

Key Points
N-RasG12D and haploinsufficient Tet2 collaborate to induce lethal and highly penetrant CMML in mice with shortened overall survival.
N-RasG12D and haploinsufficient Tet2 together promote balanced proliferation and enhanced competitiveness and self-renewal in HSPCs.
Abstract
Concurrent genetic lesions exist in a majority of patients with hematologic malignancies. Among these, somatic mutations that activate RAS oncogenes and inactivate the epigenetic modifier ten-eleven translocation 2 (TET2) frequently co-occur in human chronic myelomonocytic leukemias (CMMLs) and acute myeloid leukemias, suggesting a cooperativity in malignant transformation. To test this, we applied a conditional murine model that endogenously expressed oncogenic NrasG12D and monoallelic loss of Tet2 and explored the collaborative role specifically within hematopoietic stem and progenitor cells (HSPCs) at disease initiation. We demonstrate that the 2 mutations collaborated to accelerate a transplantable CMML-like disease in vivo, with an overall shortened survival and increased disease penetrance compared with single mutants. At preleukemic stage, N-RasG12D and Tet2 haploinsufficiency together induced balanced hematopoietic stem cell (HSC) proliferation and enhanced competitiveness. NrasG12D/+/Tet2+/− HSCs displayed increased self-renewal in primary and secondary transplantations, with significantly higher reconstitution than single mutants. Strikingly, the 2 mutations together conferred long-term reconstitution and self-renewal potential to multipotent progenitors, a pool of cells that usually have limited self-renewal compared with HSCs. Moreover, HSPCs from NrasG12D/+/Tet2+/− mice displayed increased cytokine sensitivity in response to thrombopoietin. Therefore, our studies establish a novel tractable CMML model and provide insights into how dysregulated signaling pathways and epigenetic modifiers collaborate to modulate HSPC function and promote leukemogenesis.
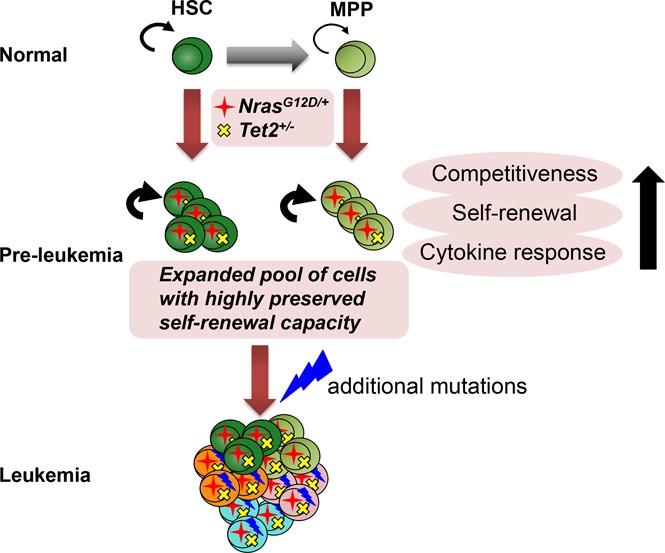
Visual Abstract
Introduction
The transformation of hematopoietic stem and progenitor cells (HSPCs) into leukemic stem cells (LSCs) requires a stepwise accumulation of mutations that dysregulate HSPCs to allow the mutant cells to outcompete the normal cells.1,2 These mutations must coordinate the balance of proliferation, differentiation, and self-renewal to sustain the competitive advantage of LSCs.3 Understanding how leukemia-associated mutations dysregulate HSPCs to transform them into LSCs is essential for developing targeted therapies to eliminate LSCs. Advanced genomic sequencing of human leukemia has identified a spectrum of key somatic mutations in genes that encode cell signaling molecules, transcription factors, epigenetic regulators, chromatin/histone modulators, and spliceosome components.4,5 Although the effects of many individual mutations on HSPCs have been extensively studied, how different mutations interact with one another and how the combined mutations modulate HSPC function remain elusive.
Concomitant mutations in epigenetic modifier TET2 and signaling molecule NRAS are frequently detected in myeloid malignancies such as chronic myelomonocytic leukemia (CMML)6,7 and acute myeloid leukemia (AML),8,9 suggesting a cooperativity of the 2 mutations. TET2 is a member of the TET family methylcytosine dioxygenases, which catalyze the conversion of 5-methyl-cytosine to 5-hydroxymethyl-cytosine and promote DNA demethylation.10 Loss-of-function mutations in TET2 are found in many human malignancies, including CMML (40%-60%).11,12 Notably, in most cases of CMML, TET2 mutations precede other genetic abnormalities and are therefore believed to establish preleukemic clonal hematopoiesis but likely acquire additional mutations to develop overt leukemia.6,13 Although most TET2 mutations in human leukemia are monoallelic,14 Tet2 haploinsufficiency in mice was not sufficient to induce leukemia in a majority of cases,15 suggesting a requirement for collaborating mutations. Complete ablation of Tet2, in contrast, drives an indolent CMML-like disease, characterized by monocytosis and extramedullary hematopoiesis.15,16 At the preleukemic stage, Tet2 deficiency increases hematopoietic stem cell (HSC) self-renewal.15-18 The effect of Tet2 deficiency on HSC proliferation has not been investigated.
The p21ras (Ras) family of signal switch molecules is essential for proliferative responses to hematopoietic growth factors.19-22 Activating RAS mutations are prevalent in human cancers, including hematologic malignancies.23,24 In CMML, oncogenic NRAS and KRAS mutations are found in ∼15% to 40% of patients4,25,26 and are associated with a more proliferative phenotype.27 RAS mutations have been detected as the initial or secondary mutations in CMML.6 In some cases of CMML, RAS mutations can persist after the patients have achieved complete disease remission.28 In murine models, endogenous N-RasG12D expression leads to a CMML-like disease21,29 and increased HSC proliferation, competitiveness, and self-renewal at the preleukemic stage.30 These data support the model that hyperactive Ras can act as either an initiating mutation to induce preleukemic clonal expansion or a collaborating mutation to promote disease progression.
The co-occurrence of NRAS and TET2 mutations in leukemia implies collaboration between the 2 in leukemogenesis. However, whether NRAS and TET2 mutations collaborate in vivo and how the 2 interact to modulate HSPC function at leukemia initiation have not been investigated. We report here that oncogenic N-RasG12D and Tet2 haploinsufficiency collaborate to dysregulate HSPCs in vivo by providing both distinct and complementary competitive advantages to HSPCs and accelerate CMML with significantly shortened overall survival and more complete disease penetrance.
Methods
Animals
The conditional mouse strains of Mx1-Cre/LSL-NrasG12D,29 Mx1-Cre/Tet2+/fl16 and transgenic Col1A1-H2B-GFP/Rosa26-M2-rtTA31 were previously described. All mice were housed in the Unit for Laboratory Animal Medicine at the University of Michigan, and protocols were approved by the University Committee on the Use and Care of Animals.
Flow cytometry analysis of hematopoietic populations
Enumeration of hematopoietic populations was performed as previously described.30 The gating strategy for SLAM HSCs (CD150+CD48−LSK), SLAM multipotent progenitors (MPPs; CD150−CD48−LSK), LSKs (Lineage−Sca1+cKit+), common myeloid progenitors (CMPs; Lin−Sca1−cKit+CD34+CD16/32−), granulocyte-macrophage progenitors (GMPs; Lin−Sca1−cKit+CD34+CD16/32+), megakaryocyte-erythroid progenitors (MEPs; Lin−Sca1−cKit+CD34−CD16/32−), monocytes (Gr-1lowMac-1+), granulocytes (Gr-1highMac-1+), T lymphocytes (CD3+), and B lymphocytes (B220+) is shown in supplemental Figure 1.
BM transplantation
Whole bone marrow (BM) or sorted cells from donor CD45.2 mice were collected in fluorescence-activated cell sorting (FACS) buffer (Hanks balanced salt solution buffer containing 2% fetal calf serum) and transplanted into lethally irradiated (1100 Gy) CD45.1 mice along with CD45.1 competitor cells. Donor chimerism was monitored for 20 weeks by peripheral blood FACS staining. At 20 weeks, mice were euthanized, and BM cells were stained for donor chimerism analysis.
In vivo BrdU incorporation assay
Mice were administered with a single dose of 100 mg/kg of 5-bromo-2′-deoxyuridine (BrdU) via intraperitoneal injection and placed on BrdU water (1 mg/mL) for 24 hours before BrdU incorporation assay.30 BM cells were stained with SLAM HSC antibodies, permeabilized and stained with anti-BrdU (APC BrdU Flow Kit; Fisher Scientific), and then analyzed by flow cytometry.
RNA sequencing
RNA sequencing libraries were prepared using the SMARTer Stranded RNA-Seq Kit (Takara), after removal of ribosomal RNA and mitochondrial RNA using the RiboGone-Mammalian Kit (Takara). Samples were multiplexed and sequenced on Illumina HiSeq 2500 with a paired-end sequencing length of 200 bp. RNA sequencing data were deposited in the Gene Expression Omnibus (accession #GSE97640).
Quantification and statistical analysis
Data are presented as mean ± standard error of the mean. Data were analyzed using Student t test to assess statistical significance. Additional experimental procedures are described in supplemental Methods.
Results
N-RasG12D and Tet2 haploinsufficiency together induce a lethal and highly penetrant CMML-like disease
To understand the functional effects of coexisting N-Ras and Tet2 mutations on leukemogenesis, we crossed Mx1-cre+/NrasLSL-G12D/+ mice29 with Tet2 conditional knockout mice.16 Single-mutant Mx1-cre+/Loxp-STOP-Loxp-NrasG12D/+ (NrasG12D/+) or Mx1-cre+/Tet2+/fl (Tet2+/−) and double-mutant Mx1-cre+/Loxp-STOP-Loxp-NrasG12D/+/Tet2+/fl (NrasG12D/+/Tet2+/−) mice were analyzed together with wild-type (WT) control mice. We chose to use heterozygous Tet2 knockout because most TET2 mutations in human leukemia are monoallelic.14 Administration of polyinosine/polycytosine (poly [I:C]) in 6-week-old sex- and age-matched mice led to activation of a single allele of NrasG12D and deletion of 1 allele of Tet2 in hematopoietic tissues.16,29 Mice were observed for a period of 600 days. All mutant groups of mice had reduced overall survival compared with control mice. Consistent with previous report,15 Tet2+/− mice showed indolent onset of disease, with median survival not reached, and only 16.7% of mice became moribund within 600 days. NrasG12D/+ mice also displayed indolent disease onset and incomplete disease penetrance (36.8%), with median survival not reached (Figure 1A). This is similar to our previous report showing a median survival of 588 days with NrasG12D/+ mice on a pure C57BL6 background.29 However, NrasG12D/+/Tet2+/− double mutants demonstrated significantly shortened median survival (439 days) with increased disease penetrance (73.3%) compared with single mutants (Figure 1A).
Figure 1.

N-RasG12Dand Tet2 haploinsufficiency induce accelerated CMML-like diseases in mice. (A) Kaplan-Meier survival curve of NrasG12D/+ (n = 33), Tet2+/− (n = 19), NrasG12D/+/Tet2+/− (n = 23), and control WT littermates (n = 30). The log-rank test was used to assess statistical significance. (B) Representative flow cytometry analysis of BM cells and splenocytes (SPs) from diseased mutant mice and age-matched WT controls. The frequency of myeloid cells is determined with markers of Mac-1 and Gr-1 (Mac-1+Gr-1low and Mac-1+Gr-1+). (C) Hematoxylin and eosin staining of spleen (scale bar represents 100 μm) and liver (scale bar represents 20 μm) sections from different groups of animals. Infiltration (IFT) of neoplastic cells is indicated. *P ≤ .05, **P ≤ .01, ***P ≤ .001.
The disease in moribund mice was best characterized as CMML-like by histopathology and immunophenotyping. Both NrasG12D/+/Tet2+/− and NrasG12D/+ mice demonstrated a dramatic expansion of myeloid cells (Mac1+Gr1+) in BM and spleen (Figure 1B; supplemental Table 1). In moribund and 600-day-old surviving mice, NrasG12D/+/Tet2+/− and NrasG12D/+ mice showed marked leukocytosis with significantly increased white blood cell counts. Monocytosis occurred in all mutant mice (supplemental Table 2). NrasG12D/+/Tet2+/− mice exhibited significantly enlarged spleen compared with WT mice. Histological examination revealed significant extramedullary hematopoiesis in moribund NrasG12D/+/Tet2+/− and NrasG12D/+ mice, with loss of normal spleen and liver architecture and extensive infiltration of mature myeloid cells (Figure 1C). No AML was detected in NrasG12D/+/Tet2+/− mice.
Although the moribund NrasG12D/+/Tet2+/− and NrasG12D/+ mice exhibited similar disease burden, more NrasG12D/+/Tet2+/− mice became sick during the observation period, indicating that Tet2 haploinsufficiency accelerates the disease. We performed further analysis at 6 months, when mutant mice demonstrated no phenotypic abnormalities. NrasG12D/+/Tet2+/− mice had lower BM cellularity (Figure 2A). Within BM, the percentage and total number of MPPs in NrasG12D/+/Tet2+/− mice were significantly higher than those of NrasG12D/+ mice, although the HSC and LSK levels were similar (Figure 2B; supplemental Figure 2A). Both NrasG12D/+/Tet2+/− and NrasG12D/+ mice showed increased numbers of CMPs, GMPs, and MEPs compared with WT mice; however, NrasG12D/+/Tet2+/− mice further enriched CMPs within total BM and the myeloid progenitor pool (Lin−Sca1−cKit+; Figure 2C; supplemental Figure 2B-C). Both NrasG12D/+/Tet2+/− and NrasG12D/+ mice displayed BM monocytosis (Figure 2D; supplemental Figure 2D). NrasG12D/+/Tet2+/− mice were characterized by marked splenomegaly with increased cellularity (Figure 2E). As a result, NrasG12D/+/Tet2+/− mice tended to have higher numbers of HSCs, MPPs, LSKs, myeloid progenitors, and mature myeloid cells in the spleen compared with NrasG12D/+ mice, although their relative percentages were similar (Figure 2F-H; supplemental Figure 2A-D). Notably, T and B lymphocytes were significantly reduced in NrasG12D/+/Tet2+/− mice, suggesting a myeloid-skewed differentiation (supplemental Figure 2D). Pathologic evaluation revealed that the spleen architecture from NrasG12D/+/Tet2+/− mice was largely effaced, whereas NrasG12D/+ spleen showed preserved architecture with moderate myeloid infiltration. Extensive myeloid infiltration was observed in the liver and lung in each single NrasG12D/+/Tet2+/− mouse, whereas this was rarely seen in single-mutant mice (Figure 2I). Taken together, NrasG12D/+/Tet2+/− mice have significantly higher degrees of extramedullary hematopoiesis and myeloid infiltration into nonhematopoietic organs, indicating that the 2 mutations collaborate to induce myeloid expansion and increase mortality.
Figure 2.

Steady-state hematopoietic differentiation in 6-month-old mice. (A) Total cellularity in the BM. (B-D) Absolute numbers of HSCs, MPPs, and LSKs (B); CMPs, GMPs, and MEPs (C); and mature granulocytes and monocytes (D) in the BM. (E) Spleen (SP) weight (g) and total cellularity. (F-H) Absolute numbers of HSCs, MPPs, and LSKs (F); CMPs, GMPs, and MEPs (G); and mature granulocytes and monocytes (H) in the SP. (I) Hematoxylin and eosin staining of SP, liver, and lung sections from different groups. IFT of neoplastic cells is indicated. The absolute numbers of BM cells were calculated using an established formula that estimates that 1 femur and 1 tibiae contain 7.5% of total nucleated BM cells. Data represent mean ± standard error of the mean. One-tailed Student t tests were used to assess statistical significance. *P ≤ .05, **P ≤ .01, ***P ≤ .001.
The CMML-like disease in NrasG12D/+/Tet2+/− mice is highly transplantable, and within 9 weeks, all secondary transplant recipients developed lethal CMML-like disease similar to phenotypes observed in primary donor mice, with significant monocytosis and enlarged spleen and liver (supplemental Figure 3). Although hemoglobin and red blood cell counts were normal in donor mice, the mice undergoing transplantation developed severe anemia and thrombocytopenia, which represent a more advanced disease stage (supplemental Tables 2 and 3). No somatic loss of the WT Nras allele or amplification of the NrasG12D mutant allele was detected in primary or diseased secondary transplant recipients (supplemental Figure 4A). Interestingly, Nras messenger RNA level was increased in diseased secondary transplant recipients, as compared with primary diseased mice (supplemental Figure 4B), which may explain the advanced disease phenotype. Taken together, oncogenic N-Ras and haploinsufficient Tet2 collaborate to induce a highly penetrative and transplantable CMML-like disease in mice.
N-RasG12D and Tet2 haploinsufficiency collaboratively enhance HSC competitiveness and self-renewal
Given that CMML is driven by dysregulated HSCs, we sought to investigate whether combining oncogenic N-RasG12D with haploinsufficient Tet2 would alter HSC function. We conducted all HSC analyses at 2 weeks post poly (I:C) injection, when the mutant alleles were fully activated and the mice showed no evidence of disease. This allowed us to investigate the early changes in mutant HSPCs that lead to the initiation of leukemia.
We first performed a competitive repopulation assay to assess HSC competitiveness. BM cells from CD45.2 Tet2+/−, NrasG12D/+, NrasG12D/+/Tet2+/−, and WT mice along with CD45.1 WT BM cells were transplanted into lethally irradiated CD45.1 mice at a 1:3 ratio of donor/recipient cells. Donor contribution (CD45.2+) to myeloid and lymphoid compartments was analyzed every 4 weeks in peripheral blood. Both N-RasG12D and Tet2 haploinsufficiency gave rise to significantly higher long-term multilineage reconstitution, consistent with previous reports.16,29 However, recipients of NrasG12D/+/Tet2+/− cells exhibited a significant further increase of donor reconstitution in all lineages over 20 weeks (Figure 3A). By 20 weeks, we assessed the percentage of donor-derived cells in HSPC compartments in the BM. Recipients of Tet2+/− cells exhibited significantly higher levels of donor-derived HSCs, MPPs, and LSKs, whereas recipients of NrasG12D/+ cells displayed similar levels of donor-derived HSCs and MPPs but significantly higher levels of donor-derived LSKs. The NrasG12D/+/Tet2+/− cells gave rise to the highest donor reconstitution across all HSPC compartments and mature lineage cells (Figure 3B). In transplant recipients, the myeloid/lymphoid ratio generated from mutant cells was balanced, without lineage skewing (supplemental Figure 5), indicating that donor HSCs are dysregulated at this early stage but not fully transformed to give rise to myeloid bias. Therefore, hyperactive N-Ras and Tet2 haploinsufficiency dysregulate HSCs at a preleukemic stage.
Figure 3.

N-RasG12Dand Tet2 haploinsufficiency collaborate to enhance HSC competitiveness and self-renewal. (A) Donor (CD45.2) BM cells (5 × 105) from Mx1-Cre/NrasG12D/+, Mx1-Cre/Tet2+/−, Mx1-Cre/NrasG12D/+/Tet2+/−, and littermate control mice at 2 weeks after poly (I:C) treatment (n = 3 donors per genotype) were transplanted into lethally irradiated recipient (CD45.1) mice (n = 15 recipients per genotype) along with 1.5 × 106 recipient BM cells. Donor-cell reconstitution in total nucleated cells and myeloid (Mac1+Gr1low and Mac1+Gr1+) and B- (B220+) and T-cell (CD3+) lineages was assessed in peripheral blood for 4 to 20 weeks after transplantation. (B) BM percentages of donor-derived HSCs (CD150+CD48−LSK), MPPs (CD150−CD48−LSK), and LSKs (Lin−Sca1+c-Kit+) and donor-derived myeloid (Mac1+Gr1low and Mac1+Gr1+) and B- (B220+) and T-cell (CD3+) lineages in primary transplant recipients (n = 8 per genotype). (C) Secondary transplantation (n = 10 recipients per genotype) of 3 × 106 BM cells from primary recipient mice in panel A (n = 2 donors per genotype). Donor-cell reconstitution in total nucleated cells and myeloid and B- and T-cell lineages was assessed. (D) Donor-derived myeloid and B- and T-cell lineages in the BM of secondary recipient mice (n = 9 per genotype). Data represent mean ± standard error of the mean. Two-tailed Student t tests were used to assess statistical significance. *P ≤ .05, **P ≤ .01, ***P ≤ .001.
To examine the effect of combined Nras and Tet2 mutations on HSC self-renewal, we performed serial transplantation assay by transplanting 3 × 106 BM cells from primary transplant recipients (Figure 3A) into lethally irradiated CD45.1 mice. In secondary transplant recipients, we continued to observe the highest long-term donor reconstitution by NrasG12D/+/Tet2+/− cells in all lineages in both peripheral blood (Figure 3C) and BM (Figure 3D).
To assess competitiveness and self-renewal of individual HSCs, we transplanted 15 FACS-purified SLAM HSCs from CD45.2 mice along with 3 × 105 CD45.1 WT BM cells into lethally irradiated CD45.1 recipients. At this dosage, WT, Tet2+/−, and NrasG12D/+ HSCs gave rise to low levels of donor reconstitution, with no significant difference between each group. However, NrasG12D/+/Tet2+/− HSCs gave rise to significantly higher levels of long-term multilineage reconstitution in transplant recipients (Figure 4A). Taken together, these results demonstrate a synergistic effect of N-RasG12D and Tet2 haploinsufficiency on HSC competitiveness and self-renewal.
Figure 4.

Purified HSCs and MPPs from NrasG12D/+/Tet2+/−mice gain enhanced competitiveness and self-renewal. (A) Fifteen donor (CD45.2) HSCs (CD150+CD48−LSK) from NrasG12D/+, Tet2+/−, NrasG12D/+/Tet2+/−, and littermate control mice (n = 2 donors per genotype) were transplanted into lethally irradiated recipient (CD45.1) mice (n = 10 recipients per genotype) along with 3 × 105 recipient BM cells. Donor-cell reconstitution in total nucleated cells and myeloid and B- and T-cell lineages was assessed in peripheral blood after transplantation. (B) Fifteen donor (CD45.2) MPPs (CD150−CD48−LSK) were transplanted into lethally irradiated recipient (CD45.1) mice (n = 10 recipients per genotype) along with 3 × 105 recipient BM cells. The number of recipient mice with long-term engraftment from 2 separate rounds of transplantation is shown for each genotype. (C) Donor-cell reconstitution was assessed in peripheral blood from mice in panel B. Data represent mean ± standard error of the mean. Two-tailed Student t tests were used to assess statistical significance. *P ≤ .05, **P ≤ .01, ***P ≤ .001.
MPPs from NrasG12D/+/Tet2+/− mice gain long-term multilineage reconstitution
Compared with HSCs, MPPs can generate all lineages of blood cells but have limited self-renewal potential and are not able to sustain long-term multilineage reconstitution in transplant recipients. Recent studies have shown that in a Tet2−/−/Flt3ITD-induced AML model, MPPs can propagate and sustain leukemia phenotypes in secondary transplant recipients.32 We next sought to assess the self-renewal potential of MPPs from NrasG12D/+/Tet2+/− mice. We transplanted 15 FACS-purified MPPs from CD45.2 mice into lethally irradiated CD45.1 recipients along with 3 × 105 CD45.1 WT BM cells. NrasG12D/+/Tet2+/− MPPs exhibited long-term donor engraftment in a majority of recipients (7 of 10), with significantly higher levels of multilineage donor reconstitution. In contrast, Tet2+/− and NrasG12D/+ single-mutant MPPs rarely gave rise to long-term reconstitution in recipients, and the level of reconstitution was much lower (Figure 4B-C). These data suggest that mutations in Nras and Tet2 collaboratively provide MPPs with stem cell–like features, including enhanced long-term reconstitution and self-renewal capacity.
Tet2 haploinsufficiency maintains HSCs and progenitors at a more quiescent state
To maintain hematopoietic homeostasis, HSCs preserve dormancy to retain long-term self-renewal capacity while also activating proliferation and differentiation to produce mature blood cells.33 Our previous study demonstrated that N-RasG12D causes overall HSC hyperproliferation without compromising self-renewal capacity through a bimodal effect.30 Here, we sought to assess how Tet2 haploinsufficiency regulates HSC proliferation, on its own and in combination with N-RasG12D. We first performed an in vivo 24-hour BrdU labeling assay (Figure 5A). Consistent with our previous report,30 NrasG12D/+ HSCs exhibited significantly increased overall BrdU incorporation compared with WT. Interestingly, Tet2+/− significantly reduced the level of BrdU incorporation in NrasG12D/+/Tet2+/− compared with NrasG12D/+ HSCs, restoring it to a level similar to the control (Figure 5B).
Figure 5.

Tet2 haploinsufficiency decreases cell division in HSPCs. (A) Scheme of BrdU incorporation by HSCs. A 24-hour pulse of BrdU was administered to mice at 2 weeks after poly (I:C) treatment. Percentages of BrdU+ HSCs were analyzed by flow cytometry. (B) BrdU incorporation by HSCs from NrasG12D/+, Tet2+/−, NrasG12D/+/Tet2+/−, and littermate control mice (n = 4 per genotype). (C) GFP expression in HSCs, MPPs, and LSKs from Mx1-Cre/Tet2+/−/Col1A1-H2B-GFP/Rosa26-M2-rtTA (Tet2+/−) mice and littermate controls after labeling followed by 6 or 18 weeks of chase without doxycycline (doxy; n = 4 pairs of mice from 3 or 4 independent experiments). All mouse pairs were age and sex matched. Data represent mean ± standard error of the mean. Two-tailed Student t tests were used to assess statistical significance. *P ≤ .05, **P ≤ .01, ***P ≤ .001. MFI, mean fluorescence intensity.
With the short pulse of BrdU labeling, Tet2+/− HSCs did not show changes in BrdU incorporation compared with WT (Figure 5B). Previous reports have indicated that at steady state, most HSCs are quiescent in G0, and only 3% are in the S/G2/M phase,34 and HSCs are heterogeneous in their rate of entering cell cycle.31,35 Therefore, the short-term BrdU labeling may not capture changes in HSC proliferation over time. To address this, we performed an H2B-GFP label retention assay by mating Mx1-Cre/Tet2+/− mice with Col1A1-H2B-GFP/Rosa26-M2-rtTA double transgenic mice.31 In this model, HSCs were first labeled with H2B-GFP during a 6-week period of doxycycline administration (2 g/L). After removal of doxycycline, HSCs dilute H2B-GFP with each round of division, which allows tracking of proliferation over time. After either a 6- or 18-week chase period, Tet2+/− HSCs retained much higher GFP levels compared with WT, indicating an overall less proliferative state of HSCs. Similarly, Tet2 haploinsufficiency led to decreased cell division in MPPs and LSKs (Figure 5C). Because quiescent HSCs maintain higher competitive advantage and self-renewal potential than fast-cycling HSCs,30,31,34 these results indicate Tet2 haploinsufficiency maintains balanced HSC proliferation and expands the HSC pool with high self-renewal and competitive potential.
Hyperactive N-Ras and Tet2 haploinsufficiency coordinate to dysregulate transcription programs in HSCs
We next performed RNA sequencing analysis in FACS-sorted SLAM HSCs to determine how Nras and Tet2 mutations interact to alter transcriptional programs at the preleukemic stage. Unsupervised correspondence analysis segregated mutant genotypes into clusters (Figure 6A), suggesting that transcription programs had been altered by single or combined mutations even at this early stage (2 weeks post–poly [I:C]). To explore downstream targets regulated by the 2 mutations, we examined differentially expressed genes in NrasG12D/+/Tet2+/− HSCs versus WT, with a fold change of >1.5 and false discovery rate of <0.1 (top-ranking genes are listed in Figure 6B). Among these genes, 3 negative regulators of the JAK/STAT pathway, Socs1 and 2 and Pim1, were significantly downregulated in NrasG12D/+/Tet2+/− HSCs. We validated the reduced expression by quantitative real-time reverse transcription polymerase chain reaction (Figure 6C).
Figure 6.

Analysis of differential gene expression in NrasG12D/+/Tet2+/−HSCs by RNA sequencing. (A) Clustering of differential genes by unsupervised correspondence analysis (COA). (B) List of the top-ranking upregulated and downregulated genes in NrasG12D/+/Tet2+/− vs WT HSCs with a fold change of >2 and false discovery rate (FDR) of <0.1 (P < .05). (C) Quantitative real-time reverse transcription polymerase chain reaction of negative regulators of JAK/STAT signaling in NrasG12D/+/Tet2+/− and WT HSCs from mice 2 weeks post–poly (I:C). Data represent mean ± standard error of the mean. Two-tailed Student t tests were used to assess statistical significance. *P ≤ .05, **P ≤ .01.
We next performed gene set enrichment analysis in single- and double-mutant HSCs as compared with WT. Consistent with the proliferation results, gene set enrichment analysis showed opposing effects of Nras and Tet2 mutations on cell proliferation–related pathways. Compared with WT, N-RasG12D upregulated cell-cycle, DNA replication, and RNA polymerase pathways, whereas Tet2+/− downregulated the cell-cycle pathway. In NrasG12D/+/Tet2+/− HSCs, cell-cycle and DNA replication pathways were downregulated compared with NrasG12D/+ HSCs (supplemental Figure 6). Again, these data support the model of balanced HSC proliferation and therefore more preserved HSC self-renewal in double-mutant mice.
Notably, DNA repair pathways were enriched in NrasG12D/+/Tet2+/− versus WT HSCs (supplemental Figure 7A). A defective DNA damage repair system has been shown to cause reduced HSC function, including impaired reconstitution potential and depletion of the HSC pool.36 In addition, many metabolic pathways were among the negatively enriched gene sets in NrasG12D/+/Tet2+/− (supplemental Figure 7A), raising the interesting possibility that hyperactive N-Ras and Tet2 haploinsufficiency together maintain HSCs in a more metabolically inactive state to preserve HSC function. Future investigations are needed to functionally validate the importance of these pathways. Notably, 1 of the top 20 gene sets positively enriched in double-mutant HSCs versus WT was the chemokine signaling pathway, which includes many signaling proteins in the JAK/STAT pathway (supplemental Figure 7B). Together with the reduced expression of JAK/STAT negative regulators (Figure 6C), these results suggest hyperactivation of JAK/STAT signaling in double-mutant HSCs.
Concurrent Nras and Tet2 mutations induce cytokine hypersensitivity in HSPCs
The RNA sequencing analysis suggested a hyperactivation of JAK/STAT signaling in NrasG12D/+/Tet2+/− HSCs. We next assessed response of HSPCs to cytokines known to support HSPC function. One hundred FACS-sorted SLAM HSCs were cultured in the presence of stem-cell factor (SCF) and thrombopoietin (TPO) for 10 days, and cell number was counted. Compared with WT, NrasG12D/+/Tet2+/− and NrasG12D/+ HSPCs required much lower concentration of cytokines to grow (Figure 7A left). To better define the response to each cytokine, 500 HSCs were sorted and cultured with either TPO or SCF. When cultured with TPO alone, NrasG12D/+/Tet2+/− HSPCs gave rise to significantly higher cell numbers compared with NrasG12D/+ or WT (Figure 7A right). SCF alone did not support HSPC growth ex vivo (data not shown). Therefore, our data suggest that combined N-RasG12D and Tet2 haploinsufficiency induce TPO hypersensitivity in HSPCs.
Figure 7.

N-RasG12Dand Tet2+/−collaborate to enhance cytokine response in HSPCs. (A) Total number of cells generated from 100 FACS-sorted HSCs after a 10-day culture (left) or from 500 HSCs after a 6-day culture (right) with SCF and/or TPO (n = 3). (B) Phosphorylated FACS analysis of STAT5 activation in LSKs. Whole BM cells were starved at 37°C for 30 minutes and then stimulated with TPO (6.25 ng/mL) for 15 minutes. The percentage of phosphorylated STAT5+ cells within the mutant LSK compartment was normalized to that of WT. Data represent mean ± standard error of the mean. Two-tailed Student t tests were used to assess statistical significance. *P ≤ .05, **P ≤ .01.
Given that STAT5 is downstream of TPO and is essential for leukemic stem-cell proliferation and self-renewal,37 we next examined whether STAT5 signaling is dysregulated in double-mutant HSPCs. Phosphorylated FACS analysis revealed that a significantly higher number of NrasG12D/+/Tet2+/− LSKs showed phosphorylated STAT5 activation compared with NrasG12D/+ and WT LSKs (Figure 7B). Taken together, these results suggest that oncogenic NrasG12D and haploinsufficient Tet2 collaborate to confer cytokine hypersensitivity and hyperactivation of cytokine signaling to HSPCs, which may contribute to their enhanced function.
Discussion
CMML is a clonal stem-cell disorder38 with poor prognosis, and in approximately 30% of patients, CMML transforms to AML.4,39 Allogeneic stem-cell transplantation remains the only curative treatment for CMML; however, it is only feasible for a minority of patients and carries significant risk of toxicity and mortality. Clinical trials for CMML are challenging given the rarity of CMML and the heterogeneous clinical presentation. Therefore, preclinical modeling of CMML is essential for interrogating the biology of the disease and testing and developing new therapeutic strategies. The novel mouse model we generated here represents significant advantages over previous models of CMML. A previous study using short hairpin RNA to knock down Tet2 in NrasG12D/+ cells demonstrated no effects on NrasG12D/+-induced CMML when transplanted into lethally irradiated recipients.40 However, this model is limited, given that the efficiency and sustained knockdown of protein or enzyme activity by short hairpin RNA are difficult to predict. In genetic mouse models, single-mutant N-RasG12D induces a CMML-like phenotype, with long latency and low disease penetrance.29 Moreover, many mice developed significant lymphoproliferation and histiocytic sarcoma in the BM, spleen, and nonhematopoietic organs.29 The disease diversity in this model complicates the study of CMML. In a previous report of Tet2-knockout models, heterozygous loss of Tet2 led to a mild disease phenotype of CMML, with disease penetrance of <8%.15 Given that TET2 and NRAS are 2 of the most frequently mutated genes in human CMML, our study provides a novel genetically accurate, highly penetrative, and transplantable murine CMML model by coexpressing NrasG12D and haploinsufficient Tet2.
In this study, we demonstrate that hyperactive N-Ras and Tet2 haploinsufficiency collaborate to facilitate HSPC transformation. First, we show that Nras and Tet2 mutations collaborate to increase HSC competitiveness and self-renewal capability through serial transplantation. These results reflect stronger HSC stemness compared with either mutation alone. Notably, enhanced HSPC stemness may have important therapeutic implications, because it correlates with drug resistance.2 Furthermore, our study demonstrates that N-RasG12D and Tet2 haploinsufficiency collaborate to enhance MPP stemness, with gained long-term reconstitution and self-renewal potential. Interestingly, similar results have been observed in primary human AML xenograft models, where MPPs propagate leukemia.41 Taken together, we propose that the enhanced competitiveness and self-renewal of double-mutant HSCs and MPPs lead to the expansion of a primed cell population, where the acquisition of additional mutations leads to clinical disease (supplemental Figure 3).
HSCs balance quiescent and proliferative states both to retain long-term self-renewal capacity and to replenish the BM with all needed progenitors.33 Previous studies have shown that Tet2 deficiency promotes HSPC proliferation; however, the analysis was performed using in vitro colony-forming unit formation assays.15 For the first time, we demonstrate in vivo that Tet2 haploinsufficiency keeps HSPCs at a more quiescent state and reverses the accelerated HSC proliferation mediated by N-RasG12D. From these data, important implications surface. Although N-RasG12D exerted a bimodal effect on HSCs by inducing a fast-proliferating and more quiescent HSC subpopulation, the fast-proliferating population lost its reconstitution capacity much quicker than the quiescent population.30 Thus, Tet2 haploinsufficiency maintains the quiescence of HSCs when combined with N-RasG12D and expands the HSC pool with preserved self-renewal capacity. This conclusion is consistent with previous reports showing that mutant Tet2 enhances HSC competitiveness and self-renewal.16 Illustratively, Tet2 mutations often occur as early events and are associated with different disease phenotypes as additional mutations are acquired. For example, murine models coexpressing Tet2 loss with signaling molecules demonstrate a broad spectrum of blood diseases. Coexistence of KitD816V and Tet2 loss leads to aggressive mastocytosis.42 Combined JAK2V617F and Tet2 loss promotes advanced myeloproliferative neoplasms.43 FLT3ITD with Tet2 loss induces a rapid onset of AML.32 It is notable that the disease phenotype reflects the prevalence of each signaling mutation in human myeloid malignancies, with RAS mutations being more frequent in CMML, KIT mutations more dominant in systemic mastocytosis, JAK2 mutations more common in myeloproliferative neoplasms, and FLT3ITD more prevalent in AML.
Lastly, our study shows that Tet2 deficiency works to enhance the cellular responses of N-Ras to provide HSPC advantages over WT cells. Tet2 haploinsufficiency together with hyperactive N-Ras promotes HSPC expansion in response to TPO through synergistic activation of STAT5. Notably, oncogenic N-Ras does not, by itself, exhibit similar effects. These findings are supported by RNA sequencing analysis showing suppressed expression of JAK/STAT negative regulators SOCS1/2 and Pim1 in double-mutant HSCs. Although Pim1 expression is largely associated with JAK/STAT activation, a recent study suggests that Pim1 acts as a negative feedback inhibitor to restrict JAK/STAT activity.44 A recent report showed Epor-mediated JAK-STAT activation in a Dnmt3a/Tet2 double-knockout mouse model,45 suggesting a role of Tet2 in signaling regulation. Thus, Tet2 haploinsufficiency and hyperactive N-Ras collaborate to promote cytokine hypersensitivity through activation of JAK/STAT, which may have implications in modulating therapeutic responses.
Taken together, our studies establish a novel, genetically accurate mouse model where hyperactive N-RasG12D and Tet2 haploinsufficiency collaborate to dysregulate HSPCs. This promotes an accelerated, transplantable CMML disease with high penetrance. We present this model as a tractable platform for mechanistic and preclinical therapeutic studies to target CMML stem cells.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the University of Michigan Unit for Laboratory Animal Medicine for mouse colony maintenance, the Flow Cytometry Core for technical assistance, and the DNA Sequencing and Bioinformatics Core for performing RNA sequencing.
This research was supported by National Institutes of Health (NIH), National Heart, Lung, and Blood Institute grant R01HL132392, American Cancer Society Research Scholar grant 125080-RSG-13-253-01-LIB, and the Gabrielle's Angel Foundation for Cancer Research Medical Research Award (Q.L.). G.N. was supported by NIH T32 (5-T32-HL007622) from the National Heart, Lung, and Blood Institute.
Authorship
Contribution: Q.L. and X.J. conceived the project and wrote the manuscript with input from all authors; X.J., T.Q., M.E.F., and Q.L. designed the experiments; X.J., M.Z., L.L., K.Y., V.N., T.H., G.N., and R.C. developed experimental protocols and performed experiments; T.Q. and M.E.F. developed computational methods; T.Q. performed bioinformatic analysis; and N.B. performed histopathological analysis.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Qing Li, University of Michigan, 109 Zina Pitcher Pl, BSRB 1520, Ann Arbor, MI 48109; e-mail: lqing@med.umich.edu.
References
- 1.Jan M, Majeti R. Clonal evolution of acute leukemia genomes. Oncogene. 2013;32(2):135-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275-291. [DOI] [PubMed] [Google Scholar]
- 3.Jude CD, Gaudet JJ, Speck NA, Ernst P. Leukemia and hematopoietic stem cells: balancing proliferation and quiescence. Cell Cycle. 2008;7(5):586-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patnaik MM, Tefferi A. Cytogenetic and molecular abnormalities in chronic myelomonocytic leukemia. Blood Cancer J. 2016;6:e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welch JS, Ley TJ, Link DC, et al. . The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itzykson R, Kosmider O, Renneville A, et al. . Clonal architecture of chronic myelomonocytic leukemias. Blood. 2013;121(12):2186-2198. [DOI] [PubMed] [Google Scholar]
- 7.Kohlmann A, Grossmann V, Klein HU, et al. . Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol. 2010;28(24):3858-3865. [DOI] [PubMed] [Google Scholar]
- 8.Patel JP, Gönen M, Figueroa ME, et al. . Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12(9):599-612. [DOI] [PubMed] [Google Scholar]
- 10.Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W. The ten-eleven translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia. 2014;28(3):485-496. [DOI] [PubMed] [Google Scholar]
- 11.Patnaik MM, Tefferi A. Chronic myelomonocytic leukemia: 2016 update on diagnosis, risk stratification, and management. Am J Hematol. 2016;91(6):631-642. [DOI] [PubMed] [Google Scholar]
- 12.Abdel-Wahab O, Mullally A, Hedvat C, et al. . Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood. 2009;114(1):144-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Itzykson R, Solary E. An evolutionary perspective on chronic myelomonocytic leukemia. Leukemia. 2013;27(7):1441-1450. [DOI] [PubMed] [Google Scholar]
- 14.Langemeijer SM, Kuiper RP, Berends M, et al. . Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41(7):838-842. [DOI] [PubMed] [Google Scholar]
- 15.Li Z, Cai X, Cai CL, et al. . Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood. 2011;118(17):4509-4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moran-Crusio K, Reavie L, Shih A, et al. . Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell. 2011;20(1):11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ko M, Bandukwala HS, An J, et al. . Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci USA. 2011;108(35):14566-14571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shide K, Kameda T, Shimoda H, et al. . TET2 is essential for survival and hematopoietic stem cell homeostasis. Leukemia. 2012;26(10):2216-2223. [DOI] [PubMed] [Google Scholar]
- 19.Braun BS, Tuveson DA, Kong N, et al. . Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci USA. 2004;101(2):597-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Liu Y, Li Z, et al. . Endogenous oncogenic Nras mutation promotes aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood. 2010;116(26):5991-6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Liu Y, Li Z, et al. . Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood. 2011;118(2):368-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Meter ME, Díaz-Flores E, Archard JA, et al. . K-RasG12D expression induces hyperproliferation and aberrant signaling in primary hematopoietic stem/progenitor cells. Blood. 2007;109(9):3945-3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3(6):459-465. [DOI] [PubMed] [Google Scholar]
- 24.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7(4):295-308. [DOI] [PubMed] [Google Scholar]
- 25.Onida F, Kantarjian HM, Smith TL, et al. . Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood. 2002;99(3):840-849. [DOI] [PubMed] [Google Scholar]
- 26.Itzykson R, Kosmider O, Renneville A, et al. . Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol. 2013;31(19):2428-2436. [DOI] [PubMed] [Google Scholar]
- 27.Ricci C, Fermo E, Corti S, et al. . RAS mutations contribute to evolution of chronic myelomonocytic leukemia to the proliferative variant. Clin Cancer Res. 2010;16(8):2246-2256. [DOI] [PubMed] [Google Scholar]
- 28.Callahan MK, Rampal R, Harding JJ, et al. . Progression of RAS-mutant leukemia during RAF inhibitor treatment. N Engl J Med. 2012;367(24):2316-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Q, Haigis KM, McDaniel A, et al. . Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood. 2011;117(6):2022-2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Q, Bohin N, Wen T, et al. . Oncogenic Nras has bimodal effects on stem cells that sustainably increase competitiveness. Nature. 2013;504(7478):143-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Foudi A, Hochedlinger K, Van Buren D, et al. . Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat Biotechnol. 2009;27(1):84-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shih AH, Jiang Y, Meydan C, et al. . Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer Cell. 2015;27(4):502-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rossi L, Lin KK, Boles NC, et al. . Less is more: unveiling the functional core of hematopoietic stem cells through knockout mice. Cell Stem Cell. 2012;11(3):302-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson A, Laurenti E, Oser G, et al. . Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118-1129. [DOI] [PubMed] [Google Scholar]
- 35.Kiel MJ, He S, Ashkenazi R, et al. . Haematopoietic stem cells do not asymmetrically segregate chromosomes or retain BrdU. Nature. 2007;449(7159):238-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moehrle BM, Geiger H. Aging of hematopoietic stem cells: DNA damage and mutations? Exp Hematol. 2016;44(10):895-901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kato Y, Iwama A, Tadokoro Y, et al. . Selective activation of STAT5 unveils its role in stem cell self-renewal in normal and leukemic hematopoiesis. J Exp Med. 2005;202(1):169-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshimi A, Balasis ME, Vedder A, et al. . Robust patient-derived xenografts of MDS/MPN overlap syndromes capture the unique characteristics of CMML and JMML. Blood. 2017;130(4):397-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Patnaik MM, Wassie EA, Lasho TL, Hanson CA, Ketterling R, Tefferi A. Blast transformation in chronic myelomonocytic leukemia: risk factors, genetic features, survival, and treatment outcome. Am J Hematol. 2015;90(5):411-416. [DOI] [PubMed] [Google Scholar]
- 40.Chang YI, Damnernsawad A, Allen LK, et al. . Evaluation of allelic strength of human TET2 mutations and cooperation between Tet2 knockdown and oncogenic Nras mutation. Br J Haematol. 2014;166(3):461-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goardon N, Marchi E, Atzberger A, et al. . Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell. 2011;19(1):138-152. [DOI] [PubMed] [Google Scholar]
- 42.De Vita S, Schneider RK, Garcia M, et al. . Loss of function of TET2 cooperates with constitutively active KIT in murine and human models of mastocytosis. PLoS One. 2014;9(5):e96209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen E, Schneider RK, Breyfogle LJ, et al. . Distinct effects of concomitant Jak2V617F expression and Tet2 loss in mice promote disease progression in myeloproliferative neoplasms. Blood. 2015;125(2):327-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peltola KJ, Paukku K, Aho TL, Ruuska M, Silvennoinen O, Koskinen PJ. Pim-1 kinase inhibits STAT5-dependent transcription via its interactions with SOCS1 and SOCS3. Blood. 2004;103(10):3744-3750. [DOI] [PubMed] [Google Scholar]
- 45.Zhang X, Su J, Jeong M, et al. . DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat Genet. 2016;48(9):1014-1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.