

Key Points
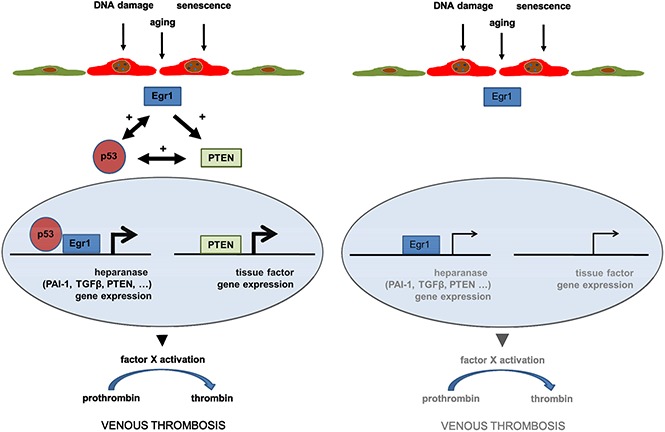
Deletion of p53 in endothelial cells prevents venous thrombosis in aged, but not in adult, mice.
Neutralization of heparanase in aged mice using TFPI2 peptides restores the thrombotic phenotype of adult mice.
Abstract
Venous thromboembolism (VTE) is a leading cause of morbidity and mortality in elderly people. Increased expression of tumor suppressor protein 53 (p53) has been implicated in vascular senescence. Here, we examined the importance of endothelial p53 for venous thrombosis and whether endothelial senescence and p53 overexpression are involved in the exponential increase of VTE with age. Mice with conditional, endothelial-specific deletion of p53 (End.p53-KO) and their wild-type littermates (End.p53-WT) underwent subtotal inferior vena cava (IVC) ligation to induce venous thrombosis. IVC ligation in aged (12-month-old) End.p53-WT mice resulted in higher rates of thrombus formation and greater mean thrombus size vs adult (12-week-old) End.p53-WT mice, whereas aged End.p53-KO mice were protected from vein thrombosis. Analysis of primary endothelial cells from aged mice or human vein endothelial cells after induction of replicative senescence revealed significantly increased early growth response gene-1 (Egr1) and heparanase expression, and plasma factor Xa levels were elevated in aged End.p53-WT, but not in End.p53-KO mice. Increased endothelial Egr1 and heparanase expression also was observed after doxorubicin-induced p53 overexpression, whereas p53 inhibition using pifithrin-α reduced tissue factor (TF) expression. Importantly, inhibition of heparanase activity using TF pathway inhibitor-2 (TFPI2) peptides prevented the enhanced venous thrombus formation in aged mice and restored it to the thrombotic phenotype of adult mice. Our findings suggest that p53 accumulation and heparanase overexpression in senescent endothelial cells are critically involved in mediating the increased risk of venous thrombosis with age and that heparanase antagonization may be explored as strategy to ameliorate the prothrombotic endothelial phenotype with age.
Visual Abstract
Introduction
Venous thromboembolism (VTE), manifested as deep vein thrombosis (DVT) or pulmonary embolism, is the third most common cardiovascular syndrome and a primary cause of mortality and morbidity in Europe, with an annual incidence of 2 to 3 cases per 1000 inhabitants per year.1,2 Predisposing factors include altered blood flow, abnormal coagulation, and endothelial dysfunction.3 Clinical studies revealed a strong, almost exponential increase of annual DVT incidence rates in the elderly,4 which cannot be simply explained by an age-associated rise in known risk factors for VTE, such as immobilization, obesity, or malignancies.5
Cellular senescence has been proposed as a mechanism underlying the age-associated structural and functional changes of the vasculature, which occur in apparently healthy subjects, are separate from atherosclerosis, and confer increased cardiovascular risk. In the arterial system, changes include increases in vessel diameter, thickness, and stiffness, and may be accompanied by endothelial dysfunction.6,7 Increased levels of apoptotic endothelial cells in the aorta and femoral arteries of old monkeys inversely correlated with endothelial cell density and vascular function.8 Age-dependent changes in the venous cross-sectional area were recently reported also in the murine venous system.9
Previous studies have shown that aging is associated with elevated levels of tumor suppressor protein 53 (p53), a key factor involved in the regulation of cell cycle arrest and apoptosis.10 Mice with a truncated p53 mutation resulting in p53 activation are protected against tumor formation, but exhibit an early-onset phenotype consistent with accelerated aging.11 Prolonged passaging (mimicking senescence) of vein endothelial cells also is associated with p53 accumulation.12 In addition to its role in DNA damage signaling, preliminary data in cells suggest that p53 may exert prothrombotic effects. For example, overexpression of p53 in endothelial cells was found to reduce the expression of nitric oxide synthase and thrombomodulin, to increase the expression of plasminogen activator inhibitor-1 (PAI-1) and to enhance ex vivo blood coagulation on an endothelial monolayer.13 However, little is known about the role of p53 in thrombosis in vivo and whether changes in endothelial p53 expression may underlie the age-related increased risk of venous thrombosis.
In this study, we examined the hypothesis that p53 expressed in endothelial cells plays a role in experimental DVT and that vascular senescence and increased levels of endothelial p53 are causally involved in the exponential increase of VTE with age.
Methods
A detailed description of all methods and material used is provided as supplemental Methods.
Experimental animals
Mice with inducible endothelial cell-specific p53 deletion (End.p53-KO) were generated, as described.14,15 Cre recombinase activity was induced with tamoxifen-containing rodent chow (TD55125; Harlan Teklad).16-18 Cre–wild-type (WT) × p53fl/fl mice fed tamoxifen were used as controls (End.p53-WT). Age- and sex-matched littermates were used throughout the study. All animal care and experimental procedures had been approved by the institutional animal research committee and the State Office for Consumer Protection and Food Safety and complied with national guidelines for the care and use of laboratory animals.
Mouse model of venous thrombosis
Venous thrombosis was induced in male mice by subtotal inferior vena cava (IVC) ligation according to Brandt et al,19 von Brühl et al,20 and Bonderman et al21 with minor modifications. Mice were anesthetized by intraperitoneal injection of a mixture of midazolam (5.0 mg/kg body weight [BW]), medetomidine (0.5 mg/kg BW), and fentanyl (0.05 mg/kg BW). A midline laparotomy incision was made and the IVC was exposed immediately below the renal veins. A 5-0 Prolene suture (Ethicon) was placed alongside the IVC as a spaceholder. A stenosis was produced by tying a 6-0 silk suture (Resorba) around the IVC. Subsequently, the spaceholder was removed to allow blood to pass the vessel occlusion. Side or back branches were not ligated. The abdominal wall was sutured using a 6-0 Ethilon suture (Ethicon). Sedation was reversed with atipamezole (0.05 mg/kg BW) and flumazenile (0.01 mg/kg BW). As analgesic, buprenorphine hydrochloride was subcutaneously injected at day 1 and day 2 postsurgery (0.075 mg/kg BW). In some experiments, 12- or 52-week-old male C57BL/6J mice (Janvier Laboratories) were subcutaneously injected with tissue factor (TF) pathway inhibitor-2 (TFPI2) peptides (2 mg/g BW in sterile phosphate-buffered saline [PBS]) or PBS alone 30 minutes before and every 24 hours after subtotal IVC ligation, according to previously described protocols.22
High-frequency ultrasound measurements
On days 1, 7, 14, and 21 after surgery, mice were anesthetized via inhalation of 2.5% isoflurane, and anesthesia was maintained with a face mask (0.5%-1.5% isoflurane). The extent of venous thrombosis was determined using high-frequency ultrasound (Vevo770 system) and a 40-MHz mouse scanhead (Visual Sonics). First, a long axis view was taken using the pulse-wave Doppler mode to visualize the blood flow in the aorta, followed by focusing on the IVC, ligation site, and/or thrombus. An optimal freeze-frame image was obtained (based on the careful assessment of real-time video sequences) and the longitudinal cross-sectional area of the clot was traced using the Vevo 770 software. Thrombus length and width were measured in triplicate in each mouse on the site of maximum cross-sectional extension of the thrombus applying B-mode and the cross-sectional area was calculated (“thrombus size”).
Histology and immunohistochemistry
Two days or 3 weeks following surgery, venous tissue starting immediately distal of the ligature until the iliac bifurcation was removed together with its surrounding tissue and fixed in 4% zinc formalin. Serial 5-µm-thick paraffin-embedded cross sections, equally spaced through the thrombosed vein segment were cut and stained with Masson trichrome (MTC) to visualize fibrin (red signal), fibrosis (blue signal), and cell nuclei (black signal). Image analysis software (Image Pro Plus; Media Cybernetics) was used to quantify the amount of red/unresolved material (count-size function) and the thrombus cross-sectional area (measure function). Per mouse, 3 sections (∼300 µm apart) were evaluated and the results averaged. Carstairs stain was used to visualize platelets (gray-blue) and erythrocytes (yellow). A schematic representation of the cutting scheme is given in supplemental Figure 1.
Immunohistochemistry was performed using monoclonal rat anti-mouse Mac2 antibodies (Biozol), rabbit anti-mouse CD3 antibodies (Abcam), rat anti-mouse CD31 antibodies (Santa Cruz Biotechnology), rat anti-mouse CD41 antibodies (EXBIO), and rat anti-mouse ELANE antibody (Abcam). Sections were counterstained with Gill's hematoxyline (Sigma), mounted in ImmuMount (Thermo Scientific), and inspected at an Olympus BX51 microscope. All morphometric analyses were performed using image analysis software (Image ProPlus, version 7.0). Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)-positive apoptotic cells were detected using the In Situ Cell Death Detection kit (Roche) and manually counted.
Confocal microscopy
Immunofluorescence staining was performed on 8-µm-thick cryosections embedded in Tissue-Tek O.C.T. (Sakura). Sections were postfixed in acetone and permeabilized in 0.05% Triton X-100 (in PBS; Roth). Sections were incubated with antibodies against CD31 (Santa Cruz Biotechnology), p53 (Abcam), heparanase-1 (Abcam), early growth response gene-1 (Egr1; Abcam), tissue factor (TF; Abcam). Cell nuclei were visualized using 4′,6-diamidine-2-phenylindole (DAPI; Roth). Sections were mounted in fluorescence mounting medium (Dako) and photographed on a confocal microscope (Leica SP8) using LAS X software. As negative control, the first antibody was omitted (a representative example is shown in supplemental Figure 2).
Tail-bleeding time
Male mice were anesthetized with 2% isoflurane, placed on a heating pad, and subjected to tail-tip amputation (∼2 mm) using surgical scissors. Tails were immediately immersed in a 50-mL tube containing 45 mL of prewarmed 0.9% NaCl solution and the time to the first and the last bleeding stop (for at least 30 seconds) recorded. At the end of the observation period of 10 minutes, the optical density of the solution was determined at 560 nm to quantify the total blood loss.
Blood cell count and plasma factor Xa activity assay
Blood was obtained from anesthetized female mice by cardiac puncture into 3.8% sodium citrate. The complete blood cell count was determined using an automated hematology analyzer (Sysmex KX21N; Sysmex). The remaining blood was centrifuged at 3000 rpm for 10 minutes; the supernatant (plasma) was removed and stored at −80°C pending analysis. Plasma factor Xa activity was measured using the Spectrozyme FXa chromogenic assay (Sekisui Diagnostics).
Cell culture studies
Human umbilical vein endothelial cells (HUVECs; PromoCell) were cultured on plates coated with 0.2% gelatin (Sigma-Aldrich) in endothelial cell growth medium (PromoCell). Cells were serially passaged until permanent growth arrest. The number of population doublings was determined according to the American Tissue Culture Collection (ATCC).23 Cells were analyzed between passage 5 and 13. Senescent cells were visualized using the Senescence Detection kit (Abcam).
Isolation of primary murine endothelial cells
Lungs were excised and diced into 1-mm-sized pieces. The tissue was digested in 1.5 mg/mL collagenase A (Worthington) at 37°C for 30 minutes, meshed through 70-µm Falcon cell strainers (Corning) and neutralized with Dulbecco modified Eagle medium (Gibco) containing 20% fetal bovine serum (Gibco). After a brief centrifugation, the cell pellet was resuspended in 1× PBS containing 0.5% fetal bovine serum and 2 mM EDTA. Nonendothelial cells were depleted using CD45 followed by selection of endothelial cells with CD31-conjugated magnetic MicroBeads and magnetic separation LS columns (Miltenyi Biotec).
Western blot analysis
Cells were resuspended in radioimmunoprecipitation assay lysis buffer (Santa Cruz Biotechnology). Equal amounts of protein were fractionated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Protran; Whatman). Membranes were blocked in 5% nonfat dry milk in Tris-buffered saline (0.1% Tween 20) followed by overnight incubation with antibodies against human or mouse CD31 (Dianova), Egr1 (Abcam), heparanase (ProSpec), p16INK4A (Abcam), p53 (Santa Cruz Biotechnology), phospho-histone 2A.X (p-H2A.X; Ser139; Cell Signaling Technology), phosphatase and tensin homolog (PTEN; Cell Signaling Technology), PAI-1 (Abcam), TF (clone EPR8986; Abcam), and transforming growth factor-β (TGFβ; Novus Biologicals). Protein bands were visualized using horseradish peroxidase–conjugated secondary antibodies (GE Healthcare) followed by SuperSignal West Pico chemiluminescent substrate (Thermo Scientific). Protein bands were quantified by densitometry and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH; HyTest Ltd).
Statistical analysis
Normal distribution was examined using the d’Agostino–Pearson omnibus normality test. When 2 groups were compared, the Student t test for unpaired means was used, if samples were normally distributed, or Mann-Whitney U test, if not. When >2 groups were compared, 1-way analysis of variance (ANOVA) followed by Bonferroni or Kruskall-Wallis followed by the Dunn multiple comparisons test were used. Nonparametric variables were examined using the Pearson χ2 test. All analyses were performed using GraphPad PRISM data analysis software (version 6.0; GraphPad Software).
Results
Aging in mice is associated with increased vein endothelial apoptosis and p53 expression
Age is associated with endothelial dysfunction and an increased risk to develop DVT. To examine the effects of age on endothelial senescence in the venous system, uninjured IVC segments were harvested from End.p53-WT mice aged 12 weeks (hereafter termed “adult”) or 12 months (hereafter termed “aged”). Histological analysis revealed no differences in total vein perimeter (Figure 1A; representative images in Figure 1D top row), but significantly lower numbers of cells were found lining the vein lumen in aged mice (Figure 1B,D middle row). The proportion of apoptotic cells lining the IVC lumen was significantly increased in aged compared with adult mice (Figure 1C-D bottom row). Endothelial cells lining the vein lumen of aged mice also exhibited a stronger p53 immunosignal (representative confocal microscopy images in Figure 1E). The age-dependent increase in endothelial apoptosis (P = .462 vs adult mice; not shown) and p53 expression (Figure 1E) was not observed in aged End.p53-KO mice.
Figure 1.

Analysis of vascular senescence. End.p53-WT mice aged 12 weeks (adult) or 12 months (aged) were histologically examined for signs of vascular aging, such as (A) the total vein perimeter and the number of (B) total cell nuclei and (C) TUNEL+, apoptotic cells lining the vein lumen. Graphs show individual values as well as the mean ± standard error of the mean (SEM) per group. *P < .05. (D) Representative images after staining with MTC (top and middle row; asterisks indicate the IVC; arrows point to cell nuclei) or TUNEL (bottom row; arrowheads point to apoptotic cells [pink signal]). Scale bars represent 200 µm (top row) or 20 µm (middle and bottom row). (E) Confocal microscopy images after dual immunofluorescence staining of the IVC from End.p53-WT and End.p53-KO mice 12-week-old (adult) or 12-month-old (aged) for CD31 and p53. Cell nuclei were visualized using DAPI. Scale bars represent 10 µm.
Aged mice exhibit increased venous thrombus formation and delayed resolution
To examine the effects of age on venous thrombosis, male adult and aged End.p53-WT mice were subjected to subtotal IVC ligation. Ultrasound measurements on day 1 after surgery detected venous thrombus formation in 12 of 26 (46%) adult and 17 of 22 (77%) aged End.p53-WT mice (P < .05 as determined by χ2 test; Figure 2A). Mean thrombus size at day 1 in mice that formed a venous thrombi was significantly larger in aged compared with adult End.p53-WT mice (Figure 2B; representative ultrasound images are shown in Figure 2C). Although significantly higher platelet counts were observed in whole blood of aged mice (supplemental Table 1), the amount of CD41+ platelets in 2-day-old venous thrombi did not differ between adult and aged mice (supplemental Figure 3A-B). Similar numbers of ELANE+ neutrophils were detected in adult and aged End.p53-WT mouse thrombi (supplemental Figure 3C-D).
Figure 2.

Thrombus formation and resolution over 3 weeks. (A) Percentage of adult and aged End.p53-WT and End.p53-KO mice, in which thrombosis after subtotal IVC ligation was observed or not. The absolute numbers per group and category (“thrombus” and “no thrombus”) are also given. Results were analyzed using χ2 test. *P < .05, ***P < .001, and ****P < .0001. (B) Thrombus size at day 1 after subtotal IVC ligation. Only mice that formed a thrombus are shown. Graphs show individual values as well as the mean ± SEM and were analyzed using 1-way ANOVA. *P < .05. (C) Representative ultrasound images of venous thrombi (encircled by broken white lines) at day 1 after subtotal IVC ligation. The calculated thrombus area is also shown. (D) Venous thrombus resolution in mice that formed a thrombus. Results are expressed as the percentage change vs thrombus size at day 1 (set at 100%) and were analyzed using 2-way ANOVA. ***P < .001 vs day 1 and #P < .05 vs adult End.p53-WT mice. (E) Serial cross sections through the thrombosed IVC segment of adult and aged End.p53-WT or End.p53-KO mice were examined 21 days after surgery and the thrombus cross-sectional area determined. Only mice that formed a thrombus are shown. *P < .05. (F) Representative Carstairs-stained cross sections of thrombosed IVC segments are shown. Scale bars represent 200 µm.
Weekly ultrasound examination of mice over 3 weeks revealed that venous thrombus size started to decrease beyond day 7 after subtotal IVC ligation in adult and, to a lesser extent, aged End.p53-WT mice (Figure 2D). Thrombi were significantly larger in aged compared with adult End.p53-WT mice at day 21. Quantitative morphometric analysis of MTC and Carstairs-stained cross-sections of 21-day-old thrombi confirmed the ultrasound findings and demonstrated a significantly larger mean cross-sectional thrombus area in aged compared with adult End.p53-WT mice (Figure 2E, representative findings in Figure 2F) and larger relative amounts of unresolved thrombotic material (red signal) lacking signs of fibrotic organization (P < .05; supplemental Figure 4). The number of Mac2-positive macrophages (Figure 3A-B) and CD3-immunopositive (Figure 3C-D) lymphocytes within 21-day-old venous thrombi was not affected by age. Similar findings were observed using antibodies against F4/80 to detect tissue macrophages (P = .417; data not shown). Thrombi of aged End.p53-WT mice exhibited a significantly lower CD31-immunopositive area compared with adult End.p53-WT mice (Figure 3E-F), suggesting that impaired thrombus vascularization may have contributed to the delayed thrombus resolution in aged mice.
Figure 3.

Immunohistochemical analysis of thrombus inflammation and angiogenesis. Thrombosed IVC segments of adult and aged End.p53-WT or End.p53-KO mice were examined 21 days after surgery. (A) Quantitative analysis of the Mac2-immunopositive area per thrombus area. (B) Representative images. Scale bars represent 200 µm. (C) Quantitative analysis of the CD3-immunopositive area per thrombus area. (D) Representative images. Scale bars represent 200 µm. (E) Quantitative analysis of the CD31-immunopositive area per thrombus area. *P < .05. (F) Representative images. Scale bars represent 50 µm. Individual values and the mean ± SEM (A) or the median ± interquartile range (C,E) are shown.
Deletion of p53 in endothelial cells protects aged mice against vein thrombosis
To determine the importance of increased endothelial p53 expression for the effects of age on venous thrombus formation, adult or aged mice with inducible, endothelial-specific p53 deletion (End.p53-KO mice) were examined. Of note, absence of p53 in endothelial cells was not associated with differences in peripheral blood cell counts (supplemental Table 1) or bleeding times (supplemental Figure 5) excluding major effects of endothelial p53 deletion on hematopoiesis and hemostasis, respectively. As shown in Figure 2, absence of p53 in endothelial cells did not affect thrombus formation and resolution in adult mice: IVC thrombosis was observed by ultrasound in 13 of 21 End.p53-KO mice (62%; P = .282 vs adult End.p53-WT; Figure 2A), and the mean thrombus size on day 1 also was similar (P = .337 vs adult End.p53-WT; Figure 2B-C). Venous thrombi resolved similarly in adult End.p53-WT and End.p53-KO mice (Figure 2D), and no differences in the thrombus cross-sectional area at day 21 after surgery were observed (Figure 2E-F). Also, no differences in the cellular composition were detected histologically between adult End.p53-WT and End.p53-KO mice (Figure 3). On the other hand, venous thrombi were not detected by ultrasound in any of the examined aged End.p53-KO mice (0 of 16 mice; Figure 2A-C), and organized thrombi were observed histologically in 2 mice at day 21 and found to be significantly smaller (Figure 2E-F).
Increased endothelial heparanase expression and plasma procoagulant activity in aged mice: effects of endothelial p53
Analyses were performed to determine the mechanisms underlying the effects of age on venous thrombus formation and the role of p53 therein. Aged End.p53-WT mice exhibited significantly higher plasma factor Xa levels compared with their adult counterparts, whereas a similar age-dependent increase was not observed in End.p53-KO mice (Figure 4A). In line with the role of heparanase in factor X activation, as previously shown by our group,24,25 western blot analysis revealed significantly increased heparanase expression in primary endothelial cells isolated from lungs of aged compared with adult WT mice (Figure 4B; representative western blot findings in Figure 4E). Protein levels of Egr1, involved in the transcriptional regulation of p5326 and heparanase,27,28 also were higher in endothelial cells of aged mice (Figure 4C,E). Expression of the endothelial cell marker CD31 did not differ between adult and aged endothelial cells (Figure 4D-E). Confocal microscopy analysis confirmed higher numbers of Egr1+ cells (86 ± 2.0 vs 56% ± 2.5% of total cells; P < .05) and increased expression of heparanase in endothelium lining the IVC of aged End.p53-WT mice (Figure 4F). Expression of TF was low in “resting” endothelium lining the IVC lumen (supplemental Figure 6A), however, a pronounced TF immunosignal was detected at the endothelium-thrombus interface of 2-day-old venous thrombi of aged, and to a lesser extent in adult End.p53-WT mice (supplemental Figure 6B). The relative number of Egr1+ endothelial cells lining the IVC lumen continued to be elevated in aged End.p53-KO mice (Figure 4F), suggesting that it is induced “upstream” of p53.29 In contrast, the age-dependent increased expression of heparanase was not observed in the absence of p53 (Figure 4G).
Figure 4.

Plasma factor Xa levels and expression of heparanase in murine endothelial cells. (A) Active factor X levels were examined in plasma of End.p53-WT and End.p53-KO mice, aged 12 weeks (adult) or 12 months (aged), as described in “Methods.” Graphs show individual values as well as the mean ± SEM per group. **P < .01 and ***P < .001. Primary endothelial cells were isolated from lungs of adult and aged WT mice and examined for the expression of (B) heparanase (Hpse), (C) Egr1, and (D) the endothelial marker Cd31 (n = 4 biological replicates per group). (E) Representative western blot membrane. Vena cava segments were obtained from adult and aged End.p53-WT and End.p53-KO mice, cryoembedded and examined for the expression of Egr1 (F) or Hpse (G; green signal) and Cd31 (red signal) using immunofluorescence. Representative confocal microscopy images are shown. Scale bars represent 10 µm.
Human vein endothelial cell senescence results in p53, Egr1, and heparanase overexpression
To further examine the mechanisms underlying our findings in aged mice or murine endothelial cells, HUVECs were cultivated for increasing population doublings to induce replicative senescence (supplemental Figure 7A). Changes in cell number (ie, reduction) and morphology (ie, enlargement) with repeated serial passaging are depicted in supplemental Figure 7B, representative findings after staining of senescence-associated β-galactosidase activity are shown in supplemental Figure 7C. Western blot analysis confirmed significantly increased protein expression of p53 (supplemental Figure 7D-E) and the senescence markers phospho-H2A.X (supplemental Figure 7D,F) and p16INK4A (supplemental Figure 7G-H) in cells at passage 13 (P13) compared with P5. Direct comparison of HUVECs at P5 and P13 (n = 6 independent experiments) confirmed significantly increased protein expression levels of p53 (Figure 5A) and heparanase (Figure 5B) in senescent HUVECs. Similar to endothelial cells from aged mice, senescent HUVECs exhibited elevated protein levels of Egr1 (Figure 5C), and other factors regulated by Egr1, such as PAI-1 (Figure 5D), TGFβ (Figure 5E), and PTEN (Figure 5F), were also expressed at higher levels. Representative western blot membranes are shown in Figure 5G.
Figure 5.

Protein expression of factors involved in thrombosis and fibrinolysis in human vein endothelial cells. HUVECs were passaged from passage 5 (P5) to 13 to induce replicative senescence and the protein expression of (A) tumor suppressor p53 (TP53), (B) heparanase (HPSE), (C) EGR1, (D) PAI-1, (E) TGFβ, and (F) PTEN was analyzed using western blot. *P < .05 vs HUVEC at P5 (set at 1); **P < .01. (G) Western blot membrane images representative for n = 6 biological replicates. GAPDH expression was examined as control for equal protein loading and is shown for the results of the same membrane. Numbers on the left indicate the molecular weight of the bands.
To further examine the role of p53 in endothelial heparanase expression, HUVECs were treated with doxorubicin to induce p53 overexpression or pifithrin-α to pharmacologically inhibit p53 activity (Figure 6). Stimulation of HUVECs with doxorubicin resulted in elevated p53 (Figure 6A), heparanase (Figure 6B), and Egr1 (Figure 6C) protein levels, and significantly lower levels were observed after pharmacological inhibition of p53 activity using pifithrin-α (representative findings in Figure 6F). Moreover, p53 inactivation significantly reduced the expression of PTEN (Figure 6D,F) and its transcriptional target TF (Figure 6E-F) in human endothelial cells. Reduced endothelial PTEN expression was also observed in IVC segments of aged End.p53-KO mice (supplemental Figure 8A) and murine endothelial cells lacking p53 (supplemental Figure 8B).
Figure 6.

Human endothelial heparanase expression in response to p53 modulation. Results after quantitative analysis of (A) tumor suppressor p53 (TP53), (B) heparanase (HPSE), (C) EGR1, (D) PTEN, and (E) TF protein expression in HUVECs following incubation with doxorubicin (500 nM for 24 hours), pifithrin-α (20 µM for 24 hours), or vehicle alone (saline or dimethyl sulfoxide [DMSO] = control). Quantitative analysis of findings in n = 4 biological replicates. *P < .05, **P < .01, and ****P < .0001. (F) Representative western blot membranes from the same experiment are shown. GAPDH was used as control for equal protein loading.
Neutralization of heparanase in aged mice restores the venous thrombosis phenotype of adult mice
Finally, and to examine the importance of increased endothelial heparanase expression observed in senescent human and murine endothelial cells and to determine its role in the prothrombotic tendency observed in aged mice, subtotal IVC ligation was induced in adult and aged C57BL/6J WT mice treated with TFPI2 peptides to antagonize heparanase activity.22,30 Confirming our findings in aged End.p53-WT mice, aged C57B/6J WT mice also developed significantly larger venous thrombi (Figure 7A; representative ultrasound findings in Figure 7B). Importantly, TFPI2 peptide injection significantly reduced venous thrombus size in aged WT mice, whereas thrombus size was not significantly altered in adult WT mice. Thrombus weights on day 2 after subtotal IVC ligation are shown in Figure 7C, images of freshly harvested venous thrombi in Figure 7D. Taken together, these analyses strongly support a causal role of increased endothelial heparanase expression in the enhanced propensity of older mice to develop venous thrombi and suggest that heparanase antagonization using TFPI2 peptides could be a useful strategy to ameliorate the prothrombotic endothelial phenotype with age.
Figure 7.

Effect of heparanase inactivation on venous thrombus formation in mice. (A) Venous thrombosis was induced by subtotal IVC ligation in C57BL/6J mice at the age of 12 weeks (adult) or 12 months (aged). Mice were injected with TFPI2 peptide (2 mg/g body weight) 30 minutes before and every 24 hours after surgery. (A) High-frequency ultrasound determination of thrombus size at day 2 after surgery. Individual values and the mean ± SEM per group are shown. *P < .05. (B) Representative ultrasound images. The thrombus circumference is highlighted by broken white lines. The calculated thrombus area is also shown. (C) Thrombus weights at tissue harvest on day 2. Individual values and the mean ± SEM per group are shown. ****P < .0001. (D) Representative macroscopic images of thrombosed vein segments immediately after removal.
Discussion
In this study, it was found that aging is associated with signs of vascular senescence in the venous system and that aged mice more frequently develop venous thrombosis when induced with the IVC stenosis model. The phenomenon of increased incidence of thrombosis with age is widely observed in the human population. Thrombi in aged mice were larger and contained greater amounts of unresolved thrombotic material (fibrin) and fewer endothelial cells. Endothelial aging was associated with elevated levels of p53 as well as increased expression of endothelium-derived factors involved in coagulation and fibrinolysis resistance, such as heparanase or PAI-1, and pharmacological modulation of p53 expression/activity in endothelial cells also altered heparanase levels. Importantly, aged mice with endothelial p53 deletion were protected from venous thrombosis, and increased thrombus formation in aged mice could be prevented by perioperative TFPI2 peptide administration to inactivate heparanase. Our findings suggest that age-associated endothelial dysfunction, in particular p53 and heparanase overexpression in senescent endothelium, critically contribute to venous thrombus formation and the increased risk of thrombosis with age.
Age is an established cardiovascular risk factor and associated with an increased risk to develop thrombosis, both in the arterial and the venous system. With regard to VTE, the relative risk for DVT significantly increases with age, rising from 1 in 10 000 at 20 years of age to 1 in 1000 at 50 years of age and to 1 in 100 at 80 years of age.2,4 Although age is a strong (and prevalent) risk factor for venous thrombosis, the molecular mechanisms underlying this association remain obscure. In addition to changes in vein anatomy or vein valves with age,9 a higher frequency of immobility, surgery, malignancies, or comorbidities are among the potential factors contributing to the increased thrombosis risk in the elderly.31 Aging is associated with alterations in the hemostatic system favoring thrombosis: increased levels of factors involved in coagulation (eg, fibrinogen, factor V, VII, VIII, IX, and X) and fibrinolysis (eg, PAI-1) have been documented in the elderly population,32 and clinical studies confirmed that increased levels of PAI-1 predispose to myocardial infarction33 and venous thrombosis.34 In mice, aging was associated with increased expression of TF35 and PAI-1,36-39 and mice transgenic for PAI-1 age-dependently developed spontaneous coronary artery thrombosis37,40 and venous occlusions.41 Increased expression of PAI-1, as reported in senescent endothelial cells in this and previous studies (reviewed in Bochenek et al7) could also underlie the slower venous thrombus resolution observed in aged mice. In addition to its role in clot resolution,42 we and others have shown that fibrinolytic factors are important for pericellular proteolysis and cell migration into the fibrin matrix.43 In the present study, larger and less organized venous thrombi were observed in aged mice containing fewer endothelial cells.
The majority of experimental studies on the cardiovascular effects of age have focused on the arterial system, whereas less attention has been paid to the impact of vascular senescence on vein thrombosis. A larger thrombus mass was reported in 11-month-old mice after IVC ligation, and findings were attributed to differences in vein wall inflammation and circulating factors, such as plasma PAI-1 and procoagulant microparticles.44 In another study, larger thrombi in 18-month-old mice following electrolytic IVC endothelial injury were associated with higher soluble P-selectin levels and increased vein wall P-selectin expression.45 Of note, elevated circulating platelet numbers in aged mice were observed in our and previous studies45,46 and may have contributed to our findings, although the amount of CD41+ platelets within 2-day-old murine thrombi was not found to differ with age. Increased platelet counts were also observed in the Bmal1-deficient mouse model of aging and associated with an increased megakaryocyte density in the bone marrow, but did not affect platelet aggregation.46 In humans, platelet counts exhibit an age-associated decrease,47 and a mild thrombocytopenia with age is a frequent finding in the general population.48,49
Disturbances of blood flow and stasis in veins as well as hypercoagulability are considered major pathogenic factors for venous thrombus formation, whereas endothelial dysfunction or damage are believed to be of greater importance during arterial thrombosis. However, previous studies have already suggested that the contribution of endothelial changes to venous thrombosis may have been underestimated in the past. For example, the hemostatic thrombophilic alterations observed in the elderly include increased levels of endothelial-derived factors, such as factor VIII–von Willebrand factor,50 and experimental studies demonstrated the importance of von Willebrand factor51 or P-selectin52 for venous thrombus formation. Endothelial dysfunction and elevated factor VII levels were reported in a mouse model of premature aging.46 Flow-mediated dilation, a surrogate marker of arterial endothelial dysfunction, was found to strongly correlate with the incidence of unprovoked DVT.53 Findings of increased plasma levels of endothelial microparticles (ie, small membrane vesicles shed from activated or apoptotic cells54) in patients with unprovoked VTE55,56 or aged mice44 point to the importance of endothelial dysfunction or injury in venous thrombosis. In this regard, analyses of mice with endothelial p53 deletion using electrolytic endothelial injury, a model that can be adapted to examine both pro- and antithrombotic phenotypes with less variation in both the incidence and size of the resulting thrombus compared with the IVC stenosis model,57,58 may be of interest.
Cellular hallmarks of vascular aging include cell loss, the net result of a reduction in the regenerative potential and decreased proliferation, but also increased cell death. In old monkeys, the decline in endothelial function was found to correlate with elevated numbers of apoptotic endothelial cells in the aorta and femoral artery.8 Interestingly, mice overexpressing the cell cycle inhibitor p16INK4A exhibited accelerated times to venous thrombosis and delayed thrombus resolution.59 Moreover, a histological study in human superficial vein segments found higher apoptosis activities in those containing thrombus.60 The diverse pathways involved in cellular senescence converge on p53, a transcription factor controlling cell cycle, growth arrest, and apoptosis.10,61 However, very little is known regarding p53 and the risk of thrombosis. Disturbed blood flow was found to induce endothelial senescence via the p53-signaling pathway.62 Other cellular stressors known to induce p53 include hypoxia.63 Low oxygen levels, such as occurring in areas of low flow behind vein valves, have been implicated in the pathophysiology of venous thrombosis (reviewed in Bovill and van der Vliet64), and mice exposed to hypoxia exhibited an increased prevalence of venous thrombosis.65 Interestingly, elevated serum levels of growth differentiation factor-15, a soluble marker of endothelial injury induced by p53, are useful for the prognostic risk assessment of patients with pulmonary embolism.66,67 Altogether, these studies point to a role for p53-mediated endothelial damage during venous thrombosis. Recently, p53 expressed in myeloid cells was shown to enhance venous thrombus resolution in mice by promoting macrophage polarization.68 In the present study, venous thrombi of adult and aged mice did not differ with respect to the relative amount of macrophages or T cells within venous thrombi, and no effects of endothelial p53 expression on both parameters were observed.
Inactivation of p53 is part of the cellular survival pathway, and replicative senescence can be delayed by inactivation of p53 (reviewed in Zhang61). Our findings suggest that genetic p53 deletion in endothelial cells may also protect against other age-associated alterations, such as the increased risk of venous thrombosis. Although loss of p53 may be associated with uninhibited growth and the development of cancer, a study in p53-deficient endothelial cells did not observe any malignant transformation.69 Similarly, endothelial cell-specific p53 deletion in mice did not affect survival up to 12 months and was not associated with increased spontaneous tumor formation in this and a previous study from our group.14
How does p53 overexpression in endothelial cells contribute to venous thrombus formation? Elevated levels of p53 and its downstream target PAI-1 are typical features of replicative senescence,70 in line with our findings in aged human and murine endothelial cells. In a previous study, overexpression of p53 was found to promote a prothrombotic endothelial cell phenotype in vitro via downregulation of Krüppel-like factor-2 and subsequent alterations in expression of nitric oxide synthase, thrombomodulin, and PAI-1,13 and reduced expression of thrombomodulin was also observed following UV B irradiation injury and p53 accumulation.71 Here, analysis of primary endothelial cells from aged mice and senescent human endothelial cells as a model system of p53 overexpression revealed elevated expression of heparanase, an endo-β-d-glucuronidase capable of degrading polymeric heparan sulfate, an inhibitor of coagulation pathway enzymes such as thrombin and factor Xa. Inflammation, diabetes or cancer are associated with elevated heparanase expression.72 Although the incidence of these conditions increase with age, the effect of age per se on heparanase expression has not been examined so far. Regarding the mechanistic link between p53 and heparanase, our findings in murine endothelial cells and HUVECs after induction of replicative senescence or pharmacological modulation of p53 activity (using doxorubicin or pifithrin-α) suggest that elevated expression of transcription factors, in particular Egr1, may have been involved in the observed increased heparanase27,28 and possible also p5326 expression. Previous studies have found that Egr1 physically associates with p53 and that both translocate to the nucleus to induce gene transcription.73 On the other hand, p53 may also induce Egr174 establishing a reciprocal interaction. Of note, in mouse embryonic fibroblasts and cancer cells, others found that p53 binds to the heparanase promoter and inhibits its activity.75 Moreover, we found aging and p53 overexpression to be associated with increased expression of PTEN, a negative regulator of protein kinase B (Akt), also induced by Egr1. Previous studies have established a role of the PI3K/Akt pathway in the suppression of TF in endothelial cells,76,77 and p53-dependent modulation of PTEN could underlie the observed changes in endothelial TF expression and possibly also the absence of venous thrombosis in aged Endp53-KO mice. On the other hand, whether genetic deletion of p53 in endothelial cells may have affected venous thrombus formation indirectly, eg, by paracrine effects on smooth muscle cells, could not be addressed in the present study.
In line with our previous findings,24,25 endothelial heparanase overexpression observed in aged mice was paralleled by elevated plasma factor Xa activity levels. Moreover, transgenic mice overexpressing human heparanase exhibited shortened time to arterial thrombosis following vascular injury and increased in-stent thrombosis.78 Importantly and strongly suggesting a causal role for heparanase overexpression in senescent endothelial cells for venous thrombosis, we now show that the prothrombotic phenotype of aged mice can be reversed by perioperative TFPI2 peptide administration and inhibition of heparanase procoagulant activity.22 From a translational viewpoint, targeting endothelial heparanase to prevent age-associated enhanced coagulation might represent a ‘safer’ and more specific alternative to p53 inhibitors.
Taken together, our findings suggest that the age-associated endothelial p53 accumulation contributes to the increased risk of venous thrombosis in the elderly and that upregulated expression of procoagulant and antifibrinolytic factors, specifically heparanase, are involved in mediating the increased tendency to venous thrombosis downstream of p53. Although inhibition or deletion of the tumor suppressor p53 in endothelium may not represent a safe therapeutic approach to prevent venous thrombosis in elderly humans, our findings point to alternative targets, whose clinical applicability should be further explored.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors are grateful to Bernd Arnold (German Cancer Research Center Deutsches Krebsforschungszentrum) for providing Tie2.ERT2-Cre mice. The authors also thank Anton Berns (The Netherlands Cancer Institute) for providing p53fl/fl mice. The authors acknowledge the expert technical assistance of Frauke Dormann, Marina Janocha, Anna Kern, and Kathrin Rost.
This work was supported by the Bundesministerium für Bildung und Forschung (01E01003) (K.S.) and the Deutsches Zentrum für Herz-Kreislauf-Forschung e.V. (Doktorandenstipendium) (T.B.).
Results shown in this study are part of the medical thesis of T.B.
Authorship
Contribution: K.S. designed the study, interpreted the findings, wrote the manuscript and acquired funding; M.L.B., T.B., and R.G. performed and analyzed experiments and interpreted the findings; Y.N. provided the TFPI2 peptides and gave critical input to the manuscript; A.M. acquired data; T.S. and L.H. performed the IVC surgery and ultrasound measurements; P.W. provided collaboration in experimental design and establishment of the IVC ligation model; B.B., P.W., T.M., and S.K. critically revised the manuscript; and all authors have read and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Katrin Schäfer, Center for Cardiology, Cardiology I, University Medical Center, Johannes Gutenberg University, Langenbeckstr 1, 55131 Mainz, Germany; e-mail: katrin.schaefer@unimedizin-mainz.de.
References
- 1.Cohen AT, Agnelli G, Anderson FA, et al. ; VTE Impact Assessment Group in Europe (VITAE). Venous thromboembolism (VTE) in Europe. The number of VTE events and associated morbidity and mortality. Thromb Haemost. 2007;98(4):756-764. [DOI] [PubMed] [Google Scholar]
- 2.Naess IA, Christiansen SC, Romundstad P, Cannegieter SC, Rosendaal FR, Hammerstrøm J. Incidence and mortality of venous thrombosis: a population-based study. J Thromb Haemost. 2007;5(4):692-699. [DOI] [PubMed] [Google Scholar]
- 3.Reitsma PH, Versteeg HH, Middeldorp S. Mechanistic view of risk factors for venous thromboembolism. Arterioscler Thromb Vasc Biol. 2012;32(3):563-568. [DOI] [PubMed] [Google Scholar]
- 4.White RH. The epidemiology of venous thromboembolism. Circulation. 2003;107(23 suppl 1):I4-I8. [DOI] [PubMed] [Google Scholar]
- 5.Anderson FA Jr, Spencer FA. Risk factors for venous thromboembolism. Circulation. 2003;107(23 suppl 1):I9-I16. [DOI] [PubMed] [Google Scholar]
- 6.Najjar SS, Scuteri A, Lakatta EG. Arterial aging: is it an immutable cardiovascular risk factor? Hypertension. 2005;46(3):454-462. [DOI] [PubMed] [Google Scholar]
- 7.Bochenek ML, Schütz E, Schäfer K. Endothelial cell senescence and thrombosis: ageing clots. Thromb Res. 2016;147:36-45. [DOI] [PubMed] [Google Scholar]
- 8.Asai K, Kudej RK, Shen YT, et al. . Peripheral vascular endothelial dysfunction and apoptosis in old monkeys. Arterioscler Thromb Vasc Biol. 2000;20(6):1493-1499. [DOI] [PubMed] [Google Scholar]
- 9.Palmer OR, Chiu CB, Cao A, Scheven UM, Diaz JA, Greve JM. In vivo characterization of the murine venous system before and during dobutamine stimulation: implications for preclinical models of venous disease. Ann Anat. 2017;214:43-52. [DOI] [PubMed] [Google Scholar]
- 10.Ullrich SJ, Anderson CW, Mercer WE, Appella E. The p53 tumor suppressor protein, a modulator of cell proliferation. J Biol Chem. 1992;267(22):15259-15262. [PubMed] [Google Scholar]
- 11.Tyner SD, Venkatachalam S, Choi J, et al. . p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415(6867):45-53. [DOI] [PubMed] [Google Scholar]
- 12.Grillari J, Hohenwarter O, Grabherr RM, Katinger H. Subtractive hybridization of mRNA from early passage and senescent endothelial cells. Exp Gerontol. 2000;35(2):187-197. [DOI] [PubMed] [Google Scholar]
- 13.Kumar A, Kim CS, Hoffman TA, et al. . p53 impairs endothelial function by transcriptionally repressing Kruppel-like factor 2. Arterioscler Thromb Vasc Biol. 2011;31(1):133-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gogiraju R, Xu X, Bochenek ML, et al. . Endothelial p53 deletion improves angiogenesis and prevents cardiac fibrosis and heart failure induced by pressure overload in mice. J Am Heart Assoc. 2015;4(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14(8):994-1004. [PMC free article] [PubMed] [Google Scholar]
- 16.Forde A, Constien R, Gröne HJ, Hämmerling G, Arnold B. Temporal Cre-mediated recombination exclusively in endothelial cells using Tie2 regulatory elements. Genesis. 2002;33(4):191-197. [DOI] [PubMed] [Google Scholar]
- 17.Hubert A, Bochenek ML, Schütz E, Gogiraju R, Münzel T, Schäfer K. Selective deletion of leptin signaling in endothelial cells enhances neointima formation and phenocopies the vascular effects of diet-induced obesity in mice. Arterioscler Thromb Vasc Biol. 2017;37(9):1683-1697. [DOI] [PubMed] [Google Scholar]
- 18.Jäger M, Hubert A, Gogiraju R, Bochenek ML, Münzel T, Schäfer K. Inducible knockdown of endothelial protein tyrosine phosphatase-1B promotes neointima formation in obese mice by enhancing endothelial senescence [published online ahead of print 1 February 2018]. Antioxid Redox Signal. doi:10.1089/ars.2017.7169. [DOI] [PubMed] [Google Scholar]
- 19.Brandt M, Schönfelder T, Schwenk M, et al. . Deep vein thrombus formation induced by flow reduction in mice is determined by venous side branches. Clin Hemorheol Microcirc. 2014;56(2):145-152. [DOI] [PubMed] [Google Scholar]
- 20.von Brühl ML, Stark K, Steinhart A, et al. . Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819-835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonderman D, Jakowitsch J, Redwan B, et al. . Role for staphylococci in misguided thrombus resolution of chronic thromboembolic pulmonary hypertension. Arterioscler Thromb Vasc Biol. 2008;28(4):678-684. [DOI] [PubMed] [Google Scholar]
- 22.Axelman E, Henig I, Crispel Y, et al. . Novel peptides that inhibit heparanase activation of the coagulation system. Thromb Haemost. 2014;112(3):466-477. [DOI] [PubMed] [Google Scholar]
- 23.Hayflick L. The cell biology of aging. Clin Geriatr Med. 1985;1(1):15-27. [PubMed] [Google Scholar]
- 24.Nadir Y, Brenner B, Zetser A, et al. . Heparanase induces tissue factor expression in vascular endothelial and cancer cells. J Thromb Haemost. 2006;4(11):2443-2451. [DOI] [PubMed] [Google Scholar]
- 25.Nadir Y, Brenner B, Fux L, Shafat I, Attias J, Vlodavsky I. Heparanase enhances the generation of activated factor X in the presence of tissue factor and activated factor VII. Haematologica. 2010;95(11):1927-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nair P, Muthukkumar S, Sells SF, Han SS, Sukhatme VP, Rangnekar VM. Early growth response-1-dependent apoptosis is mediated by p53. J Biol Chem. 1997;272(32):20131-20138. [DOI] [PubMed] [Google Scholar]
- 27.de Mestre AM, Rao S, Hornby JR, Soe-Htwe T, Khachigian LM, Hulett MD. Early growth response gene 1 (EGR1) regulates heparanase gene transcription in tumor cells. J Biol Chem. 2005;280(42):35136-35147. [DOI] [PubMed] [Google Scholar]
- 28.Ogishima T, Shiina H, Breault JE, et al. . Increased heparanase expression is caused by promoter hypomethylation and up-regulation of transcriptional factor early growth response-1 in human prostate cancer. Clin Cancer Res. 2005;11(3):1028-1036. [PubMed] [Google Scholar]
- 29.Krones-Herzig A, Mittal S, Yule K, et al. . Early growth response 1 acts as a tumor suppressor in vivo and in vitro via regulation of p53. Cancer Res. 2005;65(12):5133-5143. [DOI] [PubMed] [Google Scholar]
- 30.Crispel Y, Axelman E, Tatour M, et al. . Peptides inhibiting heparanase procoagulant activity significantly reduce tumour growth and vascularisation in a mouse model. Thromb Haemost. 2016;116(4):669-678. [DOI] [PubMed] [Google Scholar]
- 31.Engbers MJ, van Hylckama Vlieg A, Rosendaal FR. Venous thrombosis in the elderly: incidence, risk factors and risk groups. J Thromb Haemost. 2010;8(10):2105-2112. [DOI] [PubMed] [Google Scholar]
- 32.Wilkerson WR, Sane DC. Aging and thrombosis. Semin Thromb Hemost. 2002;28(6):555-568. [DOI] [PubMed] [Google Scholar]
- 33.Juhan-Vague I, Pyke SD, Alessi MC, Jespersen J, Haverkate F, Thompson SG. Fibrinolytic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. ECAT Study Group. European Concerted Action on Thrombosis and Disabilities. Circulation. 1996;94(9):2057-2063. [DOI] [PubMed] [Google Scholar]
- 34.Ridker PM, Vaughan DE, Stampfer MJ, et al. . Baseline fibrinolytic state and the risk of future venous thrombosis. A prospective study of endogenous tissue-type plasminogen activator and plasminogen activator inhibitor. Circulation. 1992;85(5):1822-1827. [DOI] [PubMed] [Google Scholar]
- 35.Yamamoto K, Shimokawa T, Yi H, et al. . Aging and obesity augment the stress-induced expression of tissue factor gene in the mouse. Blood. 2002;100(12):4011-4018. [DOI] [PubMed] [Google Scholar]
- 36.Yamamoto K, Shimokawa T, Yi H, et al. . Aging accelerates endotoxin-induced thrombosis : increased responses of plasminogen activator inhibitor-1 and lipopolysaccharide signaling with aging. Am J Pathol. 2002;161(5):1805-1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eren M, Painter CA, Atkinson JB, Declerck PJ, Vaughan DE. Age-dependent spontaneous coronary arterial thrombosis in transgenic mice that express a stable form of human plasminogen activator inhibitor-1. Circulation. 2002;106(4):491-496. [DOI] [PubMed] [Google Scholar]
- 38.Samad F, Loskutoff DJ. Tissue distribution and regulation of plasminogen activator inhibitor-1 in obese mice. Mol Med. 1996;2(5):568-582. [PMC free article] [PubMed] [Google Scholar]
- 39.Takeshita K, Yamamoto K, Ito M, et al. . Increased expression of plasminogen activator inhibitor-1 with fibrin deposition in a murine model of aging, “Klotho” mouse. Semin Thromb Hemost. 2002;28(6):545-554. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto K, Takeshita K, Shimokawa T, et al. . Plasminogen activator inhibitor-1 is a major stress-regulated gene: implications for stress-induced thrombosis in aged individuals. Proc Natl Acad Sci USA. 2002;99(2):890-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Erickson LA, Fici GJ, Lund JE, Boyle TP, Polites HG, Marotti KR. Development of venous occlusions in mice transgenic for the plasminogen activator inhibitor-1 gene. Nature. 1990;346(6279):74-76. [DOI] [PubMed] [Google Scholar]
- 42.Konstantinides S, Schäfer K, Thinnes T, Loskutoff DJ. Plasminogen activator inhibitor-1 and its cofactor vitronectin stabilize arterial thrombi after vascular injury in mice. Circulation. 2001;103(4):576-583. [DOI] [PubMed] [Google Scholar]
- 43.Schäfer K, Konstantinides S, Riedel C, et al. . Different mechanisms of increased luminal stenosis after arterial injury in mice deficient for urokinase- or tissue-type plasminogen activator. Circulation. 2002;106(14):1847-1852. [DOI] [PubMed] [Google Scholar]
- 44.McDonald AP, Meier TR, Hawley AE, et al. . Aging is associated with impaired thrombus resolution in a mouse model of stasis induced thrombosis. Thromb Res. 2010;125(1):72-78. [DOI] [PubMed] [Google Scholar]
- 45.Culmer DL, Diaz JA, Hawley AE, et al. . Circulating and vein wall P-selectin promote venous thrombogenesis during aging in a rodent model. Thromb Res. 2013;131(1):42-48. [DOI] [PubMed] [Google Scholar]
- 46.Hemmeryckx B, Van Hove CE, Fransen P, et al. . Progression of the prothrombotic state in aging Bmal1-deficient mice. Arterioscler Thromb Vasc Biol. 2011;31(11):2552-2559. [DOI] [PubMed] [Google Scholar]
- 47.Santimone I, Di Castelnuovo A, De Curtis A, et al. ; MOLI-SANI Project Investigators. White blood cell count, sex and age are major determinants of heterogeneity of platelet indices in an adult general population: results from the MOLI-SANI project. Haematologica. 2011;96(8):1180-1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biino G, Santimone I, Minelli C, et al. . Age- and sex-related variations in platelet count in Italy: a proposal of reference ranges based on 40987 subjects’ data. PLoS One. 2013;8(1):e54289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Segal JB, Moliterno AR. Platelet counts differ by sex, ethnicity, and age in the United States. Ann Epidemiol. 2006;16(2):123-130. [DOI] [PubMed] [Google Scholar]
- 50.Abbate R, Prisco D, Rostagno C, Boddi M, Gensini GF. Age-related changes in the hemostatic system. Int J Clin Lab Res. 1993;23(1):1-3. [DOI] [PubMed] [Google Scholar]
- 51.Brill A, Fuchs TA, Chauhan AK, et al. . von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood. 2011;117(4):1400-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Myers DD, Hawley AE, Farris DM, et al. . P-selectin and leukocyte microparticles are associated with venous thrombogenesis. J Vasc Surg. 2003;38(5):1075-1089. [DOI] [PubMed] [Google Scholar]
- 53.Mazzoccoli G, Fontana A, Grilli M, et al. . Idiopathic deep venous thrombosis and arterial endothelial dysfunction in the elderly. Age (Dordr). 2012;34(3):751-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aupeix K, Hugel B, Martin T, et al. . The significance of shed membrane particles during programmed cell death in vitro, and in vivo, in HIV-1 infection. J Clin Invest. 1997;99(7):1546-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Campello E, Spiezia L, Radu CM, et al. . Endothelial, platelet, and tissue factor-bearing microparticles in cancer patients with and without venous thromboembolism. Thromb Res. 2011;127(5):473-477. [DOI] [PubMed] [Google Scholar]
- 56.Chirinos JA, Heresi GA, Velasquez H, et al. . Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism. J Am Coll Cardiol. 2005;45(9):1467-1471. [DOI] [PubMed] [Google Scholar]
- 57.Banno F, Kita T, Fernández JA, et al. . Exacerbated venous thromboembolism in mice carrying a protein S K196E mutation. Blood. 2015;126(19):2247-2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Diaz JA. Animal models of VT: to change or not to change? Blood. 2015;126(19):2177-2178. [DOI] [PubMed] [Google Scholar]
- 59.Cardenas JC, Owens AP III, Krishnamurthy J, Sharpless NE, Whinna HC, Church FC. Overexpression of the cell cycle inhibitor p16INK4a promotes a prothrombotic phenotype following vascular injury in mice. Arterioscler Thromb Vasc Biol. 2011;31(4):827-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Filis K, Kavantzas N, Dalainas I, et al. . Evaluation of apoptosis in varicose vein disease complicated by superficial vein thrombosis. Vasa. 2014;43(4):252-259. [DOI] [PubMed] [Google Scholar]
- 61.Zhang H. Molecular signaling and genetic pathways of senescence: its role in tumorigenesis and aging. J Cell Physiol. 2007;210(3):567-574. [DOI] [PubMed] [Google Scholar]
- 62.Warboys CM, de Luca A, Amini N, et al. . Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler Thromb Vasc Biol. 2014;34(5):985-995. [DOI] [PubMed] [Google Scholar]
- 63.Sano M, Minamino T, Toko H, et al. . p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446(7134):444-448. [DOI] [PubMed] [Google Scholar]
- 64.Bovill EG, van der Vliet A. Venous valvular stasis-associated hypoxia and thrombosis: what is the link? Annu Rev Physiol. 2011;73(1):527-545. [DOI] [PubMed] [Google Scholar]
- 65.Brill A, Suidan GL, Wagner DD. Hypoxia, such as encountered at high altitude, promotes deep vein thrombosis in mice. J Thromb Haemost. 2013;11(9):1773-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lankeit M, Kempf T, Dellas C, et al. . Growth differentiation factor-15 for prognostic assessment of patients with acute pulmonary embolism. Am J Respir Crit Care Med. 2008;177(9):1018-1025. [DOI] [PubMed] [Google Scholar]
- 67.Osada M, Park HL, Park MJ, et al. . A p53-type response element in the GDF15 promoter confers high specificity for p53 activation. Biochem Biophys Res Commun. 2007;354(4):913-918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mukhopadhyay S, Antalis TM, Nguyen KP, Hoofnagle MH, Sarkar R. Myeloid p53 regulates macrophage polarization and venous thrombus resolution by inflammatory vascular remodeling in mice. Blood. 2017;129(24):3245-3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nishiyama T, Mishima K, Ide F, et al. . Functional analysis of an established mouse vascular endothelial cell line. J Vasc Res. 2007;44(2):138-148. [DOI] [PubMed] [Google Scholar]
- 70.Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8(8):877-884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang HC, Chang TM, Chang YJ, Wen HY. UVB irradiation regulates ERK1/2- and p53-dependent thrombomodulin expression in human keratinocytes. PLoS One. 2013;8(7):e67632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vlodavsky I, Iozzo RV, Sanderson RD. Heparanase: multiple functions in inflammation, diabetes and atherosclerosis. Matrix Biol. 2013;32(5):220-222. [DOI] [PubMed] [Google Scholar]
- 73.Liu J, Grogan L, Nau MM, Allegra CJ, Chu E, Wright JJ. Physical interaction between p53 and primary response gene Egr-1. Int J Oncol. 2001;18(4):863-870. [DOI] [PubMed] [Google Scholar]
- 74.Weisz L, Zalcenstein A, Stambolsky P, et al. . Transactivation of the EGR1 gene contributes to mutant p53 gain of function. Cancer Res. 2004;64(22):8318-8327. [DOI] [PubMed] [Google Scholar]
- 75.Baraz L, Haupt Y, Elkin M, Peretz T, Vlodavsky I. Tumor suppressor p53 regulates heparanase gene expression. Oncogene. 2006;25(28):3939-3947. [DOI] [PubMed] [Google Scholar]
- 76.Blum S, Issbrüker K, Willuweit A, et al. . An inhibitory role of the phosphatidylinositol 3-kinase-signaling pathway in vascular endothelial growth factor-induced tissue factor expression. J Biol Chem. 2001;276(36):33428-33434. [DOI] [PubMed] [Google Scholar]
- 77.Kim I, Oh JL, Ryu YS, et al. . Angiopoietin-1 negatively regulates expression and activity of tissue factor in endothelial cells. FASEB J. 2002;16(1):126-128. [DOI] [PubMed] [Google Scholar]
- 78.Baker AB, Gibson WJ, Kolachalama VB, et al. . Heparanase regulates thrombosis in vascular injury and stent-induced flow disturbance. J Am Coll Cardiol. 2012;59(17):1551-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.