

Abstract
The optimal duration of mild “therapeutic” hypothermia for neonates with hypoxic-ischemic encephalopathy is surprisingly unclear. This study assessed the relative efficacy of cooling for 48 h versus 72 h. Fetal sheep (0.85 gestation) received sham ischemia (n = 9) or 30 min global cerebral ischemia followed by normothermia (n = 8) or delayed hypothermia from 3 h to 48 h (n = 8) or 72 h (n = 8). Ischemia was associated with profound loss of electroencephalogram (EEG) power, neurons in the cortex and hippocampus, and oligodendrocytes and myelin basic protein expression in the white matter, with increased Iba-1-positive microglia and proliferation. Hypothermia for 48 h was associated with improved outcomes compared to normothermia, but a progressive deterioration of EEG power after rewarming compared to 72 h of hypothermia, with impaired neuronal survival and myelin basic protein, and more microglia in the white matter and cortex. These findings show that head cooling for 48 h is partially neuroprotective, but is inferior to cooling for 72 h after cerebral ischemia in fetal sheep. The close association between rewarming at 48 h, subsequent deterioration in EEG power and increased cortical inflammation strongly suggests that deleterious inflammation can be reactivated by premature rewarming.
Keywords: Hypoxia-ischemia, encephalopathy, fetus, hypothermia, neuroprotection
Introduction
There is now compelling clinical and experimental evidence that therapeutic hypothermia can reduce neuronal loss and improve neurological outcome after a severe hypoxic-ischemic insult in term infants.1,2 Follow-up studies of large randomized trials of infants with hypoxic-ischemic encephalopathy (HIE) confirm that treatment with therapeutic hypothermia was associated with reduced death, cerebral palsy and disability, and improved neurocognitive functioning that persisted into middle childhood.2–5
Strikingly, however, a recent clinical study found that extending the duration of hypothermia from 72 h to 120 h was not associated with any added benefit and indeed was associated with a small increase in risk of death.6 Consistent with this, 120 h of delayed hypothermia after global cerebral ischemia in term-equivalent fetal sheep was associated with slightly less neuroprotection and a small increase in inflammation compared to 72 h of hypothermia.7 Although cooling for 72 h is unequivocally highly protective,7–9 this raises the question whether a shorter interval might offer similar or better neuroprotection. There would be considerable practical appeal in reducing the duration of treatment for neonates, who require intensive monitoring with limited parental interaction during hypothermia.
It is notable that in piglets, mild hypothermia for 24 h after hypoxia-ischemia has been shown to be neuroprotective in a range of studies.10–12 In adult rats, 48 h of hypothermia after acute ischemic stroke was neuroprotective, whereas 24 h was not.13 Similarly, selective head cooling for 48 h, but not 12 h, was neuroprotective in adult rats after permanent middle cerebral artery occlusion.14 Further, graded neuroprotection was seen with 12, 24, and 48 h of systemic hypothermia after permanent middle cerebral artery occlusion in adult rats.15
These observations suggest that 24 to 48 h of cooling can provide benefit, but none of these studies directly assessed outcomes against extended cooling for 72 h. Thus, in this study, we compared the effect of 48 h and 72 h of head cooling after 30 min of global cerebral ischemia in near-term fetal sheep on recovery of brain activity and neuronal and white matter cell survival after 7 days recovery.
Methods
Fetal surgery
All procedures were approved by the Animal Ethics Committee of The University of Auckland under the New Zealand Animal Welfare Act, and the Code of Ethical Conduct for animals in research established by the Ministry of Primary Industries, Government of New Zealand. The experiment has been reported in compliance with the ARRIVE guidelines.16 In brief, 33 time-mated Romney/Suffolk fetal sheep were instrumented using sterile techniques at 124 ± 1 days gestation (term is 145). Food, but not water was withdrawn 18 h before surgery. Ewes were given long-acting oxytetracycline (20 mg/kg, Phoenix Pharm, Auckland, New Zealand) i.m. 30 min before the start of surgery. Anesthesia was induced by intravenous injection of propofol (5 mg/kg, AstraZeneca Limited, Auckland, New Zealand) and maintained using 2–3% isoflurane in O2. The depth of anesthesia, maternal heart rate, and respiration were constantly monitored by trained anesthetic staff. Ewes received a constant infusion isotonic saline drip (at an infusion rate of approximately 250 mL/h) to maintain fluid balance.
Following a maternal midline abdominal incision, the fetus was exposed and both fetal brachial arteries were catheterized with polyvinyl catheters to measure mean arterial blood pressure (MAP) and allow fetal blood sampling. An amniotic catheter was secured to the fetal shoulder. ECG electrodes (AS633–3SSF; Cooner Wire Co., Chatsworth, California, USA) were sewn across the fetal chest to record fetal heart rate (FHR). The vertebral-occipital anastomoses were ligated and inflatable occluder cuffs were placed around both carotid arteries. A 3S Transonic ultrasonic flow probe (Transonic systems, Ithaca, NY) was placed around the right carotid artery. Using a seven stranded stainless steel wire (AS633–7SSF; Cooner Wire Co.), two pairs of electroencephalogram (EEG) electrodes were placed on the dura over the parasagittal parietal cortex (10 and 20 mm anterior to bregma and 10 mm lateral) and secured with cyanoacrylate glue. A reference electrode was sewn over the occiput. A further two electrodes were sewn in the nuchal muscle to record electromyographic (EMG) activity as a measure of fetal movement. To measure cortical impedance, a third pair of electrodes (AS633-3SSF; Cooner Wire Co.) was placed over the dura 10 mm lateral to the EEG electrodes. A thermistor (Replacement Parts Industries, Inc, Chatsworth, CA, USA) was placed over the parasagittal dura 30 mm anterior to bregma to measure extradural temperature and a second thermistor was inserted into the esophagus to measure core temperature. A cooling cap made from silicon tubing (3 × 6 mm, Degania Silicone, Israel) was secured to the fetal head. The uterus was then closed and antibiotics (80 mg gentamicin, Pharmacia and Upjohn, Rydalmere, New South Wales, Australia) were administered into the amniotic sac. The maternal laparotomy skin incision was repaired and infiltrated with 10 ml 0.5% bupivacaine plus adrenaline (AstraZeneca Ltd., Auckland, New Zealand). All fetal catheters and leads were exteriorized through the maternal flank. The maternal long saphenous vein was catheterized to provide access for post-operative maternal care and euthanasia.
Post-operative care
Sheep were housed together in separate metabolic cages with access to food and water ad libitum. They were kept in a temperature-controlled room (16 ± 1℃, humidity 50 ± 10%), in a 12 h light/dark cycle. Intravenous antibiotics were administered daily for 4 days to the ewe (600 mg benzylpencillin sodium, Novartis Ltd, Auckland, New Zealand, and 80 mg gentamicin). Fetal catheters were maintained patent by continuous infusion of heparinized saline (20 U/mL at 0.2 mL/h) and the maternal catheter maintained by daily flushing.
Data recording
Fetal MAP, corrected for maternal movement by subtraction of amniotic pressure (Novatrans II, MX860; Medex Inc., Hilliard, OH, USA), CaBF, ECG, EEG, and impedance were recorded from 24 h before the start of the experiment and continued for the remainder of the experiment. The raw ECG was analogue filtered between 0.05 and 100 Hz and digitized at 1024 Hz. The EEG signal was processed with a first-order high-pass filter at 1.6 Hz and a sixth-order Butterworth low-pass filter with the cut-off frequency at 256 Hz, and then digitally stored at a sampling rate of 512 Hz. EEG intensity (power) was derived from the power spectrum between 1 and 20 Hz and log transformed (decibels (dB), 20 × log (intensity)) as this transformation gives a better approximation of the normal distribution.17 Spectral edge was defined as the frequency below which 90% of total EEG power was present, as a measure of the relative frequency of the EEG. Impedance increases as the temperature of the medium, through which the signal passes, falls. Therefore, in each fetus, the slope of impedance change at the onset of hypothermia was used to correct the impedance signal: corrected impedance = impedance − (slope × Δtemperature).18,19 Data were recorded and saved continuously to disk for off-line analysis using custom data acquisition programs (LabView for Windows, National Instruments, Austin, Texas, USA).
Arterial blood samples were taken for pre-ductal pH, blood gas, base excess (ABL800 Flex Analyser, Radiometer, Auckland, New Zealand), glucose and lactate measurements (YSI model 2300, Yellow Springs, Ohio, USA). All fetuses had normal biochemical variables for their gestational ages.
Experimental protocols
At 128 ± 1 day gestation, cerebral ischemia was induced by reversible inflation of the carotid occluder cuffs with sterile saline for 30 min. Successful occlusion was confirmed by the onset of an isoelectric EEG signal within 30 s of inflation. The carotid occluder cuffs were not inflated in sham control experiments. Fetal blood samples were drawn just before the occlusion and 2, 4, and 6 h after occlusion followed by daily sampling for the remainder of the experiment.
Randomization was stratified by cohort to control for time of year. Within each cohort, fetuses were randomized by computer program to ischemia-normothermia (n = 8), ischemia-48 h hypothermia (n = 8), ischemia-72 h hypothermia (n = 8), or sham control (n = 9) groups. Based on a pooled standard deviation of 20 for neuronal survival in the parasagittal cortex after cerebral ischemia, a minimum group size of 8 was estimated to be needed to provide 80% power to detect a difference of 30 cells per field of view or more between groups. Cooling was started 3 h after reperfusion and continued until 48 or 72 h after the onset of ischemia in the ischemia-48 h hypothermia and ischemia-72 h hypothermia groups, respectively. Cooling was performed by linking the cooling coil over the fetal scalp with a pump (TX150 Heating circulator, Grant Instruments Ltd., Cambridge, England) in a cooled water bath and circulating cold water through the cooling coil. Consistent with previous studies in near-term fetal sheep, the initial target extradural temperature was set between 31 and 33℃.7,9,18 In the ischemia-normothermia and sham control groups, the water was not circulated and the cooling coil remained in equilibrium with fetal temperature. As previously described, at the end of the cooling period, the water pump was switched off and fetuses were allowed to spontaneously rewarm over approximately 60 min.18,20 Ewes and fetuses were killed 7 days after ischemia with an overdose of sodium pentobarbitone (9 g intravenous to the ewe; Pentobarb 300; Chemstock, Christchurch, New Zealand) for immunohistochemistry.
Immunohistochemistry
Fetal brains were perfusion fixed with 10% phosphate-buffered formalin. Coronal slices (10 µm thick) were cut using a microtome (Leica Jung RM2035, Wetzlar, Germany) starting at the level of the dorsal hippocampus. Slides were dewaxed in xylene and rehydrated in decreasing concentrations of ethanol, then washed in 0.1 mol/L phosphate-buffered saline (PBS). Antigen retrieval was performed using the pressure cooker method (2100 Antigen Retriever, Aptum Biologics Ltd., Southampton, England) in citrate buffer followed by incubation in 1% H2O2 in methanol (or PBS for Olig2) to block endogenous peroxidase activity. Blocking was performed in 3% normal goat serum (NGS) for NeuN, Olig2, Ki67, and MBP and normal horse serum (NHS) for Iba1 for 1 h at room temperature. Sections were labelled with 1:200 rabbit anti-neuronal nuclei monoclonal antibody (NeuN, Abcam, Cambridge, England), 1:200 rabbit anti-Olig2 (Abcam), 1:200 mouse anti-Ki67 (Dako Limited, Sydney, Australia), 1:200 goat anti-Iba1 (Abcam), and 1:200 mouse anti-MBP (Merck Millipore, Auckland, New Zealand) overnight at 4℃. Sections were incubated for 3 h in biotin-conjugated 1:200 anti-rabbit (NeuN and Olig2, Vector Laboratories, Burlingame, USA), 1:200 anti-goat (Iba1, Vector Laboratories) or 1:200 anti-mouse antibody (Ki67 and MBP, Vector Laboratories) in 3% NGS for NeuN, Olig2, Ki67, and MBP and 3% NHS for Iba1. Slides were then incubated in ExtrAvidin® (1:200, Sigma-Aldrich Pty. Ltd, St Louis, USA) in PBS for 2 h at room temperature and then reacted in diaminobenzidine tetrachloride (Sigma-Aldrich Pty. Ltd.). The reaction was stopped by washing in PBS. Slides labelled with Ki67 were counterstained with thionine acetate (Sigma-Aldrich Pty. Ltd). Sections were dehydrated in increasing concentrations of alcohol and mounted.
Neurons in the cortex of the first and second parasagittal gyri, CA1, CA3, CA4, and dentate gyrus of the hippocampus were counted using light microscopy (Nikon Eclipse 80i, Scitech Ltd, Preston, Victoria, Australia) at 20× magnification (image size 512 µm × 384 µm) by an investigator masked to the treatment group by separate coding of the slides. Normal-appearing NeuN-positive neurons were identified morphologically by the presence of typical nuclei; cells showing condensed nuclei or fragmented appearance were not counted.21 Images were all auto-contrasted using ImageJ (National Institutes of Health, Bethesda, USA) to make individual cells easier to identify. Numbers of oligodendrocytes and microglia were assessed using light microscopy at 20× (image size 512 µm × 384 µm) magnification in the intragyral white matter of the first and second parasagittal gyri and the periventricular white matter. Microglial number in the cortex of the first parasagittal gyrus was assessed using light microscopy at 40× magnification (image size 259 µm × 193 µm). Microglia showing either an amoeboid or ramified morphology were included. Given the difficulty distinguishing separate cells in images with patchy areas of marked microglial aggregation, auto-contrast and background subtraction (separate colors) was performed on all images using ImageJ software to make individual microglia easier to identify. The area fraction of myelin basic protein was quantified as the percentage of each image showing positive labelling after converting the image to an 8-bit image and running an auto-threshold using the default setting on the ImageJ software.
Statistical analysis
Data were analyzed by an investigator who was not masked to the treatment group using ANOVA or repeated measures ANOVA, followed by the Tukey post-hoc test when a significant difference was found. Statistical significance was accepted when p < 0.05.
Results
Sex distribution, body and brain weight at post-mortem
There was no significant difference in the ratio of males to females between groups (sham controls 5:4; ischemia-normothermia 3:5; ischemia-48 h hypothermia 2:6; ischemia-72 h hypothermia 4:4). There was no significant difference in post-mortem body weight between groups (control (4930 ± 275 g); ischemia-normothermia (4575 ± 311 g); ischemia-48 h hypothermia (4263 ± 204 g); ischemia-72 h hypothermia (4720 ± 145 g)).
Ischemia-normothermia was associated with reduced post-mortem brain weight (40.1 ± 1.5 g) compared to sham controls (48.9 ± 1.9, p < 0.05). Brain weight was increased after ischemia-72 h hypothermia compared to ischemia-normothermia (p < 0.05), to sham control values (44.9 ± 0.9), but not after ischemia-48 h hypothermia (41.9 ± 1.7, p < 0.05 vs. sham controls).
Blood gas, glucose and lactate measurements
There were no significant differences in baseline blood gas, pH, glucose or lactate measurements between groups (Table 1). Ischemia was associated with transiently increased plasma lactate and glucose concentration (p < 0.05), which resolved to baseline levels by day 2 in the ischemia-normothermia and ischemia-72 h hypothermia groups and by day 3 in the ischemia-48 h hypothermia group.
Table 1.
Blood gas, pH, glucose and lactate concentrations before and after 30 min of global cerebral ischemia in the ischemia-normothermia, ischemia-48 h hypothermia, and ischemia-72 h hypothermia groups.
| Baseline | 2 h | 4 h | 6 h | 1 day | 2 days | 7 days | |
|---|---|---|---|---|---|---|---|
| pH | |||||||
| Normothermia | 7.38 ± 0.01 | 7.36 ± 0.01 | 7.38 ± 0.01 | 7.39 ± 0.01 | 7.35 ± 0.02 | 7.39 ± 0.01 | 7.44 ± 0.01 |
| 48 h hypo | 7.37 ± 0.01 | 7.34 ± 0.01 | 7.37 ± 0.01 | 7.38 ± 0.01 | 7.37 ± 0.01 | 7.38 ± 0.01 | 7.32 ± 0.01 |
| 72 h hypo | 7.38 ± 0.01 | 7.37 ± 0.01 | 7.38 ± 0.01 | 7.41 ± 0.01 | 7.40 ± 0.01 | 7.41 ± 0.01 | 7.37 ± 0.01 |
| PaCO2 (mmHg) | |||||||
| Normothermia | 48.1 ± 1.0 | 49.1 ± 1.2 | 48.6 ± 1.4 | 48.6 ± 1.4 | 50.7 ± 1.7 | 50.1 ± 0.9 | 53.9 ± 0.6 |
| 48 h hypo | 48.9 ± 1.3 | 47.5 ± 1.3 | 46.3 ± 0.8 | 47.4 ± 1.2 | 45.1 ± 1.4 | 46.8 ± 1.0 | 51.9 ± 1.4 |
| 72 h hypo | 50.9 ± 0.6 | 49.5 ± 1.4 | 47.5 ± 1.3 | 47.5 ± 1.2 | 50.8 ± 1.3 | 49.6 ± 1.2 | 52.9 ± 1.4 |
| PaO2 (mmHg) | |||||||
| Normothermia | 20.9 ± 1.5 | 20.8 ± 1.3 | 21.4 ± 1.6 | 20.0 ± 1.6 | 18.8 ± 1.5 | 21.2 ± 1.7 | 20.5 ± 1.5 |
| 48 h hypo | 21.8 ± 1.0 | 21.3 ± 1.3 | 20.6 ± 1.7 | 20.1 ± 1.5 | 21.0 ± 1.4 | 20.0 ± 2.0 | 22.3 ± 1.6 |
| 72 h hypo | 22.3 ± 0.9 | 23.1 ± 0.7 | 22.4 ± 1.1 | 22.0 ± 1.0 | 21.0 ± 1.5 | 22.1 ± 1.1 | 21.3 ± 0.8 |
| Lactate (mmol/L) | |||||||
| Normothermia | 1.3 ± 0.1 | 3.6 ± 0.6* | 2.9 ± 0.5 | 2.4 ± 0.4 | 3.9 ± 0.9* | 2.1 ± 0.6 | 1.4 ± 0.2 |
| 48 h hypo | 1.2 ± 0.0 | 2.9 ± 0.4* | 3.1 ± 0.5* | 2.5 ± 0.4* | 2.3 ± 0.4 | 2.6 ± 0.4* | 1.0 ± 0.1 |
| 72 h hypo | 1.1 ± 0.1 | 2.5 ± 0.4* | 2.5 ± 0.3* | 2.0 ± 0.3 | 1.8 ± 0.4 | 1.5 ± 0.3 | 1.0 ± 0.1 |
| Glucose (mmol/L) | |||||||
| Normothermia | 0.9 ± 0.0 | 1.3 ± 0.1* | 1.2 ± 0.1* | 1.2 ± 0.1* | 1.4 ± 0.1* | 1.2 ± 0.1 | 0.9 ± 0.1 |
| 48 h hypo | 0.8 ± 0.0 | 1.2 ± 0.1* | 1.3 ± 0.1* | 1.3 ± 0.1* | 1.1 ± 0.1 | 1.3 ± 0.1* | 0.7 ± 0.1 |
| 72 h hypo | 1.0 ± 0.0 | 1.4 ± 0.1* | 1.5 ± 0.1* | 1.4 ± 0.1* | 1.4 ± 0.1* | 1.3 ± 0.1 | 0.9 ± 0.1 |
Normothermia: ischemia-normothermia (n = 8); 48 h hypo: ischemia-48 h hypothermia (n = 8); 72 h hypo: ischemia-72 h hypothermia (n = 8).
p < 0.05 vs. baseline.
Temperature changes
Head cooling was associated with a fall in extradural temperature to similar nadirs of 32.3 ± 0.3℃ in the ischemia-48 h hypothermia group and 31.3 ± 0.2℃ in the ischemia-72 h hypothermia group (N.S.), compared to 39.5 ± 0.1℃ in the ischemia-normothermia group (p < 0.05, Figure 1). Similarly, esophageal temperature fell to between 37 and 38℃ during cooling (p < 0.05 vs. ischemia-normothermia, Figure 1). After the end of hypothermia, at either 48 or 72 h, extradural and esophageal temperature returned to baseline values after approximately 1 h.
Figure 1.

Change in extradural temperature, esophageal temperature, and carotid artery blood flow before, during, and after 30 min of global cerebral ischemia (time 0) in near-term fetal sheep, showing the ischemia-normothermia (n = 8), ischemia-48 h hypothermia (n = 8), and ischemia-72 h hypothermia (n = 8) groups. Extradural temperature was significantly reduced in both hypothermia groups during treatment (p < 0.05) and returned to baseline when cooling ended at 48 or 72 h. A significant reduction in esophageal temperature was seen with the onset of hypothermia (p < 0.05). CaBF increased in the ischemia-normothermia group between 6 and 36 h after ischemia compared to either hypothermia group (p < 0.05). Data are mean ± SEM, *p < 0.05 ischemia-72 h hypothermia vs. ischemia-normothermia, #p < 0.05 ischemia-48 h hypothermia vs. ischemia-normothermia.
Brain activity
Carotid occlusion was associated with rapid, profound suppression of EEG power (Figure 2). After release of occlusion, the EEG remained suppressed until the onset of a period of intense seizure activity from ∼8 to 48 h after ischemia. After seizures resolved, total EEG power fell below baseline levels in the ischemia-normothermia group. In contrast, the ischemia-48 h and ischemia-72 h hypothermia groups showed substantially greater recovery of EEG power than ischemia-normothermia from 24 h until the end of the experiment (p < 0.05). Rewarming at 48 h was associated with a progressive deterioration of EEG power that did not occur with rewarming at 72 h, such that the ischemia-48 h hypothermia group showed reduced EEG power compared with ischemia-72 h hypothermia for the remainder of the experiment (p < 0.05).
Figure 2.

Changes in EEG activity, spectral edge frequency, and impedance before, during, and after 30 min of global cerebral ischemia (time 0) in the ischemia-normothermia (n = 8), ischemia-48 h hypothermia (n = 8), and ischemia-72 h hypothermia (n = 8) groups. EEG activity was suppressed in all groups during/immediately after ischemia, followed by a transient increase during the seizure period (8–48 h). EEG activity in the ischemia-normothermia group was reduced for the remainder of the experiment, whilst both hypothermia groups showed a significant recovery from 24 h onward (p < 0.05). The end of hypothermia was associated with a rapid reduction in EEG power in the ischemia-48 h hypothermia group, which remained significantly lower than the ischemia-72 h hypothermia group (p < 0.05). Spectral edge was suppressed in all groups during ischemia and remained suppressed in the ischemia-normothermia group but increased significantly in both hypothermia groups between 8 and 12 h and from 84 h onward (p < 0.05). Ischemia was associated with an increase in impedance, which resolved after the end of ischemia. In the ischemia-normothermia group, impedance increased between 24 and 72 h, followed by a significant reduction from 120 h. A reduction in impedance was seen in the ischemia-48 h hypothermia group from 144 h onward compared to the ischemia-72 h hypothermia group (p < 0.05). Data are mean ± SEM, *p < 0.05 ischemia-72 h hypothermia vs. ischemia-normothermia group, #p < 0.05 ischemia-72 h vs. ischemia-48 h hypothermia group, ap < 0.05 ischemia-48 h hypothermia vs. ischemia-normothermia group.
Ischemia was associated with rapid suppression of the EEG spectral edge frequency, which remained below baseline until the end of the experiment in the ischemia-normothermia group. In contrast, both the ischemia-72 h and ischemia-48 h hypothermia groups showed significantly higher spectral edge frequency from 6 to 24 h and from 48 h until the end of the experiment (Figure 2, p < 0.05) with no significant difference between the hypothermia groups (N.S.).
No seizures were seen in sham control animals. Ischemia-normothermia was associated with a total seizure burden of 300 ± 119 min. Seizure burden was similarly reduced in the ischemia-48 h hypothermia (70 ± 39 min) and ischemia-72 h hypothermia groups (62 ± 22 min, p < 0.05 vs. ischemia-normothermia).
Ischemia-normothermia was associated with a secondary increase in cortical impedance between 24 and 72 h after ischemia compared to sham controls, ischemia-48 h and ischemia-72 h hypothermia (p < 0.05). Impedance fell below sham control levels from 72 h onward in the ischemia-normothermia group and was significantly lower than the ischemia-72 h and -48 h groups from 120 h onward (p < 0.05). After rewarming, impedance in the ischemia-48 h hypothermia group fell below baseline values and was significantly lower than the ischemia-72 h hypothermia group from 144 h onward (p < 0.05).
Physiological parameters
Carotid artery blood flow (CaBF) showed a delayed increase in the ischemia-normothermia group between 6 and 36 h after ischemia, which was suppressed in both hypothermia groups (Figure 1, p < 0.05 vs. ischemia-normothermia). There were no significant differences in MAP, fetal heart rate or nuchal EMG between groups (data not shown).
Immunohistochemistry
Ischemia was associated with reduced numbers of surviving neurons in the cortex, CA1, CA3, CA4, and dentate gyrus of the hippocampus compared to sham controls (p < 0.05, Figure 3). Neuronal survival was significantly improved in the cortex and dentate gyrus (p < 0.05) after ischemia-48 h hypothermia compared to ischemia-normothermia, and after ischemia-72 h hypothermia in the cortex, CA3, CA4, and dentate gyrus. Ischemia-48 h hypothermia was associated with reduced neuronal survival in the cortex and CA4 compared to ischemia-72 h hypothermia (p < 0.05), and in the cortex, CA1, CA3, CA4 compared with sham controls. In contrast, the ischemia-72 h hypothermia group showed similar neuronal survival to sham controls except in CA1 and CA3.
Figure 3.

Neuronal survival 7 days after 30 min of global cerebral ischemia in the sham control (n = 9), ischemia-normothermia (n = 8), ischemia-48 h hypothermia (n = 8), and ischemia-72 h hypothermia (n = 8) groups. (a) Ischemia was associated with a significant reduction in neuronal survival in the cortex, CA1, CA3, CA4, and dentate gyrus of the hippocampus compared to sham control. Neuronal survival was significantly increased in the ischemia-72 h hypothermia group in all regions except the CA1 but only in the cortex and dentate gyrus in the ischemia-48 h hypothermia group. Neuronal survival was significantly greater in the cortex and CA4 in the ischemia-72 h hypothermia group compared to the ischemia-48 h hypothermia group. *p < 0.05 vs. sham control, #p < 0.05 vs. ischemia-normothermia, ap < 0.05 vs. ischemia-48 h hypothermia. (b) Photomicrograph showing NeuN-positive staining in the cortex (A, F, K, P), CA1 (B, G, L, Q), CA3 (C, H, M, R), CA4 (D, I, N, S), and dentate gyrus (E, J, O, T) in the sham control (A–E), ischemia-normothermia (F–J), ischemia-48 h hypothermia (K–O), and the ischemia-72 h hypothermia (P–T) groups. Scale bar 200 µm.
Total Olig2-positive oligodendrocyte survival was decreased after ischemia-normothermia in the intragyral white matter of the first and second parasagittal gyri and the periventricular white matter compared to sham controls (p < 0.05, Figure 4). Both hypothermia protocols were associated with a similar, partial improvement in oligodendrocyte survival, with oligodendrocyte number being significantly higher than the ischemia-normothermia group but significantly reduced compared to sham control in all regions (p < 0.05).
Figure 4.
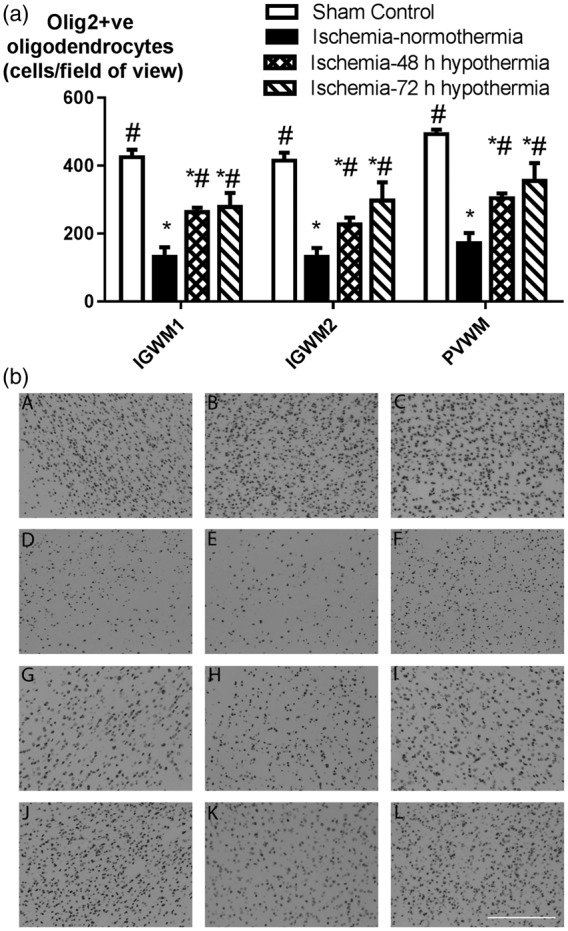
Oligodendrocyte survival in the intragyral white matter of the first and second parasagittal gyri and the periventricular white matter in the sham control (n = 9), ischemia-normothermia (n = 8), ischemia-48 h hypothermia (n = 8), and ischemia-72 h hypothermia (n = 8) groups. (a) Ischemia was associated with a significant reduction in oligodendrocyte survival in all white matter regions with a partial improvement in both hypothermia groups in all regions, which remained significantly lower than sham control. Data are mean ± SEM, *p < 0.05 vs. sham control, #p < 0.05 vs. ischemia-normothermia. (b) Photomicrograph of Olig-2 positive labelling in the intragyral white matter of the first parasagittal gyrus (A, D, G, J), the second parasagittal gyrus (B, E, H, K) and the periventricular white matter (C, F, I, L) in the sham control (A–C), ischemia-normothermia (D–F), ischemia-48 h (G–I), and ischemia-72 h (J–L) groups. Scale bar 200 µm.
Ischemia-normothermia was associated with a significant increase in the number of Ki-67 proliferating cells in the intragyral white matter of the first and second parasagittal gyri and the periventricular white matter compared to sham control (p < 0.05, Supplementary Figure 1). Both hypothermia protocols were associated with a similar profound reduction in numbers of proliferating cells compared to ischemia-normothermia in all regions (p < 0.05) and were not significantly different to sham controls.
Iba1-positive microglia were markedly increased in the intragyral white matter of the first and second parasagittal gyri and the periventricular white matter after ischemia-normothermia compared to sham controls (p < 0.05, Figure 5). Both hypothermia protocols were associated with reduced numbers of microglia in all areas compared to ischemia-normothermia (p < 0.05). However, more microglia were seen after ischemia-48 h hypothermia than ischemia-72 h hypothermia and sham controls in all white matter areas (p < 0.05).
Figure 5.

Microglial number in the intragyral white matter of the first and second parasagittal gyri, the periventricular white matter and parasagittal cortex in the sham control (n = 9), ischemia-normothermia (n = 8), ischemia-48 h hypothermia (n = 8), and ischemia-72 h hypothermia (n = 8) groups. (a) Ischemia was associated with a significant increase in microglial number in all regions, which was attenuated in both hypothermia groups but remained significantly elevated in the intragyral white matter of the first parasagittal gyrus in the ischemia-72 h hypothermia group and in all regions in the ischemia-48 h hypothermia group. Microglial number was significantly higher in the ischemia-48 h hypothermia group compared to the ischemia-72 h hypothermia group all white matter regions. (b) Photomicrograph of Iba1-positive microglial labelling in the intragyral white matter of the first parasagittal gyrus (A, D, G, J), the second parasagittal gyrus (B, E, H, K), and the periventricular white matter (C, F, I, L) in the sham control (A–C), ischemia-normothermia (D–F), ischemia-48 h (G–I), and ischemia-72 h (J–L) groups. Scale bar 200 µm. (c) Ischemia was associated with a significant increase in microglial number in the parasagittal cortex compared to sham control, which was attenuated in the ischemia-72 h hypothermia group but not the ischemia-48 h hypothermia group. (d) Photomicrograph of Iba1-positive microglial labelling in the parasagittal cortex in the sham control (A), ischemia-normothermia (B), ischemia-48 h hypothermia (C), and ischemia-72 h hypothermia (D) groups. Scale bar 100 µm. Data are mean ± SEM, *p < 0.05 vs. sham control, #p < 0.05 vs. ischemia-normothermia, ap < 0.05 vs. ischemia-48 h hypothermia.
Similarly, microglia were markedly increased in the cortex of the first parasagittal gyrus after ischemia-normothermia compared to sham controls (p < 0.05, Figure 5). Microglia were significantly suppressed after ischemia-72 h hypothermia compared to ischemia-normothermia, to sham control values (N.S.). In contrast, ischemia-48 h hypothermia was not associated with a significant effect on microglial number compared to ischemia-normothermia, with greater numbers of microglia than both sham controls and ischemia-72 h hypothermia (p < 0.05).
MBP area fraction was significantly reduced overall after ischemia-normothermia compared to sham control (p < 0.05, Figure 6). Ischemia-48 h hypothermia was associated with improved MBP area fraction compared to ischemia-normothermia in the intragyral white matter of the first parasagittal gyrus, but no other regions. Further, ischemia-48 h hypothermia was associated with significantly lower values than sham controls in all regions and than ischemia-72 h hypothermia in the intragyral white matter of the second parasagittal gyrus (p < 0.05). In contrast, ischemia-72 h hypothermia was associated with significantly greater MBP area fraction than the ischemia-normothermia group in all regions, although values were less than sham control in the intragyral white matter of the first parasagittal gyrus (p < 0.05).
Figure 6.
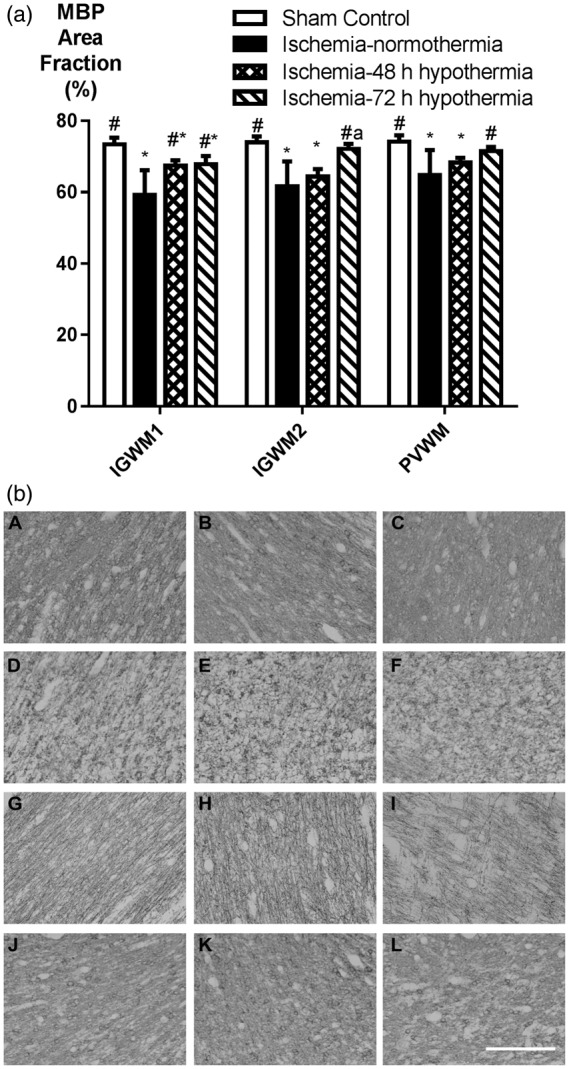
Myelin basic protein (MBP) area fraction in the intragyral white matter of the first and second parasagittal gyri and the periventricular white matter in the sham control (n = 9), ischemia-normothermia (n = 8), ischemia-48 h hypothermia (n = 8), and ischemia-72 h hypothermia (n = 8) groups. (a) MBP area fraction was significantly reduced in all regions in the ischemia-normothermia group compared to sham control. Loss of MBP was partially attenuated in the ischemia-48 h hypothermia group in the first parasagittal gyrus only, whereas in the ischemia-72 h hypothermia group, MBP loss was reduced in all regions. Data are mean ± SEM, *p < 0.05 vs. sham control, #p < 0.05 vs. ischemia-normothermia, ap < 0.05 vs. ischemia-48 h hypothermia. (b) Photomicrograph showing MBP staining in the intragyral white matter of the first parasagittal gyrus (A, D, G, J), the second parasagittal gyrus (B, E, H, K), and the periventricular white matter (C, F, I, L) in the sham control (A–C), ischemia-normothermia (D–F), ischemia-48 h (G–I), and ischemia-72 h (J–L) groups. Scale bar 200 µm.
Discussion
This study has demonstrated that although cerebral hypothermia for 48 h is partially neuroprotective in its own right, compared to 72 h of hypothermia, it was associated with less improvement in EEG power, neuronal survival and myelination, and increased microglial induction 7 days after global cerebral ischemia, particularly in the cortex. The original paradigm of extended hypothermia for 72 h arose from the observation that the phase of secondary deterioration after hypoxia-ischemia, as shown by delayed onset of seizures and secondary cytotoxic edema, resolved after approximately 72 h.18,22 Thus, it was hypothesized that it would be necessary to continue hypothermia for at least 72 h,18 to suppress ongoing delayed encephalopathic processes such as programmed cell death and inflammation.1 It could reasonably be speculated that the secondary phase might resolve earlier during cerebral hypothermia. Unfortunately, the present study demonstrates for the first time that this is not the case, since despite effective attenuation of seizures and secondary cytotoxic edema by delayed hypothermia, after rewarming at 48 h but not 72 h, EEG power progressively deteriorated over 12 h. This strongly infers that the encephalopathic processes suppressed by hypothermia can be reactivated by rewarming as late as 48 h after cerebral ischemia.
Rewarming at 48 h in the present study was not associated with rebound cytotoxic edema or increased total seizure burden, suggesting that influx of ions, such as sodium and calcium, into cells and excessive release of glutamate were not important triggers of further injury. In contrast, the finding of greater continuing microglial upregulation in the cortex and white matter after 48 h of hypothermia compared to 72 h suggests that pro-inflammatory pathways can be re-activated during rewarming after an insufficient duration of hypothermia. Broadly consistent with our findings, in adult rats, a suboptimal duration of hypothermia for 24 h after stroke was associated with only transient suppression of inflammation and necrotic pathways compared with 48 h.13 Although that study suggested that 48 h of hypothermia was effective, the reader should note that the outcome was not compared with hypothermia for 72 h. More generally, it is highly probable that the optimal duration of hypothermia is affected by maturation and the severity and nature of the insult.
In contrast with the marked effects on neuronal survival, in the present study, the duration of hypothermia had no effect on the partial improvement in numbers of oligodendrocytes after hypothermia compared to ischemia-normothermia. Given that hypothermia completely attenuated the increase in proliferation seen in the white matter in the ischemia-normothermia group, it is likely that hypothermia prevented oligodendrocyte cell death as opposed to enabling restorative proliferation of oligodendrocytes. These data are consistent with a previous study in preterm fetal sheep showing that after severe asphyxia hypothermic neuroprotection was associated with suppressed proliferation in the white matter tracts.23
Intriguingly, although there was no difference in numbers of oligodendrocytes between the hypothermia groups, there was greater improvement in myelination after 72 h hypothermia than 48 h. Potentially, this might reflect improved neuronal survival and/or axonal integrity in the ischemia-72 h hypothermia group.24 Alternatively, impaired myelination could reflect impaired differentiation of immature cells. There is evidence in preterm infants that pre-oligodendrocytes can rapidly regenerate following injury and yet fail to mature into myelinating cells.25 However, this seems an unlikely mechanism in the present study, since as above, proliferation was markedly and similarly suppressed in both hypothermia groups. Alternatively, it is possible that greater reactivation of inflammation after 48 h than 72 h of cooling, as shown by significantly greater numbers of microglia in the white matter tracts in the ischemia-48 h hypothermia group, increased damage to the myelin sheaths.
It is important to appreciate that the amount of residual microglial induction was not uniformly associated with regional outcome. For example, 48 h of hypothermia did not significantly reduce numbers of microglia in the cortex despite partial improvement in neuronal survival. However, it is now well established that the phenotype of microglia is highly labile, and varies dynamically over a wide spectrum, from a pro-inflammatory “M1” state (classical activation) to an “M2” anti-inflammatory/reparative state (alternative activation) and other variants.26 Understanding the impact of treatments such as hypothermia on changes in microglial phenotype in different regions over time is an important area of ongoing research.
In the present study, strong neuroprotection was seen after 72 h of cooling when fetuses were allowed to spontaneously rewarm over approximately 60 min, similarly to previous studies of cerebral hypothermia in fetal sheep.7,9,18 There is very limited information on how fast the brain “should” be rewarmed after therapeutic hypothermia. In randomized trials, infants were typically rewarmed at 0.5℃ per hour.27,28 Rapid rewarming (4℃ per hour) after hypothermia in neonatal piglets exposed to hypoxia-ischemia was associated with increased neural apoptosis compared to slower rewarming at 0.5℃ per hour.29 However, that study used a suboptimal duration of hypothermia (18 h) and therefore the apparently improved neuroprotection with slower rewarming may have been confounded by greater duration of hypothermia. Systematic studies of the impact of rate of rewarming after clinically relevant hypothermia protocols are vital.
More generally, the current findings strongly support the concept that even several days after hypoxia-ischemia the neuronal environment is still recovering and thus, interventions at this late time may affect the outcome of treatment. This is highly consistent with previous evidence in adults rats that late hyperthermia nullified neuroprotection from early mild hypothermia after forebrain ischemia.30 We speculate that the converse might also be true, that targeted late interventions during mild hypothermia might be beneficial. In the present study, even 72 h of hypothermia was still associated with residual microglial induction. Our previous studies have shown that extending cooling to 120 h did not further reduce this persisting inflammation.7,9 Potentially, late anti-inflammatory strategies after hypothermia, such as erythropoietin31,32 or stem cells33 might be beneficial even after the window of opportunity during normothermia. Supporting this speculation, a recent phase 2 study of combined hypothermia and erythropoietin suggested benefit despite very late treatment with erythropoietin, at a mean age of 16 h after birth.31 This finding needs to be confirmed in a large, definitive trial, but it is consistent with the hypothesis that different add-on interventions during mild hypothermia may be optimal depending on the phase of evolving injury.
Conclusion
These data demonstrate that 48 h of hypothermia is insufficient for optimal neuroprotection after global cerebral ischemia in near-term fetal sheep, and strongly support the current clinical standard of care of continuing therapeutic hypothermia for 72 h in infants with moderate to severe HIE.
Supplementary Material
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Health Research Council of New Zealand.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Joanne Davidson, Laura Bennet, and Alistair J. Gunn conceptualized and designed the study. Joanne Davidson, Simerdeep Dhillon, and Guido Wassink undertook experiments and analyzed data. Sean Whitham, Vittoria Draghi, and Guido Wassink undertook immunohistochemistry, cell quantification, analysis and preparation of figures. A.J. Gunn provided overall oversight of the research. All authors critically reviewed the manuscript and approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Supplementary material
Supplementary material for this paper can be found at the journal website: http://journals.sagepub.com/home/jcb
References
- 1.Wassink G, Gunn ER, Drury PP, et al. The mechanisms and treatment of asphyxial encephalopathy. Front Neurosci 2014; 8: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edwards AD, Brocklehurst P, Gunn AJ, et al. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta-analysis of trial data. BMJ 2010; 340: c363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azzopardi D, Strohm B, Marlow N, et al. Effects of hypothermia for perinatal asphyxia on childhood outcomes. N Engl J Med 2014; 371: 140–149. [DOI] [PubMed] [Google Scholar]
- 4.Shankaran S, Pappas A, McDonald SA, et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N Engl J Med 2012; 366: 2085–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guillet R, Edwards AD, Thoresen M, et al. Seven- to eight-year follow-up of the CoolCap trial of head cooling for neonatal encephalopathy. Pediatr Res 2012; 71: 205–209. [DOI] [PubMed] [Google Scholar]
- 6.Shankaran S, Laptook AR, Pappas A, et al. Effect of depth and duration of cooling on deaths in the NICU among neonates with hypoxic ischemic encephalopathy: a randomized clinical trial. JAMA 2014; 312: 2629–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davidson JO, Wassink G, Yuill CA, et al. How long is too long for cerebral cooling after ischemia in fetal sheep? J Cereb Blood Flow Metab 2015; 35: 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gunn AJ, Laptook AR, Robertson NJ, et al. Therapeutic hypothermia translates from ancient history in to practice. Pediatr Res 2017; 81: 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davidson JO, Yuill CA, Zhang FG, et al. Extending the duration of hypothermia does not further improve white matter protection after ischemia in term-equivalent fetal sheep. Sci Rep 2016; 6: 25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tooley JR, Satas S, Porter H, et al. Head cooling with mild systemic hypothermia in anesthetized piglets is neuroprotective. Ann Neurol 2003; 53: 65–72. [DOI] [PubMed] [Google Scholar]
- 11.Thoresen M, Penrice J, Lorek A, et al. Mild hypothermia after severe transient hypoxia-ischemia ameliorates delayed cerebral energy failure in the newborn piglet. Pediatr Res 1995; 37: 667–670. [DOI] [PubMed] [Google Scholar]
- 12.Alonso-Alconada D, Broad KD, Bainbridge A, et al. Brain cell death is reduced with cooling by 3.5℃ to 5℃ but increased with cooling by 8.5℃ in a piglet asphyxia model. Stroke 2015; 46: 275–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandu RE, Buga AM, Balseanu AT, et al. Twenty-four hours hypothermia has temporary efficacy in reducing brain infarction and inflammation in aged rats. Neurobiol Aging 2016; 38: 127–140. [DOI] [PubMed] [Google Scholar]
- 14.Clark DL, Penner M, Wowk S, et al. Treatments (12 and 48 h) with systemic and brain-selective hypothermia techniques after permanent focal cerebral ischemia in rat. Exp Neurol 2009; 220: 391–399. [DOI] [PubMed] [Google Scholar]
- 15.Clark DL, Penner M, Orellana-Jordan IM, et al. Comparison of 12, 24 and 48 h of systemic hypothermia on outcome after permanent focal ischemia in rat. Exp Neurol 2008; 212: 386–392. [DOI] [PubMed] [Google Scholar]
- 16.Kilkenny C, Browne W, Cuthill IC, et al. Animal research: reporting in vivo experiments–the ARRIVE guidelines. J Cereb Blood Flow Metab 2011; 31: 991–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams CE, Gluckman PD. Real-time spectral intensity analysis of the EEG on a common microcomputer. J Neurosci Methods 1990; 32: 9–13. [DOI] [PubMed] [Google Scholar]
- 18.Gunn AJ, Gunn TR, de Haan HH, et al. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J Clin Invest 1997; 99: 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith DC. Effects of skin blood flow and temperature on skin–electrode impedance and offset potential: measurements at low alternating current density. J Med Eng Technol 1992; 16: 112–116. [DOI] [PubMed] [Google Scholar]
- 20.Gerrits LC, Battin MR, Bennet L, et al. Epileptiform activity during rewarming from moderate cerebral hypothermia in the near-term fetal sheep. Pediatr Res 2005; 57: 342–346. [DOI] [PubMed] [Google Scholar]
- 21.Pozo Devoto VM, Chavez JC, Fiszer de Plazas S. Acute hypoxia and programmed cell death in developing CNS: Differential vulnerability of chick optic tectum layers. Neuroscience 2006; 142: 645–653. [DOI] [PubMed] [Google Scholar]
- 22.Williams CE, Gunn A, Gluckman PD. Time course of intracellular edema and epileptiform activity following prenatal cerebral ischemia in sheep. Stroke 1991; 22: 516–521. [DOI] [PubMed] [Google Scholar]
- 23.Barrett RD, Bennet L, Naylor A, et al. Effect of cerebral hypothermia and asphyxia on the subventricular zone and white matter tracts in preterm fetal sheep. Brain Res 2012; 1469: 35–42. [DOI] [PubMed] [Google Scholar]
- 24.Nave KA, Werner HB. Myelination of the nervous system: mechanisms and functions. Annu Rev Cell Dev Biol 2014; 30: 503–533. [DOI] [PubMed] [Google Scholar]
- 25.Buser JR, Maire J, Riddle A, et al. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann Neurol 2012; 71: 93–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierre WC, Smith PL, Londono I, et al. Neonatal microglia: The cornerstone of brain fate. Brain Behav Immun 2017; 59: 333–345. [DOI] [PubMed] [Google Scholar]
- 27.Gluckman PD, Wyatt JS, Azzopardi D, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet 2005; 365: 663–670. [DOI] [PubMed] [Google Scholar]
- 28.Shankaran S, Laptook AR, Ehrenkranz RA, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med 2005; 353: 1574–1584. [DOI] [PubMed] [Google Scholar]
- 29.Wang B, Armstrong JS, Lee JH, et al. Rewarming from therapeutic hypothermia induces cortical neuron apoptosis in a swine model of neonatal hypoxic-ischemic encephalopathy. J Cereb Blood Flow Metab 2015; 35: 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coimbra C, Boris-Moller F, Drake M, et al. Diminished neuronal damage in the rat brain by late treatment with the antipyretic drug dipyrone or cooling following cerebral ischemia. Acta Neuropathol (Berl.) 1996; 92: 447–453. [DOI] [PubMed] [Google Scholar]
- 31.Wu YW, Mathur AM, Chang T, et al. High-dose erythropoietin and hypothermia for hypoxic-ischemic encephalopathy: A phase II trial. Pediatrics 2016; 137: e20160191. [DOI] [PubMed] [Google Scholar]
- 32.Juul SE, Pet GC. Erythropoietin and neonatal neuroprotection. Clin Perinatol 2015; 42: 469–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennet L, Tan S, Van den Heuij L, et al. Cell therapy for neonatal hypoxia-ischemia and cerebral palsy. Ann Neurol 2012; 71: 589–600. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.