PAR4 variant. That this response was also inhibited by mAb-RC3 provides further rationale for its use as an antithrombotic approach. Although a similar trend of increased platelet procoagulant activity in response to PAR4-AP was observed with thrombin, consistent with our experiments examining platelet aggregation, the extent of any increase with thrombin was far less apparent than with PAR4-AP. This observation may explain the lack of correlation between PAR4 genotype and ex vivo thrombus formation observed here. We observed no differences in any of platelet deposition, thrombin generation, fibrin formation, or total thrombus volume between donors of different PAR4 genotypes. Whether the increased platelet sensitivity to thrombin is truly insufficient to cause effects in the more involved whole blood system will require a more thorough examination; for example, in a greater number of individuals and across a range of thrombogenic conditions. On this point, it is worth noting that the human ex vivo thrombosis assay we have used for these studies reveals considerable interindividual variation, which presumably reflects an individual’s multiple contributing pro- and antithrombotic factors. We did not measure an individual’s platelet count, hematocrit, coagulation factors, or cholesterol, nor did we genotype for anything other than the SNP at rs773902. Whether or not this intrinsic variation has masked any effect on human thrombus formation of the PAR4 sequence variant at position 120 remains to be seen and will require the study of a sizeable cohort.
Despite the lack of effect of the rs773902 SNP in PAR4 on thrombus formation, mAb-RC3 alone was sufficient to provide a marked antithrombotic effect in almost all donors examined. As predicted by our in vitro studies, this effect was largely driven by a reduction in platelet procoagulant activity and subsequent fibrin formation, although some contribution by impaired platelet deposition was also apparent. The marked antithrombotic effect provided by mAb-RC3 in blood from donors of all PAR4 genotypes suggests directly targeting the thrombin cleavage site may be required for effective and consistent PAR4 inhibition across the population. Given the efficacy and relative safety of PAR4 inhibition in animal models,7 determining the antithrombotic effects of mAb-RC3 in human blood in combination with other antiplatelet agents that target distinct aspects of thrombus formation (eg, aspirin, P2Y12 receptor antagonists) will be of interest.
In summary, we have developed a human monoclonal PAR4 antibody that is a highly selective and effective inhibitor of PAR4 cleavage and activation. This function-blocking PAR4 antibody impairs thrombin-induced PAR4 cleavage, platelet activation, and prothrombotic activity equivalently in the face of expression of either the Ala120 or the Thr120 PAR4 sequence variant. These findings reveal an approach to PAR4 inhibition that overcomes the impaired sensitivity of existing small molecule approaches targeting the second extracellular domain of the commonly expressed Thr120-PAR4 variant. These studies provide rationale for antibody-mediated blockade of PAR4 cleavage and activation for effective antithrombotic therapy in all individuals, regardless of PAR4 genotype.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors wish to thank Regeneron Pharmaceuticals for the provision of VelocImmune HumAb mice, Vivien Chen for the anti-fibrin antibody, James Moginie for invaluable contributions, and staff at Monash Micro Imaging and Monash Antibody Technology Facility for technical and project support.
This work was supported by grants from the National Health and Medical Research Council (#1047295) (J.R.H.) and the CASS Foundation (#SM16/7334) (J.R.H.), and funds to the Monash Antibody Technology Facility from Monash University (M.W.S.). J.R.H. is a Future Fellow of the Australian Research Council (#FT130100540).
Authorship
Contribution: S.L.F. designed and performed research, analyzed data, and wrote the paper; C.T. performed research and analyzed data; P.F.B., L.E.M., and A.J.M. designed research and contributed vital reagents; M.W.S. designed and performed research; and J.R.H. designed research and wrote the paper.
Conflict-of-interest disclosure: M.W.S. and J.R.H. hold patents relating to the antibody outlined in this study. L.E.M. and A.J.M. are employees of Regeneron Pharmaceuticals, the licence holder for the antibody outlined in this study. The remaining authors declare no competing financial interests.
Correspondence: Justin R. Hamilton, Australian Centre for Blood Diseases, Monash University, L2, AMREP Building, The Alfred, 89 Commercial Rd, Melbourne, VIC 3004, Australia; e-mail: justin.hamilton@monash.edu.
References
- 1.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3(8):1800-1814. [DOI] [PubMed] [Google Scholar]
- 2.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407(6801):258-264. [DOI] [PubMed] [Google Scholar]
- 3.Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103(6):879-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tricoci P, Huang Z, Held C, et al. ; TRACER Investigators. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N Engl J Med. 2012;366(1):20-33. [DOI] [PubMed] [Google Scholar]
- 5.Morrow DA, Braunwald E, Bonaca MP, et al. ; TRA 2P–TIMI 50 Steering Committee and Investigators. Vorapaxar in the secondary prevention of atherothrombotic events. N Engl J Med. 2012;366(15):1404-1413. [DOI] [PubMed] [Google Scholar]
- 6.French SL, Arthur JF, Tran HA, Hamilton JR. Approval of the first protease-activated receptor antagonist: Rationale, development, significance, and considerations of a novel anti-platelet agent. Blood Rev. 2015;29(3):179-189. [DOI] [PubMed] [Google Scholar]
- 7.Wong PC, Seiffert D, Bird JE, et al. . Blockade of protease-activated receptor-4 (PAR4) provides robust antithrombotic activity with low bleeding. Sci Transl Med. 2017;9(371):eaaf5294. [DOI] [PubMed] [Google Scholar]
- 8.French SL, Arthur JF, Lee H, et al. . Inhibition of protease-activated receptor 4 impairs platelet procoagulant activity during thrombus formation in human blood. J Thromb Haemost. 2016;14(8):1642-1654. [DOI] [PubMed] [Google Scholar]
- 9.French SL, Hamilton JR. Protease-activated receptor 4: from structure to function and back again. Br J Pharmacol. 2016;173(20):2952-2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamilton JR, Trejo J. Challenges and opportunities in protease-activated receptor drug development. Annu Rev Pharmacol Toxicol. 2017;57(1):349-373. [DOI] [PubMed] [Google Scholar]
- 11.Jacques SL, Kuliopulos A. Protease-activated receptor-4 uses dual prolines and an anionic retention motif for thrombin recognition and cleavage. Biochem J. 2003;376(Pt 3):733-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Covic L, Gresser AL, Kuliopulos A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry. 2000;39(18):5458-5467. [DOI] [PubMed] [Google Scholar]
- 13.Wilson SJ, Ismat FA, Wang Z, et al. . PAR4 (Protease-Activated Receptor 4) antagonism with BMS-986120 inhibits human ex vivo thrombus formation. Arterioscler Thromb Vasc Biol. 2018;38(2):448-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edelstein LC, Simon LM, Lindsay CR, et al. . Common variants in the human platelet PAR4 thrombin receptor alter platelet function and differ by race. Blood. 2014;124(23):3450-3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morikawa Y, Kato H, Kashiwagi H, et al. . Protease-activated receptor-4 (PAR4) variant influences on platelet reactivity induced by PAR4-activating peptide through altered Ca2+ mobilization and ERK phosphorylation in healthy Japanese subjects. Thromb Res. 2018;162:44-52. [DOI] [PubMed] [Google Scholar]
- 16.Edelstein LC, Simon LM, Montoya RT, et al. . Racial differences in human platelet PAR4 reactivity reflect expression of PCTP and miR-376c. Nat Med. 2013;19(12):1609-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mumaw MM, de la Fuente M, Noble DN, Nieman MT. Targeting the anionic region of human protease-activated receptor 4 inhibits platelet aggregation and thrombosis without interfering with hemostasis. J Thromb Haemost. 2014;12(8):1331-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mumaw MM, de la Fuente M, Arachiche A, Wahl JK III, Nieman MT. Development and characterization of monoclonal antibodies against Protease Activated Receptor 4 (PAR4). Thromb Res. 2015;135(6):1165-1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ivanciu L, Toso R, Margaritis P, et al. . A zymogen-like factor Xa variant corrects the coagulation defect in hemophilia. Nat Biotechnol. 2011;29(11):1028-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Welsh JD, Colace TV, Muthard RW, Stalker TJ, Brass LF, Diamond SL. Platelet-targeting sensor reveals thrombin gradients within blood clots forming in microfluidic assays and in mouse. J Thromb Haemost. 2012;10(11):2344-2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphy AJ, Macdonald LE, Stevens S, et al. . Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc Natl Acad Sci USA. 2014;111(14):5153-5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Macdonald LE, Karow M, Stevens S, et al. . Precise and in situ genetic humanization of 6 Mb of mouse immunoglobulin genes. Proc Natl Acad Sci USA. 2014;111(14):5147-5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.French SL, Paramitha AC, Moon MJ, Dickins RA, Hamilton JR. Humanizing the Protease-Activated Receptor (PAR) expression profile in mouse platelets by knocking PAR1 into the Par3 locus reveals PAR1 expression is not tolerated in mouse platelets. PLoS One. 2016;11(10):e0165565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munnix IC, Kuijpers MJ, Auger J, et al. . Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: regulation by transient integrin activation. Arterioscler Thromb Vasc Biol. 2007;27(11):2484-2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hua VM, Abeynaike L, Glaros E, et al. . Necrotic platelets provide a procoagulant surface during thrombosis. Blood. 2015;126(26):2852-2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu CC, Hwang TL, Liao CH, et al. . Selective inhibition of protease-activated receptor 4-dependent platelet activation by YD-3. Thromb Haemost. 2002;87(6):1026-1033. [PubMed] [Google Scholar]
- 27.Young SE, Duvernay MT, Schulte ML, Lindsley CW, Hamm HE. Synthesis of indole derived protease-activated receptor 4 antagonists and characterization in human platelets. PLoS One. 2013;8(6):e65528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.