

Abstract
A considerable proportion of tumors exhibit aneuploid karyotypes, likely resulting from the progressive loss of chromosomes after whole-genome duplication. Here, by using isogenic diploid and near-tetraploid (4N) single-cell–derived clones from the same parental cell lines, we aimed at exploring how polyploidization affects cellular functions and how tetraploidy generates chromosome instability. Gene expression profiling in 4N clones revealed a significant enrichment of transcripts involved in cell cycle and DNA replication. Increased levels of replication stress in 4N cells resulted in DNA damage, impaired proliferation caused by a cell cycle delay during S phase, and higher sensitivity to S phase checkpoint inhibitors. In fact, increased levels of replication stress were also observed in nontransformed, proliferative posttetraploid RPE1 cells. Additionally, replication stress promoted higher levels of intercellular genomic heterogeneity and ongoing genomic instability, which could be explained by high rates of mitotic defects, and was alleviated by the supplementation of exogenous nucleosides. Finally, our data found that 4N cancer cells displayed increased migratory and invasive capacity, both in vitro and in primary colorectal tumors, indicating that tetraploidy can promote aggressive cancer cell behavior.—Wangsa, D., Quintanilla, I., Torabi, K., Vila-Casadesús, M., Ercilla, A., Klus, G., Yuce, Z., Galofré, C., Cuatrecasas, M., Lozano, J. J., Agell, N., Cimini, D., Castells, A., Ried, T., Camps, J. Near-tetraploid cancer cells show chromosome instability triggered by replication stress and exhibit enhanced invasiveness.
Keywords: genomic instability, aneuploidy, lagging chromosomes, invasive front, colorectal cancer
Genetic instability, which comprises both numerical and structural chromosome abnormalities, is a hallmark of cancer. The process to establish cancer-specific aneuploid karyotypes implies certain chromosomal gains and losses that define the tumor genomic landscape (1). Further, many cancer cell populations display subpopulations of near-tetraploid (4N) cells. Although missegregation of individual chromosomes is a major mechanism of aneuploidy, it is unlikely that the rate of single chromosome gains and losses alone may explain the 4N karyotypes. Thus, an unstable intermediate state, which can arise as a result of whole-genome duplication, has been proposed as the most probable route to highly aneuploid karyotypes (2).
In addition to aneuploidy, many cancer cells display high rates of chromosomal instability (CIN) (3), by which chromosome copy number varies from cell to cell within the population. Errors in chromosome segregation during cell division resulting in unequal distribution of chromosomes between the 2 daughter cells have been acknowledged as a major cause of CIN (4). Alterations in the mitotic spindle assembly checkpoint, sister chromatid cohesion defects, or the presence of supernumerary centrosomes are well-established mechanisms of chromosome missegregation and CIN (5). In fact, extra centrosomes are sufficient to induce chromosome segregation errors by triggering the formation of merotelic attachments during a transient multipolar intermediate before a bipolar division (6, 7). Moreover, the presence of extra chromosomes has also been associated with the generation of CIN (8).
Recently, replication stress has emerged as a source of CIN in cancer cells (9); however, the link between replication stress and CIN is still largely unexplored. DNA replication is a critical step for the faithful transmission of genetic information, and DNA damage inflicted by different causes can compromise the accuracy of DNA replication. Stalled replication forks activate the DNA replication stress response by rapid coating of single-stranded DNA with replication protein A (RPA), which induces the activation of ataxia telangiectasia and rad3-related (ATR) serine/threonine kinase and, finally, results in the phosphorylation of checkpoint kinase (CHK) 1. The activation of this pathway is essential to limit the entry into mitosis in the presence of DNA damage (10).
The theory that tetraploidy is a transient state on the route to aneuploid karyotypes has also been explored in primary tumors. During the progression from Barrett esophagus to adenocarcinoma, a tetraploid genetically unstable intermediate detected in the premalignant lesions evolves to carcinoma with highly aneuploid karyotypes (11). Furthermore, subtetraploid cell populations are also associated with a higher prevalence of metastasis in in vitro models of colorectal cancer (12), primary renal cell carcinomas (13), and lung cancer (14).
Here, we unveiled significant transcriptional changes in genes linked to the replication stress response, which appear concomitantly with DNA damage and mitotic defects in 4N cells. Furthermore, these cells displayed increased migratory ability and invasiveness. Finally, 4N cells were found in the invasive fronts of primary colorectal tumors and displayed ongoing CIN.
MATERIALS AND METHODS
Cell culture, generation of clones, and primary tumors
DLD-1 and RKO cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in Rosewell Park Memorial Institute (RPMI) 1640 and DMEM–F-12 medium, respectively, supplemented with antibiotics and 10% fetal bovine serum (FBS) at 37°C in 5% CO2. The identity of the cell line was confirmed by the presence of unique chromosomal abnormalities as recorded in the SKY/CGH (Spectral Karyotyping/Comparative Genomic Hybridization) database (https://www.ncbi.nlm.nih.gov/dbvar/studies/nstd136/). To generate near-diploid (2N) and 4N clones, a suspension of bulk parental DLD-1 cells was single-cell flow-sorted by fluorescence-activated cell sorting (FACS) into 96-well plates based on size differences (i.e., forward scatter). For the RKO cell line, we treated the cells with 0.75 µM of the cytokinesis inhibitor dihydrocytochalasin D (Sigma-Aldrich, St. Louis, MO, USA) for 24 h to generate a 4N subpopulation before performing the single-cell sorting procedure. The plates were manually scrutinized to ensure that only wells harboring a single cell were used for subsequent culturing. Ploidy of individual clones was determined by karyotypic analysis of metaphase spreads. Wild-type and 3 posttetraploid clones (i.e., RPT1, RPT3, and RPT4) derived from the RPE1 cell line (kindly provided by Z. Storchova, University of Kaiserslautern, Kaiserslautern, Germany) were cultured in DMEM–F-12 supplemented with antibiotics and 10% FBS at 37°C in 5% CO2. For the experiments described in this study, we used three 2N clones and up to seven 4N clones derived from the DLD-1 cell line. In regard to the RKO cell line, we used two 2N clones and up to four 4N clones.
When required, cells were treated with 0.2 µM of the DNA polymerase inhibitor aphidicolin (Sigma-Aldrich) or exposed to 10 µM of the ATR inhibitor (ATRi) VE821 (Selleckchem, Houston, TX, USA) for 24 h. Cells were incubated with a mixture of the exogenous nucleosides deoxyadenosine (solubilized in 0.1 M NaOH), deoxyguanosine (solubilized in 1 M NH4OH), thymidine (solubilized in H2O), and deoxycytidine (solubilized in 1 M NaOH) (Sigma-Aldrich) for 48 h at a concentration range from 10 to 160 µM (15).
Colorectal primary tumors were provided by the Hospital Clínic de Barcelona. All patients signed the corresponding informed consent form, and the sample collection was approved by the local ethics committees. Sections of 4 μm from formalin-fixed, paraffin-embedded blocks were obtained, and an experienced pathologist marked the corresponding main mass of the tumor and the invasive front based on hematoxylin and eosin–stained consecutive sections for each sample.
Nucleic acid, protein, and nuclear volume quantification
In order to assess the DNA, RNA, and protein quantity after genome duplication, cells were synchronized in medium supplemented with 2% FBS and 100 µg/ml cycloheximide (Sigma-Aldrich) for 24 h, followed by 4 h of fresh medium containing 10% FBS. The same number of cells were collected for DNA, RNA, and protein with the respective extraction methods: chloroform, TRIzol, and RIPA extractions, respectively (https://ccr.cancer.gov/Genetics-Branch/thomas-ried). Measurements were made with Nanodrop (Thermo Fisher Scientific, Waltham, MA, USA), Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), and Spectramax M2 (Molecular Devices, Sunnyvale, CA, USA).
Nuclear volume was measured in synchronized G1/S cells using double thymidine block. Cells were fixed in methanol–acetic acid, stained with DAPI, and analyzed by the automatic acquisition and analysis Bioview 3.6.0.16 software (Bioview, Rehovot, Israel) on an Olympus BX63 device (Olympus, Tokyo, Japan). Five thousand nuclei were scanned for each sample. Studies were repeated in triplicate; overlapping cells were discarded. The Bioview software automatically calculated the diameter and area. The volume was measured using the following formula: V = 4/3 × A × c, where A is the area and c is the radius.
Viability, growth, and colony formation assays
One thousand cells were seeded in 96-well plates and incubated for 5 d before 1 h incubation of CellTiter-Blue (Promega, Madison, WI, USA). Fluorescent signals were measured using a SpectraMax M2 plate reader (Molecular Devices). To obtain growth curves, 70,000 cells of each clone were seeded in triplicate in 6-well plates using a Neubauer chamber every 24 h over a 5 d period. To assess for colony formation in both treated and untreated conditions, 200 cells of each clone were seeded in a 6-well plate. Cells were then cultured for 14 d and stained with crystal violet (Sigma-Aldrich). Colonies were imaged on a Zeiss Axio Observer.A1 microscope (Carl Zeiss GmbH, Jena, Germany) equipped with a ×5/0.12 NA Plan-Apochromatic objective. Digital images were captured by AxioVision software and quantified by ImageJ (Image Processing and Analysis in Java; National Institutes of Health, Bethesda, MD, USA; http://imagej.nih.gov/).
Cytokinesis-block micronucleus assay, FISH, and spectral karyotyping
Binucleated cells were generated as previously described (16). Proliferating cells were incubated with 6 µg/ml of cytochalasin B (Sigma-Aldrich) for 24 h before suspension in 0.075 KCl at 4°C and centrifuged. After centrifugation, a mixture of fixative (methanol:acetic acid, 3:1) and 37% formaldehyde was added to the cells. Finally, fixed binucleated cells were dropped on slides and air dried.
Metaphase chromosomes, and interphase and binucleate cell generation were performed and analyzed as previously described (16). Four-color FISH was performed using contigs of overlapping bacterial artificial chromosome (BAC) clones that corresponded to 4 genes: TERC (3q26), EGFR (7p12), CCND1 (11q13), and CDX2 (13q12). Nick translation was used to label each contig with Dy505 (Dyomics, Jena, Germany), Biotin-dUTP (Roche Applied Science, Penzberg, Germany), Dy415 (Dyomics), or Spectrum Orange-dUTP (Abbott Molecular, Des Plaines, IL, USA). Centromere 4 and 6, labeled with FITC and Cy3, respectively, were generated from yeast artificial chromosome. For hybridization and detection, standard FISH and SKY protocols were used (https://ccr.cancer.gov/Genetics-Branch/thomas-ried). Interphase FISH images were captured on the Metafer (MetaSystems, Boston, MA, USA) scanning system using an Olympus microscope with a ×40/1.30 NA objective.
Microarrays
Human 4 × 180K oligonucleotide-based array comparative genomic hybridization (aCGH) and 4 × 44K one-color expression arrays were conducted according to the manufacturer’s protocol (oligonucleotide array-based CGH, protocol version 4.0, June 2006, and One-Color Microarray-Based Gene Expression Analysis, protocol version 6.9, July 2015, respectively; Agilent Technologies) with minor modifications.
Segmentation for aCGH was done using R (R Foundation for Statistical Computing, Vienna, Austria; http://www.r-project.org/) package DNAcopy 1.44.0 with default options (17), and were visualized using Circos plots (18). Cutoff for defining gains and losses was set to 0.22. Gene set enrichment analysis was used with gene sets extracted from Molecular Signature Database (MSigDB v.4-0) C2 (manually curated gene sets from online pathway databases; publications in PubMed and knowledge of domain experts) (19). Only gene sets with more than 15 genes and less than 500 genes were included in the analysis. Analysis was based on a Fisher exact t test and a weighted scoring scheme with 150,000 permutations. aCGH and gene expression microarray–normalized data can be extracted from the Gene Expression Omnibus database (National Center for Biotechnology Information, Bethesda, MD, USA; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE81395.
Real-time PCR
Quantification of MYC, E2F1, and KRAS was performed using the TaqMan Assay Technology (Thermo Fisher Scientific). cDNA was generated from 1 μg of total RNA using gene-specific RT primers and the High Capacity cDNA RT Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Real-time PCR amplifications were performed in triplicate on an ABI 7900 Sequence Detection System using Universal PCR Master Mix without AmpErase Uracil N-Glycosylase (UNG; Thermo Fisher Scientific) and TaqMan made-to-order probes. The thermal cycling conditions comprised an initial denaturation step at 95°C for 10 min, followed by 45 cycles at 95°C for 15 s and 60°C for 1 min. Gene expression was normalized with GAPDH, and we used the ΔΔCt method for quantification.
Protein analysis
Cells were trypsinized and extracted with RIPA buffer (50 mM Tris-HCl, 1% NP-40, 0.5% Na-deoxycholate, 0.1% SDS, 150 mM NaCl, 2 mM EDTA, and 50 mM NaF, with protease inhibitors) to perform Western blot analysis. Protein samples were resolved by 4% to 12% SDS-PAGE gels and electroblotted onto a PVDF membrane. The membrane was blocked in 5% milk and Tris-buffered saline, 0.1% Tween 20 (TBST), for 1 h, incubated with primary antibody diluted in blocking solution overnight at 4°C, washed 3 times with TBST, incubated with secondary antibodies for 1 h at room temperature, and washed 3 times with TBST. For the detection of signals, SuperSignal West Femto (Thermo Fisher Scientific) was used. Membranes were imaged on an Image LAS400 device (GE Healthcare Life Sciences, Chicago, IL, USA). The primary antibodies and dilutions used were: rat anti-RPA32 (Cell Signaling Technology, Danvers, MA, USA), 1:2000; rabbit anti–phospho-RPA32 (S33) (Bethyl Laboratories, Montgomery, TX, USA), 1:1000; rabbit anti–minichromosome maintenance complex component 2 (MCM2; Cell Signaling Technology), 1:1000; mouse anti–ribonucleotide reductase regulatory subunit M2 (RRM2; Abnova, Taipei City, Taiwan), 1:1000; rabbit anti-CHK1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), 1:500; rabbit anti–phospho-CHK1 (Cell Signaling Technology), 1:100; rabbit anti-CHK2, 1:500; and rabbit anti–phospho-CHK2 (Thr68) (Novus Biologicals, Littleton, CO, USA), 1:500. Blots were detected using goat anti-rabbit (1:2000) and goat anti-rat (1:2000) secondary antibodies (Thermo Fisher Scientific).
For immunostaining experiments, cells were grown on sterilized 22 × 22 mm coverslips inside 6-well plates according to standard protocols. Cells were then fixed and permeabilized either using 4% paraformaldehyde for 10 min followed by 10 min in 0.5% Triton X-100 or in a single step with ice-cold methanol. Incubation with selected primary antibody was done according to the manufacturer’s recommendations. When required, cells were treated with 0.2 µM of the DNA polymerase inhibitor aphidicolin (Sigma-Aldrich) for 24 h. The primary antibodies and dilutions used in this study included: rabbit anti–γ-tubulin (Sigma-Aldrich), 1:1000; rabbit anti–tumor protein p53 binding protein 1 (53BP1; Novus Biologicals), 1:1000; mouse anti–phospho-histone 2AX (H2AX) (S139) (Millipore, Billerica, MA, USA), 1:500; rabbit anti–phospho-histone H3 (Millipore), 1:1000; mouse anti–α-tubulin (DM1A; Sigma-Aldrich), 1:500; human anti-centromere antigen (Antibodies, Atlanta, GA, USA), 1:100; and mouse anti–cyclin D1 (Abcam, Cambridge, MA, USA), 1:50. The secondary antibodies used were as follows: Rhodamine Red-X Goat Anti-Human (Jackson ImmunoResearch Laboratories, West Grove, PA, USA), 1:1000; Alexa Fluor 488 goat anti-rabbit 1:500; Alexa Fluor 594 goat anti-mouse 1:1000; and Alexa Fluor 488 Goat Anti-Mouse (Thermo Fisher Scientific), 1:1000.
To normalize the amount of DNA damage for the DNA content of each clone, the integrated density of DAPI signal and the number of γ-H2AX and/or 53BP1 foci were calculated for each individual cell using the Fiji-ImageJ software package (20).
Flow cytometry
Double analysis of DNA content and bromodeoxyuridine (BrdU) incorporation was performed to determine S-phase cell progression through the cell cycle. Cells were treated with a low concentration of aphidicolin (0.05 µM) (Sigma-Aldrich) for 30 min and with nocodazole (250 ng/ml) (Sigma-Aldrich) to arrest cells in mitosis. Cells were fixed, washed, and incubated with rat anti-BrdU (Abcam) primary antibody (1:250 dilution) for 1 h at room temperature. After washing, cells were incubated with secondary Alexa Fluor 488 Anti-Rat Conjugated Antibody (Thermo Fisher Scientific) for 45 min, washed with PBS/0.05% Tween 20 (Sigma-Aldrich), and resuspended in PBS containing 10 µg/ml propidium iodide and 0.1 mg/ml RNase A (Sigma-Aldrich) before performing FACS analysis (BD Biosciences, San Jose, CA, USA).
Migration and invasion assays
The wound-healing (scratch) assay was performed using confluent cells in a 24-well plate that was treated with mitomycin (Sigma-Aldrich) for 1 h at 37°C. A pipette tip was used to create a straight-line scratch. Migration was monitored by acquiring images every 24 h until the cells displaying fast migrating characteristics achieved a complete closure of the scratch. Images were acquired every 24 h using the AxioVision software on a Zeiss Axio Observer.A1 microscope. Digital images were analyzed by ImageJ software to determine the scratch closure area at each time point.
BioCoat inserts without Matrigel (BD Biosciences) were also used to assess migration capacity. For that, 100,000 cells were initially seeded and incubated for 48 h before evaluation. Invasion assay was performed using pore transwell polyethylene terephthalate membrane (BD Biosciences) with the appropriate concentration of Matrigel added.
Statistical analysis
The Fisher exact t test and proportions test (prop.test) were used to assess statistical significance using R packages (https://www.r-project.org/), when appropriate, unless another method has been indicated.
RESULTS
Generation and characterization of cancer diploid and 4N isogenic clones
To investigate the consequences of tetraploidization in cancer cells, we isolated 2N and 4N isogenic cell lines derived from the stable diploid colorectal cancer cell lines DLD-1 and RKO. Cytogenetic analysis suggested the existence of a 4N subpopulation in DLD-1 cells (Fig. 1A); thus, we performed single-cell flow sorting to isolate, according to differences in cell size, 2N or 4N single cells, from which we then generated individual clones and confirmed ploidy status by counting the chromosomes (Fig. 1B). For RKO cells, 4N clones were generated by cytokinesis inhibition followed by single-cell cloning. Next, we conducted proliferation assays in DLD-1 cells, which showed that 4N clones proliferated more slowly compared to their 2N counterparts (Fig. 1C). This growth impairment was further confirmed by cell viability and colony formation assays (Supplemental Fig. S1).
Figure 1.

Characterization of 2N and 4N isogenic clones. A) Percentage of FISH signals for 4 locus-specific probes on 5000 parental DLD-1 cells deciphering presence of 4N cellular subpopulation. B) Diagram showing procedure for FACS and generation of pure 2N and 4N isogenic clones. Sort window P5-selected cells have high forward scatter, and are larger and have higher probability of being 4N than cells with low forward scatter. C) Growth curve for DLD-1–derived 2N and 4N clones indicated in different colors. Statistical analysis was performed on trends by R package geepack for longitudinal data analysis. D) Comparison of total DNA, RNA, and protein amounts in seven 2N and seven 4N DLD-1 clones. Each data point represents measurement for biologic replicates (n = 4–9). E, F) Measurements for area (E, μm2) and volume (F, μm3) for ∼5000 DLD-1 cells of each 2N and 4N individual clone are represented. Data are reported as means ± sem. **P < 0.001, ***P < 0.0001.
To study the effect of whole-genome duplication on nucleic acid and protein levels, we synchronized 2N and 4N DLD-1 cells in G1/S phase and assessed their total DNA, RNA, and protein content. While 4N clones had an ∼2-fold increase in DNA and RNA content (P < 0.001), the protein quantification showed a 3-fold increase (P < 0.0001) and marked heterogeneity (Fig. 1D). Finally, we sought to investigate whether 4N cells displayed an increase in nuclear size. Automated image analysis revealed that 4N cells exhibited a 1.7-fold increase in nuclear area (Fig. 1E) and a >2-fold increase in volume (Fig. 1F).
Transcriptional profiling of 4N cancer cells reveals deregulation of genes associated with DNA replication and mitosis
In order to explore the molecular changes associated with tetraploidy, we performed genome-wide transcriptome profiling of five 2N and twelve 4N DLD-1 and two 2N and two 4N RKO clones. For both cell lines, we found a distinct gene expression difference between the 2 sets of clones by principal component analysis. In DLD-1, hierarchal gene clustering revealed a gene signature that further distinguished the 2 sets of clones (Fig. 2A). Overall, 1384 genes were differentially expressed in 4N vs. 2N clones [fold change >2.0, false discovery rate (FDR) P < 0.01]; 560 genes were up-regulated and 824 were down-regulated. On the other hand, 4N vs. 2N RKO cells showed differential regulation of 708 genes (fold change >2.0, P < 0.05); 258 genes were up-regulated and 449 were down-regulated. For the RKO comparison, the significance threshold is based on P value rather than FDR as a result of the smaller sample size. The 200 most significantly deregulated genes for both cell lines are shown in Supplemental Table S1. The complete data set can be extracted from the Gene Expression Omnibus (GSE81395). Gene set enrichment analysis revealed a significant deregulation of genes associated with the cell cycle, specifically genes involved in DNA replication or mitosis (normalized enrichment score >2.00, FDR P < 0.001) (Fig. 2B, C) in DLD-1 clones. In RKO, 4N cells showed down-regulation of genes associated with the term “DNA elongation strand” (normalized enrichment score −1.92, FDR P < 0.05). The list of the 20 most significant enrichment categories in 4N cells is provided in Supplemental Table S2. Overexpression of the replication-associated genes RRM2 and MCM2, which have previously been involved in the DNA replication, was confirmed at the protein level in DLD-1 4N clones compared to 2N clones (Fig. 2D, E).
Figure 2.

Gene expression profiling. A) Hierarchical clustering for genes that distinguished between DLD-1 derived 2N and 4N clones. Top-ranked 50 genes sorted by FDR are shown. B) Summary of gene set enrichment analysis showing most overrepresented pathways in 4N DLD-1 cells included in Molecular Signature Database. C) Gene set enrichment analysis for DNA replication and mitosis comparing 2N and 4N clones. D) Immunoblot validating up-regulation of RRM2 and phospho-MCM2 in DLD-1 4N clones. Tubulin was used as protein loading control. E) Densitometry analysis of immunoblot represented as ratio between protein of interest and loading control protein GAPDH. *P < 0.05.
4N cancer cells display increased replication stress and DNA damage
The transcriptomic profiling data revealed a deregulation of replication-related genes in 4N clones. We therefore decided to assess the levels of replicative stress in 4N compared to 2N clones. First, we measured the activation of DNA replication and DNA damage checkpoints. While we did not detect any differential activation of the Ataxia Telangiectasia mutated (ATM)/CHK2 pathway (Supplemental Fig. S2A, B), we did observe enhanced phosphorylation of CHK1 and increased levels of RPA32 and phospho-RPA32 in DLD-1 4N clones. We also determined the levels of total CHK1 and CHK2 in order to exclude an increase in the protein levels due to tetraploidization (Fig. 3A, B and Supplemental Fig. S2C, D). Moreover, phospho-CHK1 levels were even higher in response to treatment with the DNA polymerase inhibitor aphidicolin, indicating that 4N cells had a higher sensitivity to replication stress (Fig. 3C and Supplemental Fig. S2E). In support of these findings, we also observed increased phosphorylation of CHK1 and RPA32, as well as greater aphidicolin-sensitivity replication stress in the 4N clones derived from the RKO cell line (Fig. 3D–F and Supplemental Fig. S2F–H). Furthermore, we detected a larger incidence of fragile sites in DLD-1 4N clones treated with aphidicolin (Supplemental Fig. S2I).
Figure 3.

Increased replication stress in 4N cells. A) Immunoblot analysis of total CHK1 and phospho-CHK1 levels in DLD-1 2N and 4N cells. GAPDH was used as protein loading control. B) Immunoblot showing increased levels of both RPA32 and phospho-RPA32 in DLD-1 4N cells. C) Analysis of phospho-CHK1 levels after addition of aphidicolin (0.2 µM) for 24 h to both sets of DLD-1 clones. GAPDH was used as protein loading control. D) Immunoblot analysis of total CHK1 and phospho-CHK1 in RKO 2N and 4N cells. GAPDH was used as protein loading control. E) Immunoblot showing increased levels of phospho-RPA32 vs. RPA32 in RKO tetraploid cells. GAPDH was used as protein loading control. F) Analysis of phospho-CHK1 levels after addition of aphidicolin (0.2 µM) for 24 h to both sets of RKO clones. GAPDH was used as protein loading control. G) Graph depicting percentage of growth impairment of 4N compared to 2N DLD-1 cells as measured by colony formation assay after treatment with ATRi (10 µM) for 24 h (n = 3/clone). H) Immunoblot analysis of total CHK1 and phospho-CHK1 levels in RPE1 wild-type (2N) and posttetraploid (4N) clones. GAPDH was used as protein loading control. I) Immunoblot showing increased levels of phospho-RPA32 vs. RPA32 in posttetraploid RPE1 cells. GAPDH was used as protein loading control. J, K) Plots showing time-course experiments to characterize delay in cell cycle. DLD-1 cells were pulse-labeled with BrdU and analyzed by FACS every 2 h for 12 h period. 4N cells exhibit slower progression through S phase (J) and slower rate going through G2 phase (K). L) Histogram showing mitotic timing of 2N and 4N cells quantified by live cell imaging in DLD-1 (n = 50 cells/clone). M) Histogram showing mitotic index of 2N and 4N DLD-1 cells based on DAPI imaging (n = 50 cells/clone). Statistical analysis was performed on trends using R package geepack for longitudinal data analysis. Data are reported as means ± sem. **P < 0.001, ***P < 0.0001 (n.s., not significant).
To evaluate whether 4N cells showed higher levels of endogenous replication stress, sensitivity to the ATR inhibitor VE-821 was analyzed. Whereas 2N cells maintained approximately the same growth levels (based on colony formation efficiency), 4N clones displayed a significant growth impairment after VE-821 treatment (P < 0.0001) (Fig. 3G). In addition, we evaluated the expression of the oncogenes MYC, E2F1, and KRAS, which have been previously related to induce replication stress (21), and observed that whole-genome duplication led to a significant overexpression of E2F1 and KRAS in DLD-1, and MYC in RKO 4N clones (fold change >2) (Supplemental Fig. S2J). Next, we investigated whether tetraploidy itself induced replication stress in nontransformed cells. To do so, we evaluated the levels of phospho-CHK1 and phoshpo-RPA32 in RPE1 posttetraploid clones compared to wild-type RPE1 cells, and we found that both replication stress markers were significantly increased in RPE1 posttetraploid cells (Fig. 3H, I and Supplemental Fig. S2K, L). Finally, when comparing the levels of replication stress in RPE1 posttetraploid cells to the levels detected in DLD-1 and RKO cells, our results showed that the levels of phosphorylated CHK1 in cancer cells were much higher than those identified in nontransformed RPE1 posttetraploid cells (Supplemental Fig. S2M). Therefore, these results indicate that although oncogene overexpression might contribute to promote replication stress in cancer tetraploid cells, whole-genome duplication per se also triggers such a phenotype.
In order to investigate whether the growth impairment in 4N cells resulted from a delay in cell cycle progression due to replication stress, DLD-1 2N and 4N cells were BrdU pulse-labeled and analyzed at 2 h intervals. We observed a delay in S phase for the 4N clones, which resulted in a slower progression through G2 phase (Fig. 3J, K), thus confirming the association between growth impairment and replication stress-dependent S-phase delay. Given our finding that mitosis-related genes were also overexpressed in 4N cells (Fig. 2B), we investigated whether a delay similar to that observed in S phase had occurred during cell division. To this end, we evaluated the mitotic timing using live-cell imaging and determined the mitotic index by scoring cells in mitosis for both sets of clones; however, we did not observe any significant difference (Fig. 3L, M), suggesting the cell cycle delay was specific to S phase. Taken together, our data demonstrate that 4N clones experience replication stress, which appears to be kept at a level compatible with cell survival, likely due to the concerted actions of S-phase checkpoint proteins. Nevertheless, such levels of replication stress could be detrimental to 4N cells upon treatment with ATR inhibitors.
We then sought to determine whether tetraploidy resulted in increased DNA damage by immunostaining for the DNA damage markers 53BP1 and γ-H2AX, and normalizing the signal intensity by DNA content. All pairwise comparisons showed significantly higher levels of DNA damage markers in 4N cells compared to 2N cells (P < 0.0001) (Fig. 4A–D). We then aimed to determine when this damage occurred by γ-H2AX and 5-ethynyl-2′-deoxyuridine (EdU) or phospho-H3 staining in DLD-1 cells to specifically evaluate DNA damage in S and M phase. The number of γ-H2AX foci per cell normalized by DNA content was significantly higher in 4N vs. 2N clones during S phase (P < 0.0001) (Fig. 4E, F). Additionally, we identified a 2.4-fold increase in the number of γ-H2AX foci during M phase when comparing 4N and 2N cells (Fig. 4G), suggesting that not all the DNA damage occurring in S phase is repaired by the time cells reach M phase.
Figure 4.

Levels of DNA damage in 4N cells. A) Representative images of immunofluorescence for 53BP1 (red) and γ-H2AX (green). DAPI was used for nuclear counterstaining. Scale bar, 5 µm. B) Histogram depicting number of 53BP1 foci per DNA content for 2N and 4N DLD-1 cells (n > 240 cells/clone). C) Histogram showing number of γ-H2AX foci per DNA content for 2N and 4N DLD-1 cells (n > 240 cells/clone). D) Quantification of γ-H2AX foci per DNA content in RKO 2N and 4N clones (n > 100 cells/clone). E) Representative images of DAPI (blue), EdU (red), γ-H2AX (green), and merged images in both 2N and 4N DLD-1 cells. Scale bar, 5 µm. F) Plotted is normalized number of γ-H2AX foci per EdU-positive nuclei in DLD-1 cells (n > 240 cells/clone). G) Quantitative analysis of γ-H2AX foci per mitotic cell for two 2N and two 4N DLD-1 clones. For all these analyses, integrated density of DAPI signal was used to calculate DNA content of each cell. Data are reported as means ± sem. **P < 0.001, ***P < 0.0001.
4N cells display increased karyotype heterogeneity and genomic instability
Given that 4N cells overexpressed genes related to M phase (Fig. 2B) but progressed through mitosis with normal timing (Supplemental Fig. S2G, H), we wondered whether other aspects of cell division may be affected. Accordingly, we assessed karyotypic heterogeneity by counting chromosome numbers. 2N clones exhibited stable chromosome contents (modal number of 45 or 46 chromosomes in DLD-1 and 49 chromosomes in RKO). However, the 4N clones showed a wider variability in chromosome numbers in both lines, indicating a higher degree of heterogeneity (Fig. 5A, B), which was further validated by counting the number of FISH signals for 4 locus-specific probes for chromosomes 3, 7, 11, and 13 in interphase DLD-1 2N (Fig. 5C, E) and 4N (Fig. 5D, E) cells and metaphase spreads (Supplemental Fig. S3). To determine whether such heterogeneity in the chromosome number masked an underlying selection-induced karyotypic evolution of 4N cells, we performed genome profiling of early (passages 5–10) and late (passages 25–30) passage DLD-1 clones by aCGH. Although none of the 2N clones showed genomic imbalances different from those observed in the parental line, novel nonrecurrent genomic imbalances were detected in 75% of the 4N clones (Fig. 5F). Additionally, for a subset of 4N clones, we further characterized the presence of structural chromosome alterations by SKY (Fig. 5G). Our analysis revealed an average of 5 de novo structural aberrations per clone and maximum rates of instability (i.e., number of nonclonal aberrations/number of cells analyzed) (16), ranging from 0.18 to 0.3 rearrangements per cell division.
Figure 5.

Assessment of intercellular genetic heterogeneity. A, B) Dot plot depicting number of chromosomes in individual cells from six 2N and six 4N DLD-1 clones (A) and one 2N and two 4N RKO clones (B). Black lines denote modal chromosome number for each clone (n = 100 metaphases/clone). C, D) Graphs illustrate percentage of cells with corresponding FISH signals for 2N (C) and 4N (D) DLD-1 clones after total of 1000 nuclei were analyzed and show higher chromosomal variability in 4N cells. E) Representative FISH images for both sets of clones. FISH panel 1 includes fluorescent-labeled BAC clones for EGFR (7p) and CCND1 (11q), and FISH panel 2 includes fluorescent-labeled BAC clones TERC (3q) and CDX2 (13q). Scale bar, 2 µm. F) Circos plots illustrate comparative frequency of copy number detected by aCGH in early DLD-1–passaged 2N (inner histogram; n = 3) and 4N (outer histogram; n = 12) clones, as well as in late passaged 2N (inner histogram; n = 3) and 4N (outer histogram; n = 5) clones. Human chromosome ideogram is laid out at periphery of circle. Red blocks indicate copy number gains; green blocks, copy number losses. G) Representative image of SKY analysis performed in DLD-1, indicating by arrowhead presence of de novo structural chromosomal aberrations in 4N cells.
The large amount of intercellular heterogeneity and karyotypic changes observed in 4N cells suggested high levels of CIN. To assess this further, we evaluated the formation of micronuclei (MNi) and determined the rates and types of mitotic defects. We first performed a cytokinesis-block micronucleus assay to evaluate the formation of MNi in binucleated cells and found that 4N cells displayed significantly higher rates of MNi (P = 0.0001) (Supplemental Fig. S4A), which were validated in RKO 4N clones (Supplemental Fig. S4B). MNi can derive from a number of different chromosome missegregation events. Therefore, in order to better characterize the possible causes of CIN in 4N clones, we examined mitotic cells with immunostained kinetochores and microtubules, and quantified the mitotic defects during anaphase (Fig. 6A). We found that 4N cells exhibited a 4-fold increase in the levels of anaphase defects compared to their 2N DLD-1 counterparts (Fig. 6B). Specifically, our data unveiled anaphase lagging chromosomes (Fig. 6A, top row, and 6B) and multiple missegregating chromosomes (Fig. 6A, middle row, and 6B) to be the most frequent defects in anaphase cells. The latter chromosome segregation defect consisted of 2 or more chromosomes lagging behind with no evidence of merotelic attachments, and always displayed kinetochores that faced each other on the 2 sides of the spindle equator, but the intervening DNA did not appear stretched as it typically does in chromosome bridges (Fig. 6A, bottom row). However, the DNA from these missegregated chromosomes did not appear to be well separated in anaphase. Intriguingly, this defect was mainly found in 4N cells, but it was barely present in the 2N clones. When we analyzed the presence of the major anaphase defects in RKO clones, we confirmed that 4N cells exhibited greater levels of anaphase defects than 2N clones (2.5-fold, P < 0.0001), with lagging chromosomes being the most frequently observed mitotic defect, similar to what we observed in DLD-1 cells (Fig. 6C).
Figure 6.

Replication stress-associated CIN. A) Examples of mitotic defects in anaphase cells immunostained for kinetochores (green) and microtubules (red). Images show examples of most frequently observed defects, including lagging chromosomes (top row), multiple missegregating chromosomes (middle row), and chromosome bridges and acentric fragments (bottom row). Grayscale images at bottom right corners of each of DNA images are single focal planes of DAPI-stained chromosomes shown for easier visualization of mitotic defects. Arrowheads in grayscale images and merged panels point to specific mitotic defect. Scale bar, 10 µm. B, C) Frequencies of anaphase defects in normal culture conditions for 2N and 4N DLD-1 (B) and RKO (C) cells. D, E) Frequencies of anaphase defects in 2N and 4N DLD-1 (D) and RKO (E) cells treated with aphidicolin (0.2 µM) for 24 h. F, G) Frequencies of anaphase defects in 4N DLD-1 (F) and RKO (G) cells supplemented with mixture of nucleosides (30 and 100 µM, respectively) for 24 h. Data are represented as means ± sem. *P < 0.05, ***P < 0.0001.
Because anaphase lagging chromosomes can arise in cells undergoing a transient multipolar stage during prometaphase due to the presence of supernumerary centrosomes (6, 7), we sought to enumerate the number of centrosomes in G1 phase cells by coimmunostaining of γ-tubulin and cyclin D1. We found that 4N clones displayed a larger subpopulation of cells with extra centrosomes compared to 2N clones (mean 14.8 vs. 5.71, P < 0.0001 for DLD-1 cells and mean 19.7 vs. 5.66, P < 0.0001 for RKO cells) (Supplemental Fig. S4C), which can in part explain the higher rates of lagging chromosomes observed in 4N clones. Taken together, these results indicate that 4N cells display high rates of chromosome missegregation, which are responsible for the higher levels of CIN in 4N vs. 2N cells.
Replication stress triggers genomic instability
The finding that the major phenotypes of 4N cells are replication stress and genomic instability suggested a possible link between the two. To examine this, we determined the levels of CIN in response to aphidicolin treatment–induced replication stress in 2N and 4N clones. First, we evaluated the formation of MNi after aphidicolin treatment and found that it significantly increased the frequency of MNi in both 2N and 4N DLD-1 cells (P < 0.001 in both cases) (Supplemental Fig. S4A). Similarly, on aphidicolin treatment, we found an overall increase in abnormal anaphases (P < 0.0001) in these cells (Fig. 6D). Significant increases were found in lagging chromosomes (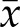 = 7.33 vs. 14.6%, P = 0.03, for 2N clones and
= 7.33 vs. 14.6%, P = 0.03, for 2N clones and 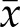 = 16.2 vs. 24.5%, P = 0.005, for 4N clones) for both 2N and 4N DLD-1 cells, indicating that such mitotic defects were not merely due to the increased amount of chromosomes in 4N cells (Fig. 6D). Intriguingly, we also found that the multiple missegregating chromosome phenotype was observed in 2N cells on aphidicolin treatment (Fig. 6D), suggesting a direct link between replication stress and this chromosome segregation defect. As expected, higher numbers of chromosome bridges and fragments were also identified (
= 16.2 vs. 24.5%, P = 0.005, for 4N clones) for both 2N and 4N DLD-1 cells, indicating that such mitotic defects were not merely due to the increased amount of chromosomes in 4N cells (Fig. 6D). Intriguingly, we also found that the multiple missegregating chromosome phenotype was observed in 2N cells on aphidicolin treatment (Fig. 6D), suggesting a direct link between replication stress and this chromosome segregation defect. As expected, higher numbers of chromosome bridges and fragments were also identified (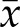 = 0 vs. 4.7%, P = 0.01, for 2N clones and
= 0 vs. 4.7%, P = 0.01, for 2N clones and 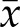 = 3.8 vs. 8.1%, P = 0.02, for 4N clones) (Fig. 6A, bottom row and 6D). When we assessed the levels of CIN in RKO clones in response to aphidicolin treatment, we also found an increased frequency of lagging chromosomes both in 2N and 4N (P < 0.02 and P < 0.01, respectively) (Fig. 6E), thus supporting the findings in DLD-1.
= 3.8 vs. 8.1%, P = 0.02, for 4N clones) (Fig. 6A, bottom row and 6D). When we assessed the levels of CIN in RKO clones in response to aphidicolin treatment, we also found an increased frequency of lagging chromosomes both in 2N and 4N (P < 0.02 and P < 0.01, respectively) (Fig. 6E), thus supporting the findings in DLD-1.
In order to support our findings, we tested whether reducing the levels of replication stress was able to alleviate the CIN phenotype. Thus, we treated DLD-1 and RKO 4N cells with a mix of exogenous nucleosides at increasing concentrations, which resulted in a 50% decrease of phospho-CHK1 levels (Supplemental Fig. S4D, E). Moreover, the treatment led to an overall rescue of the CIN phenotype, including a decrease of numerical alterations (Fig. 6F, G). Collectively, these data strengthen the hypothesis that replication stress contributes to numerical and structural CIN in 4N cells.
4N cells exhibit enhanced migratory and invasive capabilities both in cultured cells and in primary tumors
In order to investigate whether whole-genome duplication events confer sufficient physiologic changes to affect cell behavior, we assessed the migratory and the invasive capacities of 4N and 2N cells using a transwell (Boyden chamber) migration assay. The 4N DLD-1 clones displayed increased migratory capacity compared to 2N clones (P < 0.0001) (Fig. 7A). Similar results were obtained in wound-healing assays, in which the average closure area after 48 h was 13.88 vs. 28.20 μm2 for 2N and 4N DLD-1 clones, respectively (P = 0.05; Fig. 7B). At late passages, 4N cells exhibited even greater migration capabilities (P < 0.0001) (Fig. 6B). Interestingly, when a layer of Matrigel was added to the Boyden chamber in order to assess invasiveness, 4N cells displayed enhanced invasive capabilities (P = 0.04) (Fig. 7C). RKO 4N cells showed highly heterogeneous migratory capacities during the wound-healing assays, excluding the possibility that the increased size of the 4N cells was associated with mobility (data not shown). Furthermore, because extra centrosomes have been previously associated with cellular mobility (22), we sought to determine the presence of extra centrosomes (>1 centrosomes per cell) in G1-phase DLD-1 cells forming the migratory front of the wound-healing assay. Our results showed that the migratory capacity of these cells was independent of the amount of extra centrosomes (Fig. 7D).
Figure 7.

Increased migratory capabilities of 4N cells and their prevalence in invasive fronts of colorectal primary tumors. A) Boyden chamber assay representative images showing migration capability of 2N and 4N DLD-1 clones. B) Plotted is mean area of scratch closure resulting from wound-healing assay at 24 and 48 h after scratching for both early (passages 5–10) and late (passages 25–30) passaged 2N and 4N clones (P < 0.05 and P < 0.0001, respectively). C) Bar graph showing quantification of Matrigel invasiveness for 2N and 4N cells. Data are represented as mean number of invasive cells ± sem (n = 3). *P < 0.05. D) Representation of centrosome number in migrating cells during wound-healing assay. Quantification was performed by coimmunostaining of γ-tubulin and cyclin D1 (n = 250–350). E) Microscopic assessment of FISH signals using fluorescent probes for centromeres corresponding to chromosomes 4 (FITC) and 6 (Cy3) in colorectal primary tumor (top) and its associated invasive front (bottom). Single isolated nuclei are outlined with dashed circles. All 3 nuclei from primary tumor display 2 signals per probe, whereas 3 or 4 signals per probe were detected in nuclei from invasive front. F) Distribution of centromere probes signals for chromosome 4 and chromosome 6 in 9 colorectal primary tumors and their corresponding invasive fronts (P < 0.0001). G) Percentage of cells with extra centrosomes (>2 centrosomes per cell) in colorectal primary tumor samples and their corresponding invasive fronts. *P < 0.05.
On the basis of these observations, we sought to determine whether these in vitro findings could be confirmed in vivo by analyzing primary colorectal adenocarcinomas. To this end, we compared the levels of aneuploidy in the main tumor mass of colon adenocarcinomas and in their invasive fronts using centromeric FISH probes for 2 chromosomes unlikely to be involved in copy number changes in colorectal cancer (i.e., chromosomes 4 and 6). We found that cells in the invasive front displayed higher levels of aneuploidy for these 2 chromosomes (P < 0.0001) (Fig. 7E, F), and there was an enrichment of cells with 3 or more FISH signals for the 2 chromosomes under study (P < 0.0001, for the area under the curve for signals ≥4), indicating the presence of near-triploid and 4N cells. Moreover, by immunostaining with γ-tubulin, we could show that cells in the invasive front exhibited a 2.6-fold increase in the number of cells with extra centrosomes (>2 centrosomes per cell) compared to cells within the main tumor mass (P < 0.04) (Fig. 7G), therefore suggesting that both whole-genome duplication and extra centrosomes are important for invasion in primary tumors.
DISCUSSION
It is now generally accepted that CIN and aneuploidy are fundamental forces driving carcinogenesis (23). Additionally, aneuploidy per se has been shown to promote chromosome missegregation in both yeast (24, 25) and mammalian cells (26, 27). Also, tetraploidy has been proposed as a possible precursor of aneuploidy in cancer (2). Although the role of aneuploidy in cancer is well documented (28), the extent to which polyploidization affects different facets of cellular function remains unclear. Our study utilized isogenic cell lines of different ploidy states to address this question. By comparing whole transcriptome profiles of 2N and 4N clones, we identified a significant deregulation of genes involved in cell cycle (particularly M phase) and DNA replication stress response. Functionally, the consequences of polyploidization included impaired cellular growth, increased genetic heterogeneity, replication stress and associated genomic instability, and superior migratory capabilities, which were reflected in an enrichment of 4N cells in the invasive fronts of primary tumors.
Most malignant tumors display abnormal karyotypes with multiple chromosome aberrations, and it has been proposed that tetraploidy represents an important intermediate on the route to the cancerous karyotype. Despite well-established examples of 4N cells arising in phenotypically normal organs (29), this intermediate state in cancer cells is believed to be highly unstable and to rapidly evolve from 4N to near-triploid genomic content through the loss of chromosomes (30, 31). Accordingly, we found that 4N cells displayed higher levels of genomic instability compared to their 2N counterparts.
It has been previously shown that several types of mammalian cells exhibit a p53-dependent G1-phase arrest in response to cytokinesis failure-induced tetraploidization (32, 33). Substantial evidence supports the idea that a nonfunctional p53 pathway will allow more efficient and rapid proliferation of tetraploid cells (34, 35). In fact, abrogation of p53 function in tetraploid mouse mammary epithelial cells generates chromosomally unstable cell populations that are tumorigenic (36). However, a recent study demonstrated that p53 proficient polyploid cells are able to progress because of the overexpression of oncogenes, such as cyclin D2, indicating that the disruption of p53 is not essential for generating tetraploidy (37). Moreover, Ganem et al. (33) described that activation of the Hippo tumor suppressor pathway was responsible for cell cycle arrest after cytokinesis failure of nontransformed cells, and they identified the down-regulation of LATS2 in evolved tetraploids as a mechanism to overcome such arrest. In agreement with these findings, we identified the down-regulation of LATS2 in our evolved DLD-1 4N cells, and the down-regulation of another Hippo pathway component, LATS1, in our proliferating RKO 4N cells (data not shown). These observations indicate that similar mechanisms may influence the survival and proliferation of both nontransformed and cancer cells after a whole-genome duplication event.
Consistent with the idea that tetraploidy promotes genomic instability, we found increased levels of genomic instability in the 4N clones. In agreement with 2 recent studies (34, 38), our data suggest that high genomic instability may be due to increased tolerance to chromosome missegregation in 4N compared to 2N cells. In addition, we found higher rates of chromosome missegregation in 4N vs. 2N cells, indicating that there is an actual increase in rates of chromosome segregation errors and not simply a higher tolerance to the resulting aneuploidy. Some of the chromosome segregation errors we observed (i.e., anaphase lagging chromosomes) may be caused by the presence of supernumerary centrosomes and the transient assembly of a multipolar spindle, which was previously shown to promote formation of merotelic kinetochore attachment (6, 7). Because the number of cells with centrosome amplification (i.e., 2 or more centrosomes per G1 cell) in the 4N populations was rather low, this may only partially explain the occurrence of lagging chromosomes during anaphase. Moreover, some of the anaphase lagging chromosomes may also be the result of delayed spindle bipolarization, which was also shown to promote formation of merotelic kinetochore attachment (39). The large numbers of monopolar spindles in our 4N clones suggest that such a delayed spindle bipolarization may be occurring in the 4N cells. In fact, the deregulation of M-phase–related genes in 4N cells is likely linked to the mitotic defects described here.
Previous studies pointed to the relevance of replication stress at early stages of carcinogenesis through oncogene-induced acceleration of cell cycle progression (40). Furthermore, oncogene activation decreases cellular nucleoside levels and increases DNA damage, thus promoting genomic instability in newly transformed cells (41, 42). Along these lines, we observed the up-regulation of oncogenes such as E2F1, KRAS, and MYC in 4N cells, which might contribute to generate the replication stress response (43). Interestingly, despite the DLD-1 and RKO 2N cells already carry a mutation in KRAS and an amplification of MYC, respectively, a whole-genome duplication was necessary to result in a significant activation of the replication stress response. Our data revealed increased phosphorylation of RPA32 and enhanced activation of the S-phase checkpoints phospho-CHK1 in the 4N clones as well as a concerted deregulation of multiple genes implicated in DNA replication, such as several members of the MCM family. Likewise, Passerini et al. (8) described the presence of replication stress after the induction of aneuploidy, indicating that the presence of individual extra chromosomes could also lead to replication stress in cancer cells. On the other hand, nontransformed posttetraploid RPE1 cells also showed higher levels of both phospho-RPA32 and phospho-CHK1 compared to wild-type RPE1 cells, suggesting that replication stress might be a consequence rather than a cause of tetraploidy, contrary to what has been previously reported (44). Previous studies have assessed the molecular consequences of a whole-genome duplication event in nontransformed RPE1 cells. In fact, Kuznetsova et al. (38) reported transcriptional changes in chromosomally stable posttetraploid RPE1 cells compared to the parental diploid cell line; they found a significant enrichment for up-regulated genes involved in DNA replication. Nevertheless, they did not investigate the possibility that such transcriptional changes might be indicative of replication stress and DNA damage. A more recent study investigating the transcriptome of acute posttetraploid RPE1 cells revealed common signatures of activation of the tumor suppressor protein p53 and genes involved in the cellular response to DNA damage, suggesting the presence of ongoing DNA damage and the suppression of proliferation in the presence of p53 (37). We also detected increased levels of γ-H2AX and large 53BP1 foci in 4N cells, which are shown to be closely related to replication stress (45, 46). Indeed, the levels of DNA damage in S and M phase were also higher in 4N compared to 2N cells, suggesting that incomplete DNA synthesis during S phase was carried through the subsequent G1 phase (47, 48). Furthermore, the inhibition of ATR was also significantly more toxic for 4N than 2N cells, suggesting that in the absence of an active S-phase checkpoint response, the deleterious effects of replication stress are fully unleashed. Although the precise mechanism by which tetraploidy results in replication stress remains unresolved, our data strongly suggest that tetraploidy could induce replication stress through a massive deregulation of genes involved in the proper functioning of the DNA replication machinery.
Recent reports have also suggested DNA replication stress as a major driver in the generation of structural chromosomal abnormalities, as seen by the increased incidence of acentric fragments and chromosome bridges (9, 49). Furthermore, is has been described that the limitation of replication stress reduces the level of DNA damage and genomic rearrangement in induced pluripotent stem cells (50). Intriguingly, we found that chromosome bridges and acentric fragments occurred at modest frequencies in the 4N cells, despite the substantial levels of replication stress in these cells. Instead, 4N cells displayed high frequencies of anaphase lagging chromosomes and/or multiple missegregating chromosomes. Some of these lagging chromosomes may arise from transient mitotic spindle defects (monopolarity or multipolarity), as discussed earlier. However, our findings indicate that missegregation errors increase on aphidicolin treatment and are reduced after nucleoside supplementation, suggesting a novel and direct link between replication stress and mitotic defects. In fact, a functional relationship between Chk1 and Aurora B, a protein involved in the correction of merotelic attachments (51), has been recently described for abscission timing regulation (52). Therefore, the interplay between these 2 proteins might explain how replication response activation influences the frequency of numerical CIN. This is consistent with a recent report showing that activation of the DNA damage response during mitosis induced anaphase lagging chromosomes (53). In summary, our data show that replication stress is associated not only with chromosome segregation errors linked to structural defects, such as chromosome bridges and acentric fragments, but also with missegregation of whole chromosomes.
Although the functional consequences of aneuploidy and genomic instability have been thoroughly investigated, they remain a matter of debate. Numerous recent reports suggest that aneuploidy impairs proliferation (26, 54, 55). However, certain aneuploid karyotypes have been shown to confer proliferative advantages and/or adaptation to certain environments (56–58). Our own data indicate that 4N karyotypes, which are unstable by their very nature, display impaired cellular growth due to a replication stress-associated S-phase delay. Despite this proliferative disadvantage, 4N clones displayed increased migratory and invasive capacities. This is in agreement with previous studies showing that the presence of extra centrosomes disrupts migration in normal tissues but promotes invasiveness in cancer cells (59, 60). In fact, our data showed an enrichment of cells with supernumerary centrosomes in the invasive fronts of primary tumors. Nevertheless, as the percentage of cells with extra centrosomes is relatively low (∼10%), it might not be sufficient to fully explain the invasive phenotype of these near-triploid and 4N cells. Indeed, certain genes related to the invasive/migratory phenotype were found to be up-regulated in the 4N clones. The increased invasive/migratory capability of 4N clones is further reflected by the fact that within the invasive front of primary tumors, we found a high degree of CIN and increased presence of 4N cells. However, additional experiments are needed to corroborate the finding that 4N cells are more prone to migrate and invade in vivo.
In summary, our diploid–tetraploid model postulates that polyploid cells experience replication stress leading to high levels of DNA damage, which originates during S phase, is maintained through M phase, and contributes to chromosomal segregation errors and CIN. Moreover, 4N cells display high rates of chromosome missegregation and aneuploidy tolerance. This leads to high levels of intercellular heterogeneity with the potential for selection of advantageous genotypes that can drive carcinogenesis.
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
The authors thank Z. Storchova (Technische Universität Kaiserslautern, Kaiserslauter, Germany) for kindly providing cell lines; A. Bosch and M. Calvo (University of Barcelona) for assessing the data analysis; and Z. Wong [U.S. National Institutes of Health (NIH)], R. Ebner (NIH), and S. D. Rutledge (Virginia Tech) for critical reading. The authors are also grateful to B. Chen (NIH) for editorial assistance. This work was supported by the Intramural Program of the NIH, the CIBEREHD Program, and grants to J.C. from Instituto de Salud Carlos III, and cofunded by the European Regional Development Fund (ERDF) (CP13/00160 and PI14/00783), the European Commission MC-CIG (COLONGEVA), the Spanish Association Against Cancer (AECC, GCB13131592CAST), and the Agència de Gestió d’Ajuts Universitaris i de Recerca, Generalitat de Catalunya (2014 SGR 135 and 2014 SGR 903). Further support was provided by National Service Foundation (NSF) Grant MCB-1517506 to D.C. I.Q., and M.V.-C. received a Formación de Profesorado Universitario (FPU)-fellowship from Ministerio de Educación, Cultura y Deporte; K.T. received a Personal Investigador en Formació from the Universitat Autònoma de Barcelona; A.E. received a Formación de Personal Investigador (FPI) fellowship from Ministerio de Ciencia e Innovación; and Z.Y. received a fellowship from the Scientific and Technological Research Council of Turkey (TUBITAK) 2219 Program. CIBEREHD is funded by the Instituto de Salud Carlos III. The authors declare no conflicts of interest.
Glossary
- 2N
near-diploid
- 4N
near-tetraploid
- 53BP1
tumor protein p53 binding protein 1
- aCGH
array comparative genomic hybridization
- ATR
ataxia telangiectasia and rad3-related
- BrdU
bromodeoxyuridine
- CHK1/2
checkpoint kinase 1/2
- CIN
chromosomal instability
- EdU
5-ethynyl-2′-deoxyuridine
- FACS
fluorescence-activated cell sorting
- FBS
fetal bovine serum
- FDR
false discovery rate
- H2AX
histone 2AX
- MCM2
minichromosome maintenance complex component 2
- MNi
micronuclei
- RPA
replication protein A
- RRM2
ribonucleotide reductase regulatory subunit M2
- SKY
spectral karyotyping
- TBST
Tris-buffered saline and Tween 20
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
D. Wangsa, I. Quintanilla, T. Ried, and J. Camps conceived and designed the study; D. Wangsa, I. Quintanilla, K. Torabi, G. Klus, Z. Yuce, and C. Galofré performed experiments; A. Ercilla and N. Agell provided reagents and performed flow cytometry experiments; M. Vila-Casadesús and J. J. Lozano assisted with genomic and transcriptomic data analysis; D. Cimini provided reagents and assisted with phase-contrast live-cell imaging and confocal microscopy; D. Wangsa, I. Quintanilla, N. Agell, D. Cimini, T. Ried, and J. Camps conducted analysis and interpretation of the data; M. Cuatrecasas and A. Castells provided clinical samples; D. Wangsa, I. Quintanilla, D. Cimini, T. Ried, and J. Camps wrote the article; and all authors received the article for critical reading and approved the final version.
REFERENCES
- 1.Beroukhim R., Mermel C. H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J. S., Dobson J., Urashima M., Mc Henry K. T., Pinchback R. M., Ligon A. H., Cho Y.-J., Haery L., Greulich H., Reich M., Winckler W., Lawrence M. S., Weir B. A., Tanaka K. E., Chiang D. Y., Bass A. J., Loo A., Hoffman C., Prensner J., Liefeld T., Gao Q., Yecies D., Signoretti S., Maher E., Kaye F. J., Sasaki H., Tepper J. E., Fletcher J. A., Tabernero J., Baselga J., Tsao M.-S., Demichelis F., Rubin M. A., Janne P. A., Daly M. J., Nucera C., Levine R. L., Ebert B. L., Gabriel S., Rustgi A. K., Antonescu C. R., Ladanyi M., Letai A., Garraway L. A., Loda M., Beer D. G., True L. D., Okamoto A., Pomeroy S. L., Singer S., Golub T. R., Lander E. S., Getz G., Sellers W. R., Meyerson M. (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Storchova Z., Pellman D. (2004) From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 5, 45–54 [DOI] [PubMed] [Google Scholar]
- 3.Lengauer C., Kinzler K. W., Vogelstein B. (1998) Genetic instabilities in human cancers. Nature 396, 643–649 [DOI] [PubMed] [Google Scholar]
- 4.Thompson S. L., Compton D. A. (2011) Chromosome missegregation in human cells arises through specific types of kinetochore–microtubule attachment errors. Proc. Natl. Acad. Sci. USA 108, 17974–17978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicholson J. M., Cimini D. (2011) How mitotic errors contribute to karyotypic diversity in cancer. Adv. Cancer Res. 112, 43–75 [DOI] [PubMed] [Google Scholar]
- 6.Ganem N. J., Godinho S. A., Pellman D. (2009) A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silkworth W. T., Nardi I. K., Scholl L. M., Cimini D. (2009) Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One 4, e6564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Passerini V., Ozeri-Galai E., de Pagter M. S., Donnelly N., Schmalbrock S., Kloosterman W. P., Kerem B., Storchová Z. (2016) The presence of extra chromosomes leads to genomic instability. Nat. Commun. 7, 10754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burrell R. A., McClelland S. E., Endesfelder D., Groth P., Weller M. C., Shaikh N., Domingo E., Kanu N., Dewhurst S. M., Gronroos E., Chew S. K., Rowan A. J., Schenk A., Sheffer M., Howell M., Kschischo M., Behrens A., Helleday T., Bartek J., Tomlinson I. P., Swanton C. (2013) Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496 Erratum in: Nature 2013;500:490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eykelenboom J. K., Harte E. C., Canavan L., Pastor-Peidro A., Calvo-Asensio I., Llorens-Agost M., Lowndes N. F. (2013) ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep. 5, 1095–1107 [DOI] [PubMed] [Google Scholar]
- 11.Barrett M. T., Sanchez C. A., Prevo L. J., Wong D. J., Galipeau P. C., Paulson T. G., Rabinovitch P. S., Reid B. J. (1999) Evolution of neoplastic cell lineages in Barrett oesophagus. Nat. Genet. 22, 106–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Camps J., Morales C., Prat E., Ribas M., Capellà G., Egozcue J., Peinado M. A., Miró R. (2004) Genetic evolution in colon cancer KM12 cells and metastatic derivates. Int. J. Cancer 110, 869–874 [DOI] [PubMed] [Google Scholar]
- 13.Gerlinger M., Rowan A. J., Horswell S., Math M., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A., Tarpey P., Varela I., Phillimore B., Begum S., McDonald N. Q., Butler A., Jones D., Raine K., Latimer C., Santos C. R., Nohadani M., Eklund A. C., Spencer-Dene B., Clark G., Pickering L., Stamp G., Gore M., Szallasi Z., Downward J., Futreal P. A., Swanton C. (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jamal-Hanjani M., Wilson G. A., McGranahan N., Birkbak N. J., Watkins T. B. K., Veeriah S., Shafi S., Johnson D. H., Mitter R., Rosenthal R., Salm M., Horswell S., Escudero M., Matthews N., Rowan A., Chambers T., Moore D. A., Turajlic S., Xu H., Lee S. M., Forster M. D., Ahmad T., Hiley C. T., Abbosh C., Falzon M., Borg E., Marafioti T., Lawrence D., Hayward M., Kolvekar S., Panagiotopoulos N., Janes S. M., Thakrar R., Ahmed A., Blackhall F., Summers Y., Shah R., Joseph L., Quinn A. M., Crosbie P. A., Naidu B., Middleton G., Langman G., Trotter S., Nicolson M., Remmen H., Kerr K., Chetty M., Gomersall L., Fennell D. A., Nakas A., Rathinam S., Anand G., Khan S., Russell P., Ezhil V., Ismail B., Irvin-Sellers M., Prakash V., Lester J. F., Kornaszewska M., Attanoos R., Adams H., Davies H., Dentro S., Taniere P., O’Sullivan B., Lowe H. L., Hartley J. A., Iles N., Bell H., Ngai Y., Shaw J. A., Herrero J., Szallasi Z., Schwarz R. F., Stewart A., Quezada S. A., Le Quesne J., Van Loo P., Dive C., Hackshaw A., Swanton C.; TRACERx Consortium . (2017) Tracking the evolution of non–small-cell lung cancer. N. Engl. J. Med. 376, 2109–2121 [DOI] [PubMed] [Google Scholar]
- 15.Wilhelm T., Magdalou I., Barascu A., Técher H., Debatisse M., Lopez B. S. (2014) Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 111, 763–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Camps J., Ponsa I., Ribas M., Prat E., Egozcue J., Peinado M. A., Miró R. (2005) Comprehensive measurement of chromosomal instability in cancer cells: combination of fluorescence in situ hybridization and cytokinesis–block micronucleus assay. FASEB J. 19, 828–830 [DOI] [PubMed] [Google Scholar]
- 17.Venkatraman E. S., Olshen A. B. (2007) A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics 23, 657–663 [DOI] [PubMed] [Google Scholar]
- 18.Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S. J., Marra M. A. (2009) Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.-Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaillard H., García-Muse T., Aguilera A. (2015) Replication stress and cancer. Nat. Rev. Cancer 15, 276–289 [DOI] [PubMed] [Google Scholar]
- 22.Ogden A., Rida P. C. G., Aneja R. (2013) Heading off with the herd: how cancer cells might maneuver supernumerary centrosomes for directional migration. Cancer Metastasis Rev. 32, 269–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanahan D., Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 24.Sheltzer J. M., Blank H. M., Pfau S. J., Tange Y., George B. M., Humpton T. J., Brito I. L., Hiraoka Y., Niwa O., Amon A. (2011) Aneuploidy drives genomic instability in yeast. Science 333, 1026–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhu J., Pavelka N., Bradford W. D., Rancati G., Li R. (2012) Karyotypic determinants of chromosome instability in aneuploid budding yeast. PLoS Genet. 8, e1002719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheltzer J. M., Ko J. H., Replogle J. M., Habibe Burgos N. C., Chung E. S., Meehl C. M., Sayles N. M., Passerini V., Storchova Z., Amon A. (2017) Single-chromosome gains commonly function as tumor suppressors. Cancer Cell 31, 240–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicholson J. M., Macedo J. C., Mattingly A. J., Wangsa D., Camps J., Lima V., Gomes A. M., Dória S., Ried T., Logarinho E., Cimini D. (2015) Chromosome mis-segregation and cytokinesis failure in trisomic human cells. eLife 4, e05068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ried T., Hu Y., Difilippantonio M. J., Ghadimi B. M., Grade M., Camps J. (2012) The consequences of chromosomal aneuploidy on the transcriptome of cancer cells. Biochim. Biophys. Acta 1819, 784–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gentric G., Desdouets C., Celton-Morizur S. (2012) Hepatocytes polyploidization and cell cycle control in liver physiopathology. Int. J. Hepatol. 2012, 282430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baia G. S., Stifani S., Kimura E. T., McDermott M. W., Pieper R. O., Lal A. (2008) Notch activation is associated with tetraploidy and enhanced chromosomal instability in meningiomas. Neoplasia 10, 604–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zack T. I., Schumacher S. E., Carter S. L., Cherniack A. D., Saksena G., Tabak B., Lawrence M. S., Zhsng C.-Z., Wala J., Mermel C. H., Sougnez C., Gabriel S. B., Hernandez B., Shen H., Laird P. W., Getz G., Meyerson M., Beroukhim R. (2013) Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andreassen P. R., Lohez O. D., Lacroix F. B., Margolis R. L. (2001) Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol. Biol. Cell 12, 1315–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ganem N. J., Cornils H., Chiu S.-Y., O’Rourke K. P., Arnaud J., Yimlamai D., Théry M., Camargo F. D., Pellman D. (2014) Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 158, 833–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dewhurst S. M., McGranahan N., Burrell R. A., Rowan A. J., Grönroos E., Endesfelder D., Joshi T., Mouradov D., Gibbs P., Ward R. L., Hawkins N. J., Szallasi Z., Sieber O. M., Swanton C. (2014) Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 4, 175–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuffer C., Kuznetsova A. Y., Storchová Z. (2013) Abnormal mitosis triggers p53-dependent cell cycle arrest in human tetraploid cells. Chromosoma 122, 305–318 [DOI] [PubMed] [Google Scholar]
- 36.Fujiwara T., Bandi M., Nitta M., Ivanova E. V., Bronson R. T., Pellman D. (2005) Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437, 1043–1047 [DOI] [PubMed] [Google Scholar]
- 37.Potapova T. A., Seidel C. W., Box A. C., Rancati G., Li R. (2016) Transcriptome analysis of tetraploid cells identifies cyclin D2 as a facilitator of adaptation to genome doubling in the presence of p53. Mol. Biol. Cell 27, 3065–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kuznetsova A. Y., Seget K., Moeller G. K., de Pagter M. S., de Roos J. A. D. M., Dürrbaum M., Kuffer C., Müller S., Zaman G. J. R., Kloosterman W. P., Storchová Z. (2015) Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 14, 2810–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Silkworth W. T., Nardi I. K., Paul R., Mogilner A., Cimini D. (2012) Timing of centrosome separation is important for accurate chromosome segregation. Mol. Biol. Cell 23, 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bartkova J., Rezaei N., Liontos M., Karakaidos P., Kletsas D., Issaeva N., Vassiliou L.-V. F., Kolettas E., Niforou K., Zoumpourlis V. C., Takaoka M., Nakagawa H., Tort F., Fugger K., Johansson F., Sehested M., Andersen C. L., Dyrskjot L., Ørntoft T., Lukas J., Kittas C., Helleday T., Halazonetis T. D., Bartek J., Gorgoulis V. G. (2006) Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637 [DOI] [PubMed] [Google Scholar]
- 41.Bester A. C., Roniger M., Oren Y. S., Im M. M., Sarni D., Chaoat M., Bensimon A., Zamir G., Shewach D. S., Kerem B. (2011) Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145, 435–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halazonetis T. D., Gorgoulis V. G., Bartek J. (2008) An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355 [DOI] [PubMed] [Google Scholar]
- 43.Herold S., Herkert B., Eilers M. (2009) Facilitating replication under stress: an oncogenic function of MYC? Nat. Rev. Cancer 9, 441–444 [DOI] [PubMed] [Google Scholar]
- 44.Ichijima Y., Yoshioka K., Yoshioka Y., Shinohe K., Fujimori H., Unno J., Takagi M., Goto H., Inagaki M., Mizutani S., Teraoka H. (2010) DNA lesions induced by replication stress trigger mitotic aberration and tetraploidy development. PLoS One 5, e8821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szilard R. K., Jacques P.-É., Laramée L., Cheng B., Galicia S., Bataille A. R., Yeung M., Mendez M., Bergeron M., Robert F., Durocher D. (2010) Systematic identification of fragile sites via genome-wide location analysis of γ-H2AX. Nat. Struct. Mol. Biol. 17, 299–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lukas C., Savic V., Bekker-Jensen S., Doil C., Neumann B., Pedersen R. S., Grøfte M., Chan K. L., Hickson I. D., Bartek J., Lukas J. (2011) 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 13, 243–253 [DOI] [PubMed] [Google Scholar]
- 47.Chanoux R. A., Yin B., Urtishak K. A., Asare A., Bassing C. H., Brown E. J. (2009) ATR and H2AX cooperate in maintaining genome stability under replication stress. J. Biol. Chem. 284, 5994–6003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harrigan J. A., Belotserkovskaya R., Coates J., Dimitrova D. S., Polo S. E., Bradshaw C. R., Fraser P., Jackson S. P. (2011) Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 193, 97–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lamm N., Ben-David U., Golan-Lev T., Storchová Z., Benvenisty N., Kerem B. (2016) Genomic instability in human pluripotent stem cells arises from replicative stress and chromosome condensation defects. Cell Stem Cell 18, 253–261 [DOI] [PubMed] [Google Scholar]
- 50.Ruiz S., Lopez-Contreras A. J., Gabut M., Marion R. M., Gutierrez-Martinez P., Bua S., Ramirez O., Olalde I., Rodrigo-Perez S., Li H., Marques-Bonet T., Serrano M., Blasco M. A., Batada N. N., Fernandez-Capetillo O. (2015) Limiting replication stress during somatic cell reprogramming reduces genomic instability in induced pluripotent stem cells. Nat. Commun. 6, 8036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cimini D., Cameron L. A., Salmon E. D. (2004) Anaphase spindle mechanics prevent mis-segregation of merotelically oriented chromosomes. Curr. Biol. 14, 2149–2155 [DOI] [PubMed] [Google Scholar]
- 52.Mackay D. R., Ullman K. S. (2015) ATR and a Chk1–Aurora B pathway coordinate postmitotic genome surveillance with cytokinetic abscission. Mol. Biol. Cell 26, 2217–2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bakhoum S. F., Kabeche L., Murnane J. P., Zaki B. I., Compton D. A. (2014) DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov. 4, 1281–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Torres E. M., Sokolsky T., Tucker C. M., Chan L. Y., Boselli M., Dunham M. J., Amon A. (2007) Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317, 916–924 [DOI] [PubMed] [Google Scholar]
- 55.Williams B. R., Prabhu V. R., Hunter K. E., Glazier C. M., Whittaker C. A., Housman D. E., Amon A. (2008) Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322, 703–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pavelka N., Rancati G., Zhu J., Bradford W. D., Saraf A., Florens L., Sanderson B. W., Hattem G. L., Li R. (2010) Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468, 321–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rutledge S. D., Cimini D. (2016) Consequences of aneuploidy in sickness and in health. Curr. Opin. Cell Biol. 40, 41–46 [DOI] [PubMed] [Google Scholar]
- 58.Selmecki A. M., Maruvka Y. E., Richmond P. A., Guillet M., Shoresh N., Sorenson A. L., De S., Kishony R., Michor F., Dowell R., Pellman D. (2015) Polyploidy can drive rapid adaptation in yeast. Nature 519, 349–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Godinho S. A., Picone R., Burute M., Dagher R., Su Y., Leung C. T., Polyak K., Brugge J. S., Théry M., Pellman D. (2014) Oncogene-like induction of cellular invasion from centrosome amplification. Nature 510, 167–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kushner E. J., Ferro L. S., Liu J.-Y., Durrant J. R., Rogers S. L., Dudley A. C., Bautch V. L. (2014) Excess centrosomes disrupt endothelial cell migration via centrosome scattering. J. Cell Biol. 206, 257–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.