

Abstract
Fatty acid binding proteins (FABPs) serve as critical modulators of endocannabinoid signaling by facilitating the intracellular transport of anandamide and whose inhibition potentiates anandamide signaling. Our previous work has identified a novel small-molecule FABP inhibitor, α-truxillic acid 1-naphthyl monoester (SB-FI-26, 3) that has shown efficacy as an antinociceptive and anti-inflammatory agent in rodent models. In the present work, we have performed an extensive SAR study on a series of 3-analogs as novel FABP inhibitors based on computer-aided inhibitor drug design and docking analysis, chemical synthesis and biological evaluations. The prediction of binding affinity of these analogs to target FABP3, 5 and 7 isoforms was performed using the AutoDock 4.2 program, using the recently determined co-crystal structures of 3 with FABP5 and FABP7. The compounds with high docking scores were synthesized and evaluated for their activities using a fluorescence displacement assay against FABP3, 5 and 7. During lead optimization, compound 3l emerged as a promising compound with the Ki value of 0.21 μM for FABP 5, 4-fold more potent than 3 (Ki, 0.81 μM). Nine compounds exhibit similar or better binding affinity than 3, including compounds 4b (Ki, 0.55 μM) and 4e (Ki, 0.68 μM). Twelve compounds are selective for FABP5 and 7 with >10 μM Ki values for FABP3, indicating a safe profile to avoid potential cardiotoxicity concerns. Compounds 4f, 4j and 4k showed excellent selectivity for FABP5 and would serve as other new lead compounds. Compound 3a possessed high affinity and high selectivity for FABP7. Compounds with moderate to high affinity for FABP5 displayed antinociceptive effects in mice while compounds with low FABP5 affinity lacked in vivo efficacy. In vivo pain model study in mice revealed that exceeding hydrophobicity significantly affects the efficacy. Thus, among the compounds with high affinity to FABP5 in vitro, the compounds with moderate hydrophobicity were identified as promising new lead compounds for the next round of optimization, including compounds 4b and 4j. For select cases, computational analysis of the observed SAR, especially the selectivity of new inhibitors to particular FABP isoforms, by comparing docking poses, interaction map, and docking energy scores has provided useful insights.
Keywords: Fatty acid binding protein, FABP inhibitor, Anti-nociceptive agent, Anti-inflammatory agent, SAR study, Computer-aided design, Molecular docking
Graphical abstract
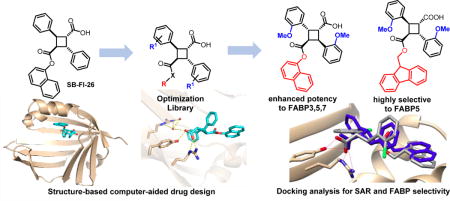
1. Introduction
The fatty acid binding proteins (FABPs) are a family of small chaperone proteins that act as cytosolic transporters for a wide variety of lipophilic substances including fatty acids, N-acylethanolamines (NAE), eicosanoids, and cannabinoids [1, 2]. FABPs are widely expressed throughout the body and play an integral role in a multitude of physiological processes such as lipid metabolism, inflammation and neuronal signaling [3]. FABPs of the mammalian central and peripheral nervous systems have been shown to facilitate the intracellular transport of NAEs, particularly the endocannabinoid arachidonoyl ethanolamide (anandamide, AEA), as well as catabolism by the endoplasmic reticulum-localized enzyme fatty acid amide hydrolase (FAAH).[4] Genetic or pharmacological inhibition of the FAAH enzyme or the FABPs results in a marked elevation of brain AEA levels, which acts upon type-1 cannabinoid receptors (CB1R) and thereby cause a suppression of pain transmission and other therapeutically beneficial effects [5–7]. As such, designing inhibitors of AEA inactivation is desirable. Over the years numerous FAAH inhibitors have been developed and have generally been well tolerated in the clinical setting [8, 9]. However, focused medicinal chemistry efforts on targeting FABPs may hold advantages over direct FAAH inhibition because unlike FAAH, which is distributed throughout the body, humans express multiple FABP isoforms that exhibit tissue-specific expression patterns. Designing small molecule inhibitors that selectively bind target FABP isoforms will allow for drugs to preferentially act upon target tissues of interest rather than systemic NAE upregulation, which may increase the likelihood of off-target adverse events. This has led to the pursuit of identifying novel compounds that are capable of selectively inhibiting the FABP isoforms that are expressed in the mammalian central and peripheral nervous systems. Three FABP isoforms have been identified in these tissues, i.e., FABP3 (heart FABP, H-FABP), FABP5 (epidermal FABP, keratinocyte FABP, E-FABP), and FABP7 (brain FABP, B-FABP) [10].
Previous work from our group has led to the identification of a novel competitive FABP inhibitor, α-truxillic acid 1-naphthyl monoester (SB-FI-26, 3) [11]. It has been shown that 3 is a potent inhibitor of FABP5 and FABP7, with sub-micromolar affinities reported in vitro (Ki = 0.9 ± 0.1 μM and 0.4 ± 0.0 μM, respectively), with weaker binding to FABP3 (Ki = 3.9 ± 0.7 μM).[6] Selective inhibition for FABP5 over FABP3 is deemed desirable, as mice bearing a knockout for FABP3 exhibited age-related cardiac hypertrophy, and thus pharmacological inhibition of this protein may have a potential to cause undesirable side effects [12].
Intriguingly, compound 3 shares the same α-truxillic acid skeleton with that of (-)-incarvillateine (Fig. 1), a natural monoterpene alkaloid isolated from the Chinese herbal Incarvillei sinensis, that has been used as a pain-reliever in traditional Eastern medicine (as the dried plant matter ‘Jiaohao’), and more recently has been shown to produce potent analgesic and anti-inflammatory effects in formalin-induced mouse models [13, 14]. Structure-antinociceptive activity studies on (−)-incarvillateine have found that the cyclobutane moiety is required for its analgesic properties [15]. To our knowledge, the mechanism of action for (−)-incarvillateine-induced analgesia has not been formally elucidated in full, though adenosine receptors likely play a role and there is some contention in the literature pertaining to the involvement of the opioid system [16, 17]. Considering the structural similarity and essentially overlapping reported pharmacological profiles of (−)-incarvillateine and 3, it is tempting to speculate a possibility that its effects are mediated, at least in part, by FABP inhibition and subsequent NAE/endocannabinoid potentiation.
Fig. 1.
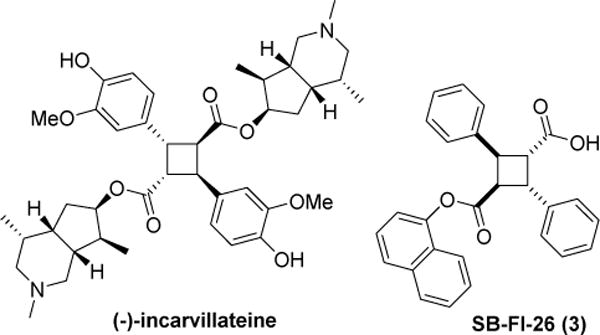
Structures of (-)-incarvillateine and 3
Compound 3 was further shown to be biologically active in a FABP and CB1R-dependent manner. The compound has a half-life of ~3 hours in vivo and is efficacious in producing anti-inflammatory and anti-nociceptive effects in rodent models of visceral, thermal, neuropathic, and inflammatory pain [6]. Furthermore, intraperitoneal (i.p.) administration of 3 up to 40 mg/kg in mice showed no conditioned place preference nor conditioned place aversion, indicating a relatively low potential for addiction [18].
Despite promising efficacy in pain models, 3 requires further preclinical optimization to improve potency, solubility, selectivity and in vivo stability. To this end, the α-truxillic acid monoester core structure was used as the scaffold for optimization in the present study. Based on the prediction using the Autodock 4.2 program [19], a series of novel 3-analogs have been designed, synthesized, and their potencies evaluated to investigate SAR for enhanced potency and selectivity.
2. Results and Discussion
2.1. Optical resolution of 3
In the previous study, we examined the potency of racemic 3. However, our recent protein X-ray structure determination of FABP5/3 as well as FABP7/3 cocrystals revealed that (S,S,S,S)-3 was incorporated into the canonical binding site. Accordingly, there is a good possibility that the (S,S,S,S)-enantiomer may be substantially more potent than the (R,R,R,R)-enantiomer. Thus, we set out to optically resolve the two enantiomers of 3.
(1R,2S)-2-amino-1,2-diphenylethanol, (S)-1-(1-naphthyl)ethylamine, and (S)-phenylalaninol (Fig. 2), were examined as resolving agents. A mixture of 3 and a resolving agent was dissolved in common lab solvents (i.e., ethanol, isopropanol, and acetonitrile) and allowed to recrystallize as diastereomeric salts. Samples showing crystal formation were acidified with 4 M HCl, extracted with ethyl acetate, and analyzed by chiral HPLC using a Chiralcel ODH column (iPrOH/hexane). The attempts using (1R,2S)-2-amino-1,2-diphenylethanol, and (S)-1-(1-naphthyl)ethylamine resulted in recovering only racemic 3. Fortunately, (S)-phenylalaninol was found to be a suitable resolving agent in combination with methanol. Thus, we were able to isolate two enantiomers, 3-A and 3-B. (See Experimental for details.)
Fig. 2.
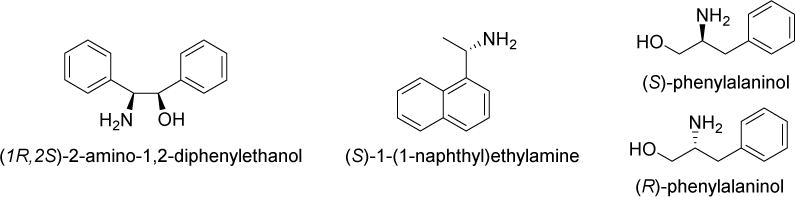
Enantiopure amines examined for optical resolution of 3
Since 3-A gave better crystals, it was subjected to an X-ray crystallographic analysis and unambiguously determined to be the (1R,2R,3R,4R)-enantiomer (Fig. 3). (See Supporting Information for crystallographic data.) Thus, 3-B was assigned to the (1S,2S,3S,4S)-enantiomer.
Fig. 3.
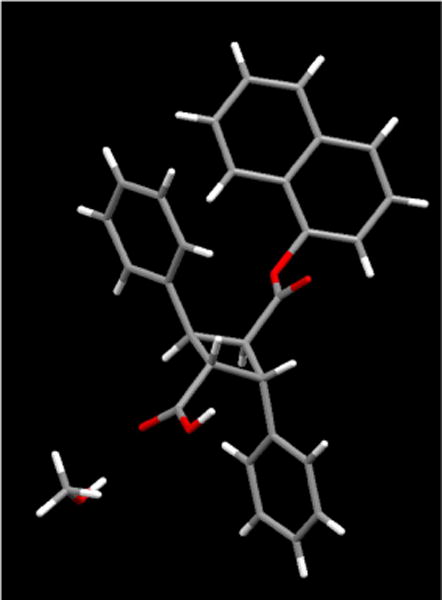
X-ray crystal structure of (R,R,R,R)-3 (3-A)
It should be noted, however, the fluorescence displacement assay of the two enantiomers showed little difference in activity. Ki values of (R,R,R,R)-3 were 0.71 ± 0.08 μM and 0.92 ± 0.22 μM for FABP5 and FABP7, respectively, while Ki values of (S,S,S,S)-3 were 0.79 ± 0.15 μM and 0.45 ± 0.01 μM for FABP5 and FABP7, respectively. The Ki values of two enantiomers for FABP5 are within an error, although there is a recognizable difference in their values for FABP7. Based on these data, we concluded that synthesis of enantiomerically pure analogs would not be necessary, and thus all new analogs in the present work were synthesized as racemic forms.
Currently we have two possible explanations why only (S,S,S,S)-3 binds to both FABP5 and FABP7 in our co-crystal structures [20]: (i) it is possible that the crystallization process may have selectively incorporated (S,S,S,S)-3 complex into the crystal lattice, leaving the R form in solution; (ii) it is also possible that (S,S,S,S)-3 may bind to the portal site more rapidly than to the canonical site, leading to an increased local concentration of (S,S,S,S)-3 for binding to the canonical site. Importantly, our computational analysis showed that both (S,S,S,S)-3 and (R,R.R.R)-3 are geometrically and energetically compatible in the binding pocket of the FABP7-3 complex [20]. Also, the docking energy scores of FABP5-(S,S,S,S)-3 and FABP5-(R,R.R.R)-3 calculated by the AutoDock 4.2 program showed only a very small difference (see Table S7, Supplementary Material). Thus, the Ki values of these two enantiomers of 3 are consistent with the computational analyses.
2.2. Design of 3-analogs as FABP inhibitors
For designing novel FABP inhibitors, AutoDock 4.2 was used to predict the binding affinity and interactions between the inhibitors and FABP proteins. The co-crystal structures of FABP5 and FABP7 with 3 (PDB ID: 5UR9 and 5URA, respectively)[20] were used as docking receptors. The structures of FABP5/3 complex (Fig. 4A) and FABP7/3 complex (Fig. 4B) provide the structural basis for computational construction of new analogs.
Fig. 4.

Structures of FABP5 and FABP7 in complex with (S,S,S,S)-3. (A) (S,S,S,S)-3 bound at the canonical site of human FABP5. (B) (S,S,S,S)-3 bound at the canonical site of human FABP7.
At the canonical-site of the FABP5/3 complex, 3 forms a salt bridge with Arg129, one H-bond with Tyr131, and four H-bonds with Arg109 via ordered water molecules, mimicking the natural substrates of FABP5 (Fig. 4A zoom). Similar interactions are also observed in the canonical-site of the FABP7/3 complex. The carboxyl group of 3 forms a salt bridge with Arg127 and a water-mediated network of hydrogen bonds with Arg127, Tyr129, Arg107, and Thr54 (Fig. 4B zoom). It should be noted that FABP5 is in an open-gate conformation as the S3-S4 and S5-S6 strands are splayed away from the H1-H2 cap as compared to those in the FABP7 structure. The computational analysis of the FABP5/3 and FABP/3 complexes revealed that the space around the two phenyl rings of 3 enables further modifications, with the purpose of generating either new H-bond or hydrophobic interactions between the inhibitor and FABP5 or FABP7. Furthermore, the 1-naphthyl group of 3 is quite exposed in the binding pocket, which indicates that it is worthwhile to replace the 1-naphthyl moiety by other groups for improved potency, selectivity and structural diversity. The binding pose was also considered as an important criterion. Thus, only compounds with good overlap with the binding pose of 3 in FABP5 or FABP7 were selected for chemical synthesis and inhibitory activity evaluation. In addition, the lipophilicity of the designed compounds was also taken into account since the partition coefficient (LogP) provides important information about bioavailability, as well as necessary drug formulation and delivery [21, 22]. Thus, the calculated LogP (cLogP) value for each designed compound was obtained using the ChemDraw 15.0 software [23]. All docking energy scores and cLogP values for the novel 3-analogs, which were designed and selected for chemical synthesis are summarized in Table S7 in the Supporting Information. Fig. 5 shown the overlay of the docked poses of representative FABP5 inhibitors, thus designed, which were selected for synthesis, wherein all those molecules possess critical canonical interactions with Tyr131 and Arg129 (AutoDock 4.2).
Fig. 5.
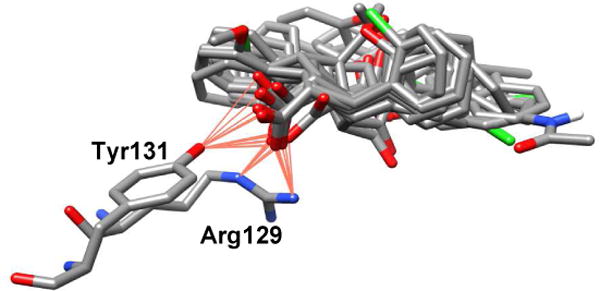
Overlay of representative FABP5 inhibitors designed (Table S7)
2.3. Chemical synthesis
The synthesis of novel 3-analogs of commenced with the preparation of the α-truxillic acid components through [2+2] photodimerization of the corresponding E-cinnamic acids in solid state [24]. A general procedure for the photochemical synthesis of α-truxillic acids and its analogs from commercially available E-cinnamic acids under the irradiation of UV light at 365 nm is shown in Scheme 1.
Scheme 1.
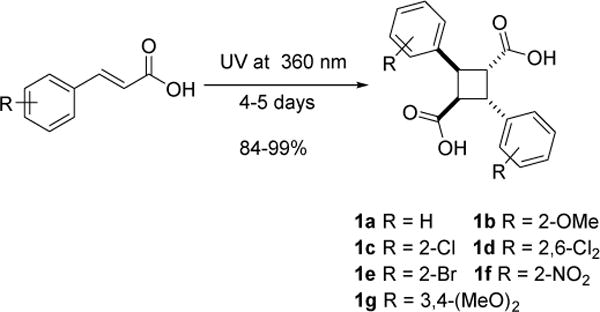
Synthesis of α-truxillic acids 1a~g via solid-state [2+2]
It was reported that direct [2+2] photodimerization of E-ferulic acid was very slow and the yield was very poor [25]. Thus, 4-OTs-E-ferulic acid was synthesized first, following Kibayashi’s procedure and subjected to photodimerization at 360 nm [25]. Under the optimized conditions, the desired product 1h was formed in 88% yield. The tosyl group was removed by NaOH in MeOH to afford 1i for FABP binding assays. In a similar manner, α-truxillic acid analog 1j bearing 4-TBDMSO-phenyl moieties was synthesized from E-4-coumaric acid in 84% yield (Scheme 2).
Scheme 2.
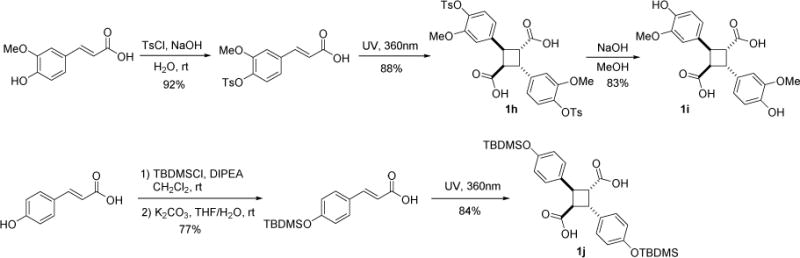
Synthesis of 1h, 1i and 1j
The resulting unsubstituted and substituted α-truxillic acids (1a~f) were reacted with thionyl chloride in the presence of a catalytic amount of dimethylformamide (DMF) under reflux to afford the corresponding diacid dichlorides (2a~f). The diacid dichlorides thus formed were further reacted with a variety of alcohols selected from the computational design to form the corresponding unsubstituted and substituted α-truxillic acid monoesters (3 and 4) as shown in Scheme 3. In some cases, the formation of α-truxillic acid diesters was inevitable (5a~e) (Scheme 3) and thus these diesters were isolated and their FABP binding examined.
Scheme 3.
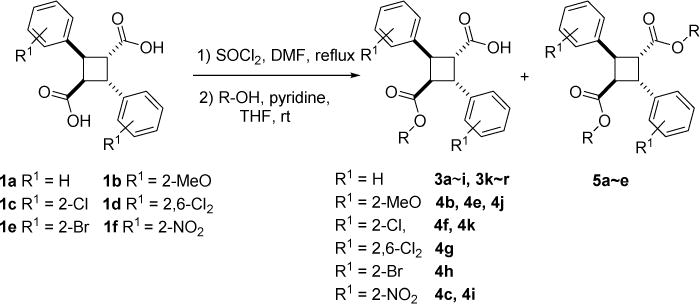
Synthesis of α-truxillic acid monoesters 3x and 4x, as well as diesters 5x (for the R group, see Table S7)
As mentioned above, (-)-incarvillateine and 3 share the α-truxillic acid core structure, we mimicked the 4-hydroxy-3-methoxy substitution pattern of (-)-incarvillateine on the phenyl groups to examine the substituent effects on the FABP binding of the corresponding 3-analog 4a. Compound 4a was synthesized by reacting 1h with 1-naphthol in the presence of EDC·HCl and DMAP, followed by the removal of the tosyl group with Na/anthracene (Scheme 4). In a similar manner, a simpler 3-analog bearing 4-hydroxyphenyl groups, 4d, was also synthesized (Scheme 4).
Scheme 4.
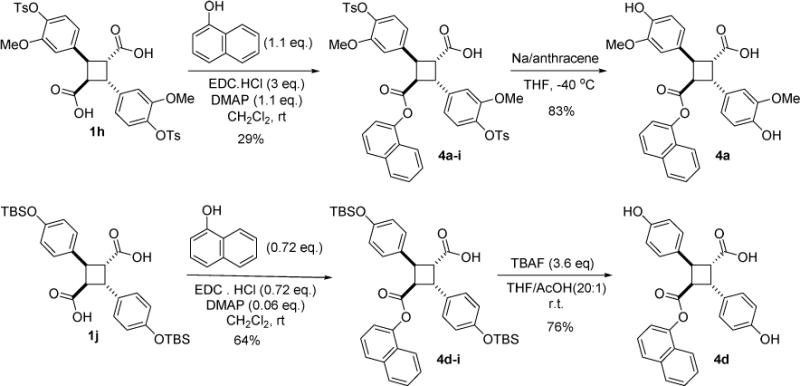
Synthesis of (-)-incarvillateine-mimicked 3-analogs, 4a and 4d
Compound 3j was synthesized in a manner similar to that described for 3a through mono-esterification of 1a, giving 3j-1, followed by deprotection of the TIPS group with tetrabutylammonium fluoride (TBAF), as shown in Scheme 5.
Scheme 5.
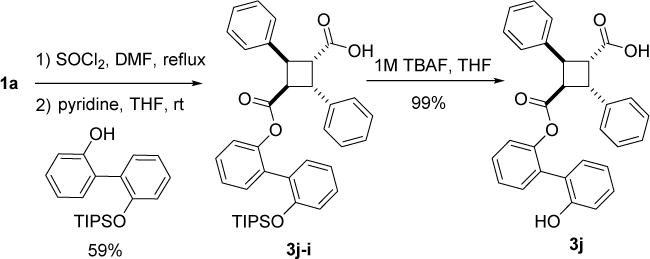
Synthesis of compound 3j.
According to our docking study on α-truxic acid monoesters, the ester moiety was found to be exposed in the FABP binding pockets. This suggests that we may be able to attach a hydrophilic group to the ester moiety without affecting binding affinity. Accordingly, we designed compound 3s, bearing a short PEG chain. The docking score of 3s for FABP5 was rather low, but favorable for FABP7 (see Table S7). Compound 3s was prepared in high yield by reacting 3f bearing an ethynyl group with azido-triethylene glycol under Cu-catalyzed click reaction conditions (Scheme 6).
Scheme 6.
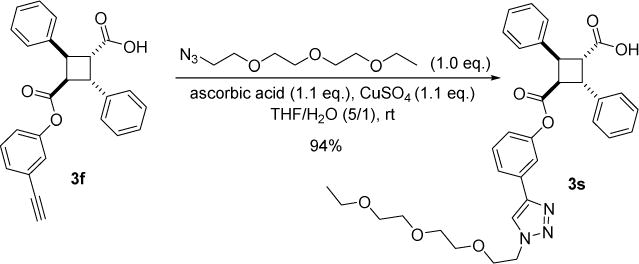
Synthesis of 3s.
Compound 3l bearing a trans-2-phenylcyclohexyl monoester gave considerably better docking scores than 3 for both FABP5 and FABP7 (see Table S7), regardless of stereochemistry. Initially, we synthesized 3l using racemic trans-2-phenylcyclohexanol for mono-esterification with 1a. However, when we used optically pure (1R,2S)-trans-2-phenylcyclohexanol, we were able to isolate single stereoisomer 3l-A by recrystallization of a diastereomer mixture 3l-A/C from EtOAc/hexanes (Scheme 7, equation a). In the same manner, 31-B/D was synthesized from 1a and (1S,2R)-trans-2-phenylcyclohexanol, and 3l-B was isolated as single isomer (Scheme 7, equation b). Thus, 3l-A and 3l-B are enantiomers. Although the absolute configuration of α-truxillic acid moiety of 3l-A or 31-C has not been determined by X-ray crystallography, molecular mechanics calculations using the ChemBio 3D Ultra 14 program [26] indicate that (1R,2R,3R,4R) configuration is favorable over (1S,2S,3S,4S) configuration (ΔE = 1.7 kcal/mol) for 3l-A. Thus, we have tentatively assigned (1R,2R,3R,4R,1’R,2’S) configuration to 3l-A and (1S,2S,3S,4S,1’R,2’S) configuration to 3l-C. Since 3l-B and 3l-D are enantiomers of 3l-A and 3l-C, respectively, the absolute configurations of these compounds should be (1S,2S,3S,4S,1’S,2’R) and (1R,2R,3R,4R,1’S,2’R), as shown in Scheme 7.
Scheme 7.
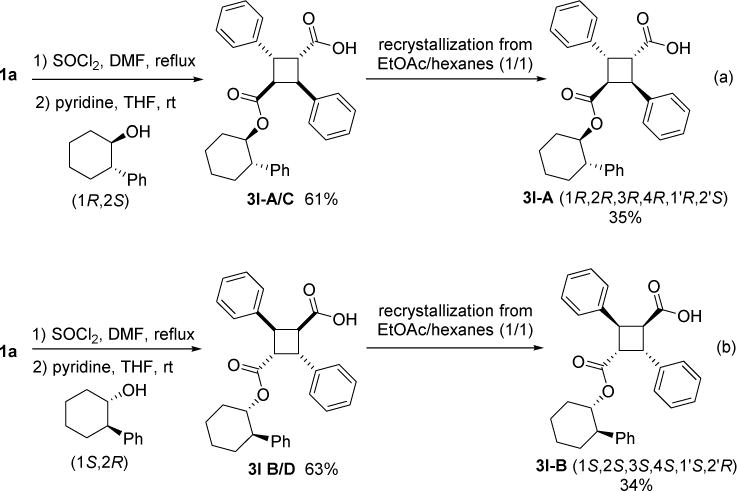
Synthesis of optically pure 3l isomers.
α-Truxillic acid monoamides 6a and 6b were synthesized from 1a through the amide coupling with an aminothiazole and an aminobiphenyl, as shown in Scheme 8. In addition to α-truxillic monoesters and monoamides, γ-truxillic acid mono ester 3o-γ and monoamide 6a-γ were synthesized from γ-truxillic anhydride (Scheme 9). γ-Truxillic anhydride (1a-γ) was readily prepared by reacting 1a with acetic anhydride in the presence of sodium acetate [11]. Then, anhydride 1a-γ was reacted with a naphthol and an aminothiazole to give the corresponding γ-monoester 3o-γ and γ-monoamide 6a-γ, respectively.
Scheme 8.
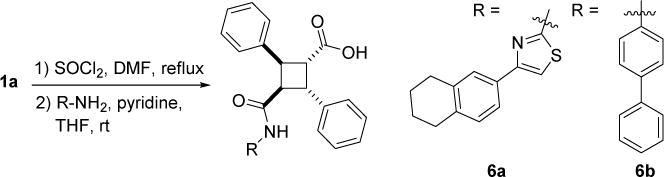
Synthesis of α-truxillic acid monoamides 6a and 6b
Scheme 9.
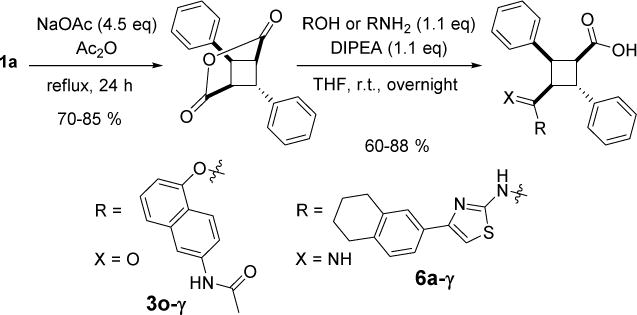
Synthesis of 3o-γ and amide 6a-γ
2.4. FABP-binding assay
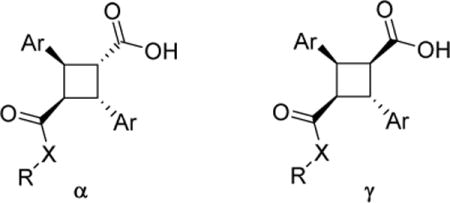
All compounds thus synthesized were evaluated for their binding affinity to human FABP3, FABP5 and FABP7 as previously described (Table 1) [11, 27].
Table 1.
In vitro affinities (Ki, μM) of α-truxillic acid and its congeners
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Compound | Ar | X | R | FABP3 Ki | FABP5 Ki | FABP7 Ki | cLogP |
| 1 | 1a | Ph | O | H | >10 | >10 | >10 | 3.07 |
| 2 | 1g | 3,4-(MeO)2Ph | O | H | >10 | >10 | >10 | 2.38 |
| 3 | 1i | 3-OH-4-(MeO)Ph | O | H | >10 | >10 | >10 | 1.43 |
| 4 | 3 (racemic) | Ph | O | 1-naphthyl | 2.70 ± 0.42 | 0.81 ± 0.09 | 0.45 ± 0.07 | 6.97 |
| 5 | (R,R,R,R)-3 | Ph | O | 1-naphthyl | 3.26 ± 0.70 | 0.78 ± 0.14 | 0.89 ± 0.24 | 6.97 |
| 6 | (S,S,S,S)- 3 | Ph | O | 1-naphthyl | 2.82 ± 0.10 | 0.80 ± 0.14 | 0.66 ± 0.16 | 6.97 |
| 7 | 3a | Ph | O | benzyl | >10 | 3.81 ± 0.53 | 0.53 ± 0.12 | 5.28 |
| 8 | 3b | Ph | O | 4-MeO-benzyl | >10 | 2.15 ± 0.10 | 1.14 ± 0.06 | 5.20 |
| 9 | 3c | Ph | O | 4-F-benzyl | >10 | 2.42 ± 0.18 | 1.65 ± 0.21 | 5.42 |
| 10 | 3d | Ph | O | 4-Br-benzyl | >10 | 1.58 ± 0.16 | 1.25 ± 0.03 | 6.13 |
| 11 | 3e | Ph | O | 2-iodophenyl | 1.18 ± 0.10 | 1.34 ± 0.21 | 0.94 ± 0.34 | 5.77 |
| 12 | 3f | Ph | O | 3-ethynylphenyl | >10 | 0.89 ± 0.15 | 0.78 ± 0.12 | 5.06 |
| 13 | 3g | Ph | O | biphenyl-2-yl | 0.70 ± 0.42 | 0.77 ± 0.08 | 0.35 ± 0.12 | 6.12 |
| 14 | 3h | Ph | O | biphenyl-3-yl | 9.75 ± 0.79 | 0.85 ± 0.22 | 0.74 ± 0.17 | 6.68 |
| 15 | 3i | Ph | O | biphenyl-4-yl | 3.93 ± 0.16 | 2.52 ± 0.36 | 2.27 ± 0.03 | 6.68 |
| 16 | 3j | Ph | O | 2′-HO-biphenyl-2-yl | 3.52 ± 0.53 | 1.59 ± 0.43 | 0.54 ± 0.18 | 5.19 |
| 17 | 3k | Ph | O | 2,4,5-trichlorophenyl | 2.98 ± 0.85 | 0.80 ± 0.11 | 0.54 ± 0.02 | 6.67 |
| 18 | 3l | Ph | O | trans-2-phenylcyclohex-1-yl | 1.08 ± 0.37 | 0.21 ± 0.02 | 0.40 ± 0.03 | 7.17 |
| 19 | 3l-A | Ph | O | (1R,2S)-2-phenylcyclohex-1-yl | 0.83 ± 0.15 | 0.21 ± 0.02 | 0.33 ± 0.05 | 7.17 |
| 20 | 3l-B | Ph | O | (1S,2R)-2-phenylcyclohex-1-yl | 0.88 ± 0.14 | 0.20 ± 0.03 | 0.25 ± 0.12 | 7.17 |
| 21 | 3l-A/C | Ph | O | (1R,2S)-2-phenylcyclohex-1-yl | 0.64 ± 0.16 | 0.18 ± 0.03 | 0.33 ± 0.15 | 7.17 |
| 22 | 3l-B/D | Ph | O | (1S,2R)-2-phenylcyclohex-1-yl | 0.82 ± 0.09 | 0.21 ± 0.02 | 0.15 ± 0.02 | 7.17 |
| 23 | 3m | Ph | O | Indan-2-yl | >10 | 1.57 ± 0.15 | 2.41 ± 0.09 | 5.56 |
| 24 | 3n | Ph | O | CF3CH2- | >10 | >10 | 1.59 ± 0.24 | 3.87 |
| 25 | 3o | Ph | O | 6-acetamidonaphth-1-yl | 2.82 ± 0.18 | 0.97 ± 0.18 | 1.12 ± 0.45 | 5.10 |
| 26 | 3o-γ | Ph | O | 5-ethynylnaphth-1-yl | 3.56 ± 0.58 | 7.08 ± 0.44 | 7.43 ± 1.11 | 6.23 |
| 27 | 3p | Ph | O | 9-fluorenylmethyl | 4.94 ± 0.31 | 3.92 ± 0.75 | 1.03 ± 0.22 | 7.10 |
| 28 | 3q | Ph | O | cyclohexyl | >10 | 2.56 ± 0.16 | 2.70 ± 0.62 | 5.61 |
| 29 | 3r | Ph | O | 3-[1-(3,6,9-trioxadodecanyl)-1,2,3- triazol-4-yl]phenyl | >10 | 2.17 ± 0.32 | 0.50 ± 0.11 | 4.49 |
| 30 | 3s | Ph | O | 6-acetamidonaphth-1-yl | >10 | >10 | 1.06 ± 0.07 | 5.10 |
| 31 | 4a | 3-MeO-4-HO-Ph | O | 1-naphthyl | 1.06 ± 0.19 | >10 | 2.12 ± 0.19 | 4.33 |
| 32 | 4b | 2-MeO-Ph | O | 1-naphthyl | 0.69 ± 0.17 | 0.55 ± 0.05 | 0.67 ± 0.04 | 5.00 |
| 33 | 4c | 2-O2N-Ph | O | 1-naphthyl | >10 | >10 | >10 | 5.29 |
| 34 | 4d | 4-HO-Ph | O | 1-naphthyl | 2.30 ± 0.47 | >10 | 1.06 ± 0.34 | 4.63 |
| 35 | 4e | 2-MeO-Ph | O | (1R,2S)-2-phenylcyclohex-1-yl | 0.40 ± 0.08 | 0.68 ± 0.06 | 0.40 ± 0.03 | 6.20 |
| 36 | 4f | 2-Cl-Ph | O | (1R,2S)-2-phenylcyclohex-1-yl | >10 | 1.70 ± 0.33 | >10 | 8.59 |
| 37 | 4g | 2,6-Cl2−Ph | O | (1R,2S)-2-phenylcyclohex-1-yl | >10 | 1.23 ± 0.18 | 6.32 ± 0.96 | 10.01 |
| 38 | 4h | 2-Br-Ph | O | (1R,2S)-2-phenylcyclohex-1-yl | >10 | 2.76 ± 0.16 | >10 | 8.89 |
| 39 | 4i | 2-O2N-Ph | O | (1R,2S)-2-phenylcyclohex-1-yl | >10 | >10 | >10 | 6.49 |
| 40 | 4j | 2-MeO-Ph | O | 9-fluorenylmethyl | >10 | 1.72 ± 0.12 | >10 | 6.14 |
| 41 | 4k | 2-Cl-Ph | O | 9-fluorenylmethyl | >10 | 0.89 ± 0.05 | 3.54 ± 0.77 | 8.52 |
| 42 | 4l | 2-MeO-Ph | O | quinolin-5-yl | >10 | 3.93±0.51 | >10 | 4.89 |
| 43 | 5a | Ph | O | benzyl | >10 | >10 | >10 | 7.50 |
| 44 | 5b | Ph | O | 4-MeO-benzyl | >10 | >10 | >10 | 7.33 |
| 45 | 5c | Ph | O | 4-F-benzyl | >10 | >10 | >10 | 7.78 |
| 46 | 5d | Ph | O | tetrahydropyran-4-ylmethyl | >10 | >10 | >10 | 3.97 |
| 47 | 5e | Ph | O | biphenyl-3-yl | N.D.** | N.D.** | N.D.** | 10.29 |
| 48 | 6a | Ph | NH | 4-(5,6,7,8-tetrahydronaphth-2-yl)thiazol-2-yl | >10 | >10 | >10 | 7.48 |
| 49 | 6a-γ | Ph | NH | biphenyl-4-yl | >10 | >10 | >10 | 7.48 |
| 50 | 6b | Ph | NH | 4-(5,6,7,8-tetrahydronaphth-2-yl)thiazol-2-yl | >10 | >10 | >10 | 6.46 |
Ki values represent an average ± S.E. of at least three independent experiments. ** No data were obtained due to poor solubility.
2.5. Structure-Activity Relationship (SAR) Analysis
Based on the Ki values obtained, the SAR of a series of new α-truxillic acid monoesters and their congeners can be analyzed. As mentioned above, (S,S,S,S)-3 and (R,R,R,R)-3 are found to have the same binding affinity (Ki) to all three FABPs within the standard deviation. Some insight into this result was discussed in our recent structural biology paper [20]. Our computational and structural biology studies have clearly indicated that the canonical interaction of the carboxylic acid moiety of 3 with Arg129 (FABP5) and Arg127 (FABP7) is essential for its binding to FABPs [11, 20]. Accordingly, we hypothesized that (-)-incarvillateine would be hydrolyzed to the corresponding monoester, which is the putative active form of this drug and binds to FABPs. Thus, a simplified mimic of this putative monoester such as 4a should show a good affinity to FABPs. As Table 1 shows, 4a exhibits fairly good affinity to FABP3 (Ki 1.06 μM) and FABP7 (Ki 2.12 μM), but not to FABP5 (Ki >10 μM) (entry 31). The results suggest that the 4-hydroxy-3-methoxyphenyl group in 4a somewhat compromises affinity as compared to the simple phenyl group in 3, probably because of its bulkiness. Since the 4-hydroxyl group could serve as a hydrogen bond donor and the methoxy group at the 3-position might cause a steric clash, compound 4d, bearing just 4-hydroxylphenyl group, is an interesting compound to examine. However, 4d shows comparable affinity and selectivity to 4a for the three FABPs (entry 34), which indicates that the 4-hydroxyl group does not bring in additional favorable interactions and rather compromises binding to FABPs, especially to FABP5. These results indicate that the substituents at the meta- and para-positions of α-truxillic acid core may cause unfavorable van der Waals interactions with amino acid residues in the binding sites, especially for FABP5.
Since α-truxillic acid, its diesters and their congeners were reported to show antinociceptive activities [14, 28, 29], we examined the affinity of α-truxillic acid (1a), its 3,4-dimethoxyphenyl derivative (1g) and 4-hydroxyphenyl derivative (1i) to the three FABPs. However, none of these dicarboxylic acids show appreciable affinity to FABPs (Ki >10 μM) (entries 1-3). Also, diesters 5a~d, obtained as by-products in the synthesis of the corresponding α-truxillic acid monoesters, do not show appreciable affinity to FABPs (Ki >10 μM) (entries 43- 46). These results unambiguously confirm that the α-truxillic acid monoester scaffold is essential for strong binding to FABPs.
Among the new unsubstituted and substituted α-truxillic acid derivatives, compound 3l shows substantially higher affinity to FABP5 than the parent compound 3 by a factor of 4 (entry 18). The Ki values of 3l for FABP5 and FABP7 are 0.21 μM and 0.40 μM, respectively. Besides 3l, 8 compounds (3f, 3g, 3h, 3k, 3o, 4b, 4e 4k) exhibit better or similar affinity towards FABP5 (Ki 0.55-0.97 μM) as compared to 3, while 6 compounds (3a, 3g, 3j, 3k, 4b and 4e) show high affinities to FABP7 (Ki 0.35-0.67 μM). For the stereoisomers of 3l (entries 19-22), all isomers exhibit the same Ki value within standard deviation for FABP5 (0.20 μM) and FABP3 (0.80 μM). For FABP7, 3l-B/D showed a little higher affinity that 3l-A, 3l-B and 3l-A/C (0.16 μM vs. 0.30 μM), suggesting that 3l-D (1R,2R,3R,4R,1’S,2’R) may have better affinity than other isomers.
The selective affinity to FABP5 would be beneficial since this FABP possesses significant potential as a drug target [30–32]. FABP7 does not seem to have particular clinical significance at present and its expression in the adult brain is substantially lower than that of FABP5 [10], the selectivity to this FABP may not be of medicinal importance in terms of pain management. Nevertheless, selective FABP7 inhibitors would be useful as research tools. Since FABP3 is primarily expressed in the heart and its inhibition might cause cardiotoxicity [12], low affinity to FABP3 appears desirable.
The most selective FABP5 inhibitors appear to be 4f (Ki 1.70 μM) and 4j (Ki 1.70 μM), which practically do not bind to both FABP3 (Ki>10 μM) and FABP7 (Ki >10 μM) (entries 36 and 40). Another FABP inhibitor 4k (Ki 0.89 μM) exhibits higher affinity to FABP5 than 4f and 4k and does not bind to FABP3 (Ki >10 μM), while binds modestly to FABP7 (Ki 3.54 μM) (entry 41). Compounds 4j and 4k bear 9-fluorenylmethyl as the ester moiety and 2-methoxyphenyl and 2-chlorophenyl groups in the α-truxillic acid core, respectively. Another compound, 3q (entry 28) also bears the 9-fluorenylmethyl ester moiety and the Ki value for FABP3 is >10 μM. Thus, it is deduced that this rather bulky ester group is blocking the binding to FABP3. The ortho substitution on the phenyl group also exerts substantial effects on the FABP selectivity. Thus, the 2-methoxy group in 4j blocks the binding to FABP7 (Ki >10 μM), while hydrogen (3q, Ki 2.70 μM) and 2-chloro group (4k, Ki 3.54 μM) are moderately tolerated. Then, the 2-chlorophenyl group is best tolerated among the three for FABP5 binding, realizing very good affinity to FABP5. However, the 2-chlorophenyl group is not always well tolerated. Thus, when the ester group is (1R,2S)-2-phenylcyclohex-1-yl (4f), 2-chlorophneyl group reduces the affinity to FABP5 (Ki 1.70 μM) (compare entry 18 and entry 36) although it is clear that this group blocks the binding to FABP3 and FABP7 and brings about selectivity towards FABP5.
When the ester moieties are a good fit to FABP binding sites, the 2-methoxyphenyl group increases the affinity to FABP3. Thus, 4b (entry 32), an analog of 3, and 4e (entry 35), an analog of 3l, bearing 2-methoxyphenyl groups in place of phenyl groups at the α-truxillic acid core, exhibit high affinity to all three FABPs (Ki 0.40-0.69 μM), especially to FABP3 (Ki: 4b, 0.69 μM; 4e, 0.40 μM), implying a possible introduction of additional hydrogen bonding.
Other ortho substituted phenyl groups, i.e., 2,6-dichlorophenyl (4g), 2-bromophenyl (4h) and 2-nitrophenyl (4i) groups, are fairly tolerated by FABP5, but none of the compounds bearing those groups show Ki values less than 1 μM (entries 37~39). These groups clearly block the binding to FABP3. 2-Bromophenyl and 2-nitrophenyl groups also block the binding to FABP7. Interestingly, a quite bulky 2,6-dichlorophenyl group (4g) is moderately tolerated by FABP7.
Two compounds, 3f and 3h, exhibit high selectivity to FABP5 (Ki 0.89 and 0.85 μM, respectively) and FABP7 (Ki 0.78 and 0.74 μM, respectively), but practically do not bind to FABP3 (Ki >10 and 9.75 μM, respectively) (entries 12 and 14). Compounds 3f and 3h bear 3-ethynylphenyl and biphenyl-3-yl groups, respectively, as their ester moieties and the substituents are placed at the meta position of the phenyl moiety in both cases. The substitution pattern, i.e., ortho, meta and para, on the phenyl group is found to be critical to the affinity and selectivity to FABPs. Thus, 3g, 3h and 3i all bear biphenyl groups as their ester moieties, wherein 3g has a biphenyl-2-yl (ortho) group, 3h a biphenyl-3-yl (meta) group and 3i a biphenyl-4-yl group (para). Then, 3g exhibits high affinity to all three FABPs (Ki for FABP3, 5 and 7: 0.70, 0.77 and 0.35 μM, respectively) (entry 13). It should be noted that 3g is a good structural mimic of 3l. Apparently, the para-substitution is not well tolerated as evident from the Ki values of 3i (for FABP3, 5 and 7: 3.93, 2.52 and 2.27 μM, respectively) and thus the affinity to all three FABPs are compromised by the substitution at this position (entry 15).
Previously, we reported that γ-isomer of 3, i.e., SB-FI-49 (3-γ) [11], showed a slightly better Ki than that of 3, although the solubility of 3-γ was very poor, which prevented us from selecting it as a lead. In the present study, we examined a pair of 3-analogs, 3o and 3o-γ, which bear the 6-acetamidonaphth-1-yl groups as the ester moieties. Although 3o keeps comparable affinities to those of 3 for the three FABPs, 3o-γ clearly shows substantially weaker affinities, especially to FABP5 and FABP7 (entry 26). Thus, the α-isomer series appears to be better than the γ-isomer series so far, although there might be exceptions.
We also reported previously that the amide analog of 3, i.e., SB-FI-60 (6) [11], showed very good selectivity to FABP7 with high affinity (Ki, 0.3 μM), fairly good affinity to FABP5 (Ki 1.6 μM), but did not bind to FABP3 (Ki >10 μM) [6]. In the present study, we examined 4-(5,6,7,8-tetrahydronaphth-2-yl)thiazol-2-yl amide with α- and γ-truxillic acid cores, 6a and 6a-γ, as well as biphenyl-4-yl amide, 6b. However, none of them exhibit appreciable affinity to FABPs (i.e., Ki >10 μM) (entries 48~50). The affinities of 6b were much weaker than the correspond ester 3i. It appears that these rather long amide moieties do not fit to the binding sites of FABPs. Also, the inherent rigidity of amide as compared to ester may be the reason for the difference. Although an amide could add hydrogen bonding donor capability for additional interactions in the binding site, it does not seem to provide beneficial effects in these systems.
2.6. Computational screening and analysis of SAR
In general, the computational analysis for selecting promising compounds for chemical synthesis and biological assays among the truxillic acid congeners designed was found to be useful in the majority of cases, but with clear exceptions. The most notable failures are monoamide 6a, 2-nitrophenyl analogs 4c and 4i, wherein docking scores are quite favorable (for FABP5: 6a, −9.60 kcal/mol; 4c, −9.37 kcal/mol; 4i: −10.01 kcal/mol), but Ki values are >10 μM.
In spite of natural limitations in the accuracy of computational screening based only on docking scores, computational SAR analysis for representative cases has provided critical insight into the affinity and selectivity of selected FABP inhibitors as described below. [Note: We used the co-crystal structures of FABP5/3 (PDB ID: 5UR9)[20] and FABP7/3 (PDB ID: 5URA)[20] for these docking analyses (See Experimental Section). For FABP3, however, no co-crystal structure with 3 was available. Thus, we used the high-resolution crystal structure of FABP3 apoprotein (PDB ID: 6AQ1) and the best docking pose of 3 with canonical interaction as the standard (see Supplementary Data).
The first analysis is for 4-hydroxyphenyl analog 4d. Although 4d differs from 3 only with 4-hydroxyl groups at the phenyl moieities of the α-truxillic acid core, this small change exerted detrimental effects on the affinity of 4d to FABP5, although 4d keeps moderate affinities to FABP3 and FABP7. Docking study of 4d using the local energy minimization algorithm of AutoDock Vina to find a binding pose starting from the pose of 3 in the co-crystal structure [20], 4d was found to be 1.29 kcal/mol less favorable than 3 (Fig. 6A). The interaction map analysis using AutoDock Vina Tool revealed that one of the 4-hydroxyphenyl group of 4d causes a steric clash with the hydrophobic alkyl moiety of Lys61 (Fig. 6B), which did not exist in the case of 3. When 4d is docked to FABP5 from the FABP5/3 co-crystal structure [20], the binding pose makes a substantial change, wherein the critical canonical interaction of the carboxylate group with Arg129 and Tyr131 is substantially weakened, and also the 1-naphthyl group occupies a very different space than that of 3 (Fig. 6C) to avoid the steric clash with Lys61. Accordingly, the computational analysis of 4d binding to FABP5 provides clear explanation for the poor binding of 4d to FABP5 observed. Also, it has turned out that the local energy minimization approach and docking approach give complementary analyses, leading to the same conclusion.
Fig. 6.
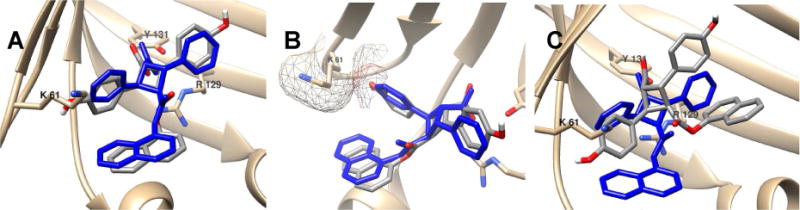
Docking analysis of 4d in FABP5. (A) Binding pose of 4d (gray) through the local search with AutoDock 4.2 in comparison with 3 (blue). (B) Steric clash of 4-hydroxyphenyl group with Lys61 visualized by AutoDock Tool. (C) Docking pose of 4d (gray) by AutoDock 4.2 in comparison with 3. The binding pose of 3 is from the co-crystal structure.13
As described above, 4k is found to be one of the most FABP5-selective inhibitors (4f and 4j are even more selective, but less potent than 4k) among the 3-analogs examined in the present study. The computational analysis of 4k-binding to FABPs has provided clear rationale for the observed selectivity. The docking poses of 4k in FABP3, FABP5 and FABP7 using AutoDock Vina are shown in Fig. 7. In the docking pose in FABP3 (Fig. 7A), the critical canonical interaction of the carboxylate group is substantially weakened and the location of the 9-fluorenylmethyl group is also very different from the 1-napthyl group of 3. Thus, this analysis indicates very weak affinity of 4k to FABP3. In fact, the observed affinity was >10 μM. In the case of FABP5 (Fig. 7B), however, the canonical interaction is almost completely kept in contrast and an excellent overlay with 3 is observed, which indicates very strong affinity to FABP5. Indeed, the observed affinity of 4k to FABP5 was 0.89 μM. For FABP7, 4k shows moderate overlay with 3, as well as reasonable preservation of the canonical interaction, suggesting moderate affinity to FABP7. As anticipated, the observed affinity was 3.54 μM.
Fig. 7.
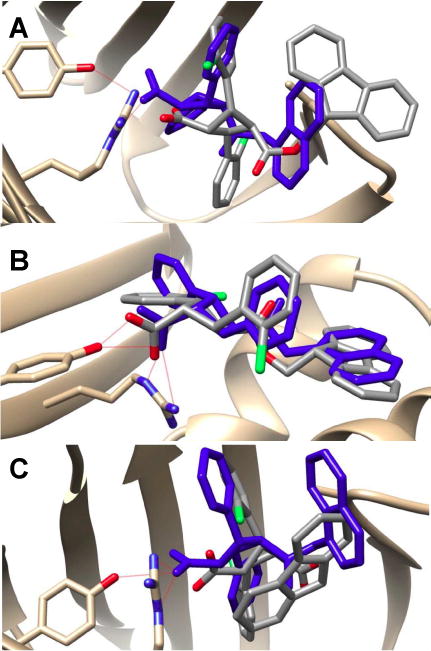
Docking poses of 4k (gray) in FABPs as compared to those of 3 (blue). (A) FABP3, (B) FABP5, (C) FABP7. The binding poses of 3 in FABP5 and FABP7 are from co-crystal structures,13 while that in FABP3 is predicted by the AutoDock 4.2.
Finally, the analysis of inhibitor 4b shows that it binds equally well to all three FABPs. As Fig. 8 illustrates, 4b shows very good overlay with 3 in FABP3 (Fig. 8A), FABP5 (Fig. 8B) and FABP7 (Fig. 8C), suggesting high affinities of 4b to all three FABPs. In fact, 4b exhibited high affinities to FABPs (Ki 0.55~0.69 μM) and thus not selective to a particular FABP.
Fig. 8.
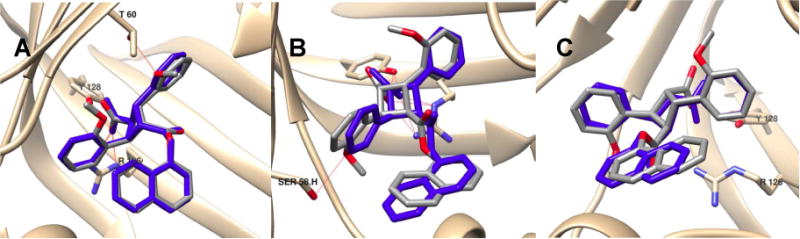
Docking poses of 4b in FABPs in comparison with 3 (blue). (A) FABP3; (B) FABP5; (C) FABP7. The binding poses of 3 in FABP5 and FABP7 are from co-crystal structures,13 while that in FABP3 is predicted by the AutoDock 4.2.
Consequently, those results demonstrate that the structure-based computational analysis and design are reasonably reliable and can be employed for further optimization of lead compounds that were identified in the present SAR study.
2.7. In Vivo Efficacy Evaluations
A subset of the 3-analogs were administered to mice (40 mg/kg, i.p.) to probe for antinociceptive activity using the complete Freund’s adjuvant (CFA) model of inflammatory pain and measurement of thermal hyperalgesia via the Hargreaves plantar apparatus [33]. In agreement with previous reports, 3 displayed potent antinociceptive effects in this model (Fig. 9A).[6] Administration of 3f and 3l-Aresulted in antinociceptive effects (Fig. 9A). Administration of 4a and 4d (which lack affinity for FABP5) was without effect (Fig. 9A). Compound 1a also did not produce antinociceptive effects, consistent with its low affinity for FABPs, confirming that the α-truxillic acid scaffold lacks efficacy in this pain model.
Fig. 9.
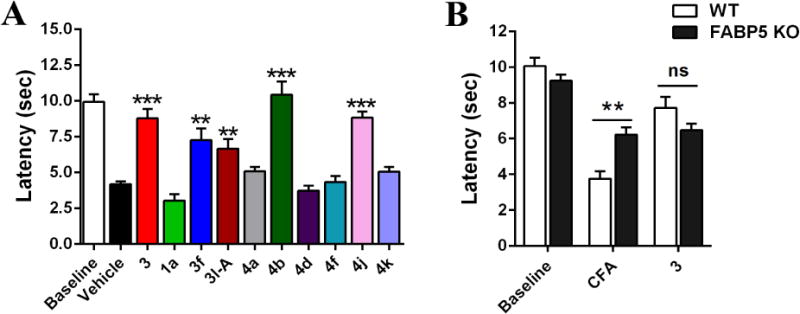
In Vivo Efficacy of Select Compounds. (A) Hyperalgesia was induced by intraplantar injection of CFA and thermal latencies were measured in mice following inhibitor administration (20 mg/kg, i.p.). n = 6-9 per group. **p < 0.01, ***p < 0.001 versus vehicle controls by one-way ANOVA with Dunnett post-hoc test. (B) Thermal latencies of wild-type (WT) and FABP5 KO mice following administration of 3 (40 mg/kg, i.p.). n = 6. ns indicates no significant difference, **p < 0.01 by two-ANOVA with Bonferonni post-hoc test. All data are presented as mean ± standard error.
These observations are in agreement with those reported in our previous work, wherein antinociceptive effects following pharmacological FABP inhibition are correlated with inhibitor affinity for FABP5, while FABP3 or FABP7 inhibition are not predictors of in vivo efficacy [6]. Furthermore, mice bearing a genetic knockout for FABP5 (FABP5-KO) display an analgesic phenotype and no further antinociceptive effects were observed in these animals following administration of 3 (Fig. 9B).
Recent work indicates that FABP4 can modulate inflammation [34] and its inhibition could contribute to the antinociceptive effects of our FABP inhibitors. However, this is unlikely because FABP4 KO mice display normal pain responses [35] and inhibitors that produce antinociceptive effects (e.g., 3f, 4b and 4j) do not appreciably inhibit FABP4 (see Fig. S5). Taken together these data lend further support to the model that FABP5 is the principal cytosolic AEA transporter and the relevant FABP isoform for the development of inhibitors to treat pain and inflammation. Select compounds, exhibiting high affinity and/or selectivity to FABP5 in vitro, were examined for their efficacy in vivo in the same mouse model as that described above. As anticipated, 4b (FABP5 Ki 0.55 μM) exhibited strong antinociceptive effects, but 4b is non-selective to FABP5, i.e., 4b binds to FABPs 3,5, and 7 non-discriminatively. The most FABP5-selective compound 4j (FABP5 Ki 1.72 μM; FABPs 3 and 7 Ki >10 μM) showed potent antinociceptive effects as observed with 3, despite a weaker affinity to FABP5 than that of 3 (FABP5 Ki 0.81 μM). Rather surprisingly, however, 4k (FABP5 Ki 0.89 μM), showing high selectivity to FABP5 (FABP3 >10 μM; FABP7 3.54 μM), did not exhibit good efficacy in vivo. This result may indicate that exceedingly high lipophilicity causes unfavorable biodistribution problems in vivo even though the compounds exhibit high affinity/potency in vitro. The estimated cLogP values for 4b and 4j are 5.00 and 6.14, respectively, while those for 4f and 4k are 8.59 and 8.52, respectively, which are exceedingly high due to two 2-chlorophenyl moieties in these molecules. A similar observation was made for the in vivo efficacy of 3l (FABP5 Ki 0.21 μM; cLogP 7.17), wherein its in vivo efficacy appears to be compromised by its high hydrophobicity despite much stronger affinity to FABP5 in vitro.
Consequently, based on the in vivo efficacy, we have selected 4b and 4j as new lead compounds for further optimization. In particular, 4j is a highly promising lead compound since it has almost exclusive selectivity to FABP5 with no appreciable affinity to FABP3 and FABP7.
3. Conclusions
In conclusion, we designed and synthesized a library of new FABP inhibitors based on the α-truxillic acid monoester scaffold, which are analogs of previously identified lead compound, 3 (SB-FI-26), as well as some diesters, monoamides, γ-isomers and substituted α-truxillic acids. The SAR study clearly shows that the α-truxillic acid monoester scaffold is essential for high affinity binding to FABPs, because the canonical interaction of the carboxylate moiety with Arg/Tyr residues is crucial for the inhibitor binding. The critical importance of this canonical interaction is strongly supported by the in-depth computational analysis of the binding poses of new FABP inhibitors for their affinity and selectivity to a particular FABP isoform. Among these new FABP inhibitors, compound 3l was found to possess 4 times higher affinity to FABP5 than 3. However, 3l also exhibits high affinity to FABP3 and FABP7. In this category, compounds 3g, 4b and 4e are highly potent as well. Compounds 4f and 4j are found to be exclusively selective to FABP5, albeit their Ki values are not submicromolar (1.70 and 1.72 μM, respectively).
Compound 4k was found to be a highly selective inhibitor to FABP5, which practically does not bind to FABP3, and possesses only modest affinity to FABP7. Thus, 4f, 4j and 4k appear to be promising FABP5-selective compounds. As noted above, the natural product incarvillateine produces antinociceptive effects in rodents. Our observation that 4a, a structural mimic of incarvillateine, does not bind FABPs or produce antinociceptive effects indicates that FABP inhibition may not account for the in vivo efficacy of incarvillateine, unless it acts as a prodrug that is activated by metabolism. Not surprisingly, in vivo efficacy evaluation of select compounds revealed that the high affinity to FABP5 in vitro does not necessarily be translated to high efficacy in vivo. It has turned out that exceedingly high hydrophobicity of these compounds significantly affects their in vivo efficacy. Thus, compounds with moderate hydrophobicity, such as 4b and 4j (ClogP 5.00 and 6.14, respectively) exhibited highly promising in vivo efficacy, while those with exceedingly high hydrophobility, such as 4f and 4k (ClogP 8.59 and 8.52, respectively) only showed low efficacy in spite of their high affinity to FABP5 in vitro. Based on the in vivo efficacy study, we were able to select 4b and 4j as new and highly promising lead compounds. Although the design of new analogs based on simple docking has limitations, the computational analysis has provided insight into the selectivity of certain compounds to the different FABP isoforms.
4. Experimental Section
4.1. General Methods
Melting points were measured on a Thomas Hoover Capillary melting point apparatus and are uncorrected. NMR spectra were recorded on a Bruker Ascend 700 spectrometer operating at 700 MHz for 1H and 175 MHz for 13C, a Bruker 500 Advance spectrometer operating at 500 MHz and 125 MHz for 1H and 13C, respectively, or a Bruker 400 Nanobay spectrometer operating at 400 MHz, 100 MHz, and 376 MHz for 1H, 13C, and 19F, respectively. Chemical shifts were referenced to the residual proton and carbon-13 peaks of solvents used for 1H and 13C NMRs, respectively (1H: CDCl3, δ 7.26; (CD3)2SO, δ 2.50; CD3OD, δ 3.31; CD3CN, δ 1.94; acetone-d6; δ 2.05; 13C: CDCl3, δ 77.16; DMSO-d6, δ 39.52; CD3OD, δ 49.00); acetone-d6 δ 29.84, 206.26. Signals are listed in ppm, and multiplicity identified as s = singlet, br = broad, d = doublet, t = triplet, q = quartet, m = multiplet; J-coupling constants in Hz, and integration. High resolution mass spectrometry analysis was carried out on an Agilent LC-UV-TOF mass spectrometer at the Institute of Chemical Biology and Drug Discovery, Stony Brook, NY or on a Waters Q-TOF Ultima ESI mass spectrometer at the Mass Spectrometry Laboratory, University of Illinois at Urbana− Champaign, Urbana, IL. Purity of synthesized compounds was determined by a Shimadzu LC-2010A HT series HPLC assembly or Agilent 1100 series HPLC assembly. Purities of new compounds were all >95%. Three analytical conditions were used for synthesized compounds (25°C, 220 and 254 nm): (1) Chiralcel ODH, 250 × 4.6 mm column, isopropanol (65%) and hexanes (35%), flow rate: 1 mL/min, isocratic, at 220 and 254 nm; (2) Kinetex PFP, 2.6 μm, 4.6 mm × 100 mm column, methanol and water, flow rate: 0.3 mL/min, t = 0-30 min, gradient of 40-95% MeOH; (3): Kinetex PFP, 2.6 μm, 4.6 mm × 100 mm column, solvent A of water/acetonitrile 95:5 (25 mM ammonium acetate, pH 6.5), solvent B of water/acetonitrile, 5:95 (25mM ammonium acetate, pH 6.5), flow rate: 1.0 mL/min, t = 0-15 min, gradient of 20-95% solvent B.
Single crystal X-ray crystallographic analysis was carried out on an Oxford Gemini A Enhance diffractometer. The 2+2 photo-catalyzed cyclization was carried on Electro-Cure 4001 from Electro-Lite Corporation.
4.2. Materials
All air- and moisture-insensitive reactions were carried out under an ambient atmosphere, magnetically stirred, and monitored by thin layer chromatography (TLC) using Agela Technologies TLC plates pre-coated with 250 μm thickness silica gel 60 F254 plates and visualized by fluorescence quenching under UV light. Flash chromatography was performed on SiliaFlash® Silica Gel 40-63μm 60Å particle size using a forced flow of eluent at 0.3-0.5 bar pressure. All air- and moisture-sensitive manipulations were performed using oven-dried glassware using the standard Schlenk and glovebox techniques under nitrogen. Diethyl ether and THF were distilled from deep purple sodium benzophenone ketyl. Dichloromethane, chloroform and acetonitrile were dried over CaH2 and distilled. Dichloromethane was degassed via three freeze-pump-thaw cycles. All other chemicals were used as received. All deuterated solvents were purchased from Cambridge Isotope Laboratories. Compound 3 (racemic) was synthesized, following the procedure previously reported by us [11]. α-2, 4-Di(3-methoxy-4-tosyloxyphenyl)-cyclobutane-1,3-dicarboxylic acid (1h) was prepared by the literature method [25]. Compound 5f was prepared by the method reported previously from our laboratories [36]. γ-Truxillic anhydride was prepared by the literature method [11]. 12-NBD-stearate [12-N-methyl-(7-nitrobenz-2-oxa-1,3-diazo)aminostearic acid] was purchased from Avanti Polar Lipids (Alabaster, AL, USA). 11-[5-(Dimethylamino)-1-naphthalenesulfonyl-amino]undecanoic acid (DAUDA) was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). 1-Anilinonaphthalene-8-sulfonic Acid (ANS) were purchased from Cayman Chemical Company (Ann Arbor, MI, USA).
4.3. Interference Compound Analysis
All 57 compounds in the present work were analyzed for their potential as pan assay interference compounds (PAINS) using the publicly available ZINC15 web tool.[37] None of the compounds were identified to contain PAINS patterns, nor were there any alerts for predicted high risk of aggregation. Additionally, dynamic light scattering (DLS) was used to experimentally probe two of the compounds in the series, 3 or 3l (20 μM), and there was no evidence of colloidal aggregation. Furthermore, the lack of efficacy of SB-FI-26 in FABP5-KO mice shown in Fig. 8, and the recent x-ray structures of 3 with FABP5 and FABP7 provides strong evidence that the activity of this compound (and related analogs) are a result of specificity for the target [20].
4.4. Chemistry
4.4.1. Optical resolution of racemic 3
To a solution of α-truxillic acid mono-1-naphthyl ester (3) (100 mg, 0.24 mmoles) in acetone (3 mL) was added (S)-phenylalaninol (36 mg, 0.24 mmoles) and heated until the reaction mixture was clear and homogeneous. The solution was allowed to cool to room temperature and left on a benchtop overnight. After crystal formation, the mother liquor was removed, and the crystals were washed with cold methanol (3 mL). Hydrochloric acid (12 N) (1.0 mL) was added to the crystals and extracted thrice with ethyl acetate (5 mL X 3). The organic layer was collected, dried over magnesium sulfate, and analyzed by chiral HPLC on a Chiralcel ODH column using 65% isopropanol and 35% hexanes as the eluent.
After the first resolution in methanol, the resulting 3 showed an optical purity of 89% ee (er = 94.5:5.5). The sample was resolved again using the same procedure, which gave 3-A with 99% ee (36 mg, 36% yield). The absolute configuration of this enantiomer 3-A was unambiguously determined to be (R,R,R,R) by single crystal X-ray crystallographic analysis (vide infra).
In the same manner, but using (R)-phenylalaninol instead of (S)-phenylalaninol, the other enantiomer, 3-B, i.e., (S,S,S,S)-3, was obtained with 95% ee (65 mg, 65% yield). When acetone was used in place of methanol in the same procedure, 3-B was obtained with 93% ee after the first resolution. Thus, acetone appears to be a better solvent for the optical resolution of 3.
4.4.2. Single Crystal X-ray diffraction analysis of 3-A (R,R,R,R)
Crystals of 3-A were selected and mounted on glass fibers using epoxy adhesive. Each crystal was centered, and the X-ray intensity data were measured on an Oxford Gemini A Enhance diffractometer by using graphite-monochromated Cu radiation. The data was collected using the Crysalis Pro 38.41 software [38], Wingx 2014.1 [39], Olex2 1.2 [40], and SHELX 2013 [41]. The crystal data and structure refinement parameters are summarized in Table S1 (Supporting Information). Fractional atomic coordinates and equivalent isotropic displacement parameters (Table S2), anisotropic displacement parameters (Table S3), bond lengths (Table S4), bond angles (Table S5), and hydrogen atom coordinates and isotropic displacement parameters (Table S6) are also shown in the Supporting Information. A ball and stick structure of 3-A, i.e., (R,R,R,R)-3, is shown in Fig. 2 and the ORTEP structure is shown in Fig. S1 (Supporting Information). The CIF for 3-A is also available as Supporting Information.
4.4.3. Chemical Synthesis
Note: For the nomenclatures of α- and γ-truxillic acid, their derivatives and their monoesters, IUPAC nomenclatures are used, but with the sign “α-“ or “γ-“ designations, wherein “α-“ stands for (1S*,2S*,3S*,4S*) or (1R*,2R*,3R*,4R*) stereochemistry, i.e., racemic with all four carbon centers bearing the same S or R configuration, while “γ-“ stands for (1S*,2R*,3S*,4S*) or (1R*,2S*,3R*,4R*) stereochemistry, i.e., racemic with only C2 carbon center bearing opposite configuration.
4.4.3.1. α-2,4-Diphenylcyclobutane-1,3-dicarboxylic acid (1a)
(E)-Cinnamic acid (1.0 g, 6.7 mmol) was placed in a pyrex dish and exposed to light at 360 nm and an intensity of 280 nW/cm2 for 5 days with periodic shaking. This process was performed in the solid state and monitored by 1H NMR. After completion of the photoreaction, the white solid was washed with diethyl ether (20 mL) to give α-truxillic acid (0.99 g, 99 % yield) as a white solid. m.p. >230°C; 1H NMR (300 MHz, acetone-d6) δ 3.99 (dd, J = 10.0, 7.6 Hz, 2 H), 4.44 (dd, J = 10.0, 7.6 Hz, 2 H), 7.23 (t, J = 7.1 Hz, 2H), 7.33 (t, J = 7.4 Hz, 4H), 7.42 (d, J = 7.4 Hz, 4H), 10.62 (s, 1H); FIA (negative mode), calcd C18H16O4 296.1, found (M-H)- 295.0. The analytical data was consistent with literature values [11].
In the same manner, other mono ester 1b to 1i were synthesized and characterized, starting from corresponding substituted (E)-cinnamic acids.
4.4.3.2. α-2,4-Di(2-methoxylphenyl)cyclobutane-1,3-dicarboxylic acid (1b)
White solid; 92% yield; m.p. >230°C; 1H NMR (700 MHz, acetone-d6) δ 3.74 - 3.79 (m, 8 H), 4.42 (dd, J = 10.1, 7.6 Hz, 2 H), 7.20 - 7.24 (m, 2 H), 7.25 - 7.28 (m, 2 H), 11.90 (s, 2 H). 13C NMR (175 MHz, acetone-d6) δ 36.0, 44.6, 55.4, 110.5, 120.1, 127.1, 127.5, 127.9, 157.2, 173.4; HRMS (ESI) m/z: calcd for C20H O + 20 6H, 357.1338, found, 357.1339 (Δ = 0.28 ppm).
4.4.3.3. α-2,4-Di(2-chlorophenyl)cyclobutane-1,3-dicarboxylic acid (1c)
White solid; 91 % yield; m.p. 212-213°C;1H NMR (700 MHz, acetone-d6) δ 3.89 (d, J = 6.3 Hz, 2 H), 4.63 (d, J = 6.3 Hz, 2 H), 7.08 (td, J = 7.6, 1.3 Hz, 2 H), 7.12 (t, J = 7.0 Hz, 2 H), 7.19 - 7.24 (m, 2 H), 7.33 (d, J = 7.6 Hz, 2 H), 12.58 (s, 2 H); 13C NMR (125 MHz, acetone-d6) δ 41.85, 42.64, 126.48, 128.01, 128.68, 129.11, 134.19, 136.53, 172.74; HRMS (ESI) m/z: calcd for C18H14O4Cl2H+, 365.0347, found, 365.0346 (Δ = 0.27 ppm).
4.4.3.4. α-2,4-Di(2,6-dichlorophenyl)cyclobutane-1,3-dicarboxylic acid (1d)
White solid; 93 % yield; m.p. decomposed at 210°C; 1H NMR (300 MHz, acetone-d6) δ 4.80 (dd, J = 4.6, 2.7 Hz, 2 H), 5.31 (dd, J = 4.6, 2.7 Hz, 2 H), 7.23 (m, J = 9.0, 2.2 Hz, 6 H), 11.18 (s, 2H); 13C NMR (126 MHz, acetone-d6) δ 42.40, 42.74, 128.87, 129.31, 134.33, 135.52, 172.83; HRMS (ESI) m/z: calcd for C18H12Cl4O4H+, 432.9568, found, 432.9562 (Δ = 1.4 ppm).
4.4.3.5. α-2,4-Di(2-bromophenyl)cyclobutane-1,3-dicarboxylic acid (1e)
White solid; 91% yield; m.p. 218-220°C; 1H NMR (700 MHz, DMSO-d6) δ 3.86 (dd, J = 4.0, 2.4 Hz, 2 H), 4.63 (d, J = 6.4 Hz, 2 H), 6.99 - 7.04 (m, 2 H), 7.16 (t, J = 7.5 Hz, 2 H), 7.33 (d, J = 7.7 Hz, 2 H), 7.40 (d, J = 7.7 Hz, 2 H), 12.58 (s, 2 H); 13C NMR (175 MHz, DMSO-d6) δ 42.6, 43.9, 124.7, 127.1, 128.5, 129.1, 132.3, 137.8, 173.6; HRMS (ESI) m/z: calcd for C18H14Br2O4H+, 451.9259, found, 451.9261; (Δ = 0.44 ppm).
4.4.3.6. α-2,4-Di(2-nitrophenyl)cyclobutane-1,3-dicarboxylic acid (1f)
White solid; 55% yield; m.p. 206-208°C; 1H NMR (300 MHz, MeOD) δ 3.20 (m, 2 H), 3.81 (m, 2 H), 7.15 (m, 2 H), 7.29 (m, 4 H), 7.53 (m, 2 H); 13C NMR (125 MHz, MeOD) δ 40.90, 44.06, 123.13, 127.93, 129.13, 132.26, 133.74, 149.52; HRMS (ESI) m/z: calcd for C18H14N2O8H+, 387.0825, found, 387.0823 (Δ = 0.05 ppm).
4.4.3.7. α-2,4-Di(3,4-dimethoxyphenyl)cyclobutane-1,3-dicarboxylic acid (1g)
White solid; 90% yield; 1H NMR (300 MHz, acetone-d6) δ 3.69 - 3.71(m, 2 H), 3.73 (s, 6 H), 3.76 (s, 6 H), 4.16 - 4.25 (m. 2 H), 6.80 - 6.96 (m, 6H), 12.06 (s, 2H); 13C NMR (176 MHz, acetone-d6) δ 41.10, 47.05, 55.89, 55.92, 111.88, 112.30, 119.92, 132.41, 148.08, 148.75, 173.58; HRMS (ESI) m/z: calcd for C22H24O8H+, 417.1544, found, 417.1548 (Δ = 0.96 ppm).
4.4.3.8. α-2,4-Di(3-methoxy-4-tosyloxyphenyl)cyclobutane-1,3-dicarboxylic acid (1h)
To a solution of (E)-ferulic acid (7.00 g, 36.0 mmol) in H2O (15 mL) was added NaOH (7.21 g, 180 mmol) and Tosyl chloride (21.75 g, 114 mmol) portions. The reaction mixture was stirred overnight and was quenched with 6 M HCl (10 mL) and diluted with water (50 mL). The precipitation was collected by vacuum filtration and the solid was washed with water. The crude solid was dried over phosphorus pentoxide in vacuo. The product was purified by recrystallization in EtOH/H2O (9:1) to afford 3-methoxy-4-tosyloxy-(E)-cinnamic acid (11.55 g, 92%) as a light yellow crystal. m.p. 201–202°C; 1H NMR (300 MHz, CDCl3) δ2.47 (s, 3 H), 3.63 (s, 3 H), 6.40 (d, J = 16.0 Hz, 1 H), 7.01 (s, 1 H), 7.10 (d, J = 8.2 Hz, 1 H), 7.20 (d, J = 8.2 Hz, 1 H), 7.33 (d, J = 8.2 Hz, 2 H), 7.71 (d, J = 16.0 Hz, 1 H), 7.78 (d, J = 8.2 Hz, 2 H); FIA m/z: calcd for C17H16O6SH+, 349.1, found, 349.1. The analytical data was consistent with literature values.[25]
3-Methoxy-4-tosyloxy-(E)-cinnamic acid (0.600 g, 0.86 mmol) was placed in a pyrex dish and exposed to light at 350 nm and an intensity of 280 nW/cm2 for 4 days with periodic shaking. This process was performed in the solid state and monitored by 1H NMR. After completion of the photoreaction, the white solid was washed with diethyl ether (20 mL) to give 1h (0.600 g, 88% yield) as a white solid. m.p.: >230°C; 1H NMR (300 MHz, acetone-d6) δ 2.45 (s, 6 H), 3.51 (s, 6 H), 3.98 (dd, J = 10.5, 7.1 Hz, 2H), 4.41 (dd, J = 10.5, 7.1 Hz, 2 H), 6.93 – 7.11 (m, 6 H), 7.40 –7.48 (m, 4 H), 7.62 – 7.73 (m, 4 H). 13C NMR (126 MHz, acetone-d6) δ 21.63, 42.24, 47.25, 55.97, 113.62, 120.40, 124.41, 129.39, 130.47, 134.16, 138.09, 140.88, 146.32, 152.43, 173.09; FIA m/z: calcd for C34H32O12S2H+, 697.1, found, 697.1. The analytical data was consistent with literature values [25].
4.4.3.9. α-2,4-Di(4-hydroxy-3-methoxyphenyl)cyclobutane-1,3-dicarboxylic acid (1i)
To a solution of 1h (1.00 g, 1.44 mmol) in MeOH (20 mL) was added KOH (0.966 g, 17.3 mmol) and the reaction mixture was stirred at room temperature overnight. The reaction was quenched with 6 M HCl (pH 1) to form precipitate. The product was collected by filtration and dried over P2O5 in vacuo to afford 1i as a white solid (0.518 g, 83%): m.p. >240°C; 1H NMR (300 MHz, acetone-d6) δ 3.32 (d, J = 9.2 Hz, 2 H), 3.85 (m, 8 H), 4.39 – 4.28 (m, 2 H), 6.72 – 6.86 (m, 4 H), 6.98 (m, 2 H); 13C NMR (125 MHz, acetone-d6) δ41.29, 46.96, 55.35, 111.59, 114.59, 120.21, 130.98, 145.54, 147.13, 172.56; FIA m/z: calcd for C20H20O8H+, 389.1, found, 389.1. These data are consistent with literature values [42].
4.4.3.10. α-2,4-Di(4-tert-butyldimethylsiloxylphenyl)cyclobutane-1,3-dicarboxylic acid (1j)
To a solution of (E)-4-hydroxycinnamic acid (2.00 g, 12.2 mmol) in dichloromethane (20 mL) at 0°C was added diisopropylethylamine (DIPEA, 4.72 g, 36.6 mmol) and tert-butyldimethylsilyl chloride (TBDMSCl, 4.59 g, 30.5 mmol) dropwise. The reaction mixture was slowly warmed up to room temperature and stirred overnight. The reaction mixture was diluted with EtOAc (50 mL) then quenched with water (20 mL). The separated organic layer was washed with 1 M HCl (2 × 30 mL), brine (30 mL), then dried over MgSO4 and concentrated in vacuo. To the resulting residue in THF (15 mL) was added a solution of K2CO3 (2.53 g, 18.3 mmol) in water (5 mL) and the reaction mixture was stirred for 4 h. Upon completion, the reaction mixture was diluted with EtOAc (60 mL) and quenched with slow addition of 1 M HCl (15 mL). The layers were separated and the organic layer was washed with 1 M HCl (2 × 20 mL), brine (30 mL), then dried over MgSO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel to afford (E)-4-tert-butyldimethylsilyloxycinnamic acid as a white solid (2.624 g, 77%): m.p. 127-128°C 1H NMR (300 MHz, CDCl3) δ0.24 (s, 6 H), 1.00 (s, 9 H), 6.33 (d, J = 15.9 Hz, 1 H), 6.87 (d, J = 8.5 Hz, 2 H), 7.47 (d, J = 8.6 Hz, 2 H), 7.76 (d, J = 15.9 Hz, 1H); FIA m/z: calcd for C15H22O3SiH+, 279.1, found, 279.1. These data are consistent with literature values [42].
(E)-4-Tert-butyldimethylsilyloxycinnamic acid (1.00 g, 3.59 mmol) was placed in a pyrex dish and exposed to light at 360 nm and an intensity of 280 nW/cm2 for 4 days with periodic shaking. This process was performed in the solid state and monitored by 1H NMR. After completion of the photoreaction, the reaction mixture was purified by column chromatography to afford 1j as white solid (0.846 g, 84%): m.p. >230°C; 1H NMR (500 MHz, acetone-d6) δ 0.22 (s, 12 H), 1.01 (s, 18 H), 3.90 (dd, J = 10.4, 7.3 Hz, 2 H), 4.36 (dd, J = 10.4, 7.3 Hz, 2 H), 6.87 – 6.80 (m, 4 H), 7.30 (d, J = 8.5 Hz, 4 H). 13C NMR (126 MHz, acetone-d6) δ -5.18, 17.86, 25.14, 40.88, 46.74, 119.61, 128.86, 132.41, 154.48, 172.36; HRMS (ESI) m/z calculated for C30H44O6Si2H+: 557.2749, found 557.2952 (Δ = 0.54 ppm).
4.4.3.11. 1-Benzyl α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3a) and 1,3-dibenzyl α-2,4-diphenylcyclobutane-1,3-dicarboxylate (5a)
To α-truxillic acid (1a, 200 mg, 0.66 mmol) suspended in thionyl chloride (3 mL) was added three drops of DMF and the mixture was heated to reflux for 3 h with stirring. Excess thionyl chloride and DMF were removed in vacuo to give the corresponding α-truxillic acid dichloride (2a) as light yellow solid, which was used directly in the subsequent reaction. To a solution of 2a in THF (10 mL) were added benzyl alcohol (0.53 mmol, 0.8 eq) and pyridine (4.0 mmoles), and the reaction mixture was stirred for 2 h. The reaction was quenched by adding distilled water (10 mL) with stirring for 30 min. The resulted solution was diluted with ethyl acetate (15 mL) and washed with aqueous copper sulfate (5 mL × 3) and water (5 mL × 3). The organic layer was collected, dried over MgSO4, and concentrated in vacuo. The crude mixture was purified by flash chromatography on silica gel (hexanes/AcOEt/AcOH = 79/20/1) as the eluent to give mono-ester 3a and diester 5a.
3a: White solid; 61% yield; m.p. 143 -144°C; 1H NMR (500 MHz, acetone-d6) δ 3.98 – 4.06 (m, 2 H), 4.48 (m, 2 H), 4.66 (d, J = 12.3 Hz, 1 H), 4.82 (d, J = 12.3 Hz, 1 H), 7.04 (m, 2 H), 7.2 –7.30 (m, 5 H), 7.36–7.30 (m, 4 H), 7.41 (m, 4 H); 13C NMR (125 MHz, acetone-d6) δ41.47, 41.76, 46.24, 46.68, 65.91, 126.80, 126.94, 127.65, 127.76, 127.85, 128.08, 128.20, 128.26, 128.26, 128.27, 128.30, 128.31, 135.99, 139.34, 139.39, 171.54, 172.20; HRMS (ESI) m/z calculated for C25H22O4H+: 387.1591, found 387.1593 (Δ = 0.52 ppm).
5a: White solid; 13% yield; m.p. 161-163°C; 1H NMR (500 MHz, acetone-d6) δ 3.99 - 4.17 (m, 2 H), 4.52 (dd, J = 10.38, 7.32 Hz, 2 H), 4.64 (d, J = 12.21 Hz, 2 H), 4.81 (d, J = 12.51 Hz, 2 H), 6.98 - 7.08 (m, 4 H), 7.23 - 7.30 (m, 7 H), 7.30 - 7.36 (m, 4 H), 7.36 - 7.45 (m, 4 H); 13C NMR (126 MHz, acetone-d6) δ 42.6, 47.5, 66.9, 128.0, 128.7, 128.8, 129.1, 129.2, 129.3, 136.9, 140.1, 172.3; HRMS (ESI) m/z calculated for C32H28O4H+: 477.206, found 477.2059 (Δ = 0.34 ppm).
In the same manner, other mono esters 3b to 3i, 3k to 3r and di-esters 5b to 5e were synthesized and characterized, starting from α-2,4-diphenylcyclobutene-1,3-dicarboxylic acid 1a.
4.4.3.11. 1-(4-Methoxybenzyl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3b) and 1,3-di(4-methoxy)benzyl α-2,4-diphenylcyclobutane-1,3-dicarboxylate (5b)
3b: White solid; 40% yield; m.p. 99 - 102°C; 1H NMR (500 MHz, acetone-d6) δ 3.78 (s, 3 H), 3.93 - 4.07 (m, 2 H), 4.40 - 4.50 (m, 2 H), 4.58 (d, J = 11.9 Hz, 1 H), 4.74 (d, J = 11.9 Hz, 1 H), 6.79 - 6.87 (m, 2 H), 6.93 - 7.03 (m, 2 H), 7.21 - 7.39 (m, 8 H), 7.41 (d, J = 7.32 Hz, 2 H); 13C NMR (126 MHz, acetone-d6) δ 42.4, 42.7, 47.1, 47.6, 55.6, 66.7, 114.6, 127.7, 127.8, 128.6, 128.7, 129.1, 129.2, 130.9, 140.3, 160.6, 172.5, 173.1; HRMS (ESI) m/z calcd for C26H24O5NH4+: 434.1962, found 434.1964 (Δ = 0.42 ppm).
5b: White solid; 12% yield; m.p. 145-148°C; 1H NMR (500 MHz, acetone-d6) δ 3.78 (s, 6 H), 3.97 - 4.08 (m, 2 H), 4.47 (dd, J = 10.38, 7.32 Hz, 2 H), 4.56 (d, J = 12.2 Hz, 2 H), 4.73 (d, J = 12.2 Hz, 2 H), 6.78 - 6.86 (m, 4 H), 6.93 - 7.01 (m, 4 H), 7.23 - 7.30 (m, 2 H), 7.30 - 7.40 (m, 8 H); 13C NMR (125 MHz, acetone-d6) δ 42.5, 47.5, 55.6, 66.7, 114.6, 127.9, 128.6, 129.3, 130.9, 140.1, 160.6, 172.3; HRMS (ESI) m/z calcd for C34H32O6NH4+ 554.2536, found 544.2537 (Δ = 0.2 ppm).
4.4.3.12. 1-(4-Fluorobenzyl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-dicarboxylate (3c) and di(4-fluorobenzyl) α-2,4-diphenylcyclobutane-1,3-dicarboxylate (5c)
3c: White solid; 34% yield; m.p. 145-148°C; 1H NMR (500 MHz, acetone-d6) δ 4.03 (dd, J = 10.38, 7.32 Hz, 2 H), 4.35 - 4.58 (m, 2 H), 4.68 (d, J = 12.21 Hz, 1 H), 4.80 (d, J = 12.21 Hz, 1 H), 6.93 - 7.14 (m, 4 H), 7.21 - 7.45 (m, 10 H), 10.63 (br. s., 1 H); 13C NMR (125 MHz, acetone-d6) δ 42.4, 42.7, 47.2, 47.6, 66.1, 115.8, 116.0, 127.8, 127.9, 128.6, 128.7, 129.2, 129.3, 131.2, 131.3, 133.1, 140.2, 140.3, 162.4, 164.3, 172.4, 173.0; HRMS (ESI) m/z calculated for C25H21FO4(M+H)+: 405.1497, found 405.1502 (Δ = 1.3 ppm).
5c: White solid; 15% yield; m.p. 103-105°C; 1H NMR (500 MHz, acetone-d6) δ 4.06 (dd, J = 10.53, 7.48 Hz, 2 H), 4.51 (dd, J = 10.38, 7.32 Hz, 2 H), 4.66 (d, J = 12.36 Hz, 2 H), 4.78 (d, J = 12.36 Hz, 2 H), 6.93 - 7.17 (m, 8 H), 7.20 - 7.44 (m, 10 H); 13C NMR (125 MHz, acetone-d6) δ 42.5, 47.4, 66.1, 115.8, 116.0, 128.0, 128.6, 129.3, 131.3, 131.3, 133.1, 133.1, 140.0, 162.4, 164.3, 172.2; HRMS (ESI) m/z calcd for C32H26F2O4H+ 513.1872, found 513.1882 (Δ = 2.0 ppm).
4.4.3.13. 1-(4-Bromobenzyl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3d)
White solid; 44% yield; m.p. 175-177°C; 1H NMR (500 MHz, acetone-d6) δ 3.92 - 4.11 (m, 2 H), 4.40 - 4.57 (m, 2 H), 4.69 (d, J = 12.66 Hz, 1 H), 4.78 (d, J = 12.66 Hz, 1 H), 6.96 (d, J = 8.54 Hz, 2 H), 7.20 - 7.29 (m, 2 H), 7.32 (t, J = 7.48 Hz, 4 H), 7.35 - 7.40 (m, 2 H), 7.40 - 7.49 (m, 4 H); 13C NMR (125 MHz, acetone-d6) δ 42.4, 42.7, 47.2, 47.5, 66.0, 122.3, 127.8, 127.9, 128.6, 128.7, 129.2, 129.3, 131.0, 132.3, 136.4, 140.2, 140.3, 172.4, 173.1; HRMS (ESI) m/z calcd for C25H21BrO4H+ 465.0696, found 465.0697 (Δ = 0.27 ppm).
4.4.3.14. 1,3-Bis(tetrahydropyran-4-methyl) α-2,4-diphenylcyclobutane-1,3-dicarboxylate (5d)
White solid; 21% yield; m.p. 143-144°C; 1H NMR (500 MHz, acetone-d6) δ 0.91 - 1.10 (m, 4 H), 1.16 - 1.31 (m, 4 H), 1.38 - 1.55 (m, 2 H), 3.17 (m, 4 H), 3.58 (d, J = 6.41 Hz, 4 H), 3.67 - 3.81 (m, 4 H), 3.90 - 4.08 (m, 2 H), 4.48 (m, 2 H), 7.19 - 7.31 (m, 2 H), 7.31 - 7.46 (m, 8 H); 13C NMR (125 MHz, acetone-d6) δ 35.2, 42.5, 47.7, 67.8, 67.8, 69.4, 127.9, 128.7, 129.3, 140.3, 172.3; HRMS (ESI) m/z calculated for C30H36O6H+: 493.2585, found 493.2591 (Δ = 1.24 ppm).
4.4.3.15. 1-(3-Ethynylphenyl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3f)
White solid; 67 % yield; m.p. 168-169°C; 1H NMR (700 MHz, CDCl3) δ 3.06 (s, 1 H). 4.12 – 4.07 (m, 1H), 4.19 (m, 1H), 4.53 - 4.60 (m, 2H), 6.32 (d, J = 8.1 Hz, 1 H), 6.39 (s, 1 H), 7.14 (t, J = 8.1 Hz, 1H), 7.25 (d, J = 7.7 Hz, 1H), 7.28 (s, 2H), 7.33 – 7.46 (m, 9 H). 13C NMR (175 MHz, CDCl3) δ 41.30, 41.80, 46.04, 46.93, 77.83, 82.41, 122.15, 123.22, 125.08, 127.43, 127.49, 127.73, 127.87, 128.63, 128.83, 129.12, 129.60, 138.09, 149.92, 170.38; HRMS (ESI) m/z calculated for C26H20O4H+: 397.1434, found 397.1446 (Δ = 3.02 ppm).
4.4.3.16. 1-(Biphenyl-2-yl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3g)
White solid; 21% yield; m.p. 195-196°C; 1H NMR (500 MHz, acetone-d6) δ 3.81 (dt, J = 11.67, 10.49 Hz, 2 H), 4.39 (t, J = 10.07 Hz, 1 H), 4.68 (t, J = 10.68 Hz, 1 H), 5.99 (dd, J = 8.09, 1.07 Hz, 1 H), 7.11 - 7.19 (m, 3 H), 7.20 - 7.28 (m, 2 H), 7.28 - 7.40 (m, 11 H), 7.40 - 7.46 (m, 2 H); 13C NMR (126 MHz, acetone-d6) δ 42.3, 45.4, 46.8, 47.0, 123.4, 127.1, 127.4, 128.1, 128.4, 129.2, 129.3, 129.6, 129.8, 131.3, 135.8, 138.5, 139.2, 143.2, 148.7, 170.6, 172.9; HRMS (ESI) m/z calcd for C30H24O4H+: 449.1747, found 449.1754 (Δ = 1.55 ppm).
4.4.3.17. 1-(Biphenyl-3-yl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3h) and 1,3-di(biphenyl-3-yl) α-2,4-diphenylcyclobutane-1,3-dicarboxylate (5e)
3h: white solid;; 42 % yield; m.p. 208-209°C; 1H NMR (500 MHz, acetone-d6) δ 4.14 (dd, J = 10.8, 7.2 Hz, 1 H), 4.29 - 4.35 (m, 1 H), 4.61 (dd, J = 10.8, 7.2 Hz, 1 H), 4.67 (dd, J = 10.8, 7.2 Hz, 1 H), 6.53 (t, J = 1.9 Hz, 1 H), 6.55 - 6.57 (m, 1 H), 7.29 (d, J = 7.4 Hz, 1 H), 7.33 (d, J = 7.9 Hz, 1 H), 7.36 - 7.41 (m, 4 H), 7.44 - 7.55 (m, 9 H), 7.59 (d, J = 7.5 Hz, 2 H); 13C NMR (175 MHz, CDCl3) δ41.34, 41.79, 45.78, 46.95, 76.86, 77.04, 77.22. 120.10, 120.12, 124.55, 127.16, 127.49, 127.56, 127.62, 127.72, 127.99, 128.67, 128.83, 129.50, 138.14, 138.18, 139.91, 142.55, 150.58, 170.65, 174.19; HRMS (ESI) m/z calcd for C30H24O4H+: 449.1747, found 449.1753 (Δ = 1.33 ppm).
5e: white solid; 15 % yield; m.p. 184-185°C; 1H NMR (500 MHz, acetone-d6) δ4.45 (dd, J = 10.8, 7.3 Hz, 2 H), 4.81 (dd, J = 10.8, 7.3 Hz, 2 H), 6.53 (m, 2 H), 6.54 - 6.58 (m, 2 H), 7.34 (t, J = 7.6 Hz, 2 H), 7.39 (m, 4 H), 7.43 - 7.55 (m, 14 H), 7.65 (d, J = 7.6 Hz, 4 H); 13C NMR (175 MHz, acetone-d6) δ 41.60, 46.38, 120.06, 120.52, 124.02, 126.86, 127.50, 127.72, 128.31, 128.62, 128.83, 129.53, 139.03, 139.62, 142.16, 151.21, 170.29; HRMS (ESI) m/z calcd for C42H32O4H+ 601.2373, found 601.2371 (Δ = 0.33 ppm).
4.4.3.18. 1-(Biphenyl-4-yl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3i)
White solid; 40% yield; m.p. 190°C (decomp.); 1H NMR (700 MHz, acetone-d6) 3.97 (t, J = 10.5 Hz, 1 H), 4.17 (t, J = 10.5 Hz, 1 H), 4.52 (t, J = 10.5 Hz, 1H), 4.78 (t, J = 10.5 Hz, 1H), 6.50 (d, J = 8.5 Hz, 2 H), 7.29 (t, J = 7.3 Hz, 1 H), 7.33 - 7.46 (m, 8H), 7.48 - 7.53 (m, 5 H), 7.59 (d, J = 7.3 Hz, 2 H), δ 10.78 (s, 1 H). 13C NMR (175 MHz, acetone-d6) δ 42.10, 44.52, 46.03, 46.28, 121.93, 126.67, 126.78, 127.24, 127.30, 127.54, 128.38, 128.45, 128.81, 128.85, 138.47, 138.50, 140.07, 142.22, 150.16, 170.09, 171.76: HRMS (ESI) m/z calcd for C30H24O4H+: 449.1747, found 449.1752 (Δ = 1.11 ppm).
4.4.3.19. 1-(2′-Hydroxybiphenyl-2-yl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3j)
2′-TIPSO-biphenyl-2-yl α-2, 4-diphenylcyclobutane-1, 3-dicarboxylate (3j-i) was prepared in the same manner as that for 3a. The crude product was purified by column chromatography on silica gel to afford 3j-i as white solid (59% yield). The TIPS protecting group of 3j-i was removed by 1 M tetrabutylammonium fluoride (TBAF) in THF at room temperature to give 3j as a white solid (99% yield): m.p. 55-57°C; 1H NMR (500 MHz, acetone-d6) δ 3.99 (dd, J = 10.5, 7.0 Hz, 1H), 4.11 (dd, J = 10.5, 7.0 Hz, 1H), 4.37 (dd, J = 10.5, 7.0 Hz, 1 H), 4.51 (dd, J = 10.5, 7.0 Hz, 1 H), 6.15 (m, 1 H), 6.93 - 6.87 (m, 1 H), 7.03 (d, J = 8.1 Hz, 1 H), 7.11 (m, 1 H), 7.26 (m,, 12 H), 7.43 (t, J = 7.5 Hz, 2 H), 7.48 (d, J = 7.5 Hz, 2 H), 10.67 (s, 1H), 8.21 (s, 1 H); 13C NMR (175 MHz, CDCl3) δ 41.10, 41.54, 45.88, 46.34, 116.27, 120.57, 122.47, 123.75, 125.32, 126.69, 126.90, 127.07, 127.46, 127.55, 128.25, 128.30, 128.43, 128.74, 129.06, 129.55, 129.65, 129.91, 130.10, 130.57, 131.58, 138.03, 138.35, 148.24, 152.88, 170.41; HRMS (ESI) m/z: calcd for C30H25O5H+, 465.1697, found, 465.1699 (Δ = 0.43 ppm).
4.4.3.20. 1-(2,4,5-Tricholrobenzyl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3k)
White solid; 70% yield; m.p. 191-193°C; 1H NMR (700 MHz, acetone-d6) δ 4.14 (dd, J = 10.7, 7.2 Hz, 1 H), 4.38 (dd, J = 10.7, 7.2 Hz, 1 H), 4.60 (dd, J = 10.7, 7.2 Hz, 1 H), 4.68 (dd, J = 10.7, 7.2 Hz, 1 H), 6.03 (s, 1 H), 7.27 (t, J = 7.4 Hz, 1 H), 7.36 (t, J = 7.6 Hz, 2 H), 7.41 (t, J = 7.3 Hz, 1 H), 7.48 (dt, J = 15.1, 9.8 Hz, 4 H), 7.57 (d, J = 7.5 Hz, 2 H), 7.70 (d, J = 7.6 Hz, 1 H), 10.75 (s, 1 H); 13C NMR (175 MHz, acetone-d6) δ 41.2, 42.0, 45.9, 46.5, 125.0, 126.3, 127.0, 127.5, 127.8, 128.8, 129.0, 129.8, 130.7, 130.9, 138.8, 139.1, 145.9, 169.2, 171.9; HRMS (ESI) m/z calcd for C24H18Cl3O4H+: 475.0265; found, 475.0264 (Δ = 0.21 ppm).
4.4.3.21. 1-[(1R,2S)-2-Phenylcyclohexyl] α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3l)
White solid; 61% yield; m.p. 192-193°C; 1H NMR (700 MHz, CDCl3) δ 0.80 (m, 1 H), 1.22 - 1.39 (m, 2 H), 1.47 - 1.59 (m, 2 H), 1.64 - 1.76 (m, 2 H), 1.90 (d, J = 13.4 Hz, 1 H), 2.57 (m, 1 H), 3.65 (m, 2 H), 3.96 (m, 1 H), 4.24 - 4.32 (m, 1 H), 4.69 (m, 1 H), 7.00 (d, J = 7.0 Hz, 2 H), 7.07 (d, J = 7.0 Hz, 2 H), 7.15 (d, J = 7.0 Hz, 2 H), 7.20 - 7.29 (m, 7 H), 7.31 (m, 2 H), 10.64 (s, 1 H). 13C NMR (175 MHz, CDCl3) δ 24.7, 25.8, 31.4, 33.7, 40.6, 42.3, 46.1, 47.3, 49.9, 76.9, 126.7, 126.8, 127.1, 127.3, 127.6, 128.5, 128.6, 138.6, 139.2, 143.2, 171.0, 177.7; HRMS (ESI) m/z: calcd for C30H31O4H+: 455.2217; found, 455.2221 (Δ = 0.81 ppm).
4.4.3.22. 1-(Indan-2-yl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3m)
White solid; 34% yield; m.p. 174-175°C; 1H NMR (500 MHz, acetone-d6) δ 2.05 (dt, J = 4.35, 2.25 Hz, 1 H), 2.10 (dd, J = 17.09, 2.44 Hz, 1 H), 2.74 (dd, J = 16.94, 2.59 Hz, 1 H), 2.87 (dd, J = 16.94, 6.26 Hz, 1 H), 3.11 (dd, J = 17.09, 6.41 Hz, 1 H), 3.90 (dd, J = 10.68, 6.71 Hz, 1 H), 3.97 - 4.07 (m, 1 H), 4.36 - 4.48 (m, 2 H), 5.14 - 5.21 (m, 1 H), 7.05 - 7.18 (m, 4 H), 7.19 - 7.25 (m, 1 H), 7.27 - 7.37 (m, 7 H), 7.40 (d, J = 7.32 Hz, 2 H); 13C NMR (125 MHz, acetone-d6) δ 39.8, 40.0, 42.3, 42.6, 46.9, 47.5, 76.0, 125.3, 125.5, 127.3, 127.4, 127.7, 127.9, 128.7, 128.7, 129.1, 129.2, 140.1, 140.4, 141.4, 173.1; HRMS (ESI) m/z calcd for C27H24O4H+: 413.1747, found 413.1749 (Δ = 0.43 ppm).
4.4.3.23. 1-(2,2,2-Trifluoroethyl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3n)
White solid, 51% yield; mp 158-159°C; 1H NMR (500 MHz, CDCl3) δ3.84 (m, 1 H), 4.00– 4.11 (m, 2 H), 4.19 (m, 1 H), 4.47 (m, 2 H), 7.24–7.39 (m, 10 H); 13C NMR (125 MHz, CDCl3) δ41.32, 41.53, 46.00, 46.44, 60.04, 60.33, 60.63, 60.92, 127.28, 127.33, 127.47, 127.55, 128.62, 128.65, 137.61, 137.69, 170.35, 176.49; HRMS (ESI) m/z: calcd for C20H17O4F3H+379.1152; found 379.1155 (Δ = 0.79 ppm).
4.4.3.24. 1-(6-Acetamidonaphth-1-yl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3o)
White solid; 37% yield; m.p. 131-132°C; 1H NMR (500 MHz, MeOD) δ 2.18 (s, 3 H), 4.08 (dd, J = 10.5, 7.0 Hz, 1 H), 4.49 (dd, J = 10.5, 7.0Hz, 1 H), 4.65 (m, 2 H), 6.32 (d, J = 7.5 Hz, 1 H), 6.96 (d, J = 9.1 Hz, 1 H), 7.35 – 7.23 (m, 3 H), 7. 37 – 7.53 (m, 7 H), 7.58 (d, J = 7.0 Hz, 3 H), 8.20 (m, 1 H); 13C NMR (176 MHz, acetone-d6) δ 24.42, 42.59, 43.08, 47.72, 115.76, 117.38, 121.02, 123.03, 124.33, 126.14, 126.74, 127.84, 128.28, 128.79, 129.20, 129.21, 129.67, 136.11, 138.50, 140.21, 140.48, 147.72, 169.29, 171.61; HRMS (ESI) m/z calcd for C27H24O4H+: 480.1811, found 480.1809 (Δ = 0.42 ppm).
4.4.3.25. 1-(6-Acetamidonaphth-1-yl) γ-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3o-γ)
To a solution of γ-truxillic anhydride (100 mg, 0.36 mmol) and 6-acetamido-1-naphthol (80 mg, 0.40 mmol) in THF (2.0 ml), was added diisoproplethylamine (0.06 ml, 0.40 mmol) dropwise. The reaction mixture was stirred at room temperature overnight. Upon completion, the reaction mixture was diluted with water (2 mL) and extracted thrice with dichloromethane (10 mL × 3). The crude mixture was purified by flash chromatography on silica gel to give 3o-γ (36 mg, 82% yield) as a white solid. m.p. decompose at 165°C; 1H NMR (500 MHz, acetone-d6) δ 2.04 (s, 3 H), 3.90 (t, J = 9.8 Hz, 1 H), 4.28 (t, J = 9.8 Hz, 1 H), 4.52 (t, J = 9.8 Hz, 1 H), 4.74 (t, J = 9.8 Hz, 1 H), 6.30 (d, J = 7.6 Hz, 1H), 6.89 (d, J = 9.0 Hz, 1 H), 7.21 (dd, J = 14.6, 7.0 Hz, 2H), 7.37 - 7.26 (m, 6 H), 7.43 - 7.55 (m, 5 H), 8.27 (s, 1 H), δ 9.24 (s, 1H); 13C NMR (176 MHz, acetone-d6) δ23.46, 42.27, 44.65, 46.32, 46.43, 114.76, 116.40, 120.03, 121.92, 123.36, 125.18, 125.80, 126.70, 126.72, 126.73, 127.43, 128.48, 128.59, 129.06, 135.17, 137.56, 138.60, 142.18, 146.71, 168.24, 170.20, 171.70; HRMS (ESI) m/z calcd for C30H26NO5H+: 480.1811, found 480.1809 (Δ = 0.45 ppm).
4.4.3.26. 1-(5-Ethynylnaphth-1-yl) α-2,4-3-hydroxycarbonyl-diphenylcyclobutane-1-carboxylate (3p)
White solid; 44% yield; m.p. 220-221°C; 1H NMR (500 MHz, acetone-d6) δ 4.06 (s, 1 H), 4.11 (dd, J = 10.7, 7.2 Hz, 1 H), 4.53 (dd, J = 10.7, 7.2 Hz, 1 H), 4.64 (dd, J = 10.7, 7.2 Hz, 1 H), 4.68 (dd, J = 10.7, 7.2 Hz, 1 H), 6.48 (d, J = 7.5 Hz, 1 H), 7.26 (m, 3 H), 7.34 (t, J = 7.6 Hz, 2 H), 7.42 (m, 4 H), 7.51 (d, J = 7.5 Hz, 2 H), 7.60 (d, J = 7.2 Hz, 2 H), 7.67 (d, J = 6.8 Hz, 1 H), 8.11 (d, J = 8.4 Hz, 1 H), 10.70 (s, 1 H); 13C NMR (176 MHz, acetone-d6) δ 41.61, 42.11, 46.41, 46.77, 80.96, 83.68, 118.80, 119.77, 122.72, 123.45, 125.70, 126.49, 126.72, 126.92, 127.46, 127.85, 128.26, 128.27, 128.80, 131.41, 134.15, 139.18, 139.47, 147.18, 170.69, 172.12; HRMS (ESI) m/z: calcd for C30H22O4H+ 447.1596; found 447.1591, (Δ = 1.1 ppm).
4.4.3.27. 1-(9-Fluorenylmethyl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3q)
White solid; 35% yield; m.p. 180-181°C; 1H NMR (700 MHz, CDCl3) δ 3.85 (t, J = 6.7 Hz, 1 H), 4.00 (m, 2 H), 4.08 (m, 2 H), 4.37 (m, 1 H), 4.47 (m, 1 H), 7.25 - 7.39 (m, 12 H), 7.40- 7.48 (m, 4 H), 7.80 (d, J = 7.5 Hz, 2 H); 13C NMR (175 MHz, CDCl3) δ 41.6, 41.8, 46.3, 46.7, 47.2, 66.6, 120.1, 125.0, 125.1, 127.2, 127.4, 127.5, 128.6, 128.7, 138.3, 138.4, 141.3, 141.4, 143.6, 144.1, 171.9, 177.2; HRMS (ESI) m/z: calcd for C32H27O4H+ 475.1904; found, 475.1907 (Δ = 0.63 ppm).
4.4.3.28. 1-(2,2,2-Trifluoroethyl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3n)
White solid, 51% yield; m.p. 158-159°C; 1H NMR (300 MHz, (acetone-d6) δ3.98 - 4.22 (m, 3 H), 4.29 - 4.64 (m, 3 H), 7.16 - 7.55 (m, 10 H); 13C NMR (75 MHz, acetone-d6) δ 41.4, 41.7, 46.0, 46.2, 59.4, 59.7, 60.1, 60.4, 121.9, 124.6, 126.9, 127.0, 127.4, 127.6, 128.2, 128.3, 138.7, 138.9, 170.3, 172.0; HRMS (ESI) m/z: calcd for C20H17O4F3H+ 379.1152; found 379.1155 (Δ = 0.79 ppm).
4.4.3.29. 1-Cyclohexyl α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3r)
White solid; 68% yield; m.p. 152-153°C; 1H NMR (500 MHz, CDCl3) δ 0.83 (m, 1 H), 1.09–1.25 (m, 5 H), 1.38–1.49 (m, 2 H), 1.59 (m, 2 H), 3.90 (dd, J = 10.6, 6.8 Hz, 1 H), 4.01 (dd, J = 10.6, 7.8 Hz, 1 H), 4.40 (m, 1 H), 4.50–4.42 (m, 2 H), 7.21–7.37 (m, 11 H); 13C NMR (125 MHz, CDCl3) δ 23.53, 23.60, 25.25, 30.92, 31.41, 41.45, 41.58, 46.25, 46.98, 72.90, 127.18, 127.47, 127.59, 128.41, 128.50, 138.50, 138.52, 171.32, 176.77; HRMS (ESI) m/z calcd for C24H27O4H+: 379.1909, found 379.1909 (Δ = 0 ppm).
4.4.3.30. 1-{3-[1-(3,6,9-Trioxadodecanyl)-1,2,3-triazol-4-yl]phenyl} α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxylate (3s)
To a solution of 3f in THF (8 mL) and water (1.5 mL), was added cupric sulfate pentahydrate (81 mg, 0.32 mmoles), ascorbic acid (57 mg, 0.32 mmoles), and 1-azido-3,6,9-dodecane (60 mg, 0.30 mmoles) and the reaction mixture was stirred at room temperature overnight. Upon completion, the reaction mixture was diluted with water (20 mL) and extracted thrice with dichloromethane (25 mL × 3). The crude mixture was purified by flash chromatography on silica gel with 3.5% methanol in dichloromethane as the eluent to give 3s (180 mg, 94% yield) as a white solid: m.p. 93-95°C; 1H NMR (500 MHz, acetone-d6) δ 1.07 (t, J = 7.02 Hz, 6 H), 3.39 (q, J = 7.02 Hz, 4 H), 3.43 - 3.49 (m, 4 H), 3.49 - 3.55 (m, 4 H), 3.55 - 3.61 (m, 4 H), 3.61 - 3.71 (m, 4 H), 3.93 (t, J = 5.19 Hz, 4 H), 4.06 - 4.19 (m, 2 H), 4.27 - 4.36 (m, 2 H), 4.54 - 4.72 (m, 8 H), 6.28 - 6.43 (m, 2 H), 6.96 (t, J = 1.83 Hz, 2 H), 7.21 - 7.32 (m, 4 H), 7.33 - 7.44 (m, 6 H), 7.44 - 7.54 (m, 8 H), 7.57 (d, J = 7.32 Hz, 4 H), 7.69 (d, J = 7.93 Hz, 2 H), 8.22 (s, 2 H); 13C NMR (126 MHz, acetone-d6) δ 15.6, 42.3, 42.9, 47.0, 47.6, 51.0, 66.8, 70.1, 70.6, 71.2, 71.3, 119.5, 121.7, 122.4, 123.4, 127.8, 128.3, 128.7, 129.0, 129.2, 129.5, 130.4, 133.7, 140.1, 140.2, 146.8, 152.1, 171.3, 173.1; HRMS (ESI) m/z calculated for C34H37N3O7H+: 600.2704, found 600.2705 (Δ = 0.13 ppm).
4.4.3.31. 1-Naphthyl α-3-hydroxycarbonyl-2,4-di(3-methoxy-4-tosyloxy)cyclobutane-1-carboxylate (4a-i)
To 1h (105 mg, 0.15 mmol) in dichloromethane (1.5 mL), was added EDC. HCl (87 mg, 0.45 mmol), DMAP (22 mg, 0.16 mmol) and 1-naphthol (26 mg, 0.16 mmol). The reaction mixture was stirred at room temperature overnight. Upon completion, the reaction mixture was diluted with water (2 mL) and extracted thrice with dichloromethane (5 mL × 3). The crude mixture was purified by flash chromatography on silica gel to give 4a-i (36 mg, 29% yield) as a white solid: m.p. 97 - 99°C; 1H NMR (500 MHz, acetone-d6) δ 2.43 (s, 3H), 2.48 (s, 3 H), 3.55 (s, 3 H), 3.58 (s, 3 H), 4.13 (dd, J = 10.5, 7.5 Hz, 1 H), 4.58 (dd, J = 10.5, 7.5 Hz, 1 H), 4.68 (m, 2 H), 6.56 (m, 1 H) 7.22 – 7.09 (m, 5H), 7.28 (m, 1 H), 7.34 – 7.54 (m, 8 H), 7.69 – 7.74 (m, 4 H), 7.78 (d, J = 8.3 Hz, 1 H), 7.89 - 7.94 (m, 1 H); 13C NMR (126 MHz, acetone-d6) δ20.64, 20.69, 41.34, 41.83, 46.51, 46.53, 55.10, 55.26, 112.84, 113.36, 117.99, 119.59, 119.98, 121.24, 123.55, 123.93, 125.59, 125.79, 126.30, 126.42, 126.77, 127.80, 128.40, 128.48, 129.57, 129.58, 133.31, 133.53, 134.56, 137.31, 137.75, 139.61, 139.71, 145.41, 146.66, 151.59, 152.09, 170.40, 171.93; HRMS (ESI) m/z calculated for C44H38O12S2H+: 823.1867, found 823.1877 (Δ = 1.21 ppm).
4.4.3.32. 1-Naphthyl α-3-hydroxycarbonyl-2,4-di(3-methoxy-4-hydroxy)cyclobutane-1-carboxylate (4a)
To a solution of 4a-i (71 mg, 0.085 mmoles), in MeOH (9.0 ml), was added 20% sodium amalgam (283 mg) and the reaction mixture was stirred at room temperature for 4 h. The reaction was quenched by 12 ml saturated aqueous NaHCO3, and the mixture was extracted with AcOEt (5 × 20 ml). The combined organic layers were washed with brine (5 ml), dried over MgSO4, and concentrated in vacuo. The crude mixture was purified using flash column chromatography on silica gel with 2% methanol in dichloromethane as the eluent to give 4a (18 mg, 40 % yield) as a white solid: m.p. >230°C; 1H NMR (500 MHz, acetone-d6) δ 3.77 (s, 3 H), 3.89 (s, 3 H), 3.90 – 3.94 (m, 1 H), 4.28 (t, J = 10.0 Hz, 1H), 4.51 (t, J = 10.0 Hz, 1 H), 4.73 (t, J = 10.0 Hz, 1 H), 6.72 (d, J = 7.5 Hz, 1H), 6.86 (m, 2 H), 6.95 – 7.03 (m, 3 H), 7.13 (m, 1 H), 7.18 (m, 1 H), 7.33 – 7.27 (m, 1H), 7.37 (t, J = 7.5 Hz, 1 H), 7.46 (t, J = 7.5 Hz, 1 H), 7.50 (s, 1 H), 7.70 (s, 1 H), 7.74 (d, J = 8.0 Hz, 1 H), 7.87 (d, J = 8.0 Hz, 1 H); 13C NMR (125 MHz, acetone-d6) δ 41.91, 44.34, 46.89, 47.22, 55.39, 110.40, 112.65, 114.83, 115.03, 118.01, 119.19, 121.44, 122.13, 125.31, 125.67, 126.11, 126.27, 126.93, 127.57, 134.50, 144.80, 146.12, 146.97, 147.16, 170.48, 171.89; HRMS (ESI) m/z calculated for C30H26O8H+: 515.1706, found 515.1717 (Δ = − 2.14 ppm).
In a manner similar to that described for the synthesis of 4a-i, 4d-i was synthesized from the corresponding α-truxillic acids 1j and characterized.
4.4.3.33. 1-Naphthyl α-3-hydroxycarbonyl-2,4-di(4-tert-butyldimethylsilylphenyl)- cyclobutane-1-carboxylate (4d-i)
To 1j (200 mg, 0.36 mmol) in DCM (15 mL) was added DMAP (3 mg, 0.026 mmol) and EDC (50 mg, 0.26 mmol), and the reaction was stirred at room temperature for 15 min. 1-naphthol in DCM (5 mL) was added to the solution by syringe pump at 2 ml/hr and the reaction was stirred at room temperature for overnight. The reaction mixture was then concentrated in vacuo. The crude mixture was purified using flash column chromatography on silica gel with 30% ethyl acetate in hexanes as the eluent to give 4d-i (94 mg, 64%) as a white solid; m.p. 84-86°C; 1H NMR (500 MHz, CDCl3) δ0.19 (s, 6H), 0.23 (d, J = 4.1 Hz, 6 H), 0.99 (s, 9 H), 4.05 (dd, J = 10.5, 7.5 Hz, 1 H), 4.32 (dd, J = 10.5, 7.5 Hz, 1 H), 4.52 - 4.63 (m, 2 H), 6.42 (d, J = 7.5 Hz, 1 H), 6.88 (m, 4 H), 7.31 – 7.23 (m, 5 H), 7.36 (m, 3 H), 7.44 (t, J = 7.5 Hz, 1 H), 7.65 (d, J = 8.2 Hz, 1 H), 7.80 (d, J = 8.2 Hz, 1 H). 13C NMR (125 MHz, CDCl3) δ-4.41, -4.36, 18.22, 25.70, 40.99, 41.23, 46.87, 47.46, 117.72, 120.25, 120.32, 121.17, 125.20, 125.85, 126.28, 126.30, 126.59, 127.72, 128.67, 129.19, 130.92, 134.42, 146.36, 154.99, 155.34, 170.79, 175.87, 207.48; HRMS (ESI) m/z: calcd for C40H50O6Si2H+, 683.3219, found, 683.3215 (Δ = 0.59 ppm).
4.4.3.34. 1-Naphthyl α-3-hydroxycarbonyl-2,4-di(4-hydroxylphenyl)cyclobutane-1-carboxylate (4d)
To a solution of 4d-i (94 mg, 0.14 mmol) in THF (10 mL) and acetic acid (0.5 mL) was added 1 M TBAF (0.5 mL) and the mixture was stirred overnight at room temperature. The reaction mixture was concentrated and purified by flash chromatography 25% ethyl acetate and 0.1% acetic acid in hexanes as eluent to give 4d (48 mg, 76 %) as white solid; m.p. >230°C; 1H NMR (300 MHz, acetone-d6) δ 4.00 (dd, J= 10.0, 7.5 Hz, 1H), 4.40 (dd, J= 10.0, 7.5 Hz, 1H), 4.62-4.51 (m, 2H), 6.61 (d, J= 7.5 Hz, 1H), 6.88 (q, J= 7.6 Hz, 4H), 7.15 (d, J= 8.5 Hz, 1H), 7.49-7.43 (m, 7H), 7.73 (d, J= 8.1 Hz, 1H), 7.87 (d, J= 8.2 Hz, 1H), 8.30 (s, 1H), 8.49 (s, 1H); 13C NMR (101 MHz, acetone-d6) δ 40.97, 41.40,47.03, 47.40, 115.06, 115.54, 118.05, 121.56, 125.25, 125.62, 126.14, 126.24, 126.93, 127.56, 128.99, 129.42, 129.99, 130.25, 134.47, 146.95, 156.45, 156.94, 170.85, 172.27; HRMS (ESI) m/z: calcd for C28H22O6H+, 455.1489, found, 455.1491 (Δ = 0.43 ppm).
In a manner similar to that described for the synthesis of 3a, the following α-truxillic acid mono esters bearing substituted phenyl moieties, 4b, 4c and 4e ~ 4k, were synthesized from the corresponding α-truxillic acids and characterized.
4.4.3.35. 1-Naphthyl α-3-hydroxycarbonyl-2,4-di(2-methoxylphenyl)cyclobutane-1-carboxylate (4b)
White solid; 35% yield; m.p. 184-185°C; 1H NMR (500 MHz, acetone-d6) δ 3.90 (s, 1 H), 4.16 - 4.21 (dd, J = 8.00, 10.50 Hz, 1 H), 4.47 (dd, J = 8.00, 10.50 Hz, 1 H), 4.86 (dd, J = 8.00, 10.50 Hz, 1 H), 4.86 (dd, J = 8.00, 10.50 Hz, 1 H), 4.93 (dd, J = 8.00, 10.50 Hz, 1 H), 6.47 (d, J = 7.5 Hz, 1 H), 7.01 - 7.04 (m, 2 H), 7.07 - 7.12 (m, 2 H), 7.28 - 7.31 (td, J = 4.45, 24 Hz, 1 H), 7.33 - 7.36 (t, J = 7.70 Hz, 1 H), 7.39 - 7.41(m, 3H), 7.47 - 7.52 (m, 2 H), 7.54 - 7.56 (d, J = 7.00 Hz, 1 H), 7.72 - 7.74 (d, J = 8.00 Hz, 1 H), 7.88 - 7.89 (d, J = 8.00 Hz, 1 H); 13C NMR (125 MHz, CDCl3) δ 37.33, 37.62, 44.32, 45.46, 55.12, 55.49, 110.32, 110.48, 117.59, 120.74, 125.30, 134.41, 146.62, 157.55, 157.78, 171.30, 177.82. HRMS (ESI) m/z: calcd for C30H26O6H+, 483.1802; found, 483.1807 (Δ = 1.03 ppm).
4.4.3.36. 1-Naphthyl α-3-hydroxycarbonyl-2,4-di(2-nitrophenyl)cyclobutane-1-carboxylate (4c)
White solid; 38% yield; m.p. 189-190°C; 1H NMR (500 MHz, DMSO-d6) δ 4.37 (s, 1 H). 4.68 (s, 1 H), 4.89 (s, 1 H), 5.04 (s, 1 H), 7.79 (m, 15 H), 13.23 (s, 1 H); 13C NMR (125 MHz, DMSO-d6) δ 40.3, 40.7, 42.2, 43.0, 118.1, 121.3, 124.5, 125.7, 126.4, 128.0, 129.6, 132.2, 132.4, 133.0, 146.3, 148.9, 171.1, 173.6; HRMS (ESI) m/z: calcd for C28H20N2O8H+, 513.1292; found, 513.1302 (Δ = 1.84 ppm).
4.4.3.37. (1R,2S)-2-Phenylcyclohexyl α-3-hydroxycarbonyl-2,4-di(2-methoxylphenyl)-cyclobutane-1-carboxylate (4e)
White solid; 48 % yield; m.p. 163-165°C; 1H NMR (500 MHz, CDCl3) δ 0.72 (m, 1 H), 1.27 (m, 2 H), 1.39 - 1.56 (m, 2 H), 1.66 (m, 2 H), 1.82 (m, 1 H), 2.51 - 2.40 (m, 1 H), 3.60 (s, 3 H), 3.67 (s, 4 H), 3.81 (dd, J = 10.5, 6.2 Hz, 1 H), 3.85 - 3.93 (m, 1 H), 4.39 - 4.29 (m, 1 H), 4.69 (dd, J = 10.7, 6.6 Hz, 1 H), 6.71 (d, J = 8.1 Hz, 1 H), 6.76 (d, J = 8.1 Hz, 1 H), 6.82 (t, J = 7.4 Hz, 1 H), 6.87 (d, J = 6.8 Hz, 1 H), 6.94 (t, J = 7.4 Hz, 1 H), 7.09 (dd, J = 16.6, 7.2 Hz, 3 H), 7.13 - 7.24 (m, 5 H). 13C NMR (125 MHz, CDCl3) δ 24.7, 25.8, 31.4, 37.2, 44.5, 45.8, 54.9, 55.2, 76.0, 110.0, 110.1, 120.2, 126.4, 127.5, 128.0, 143.2, 157.3, 171.6, 178.5. HRMS (ESI) m/z: calcd for C32H35O6H+, 515.2428, found, 515.2434 (Δ = 1.2 ppm).
4.4.3.38. (1R,2S)-2-Phenylcyclohexyl α-3-hydroxycarbonyl-2,4-di(2-chlorophenyl)-cyclobutane-1-carboxyliate (4f)
White solid; 30% yield; m.p. 163-165°C; 1H NMR (500 MHz, CDCl3) δ1.32 - 1.6 (m, 4 H), 1.80 (d, J = 12.7 Hz, 1 H), 1.85 - 2.02 (m, 2 H), 2.15 - 2.33 (m, 1 H), 2.64 - 2.81 (m, 1 H), 3.52 (t, J = 8.4 Hz, 1 H), 3.50 - 3.77 (m, 2 H), 4.48 (dd, J = 16.4, 9.8 Hz, 1 H), 4.75 - 4.89 (m, 1 H), 5.16 (dt, J = 17.2, 7.6 Hz, 1 H), 6.78 - 6.96 (m, 2 H), 6.99 (t, J = 7.5 Hz, 1 H), 7.02 - 7.11 (m, 3 H), 7.13 - 7.31 (m, 7 H). 13C NMR (125 MHz, CDCl3) δ 24.9, 25.9, 32.2, 41.5, 49.7, 49.9, 126.2, 126.6, 127.5, 127.7, 128.0, 128.5, 129.7, 134.7, 143.0, 171.0. HRMS (ESI) m/z: calcd for C30H29Cl2O4H+, 523.1437, found, 523.1443 (Δ = 0.10 ppm).
4.4.3.39. (1R,2S)-2-Phenylcyclohexyl α-3-hydroxycarbonyl-2,4-di(2,6-dichlorophenyl)-cyclobutane-1-carboxylate (4g)
White solid; 30% yield; m.p. 209-210°C; 1H NMR (700 MHz, CDCl3) δ1.40 (dd, J = 14.4, 11.3 Hz, 1 H), 1.44 - 1.61 (m, 3 H), 1.79 (d, J = 13.2 Hz, 1 H), 1.88 (d, J = 10.1 Hz, 1 H), 1.97 (d, J = 13.4 Hz, 1 H), 2.18 - 2.26 (m, 1 H), 2.76 (td, J = 12.1, 3.6 Hz, 1 H), 4.45 - 4.52 (m, 1 H), 4.70 - 4.78 (m, 1 H), 5.04 (dd, J = 11.6, 9.3 Hz, 1 H), 5.21 (td, J = 10.4, 4.2 Hz, 1 H), 5.27 (dd, J = 11.8, 8.1 Hz, 1 H), 6.95 (t, J = 8.0 Hz, 1 H), 6.99 (t, J = 8.0 Hz, 1 H), 7.04 (d, J = 8.0 Hz, 2 H), 7.12 (d, J = 7.5 Hz, 2 H), 7.20 (t, J = 7.1 Hz, 1 H), 7.30 - 7.25 (m, 5 H). 13C NMR (175 MHz, CDCl3) δ 25.0, 25.9, 32.1, 42.3, 49.8, 76.7, 126.6, 127.8, 128.1, 128.5, 129.0, 134.1, 134.3, 143.3, 171.5, 177.5. HRMS (ESI) m/z: calcd for C30H27Cl4O4H+ 591.0658, found 591.0661 (Δ = 0.50 ppm).
4.4.3.40. (1R,2S)-2-Phenylcyclohexyl α-3-hydroxycarbonyl-2,4-di(2-bromophenyl)-cyclobutane-1-carboxylate (4h)
White solid; 29% yield; m.p. 136-138°C; 1H NMR (500 MHz, acetone-d6) δ 1.33 - 1.44 (m, 1 H), 1.44 - 1.54 (m, 2 H), 1.62 (dd, J = 12.9, 3.1 Hz, 1 H), 1.76 (d, J = 12.9 Hz, 1 H), 1.86 (dd, J = 28.6, 11.6 Hz, 2 H), 2.09 (s, 1 H), 2.16 (d, J = 9.6 Hz, 1 H), 2.71 (td, J = 12.2, 3.6 Hz, 1 H), 3.61 (dd, J = 10.1, 7.5 Hz, 1 H), 3.74 (dd, J = 10.0, 7.6 Hz, 1 H), 4.44 – 4.54 (m, 1 H), 4.82 (dd, J = 9.6, 7.7 Hz, 1 H), 5.10 (td, J = 10.2, 4.0 Hz, 1 H), 7.01 (dd, J = 12.9, 7.1 Hz, 3 H), 7.11 (dt, J = 15.3, 7.4 Hz, 4 H), 7.21 (d, J = 3.9 Hz, 4 H), 7.34 (d, J = 7.7 Hz, 1 H), 7.41 (d, J = 7.8 Hz, 1 H); 13C NMR (125 MHz, acetone-d6) δ 25.4, 26.5, 32.9, 34.7, 43.4, 50.4, 76.9, 125.7, 125.9, 127.2, 127.9, 128.4, 129.1, 139.0, 144.1, 171.9, 173.2; HRMS (ESI) m/z: calcd for C30H29Br2O4H+, 611.0427, found, 611.0424 (Δ = 0.49 ppm).
4.4.3.41. (1R,2S)-2-Phenylcyclohexyl α-3-hydroxycarbonyl-2,4-di(2-nitrophenyl)-cyclobutane-1-carboxylate (4i)
White solid; 36% yield; m.p. 200-202°C; 1H NMR (500 MHz, acetone-d6) δ 1.35 - 1.45 (m, 1 H), 1.49 (dd, J = 21.7, 11.8 Hz, 2 H), 1.64 (dd, J = 12.9, 2.9 Hz, 1 H), 1.77 (d, J = 12.9 Hz, 1 H), 1.86 (dd, J = 25.5, 11.5 Hz, 2 H), 1.96 (s, 1 H), 2.70 (td, J = 12.2, 3.4 Hz, 1 H), 3.73 (dd, J = 10.4, 6.0 Hz, 1 H), 3.92 (t, J = 9.6 Hz, 1 H), 4.76 (t, J = 9.4 Hz, 1 H), 4.85 (dd, J = 10.0, 6.0 Hz, 1 H), 5.01 - 5.13 (m, 1 H), 7.14 (d, J = 7.6 Hz, 2 H), 7.20 (d, J = 4.1 Hz, 4 H), 7.34 - 7.25 (m, 2 H), 7.37 (t, J = 7.4 Hz, 2 H), 7.52 (t, J = 7.5 Hz, 1 H), 7.68 (d, J = 7.8 Hz, 1 H), 7.72 (d, J = 8.0 Hz, 1 H); 13C NMR (125 MHz, acetone-d6) δ 25.4, 26.5, 32.8, 34.4, 40.9, 50.5, 77.3, 125.2, 125.5, 127.2, 128.5, 128.8, 129.1, 130.1, 130.2, 134.0, 144.0, 149.7, 151.0, 171.4; HRMS (ESI) m/z: calcd for C30H29N2O8H+, 545.1918, found, 545.1921 (Δ = 0.55 ppm).
4.4.3.42. 9-Fluorenylmethyl α-3-hydroxycarbonyl-2,4-di(2-methoxylphenyl)cyclobutane-1-carboxylate (4j)
White solid; 28% yield; m.p. 160-162°C; 1H NMR (500 MHz, CDCl3) δ 3.76 (d, J = 11.6 Hz, 4 H), 3.80 (s, 3 H), 3.96 (dd, J = 10.6, 8.0 Hz, 1 H), 4.06 (dd, J = 10.4, 7.8 Hz, 1 H), 4.17 – 4.10 (m, 2 H), 4.66 (dd, J = 10.5, 7.1 Hz, 1 H), 4.72 (dd, J = 10.4, 7.8 Hz, 1 H), 6.82 (d, J = 8.1 Hz, 1 H), 6.86 (d, J = 8.1 Hz, 1 H), 7.00 (q, J = 7.1 Hz, 2 H), 7.24 (t, J = 7.2 Hz, 1 H), 7.36 – 7.27 (m, 5 H), 7.41 (t, J = 6.9 Hz, 3 H), 7.51 (d, J = 7.5 Hz, 1H), 7.77 (d, J = 7.5 Hz, 2 H). 13C NMR (125 MHz, CDCl3) δ 37.1, 37.7, 44.6, 46.9, 55.2, 55.3, 66.4, 110.1, 110.3, 120.0, 125.1, 127.1, 127.7, 141.2, 141.3144.0, 144.4, 157.5, 157.6, 172.6, 178.6. HRMS (ESI) m/z: calcd for C34H30O6H+, 535.2115; found, 535.2115 (Δ = 0.05 ppm).
4.4.3.43. 9-Fluorenylmethyl α-3-hydroxycarbonyl-2,4-di(2-chlorophenyl)cyclobutane-1-carboxylate (4k)
White solid; 55% yield; m.p. 187-188°C; 1H NMR (500 MHz, acetone-d6), δ 4.01 (dd, J = 9.7, 7.0 Hz, 1 H), 4.10 - 4.20 (m, 1 H), 4.23 (t, J = 7.0 Hz, 1 H), 4.38 (p, J = 10.6 Hz, 2 H), 4.60 - 4.70 (m, 1 H), 4.70 - 4.82 (m, 1 H), 7.04 - 7.20 (m, 4 H), 7.26 (dt, J = 24.3, 8.7 Hz, 4 H), 7.34 (d, J = 6.2 Hz, 1 H), 7.37 - 7.46 (m, 3H), 7.62 (t, J = 7.5 Hz, 2H), 7.87 (dd, J = 7.1, 3.2 Hz, 2 H), 12.74 (s, 1 H); 13C NMR (125 MHz, acetone-d6) δ 41.2, 41.6, 42.5, 46.2, 66.2, 120.2, 125.2, 133.5, 140.7, 143.4, 143.6, 172.1, 173.7. HRMS (ESI) m/z: calcd for C32H25Cl2O4H+, 543.1124, found, 543.1123 (Δ = 0.18 ppm).
4.4.3.44. Quinolin-5-yl α-3-hydroxycarbonyl-2,4-di(2-methoxylphenyl)cyclobutane-1-carboxylate (4l)
White solid; m.p. >230°C; 1H NMR (500 MHz, acetone-d6) δ 3.95 (dd, J = 10.4, 8.0 Hz, 1H), 4.41 (dd, J = 10.4, 8.0 Hz, 1 H), 4.65 (dd, J = 10.4, 8.0 Hz, 1 H), 4.74 (dd, J = 10.4, 8.0 Hz, 1H), 6.53 (d, J = 7.5 Hz, 1H), 7.00 – 7.03 (m, 2H), 7.06 (t, J = 7.5 Hz, 1H), 7.10 (d, J = 8.4 Hz, 1H), 7.28 (m, 1 H), 7.35 – 7.44 (m, 3 H), 7.49 (t, J = 7.0 Hz, 2 H), 7.61 (t, J = 8.3 Hz, 1 H), 7.85 (d, J = 8.3 Hz, 1 H), 8.85 (d, J = 5.0, 1 H), 12.06 (s, 1 H); 13C NMR (175 MHz, acetone-d6) δ 36.64, 36.72, 44.87, 45.34, 55.89, 56.08, 111.12, 111.42, 118.73, 120.65, 121.09, 122.12, 127.25, 127.50, 127.53, 127.84, 127.95, 128.60, 129.11, 129.35, 130.24, 146.35, 148.51, 151.40, 157.71, 157.93, 171.64, 173.52; HRMS (ESI) m/z calcd for C29H25NO6H+: 484.1755, found 484.1763 (Δ = 1.66 ppm).
4.4.3.45. 1-[4-(5,6,7,8-tetrahydronaphth-2-yl)thiazol-2-yl] α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxamide (6a)
White solid; 43% yield; m.p. >230°C; 1H NMR (500 MHz, acetone-d6) δ1.31 (s, 1 H), 1.79 (m, 4 H), 2.79 - 2.72 (m, 4 H), 4.12 (dd, J = 10.5, 7.4 Hz, 1 H), 4.37 (dd, J = 10.5, 6.8 Hz, 1H), 4.59 (dd, J = 10.5, 7.4 Hz, 1H), 4.66 (dd, J = 10.5, 6.8 Hz, 1 H), 7.04 (d, J = 8.5 Hz, 1H), 7.13 (t, J = 7.4 Hz, 1H), 7.26 (m, 4 H), 7.36 (m, 2 H), 7.43 (m, 2 H), 7.48 (m, 2 H), 7.54 (m, 2 H), 10.98 (s, 1H); 13C NMR (126 MHz, acetone-d6) δ23.07, 23.09, 40.64, 42.32, 46.45, 47.46, 106.20, 123.02, 126.45, 126.77, 126.80, 127.69, 127.79, 128.17, 128.23, 129.16, 132.05, 136.48, 136.87, 139.02, 139.66, 149.67, 157.33, 169.46, 172.35; HRMS (ESI) m/z calcd for C31H28N2O3SH+, 509.1893, found 509.1092 (Δ = 1.61 ppm).
4.4.3.46. 1-N-(Biphenyl-4-yl) α-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxamide (6b)
White solid; 40% yield; m.p. > 230°C; 1H NMR (700 MHz, acetone-d6) δ 3.84 (t, J = 10.4 Hz, 1 H), 3.88 (t, J = 10.4 Hz, 1 H), 4.42 (t, J = 10.4 Hz, 1 H), 4.92 (t, J = 10.4 Hz, 1 H), 7.14 (t, J = 7.5 Hz, 1H), 7.24 (t, J = 7.5 Hz, 3 H), 7.30 (t, J = 7.3 Hz, 1 H), 7.36 (t, J = 7.7 Hz, 2 H), 7.41 (t, J = 7.7 Hz, 3 H), 7.48 - 7.43 (m, 9 H), 7.50 (s, 1 H), 7.58 (d, J = 7.0 Hz, 2 H), 10.67 (s, 1H); 13C NMR (175 MHz, acetone-d6) δ40.66, 44.77, 45.65, 48.27, 119.66, 119.76, 119.81, 126.30, 126.41, 126.60, 126.83, 127.87, 128.31, 128.74, 128.94, 135.70, 138.06, 138.46, 140.50, 143.17, 168.72, 172.04; HRMS (ESI) m/z calcd for C30H25NO3H+: 448.1913, found 448.1914 (Δ = 0.22 ppm).
4.4.3.47. 1-[4-(5,6,7,8-tetrahydronaphth-2-yl)thiazol-2-yl] γ-3-hydroxycarbonyl-2,4-diphenylcyclobutane-1-carboxamide (6a-γ)
To a solution of γ-truxillic anhydride[11] (100 mg, 0.36 mmol) and 6-acetamido-1-naphthol (80 mg, 0.40 mmol) in THF (2.0 ml), was added diisoproplethylamine (DIPEA) (0.06 ml, 0.40 mmol) dropwise. The reaction mixture was stirred at room temperature overnight. Upon completion, the reaction mixture was diluted with water (2 mL) and extracted thrice with dichloromethane (10 mL x 3). The crude mixture was purified by flash chromatography on silica gel (CH2Cl2/MeOH, 95/5) to give 3o-γ (36 mg, 82% yield) as a white solid: m.p. 165°C (decomp.); 1H NMR (500 MHz, acetone-d6) δ 2.04 (s, 3 H), 3.90 (t, J = 9.8 Hz, 1 H), 4.28 (t, J = 9.8 Hz, 1 H), 4.52 (t, J = 9.8 Hz, 1 H), 4.74 (t, J = 9.8 Hz, 1 H), 6.30 (d, J = 7.6 Hz, 1H), 6.89 (d, J = 9.0 Hz, 1 H), 7.21 (dd, J = 14.6, 7.0 Hz, 2H), 7.37 - 7.26 (m, 6 H), 7.43 - 7.55 (m, 5 H), 8.27 (s, 1 H), δ 9.24 (s, 1H); 13C NMR (176 MHz, acetone-d6) δ23.46, 42.27, 44.65, 46.32, 46.43, 114.76, 116.40, 120.03, 121.92, 123.36, 125.18, 125.80, 126.70, 126.72, 126.73, 127.43, 128.48, 128.59, 129.06, 135.17, 137.56, 138.60, 142.18, 146.71, 168.24, 170.20, 171.70; HRMS (ESI) m/z calcd for C30H26NO5H+: 480.1811, found 480.1809 (Δ = 0.45 ppm).
4.4.4. Computational Study
In order to optimize the activity of 3-analogs, the docking program AutoDock 4.2 was used. Analogs were drawn in the Perkin Elmer program ChemDraw and then had their internal energies optimized by the program Avogadro[43]. The force field chosen for this was the Merck Molecular Force Field (MMFF94) [44]. Docking was performed with the default parameters based on the Lamarckian genetic algorithm of AutoDock 4.2. Analysis of predicted binding poses and their associated scores was performed in the program UCSF Chimera [45]. Analogs with high similarity in predicted binding mode to 3 were prioritized for synthesis. Analysis of the selectivity of truxillic acid congeners for FABP isoforms additionally employed the local search algorithm of AutoDock 4.2 [46, 47].
Crystal structures (PDB ID: 5UR9 for FABP5/3; 5URA for FABP7/3; 6AQ1 for FABP3 apoprotein) were processed by first adding polar hydrogens to reflect a pH of 7.4 via AutoDock Tools. The Gasteiger function was applied to assign partial atomic charges to a rigid receptor. Additionally, all water molecules and ligands were deleted from the binding site model. The portion of the binding site chosen for sampling was based on determining a region that would result in low RMSD to the crystal structure binding pose, or in the case of FABP3 apoprotein, the region that produced results with the canonical interaction as the top score. The AutoDock program autogrid was then used to generate an energy potential map of various atoms in the binding site. The grid files were used for docking calculations.
4.4.5. Biological Evaluations
4.4.5.1. Protein purification
Recombinant human FABP3, FABP4, FABP5, and FABP7 were expressed as N-terminal His-tagged proteins using a pET-28a vector (Novagen, Madison, WI, USA). The protein purification and delipidation was performed as previously described [1].
4.4.5.2. Direct ligand binding assay
Measurement of direct binding of the fluorescent probes to purified FABPs was performed as described previously.[11, 48] NBD-stearate affinity towards FABP3, FABP5, and FABP7 was determined in a previous study (Kd = .018 μM, 0.16 μM, and 0.22 μM, respectively).[11] Incubation of NBD-stearate with a subset of tested compounds (3k, 3p, 3q, 4j and 4k) was found to yield unexpectedly high non-specific fluorescence in the absence of protein, therefore the alternative fluorophores DAUDA (for FABP3 and FABP5; Kd = 2.04 μM and 2.56 μM, respectively) and ANS (FABP7; Kd = 1.01 μM) were utilized.
4.4.5.3. Fluorescence displacement binding assay
Purified FABPs (3 μM) were incubated with fluorescent probe (500 nM) in 30 mM Tris, 100 mM NaCl buffer (pH 7.5). Compounds to be tested were then added to the wells (0.01-50 μM) and the system was allowed to reach equilibrium by incubating in the dark at room temperature for 20 minutes. Each independent assay included wells containing a strong competitive ligand (arachidonic acid, 10 μM) as a positive control for probe displacement. Loss of fluorescence intensity was monitored with a F5 Filtermax Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA, USA) using excitation and emission wavelengths appropriate for each respective probe (NBD-stearate ex./em. = 465/535 nm, DAUDA ex./em. = 345/535 nm, ANS ex./em. = 370/465 nm). Following background subtraction, the fluorescence intensity values were normalized and fit to a one-site binding analysis using the GraphPad Prism software (Prism version 7.0 for Mac OS, Graphpad Software Inc., La Jolla, CA, USA) to determine the Ki of the tested compounds from the equation Ki = IC50/(1 + ([Probe]/Kd).
4.4.5.4. Dynamic Light Scattering
Small molecule colloidal aggregation was measured by Protein Solutions DynaPro 99 dynamic light-scattering instrument (Wyatt Technology, Santa Barbara, CA) at 24°C. Compounds were dissolved to 10 mM in DMSO and diluted with filtered PBS to a final concentration of 20 μM. Data were analyzed with the Dynamics V5.25.44 program supplied by Wyatt Technology.
4.4.5.5. CFA-induced paw edema and thermal hyperalgesia
All procedures involving vertebrate animals were approved by the Institutional Animal Care and Use Committee (IACUC) at Stony Brook University. The experiments were conducted on male wild-type C57BL/6 mice or FABP5-KO mice (bred on a C57BL/6 background). Mice were acclimated to the testing room, equipment, and experimenter for at least two days before behavioral testing. Paw edema was induced by injecting complete Freund’s adjuvant (CFA) (20 μl, in sterile saline) into the plantar surface of the right hind paw using a 30-gauge needle attached to a tuberculin syringe (Becton, Dickinson and Company). On day 5, thermal hyperalgesia was measured using the Hargreaves plantar apparatus (Ugo Basile) as previously reported.[11, 33] Measurements were taken prior to the administration of drug, and at 90 minutes post-administration. 3, 3a, 4a, and 3l-A were dissolved in 1:1:8 DMSO: Cremophor-EL:saline. Compound 5f was dissolved in 1:2:7 DMSO:Solutol:saline with gentle heating. Drugs were administered via the intraperitoneal route at a dose of 40 mg/kg in a volume of 10 μl/g, with the exception of 3l-A which was administered at 20 mg/kg.
Supplementary Material
Highlights for review.
-
➢
Novel anti-nociceptive and anti-inflammatory agents with high potency.
-
➢
Fatty acid binding proteins (FABPs) as target.
-
➢
Novel α-truxillic acid mono esters as promising inhibitors of FABPs.
-
➢
Computer-aided drug design based on FABP-inhibitor cocrystal structure.
-
➢
SAR analysis of FABP inhibitors based on in vitro and in vivo assays.
Acknowledgments
This work was supported by a grant (DA035923) from the National Institutes of Health. The authors thank Dr. Bela Ruzsicska, Institute of Chemical Biology and Drug Discovery, and Robert Rieger, Proteomics Center, of the Stony Brook University for mass spectrometry analyses. The authors thank Mr. Glenn Yu and Ms. Sarah Lee and Mr. Alby Joseph for their technical assistance for docking studies as well as chemical synthesis. The authors also thank Dr. Gökhan Hotamisligil for graciously providing the FABP5-KO mice.
Abbreviations
- FABP
fatty acid binding protein
- FABP3
heart fatty acid binding protein
- FABP5
epidermal fatty acid binding protein
- FABP7
brain fatty acid binding protein
- FAAH
fatty acid amide hydrolase
- NAE
N-acyl ethanolamines
- AEA
arachidonoyl ethanolamide
- CB1R
cannabinoid receptor type 1
- TBAF
tetra-n-butylammonium fluoride
- EDC
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride
- CFA
complete Freund’s adjuvant
Appendix A: Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.ejmech.2018.04.050.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kaczocha M, Vivieca S, Sun J, Glaser ST, Deutsch DG. Fatty acid-binding proteins transport N-acylethanolamines to nuclear receptors and are targets of endocannabinoid transport inhibitors. J Biol Chem. 2012;287:3415–3424. doi: 10.1074/jbc.M111.304907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elmes MW, Kaczocha M, Berger WT, Leung K, Ralph BP, Wang L, Sweeney JM, Miyauchi JT, Tsirka SE, Ojima I, Deutsch DG. Fatty acid-binding proteins (FABPs) are intracellular carriers for Delta9-tetrahydrocannabinol (THC) and cannabidiol (CBD) J Biol Chem. 2015;290:8711–8721. doi: 10.1074/jbc.M114.618447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deutsch DG. A personal retrospective: elevating anandamide (AEA) by targeting fatty acid amide hydrolase (FAAH) and the fatty acid binding proteins (FABPs) Front Pharmacol. 2016;7:370. doi: 10.3389/fphar.2016.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaczocha M, Glaser ST, Deutsch DG. Identification of intracellular carriers for the endocannabinoid anandamide. Proc Natl Acad Sci. 2009;106:6375–6380. doi: 10.1073/pnas.0901515106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain. 2004;109:319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 6.Kaczocha M, Rebecchi MJ, Ralph BP, Teng YH, Berger WT, Galbavy W, Elmes MW, Glaser ST, Wang L, Rizzo RC, Deutsch DG, Ojima I. Inhibition of fatty acid binding proteins elevates brain anandamide levels and produces analgesia. PLoS One. 2014;9:e94200. doi: 10.1371/journal.pone.0094200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schlosburg JE, Kinsey SG, Lichtman AH. Targeting fatty acid amide hydrolase (FAAH) to treat pain and inflammation. AAPS J. 2009;11:39–44. doi: 10.1208/s12248-008-9075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li GL, Winter H, Arends R, Jay GW, Le V, Young T, Huggins JP. Assessment of the pharmacology and tolerability of PF-04457845, an irreversible inhibitor of fatty acid amide hydrolase-1, in healthy subjects, Br. J Clin Pharmacol. 2012;73:706–716. doi: 10.1111/j.1365-2125.2011.04137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Otrubova K, Ezzili C, Boger DL. The discovery and development of inhibitors of fatty acid amide hydrolase (FAAH) Bioorg Med Chem Lett. 2011;21:4674–4685. doi: 10.1016/j.bmcl.2011.06.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Owada Y, Yoshimoto T, Kondo H. Spatio-temporally differential expression of genes for three members of fatty acid binding proteins in developing and mature rat brains. J Chem Neuroanat. 1996;12:113–122. doi: 10.1016/s0891-0618(96)00192-5. [DOI] [PubMed] [Google Scholar]
- 11.Berger WT, Ralph BP, Kaczocha M, Sun J, Balius TE, Rizzo RC, Haj-Dahmane S, Ojima I, Deutsch DG. Targeting fatty acid binding protein (FABP) anandamide transporters - a novel strategy for development of anti-inflammatory and anti-nociceptive drugs. PLoS One. 2012;7:e50968. doi: 10.1371/journal.pone.0050968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Binas B, Danneberg H, McWhir J, Mullins L, Clark AJ. Requirement for the heart-type fatty acid binding protein in cardiac fatty acid utilization. FASEB J. 1999;13:805–812. doi: 10.1096/fasebj.13.8.805. [DOI] [PubMed] [Google Scholar]
- 13.Nakamura M, Chi YM, Yan WM, Nakasugi Y, Yoshizawa T, Irino N, Hashimoto F, Kinjo J, Nohara T, Sakurada S. Strong antinociceptive effect of incarvillateine, a novel monoterpene alkaloid from Incarvillea sinensis. J Nat Prod. 1999;62:1293–1294. doi: 10.1021/np990041c. [DOI] [PubMed] [Google Scholar]
- 14.Chi YM, Nakamura M, Yoshizawa T, Zhao XY, Yan WM, Hashimoto F, Kinjo J, Nohara T, Sakurada S. Anti-inflammatory activities of a-truxillic acid derivatives and their monomer components. Biol Pharm Bull. 2005;28:1776–1778. doi: 10.1248/bpb.28.1776. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura M, Chi YM, Yan WM, Yonezawa A, Nakasugi Y, Yoshizawa T, Hashimoto F, Kinjo J, Nohara T, Sakurada S. Structure-antinociceptive activity studies of incarvillateine, a monoterpene alkaloid from Incarvillea sinensis. Planta Med. 2001;67:114–117. doi: 10.1055/s-2001-11512. [DOI] [PubMed] [Google Scholar]
- 16.Wang ML, Yu G, Yi SP, Zhang FY, Wang ZT, Huang B, Su RB, Jia YX, Gong ZH. Antinociceptive effects of incarvillateine, a monoterpene alkaloid from Incarvillea sinensis, and possible involvement of the adenosine system. Sci Rep. 2015;5:16107. doi: 10.1038/srep16107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chi YM, Nakamura M, Yoshizawa T, Zhao XY, Yan WM, Hashimoto F, Kinjo J, Nohara T, Sakurada S. Pharmacological study on the novel antinociceptive agent, a novel monoterpene alkaloid from Incarvillea sinensis. Biol Pharm Bull. 2005;28:1989–1991. doi: 10.1248/bpb.28.1989. [DOI] [PubMed] [Google Scholar]
- 18.Thanos PK, Clavin BH, Hamilton J, O’Rourke JR, Maher T, Koumas C, Miao E, Lankop J, Elhage A, Haj-Dahmane S, Deutsch D, Kaczocha M. Examination of the Addictive and Behavioral Properties of Fatty Acid-Binding Protein Inhibitor SBFI26. Front Psychiatry. 2016;7:54. doi: 10.3389/fpsyt.2016.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.AutoDock 4.2. http://autodock.scripps.edu/
- 20.Hsu HC, Tong S, Zhou Y, Elmes MW, Yan S, Kaczocha M, Deutsch DG, Rizzo RC, Ojima I, Li H. The antinociceptive agent SBFI-26 binds to anandamide transporters FABP5 and FABP7 at two different sites. Biochemistry. 2017;56:3454–3462. doi: 10.1021/acs.biochem.7b00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards MP, Price DA. Role of physicochemical properties and ligand lipophilicity efficiency in addressing drug safety risks. Annu Rep Med Chem. 2010;45:380–391. [Google Scholar]
- 22.Leeson PD, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov. 2007;6:881. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- 23.ChemDraw 15. http://www.cambridgesoft.com/news/details/?News=191.
- 24.Cohen M, Schmidt G. 383. Topochemistry. Part I. A survey. J Chem Soc. 1964:1996–2000. [Google Scholar]
- 25.Ichikawa M, Takahashi M, Aoyagi S, Kibayashi C. Total synthesis of (−)-incarvilline,(+)-incarvine C, and (−)-incarvillateine. J Am Chem Soc. 2004;126:16553–16558. doi: 10.1021/ja0401702. [DOI] [PubMed] [Google Scholar]
- 26.ChemBio 3D Ultra 14.0. https://www.cambridgesoft.com/Ensemble_for_Chemistry/details/Default.aspx?fid=13&pid=668.
- 27.Chuang S, Velkov T, Horne J, Porter CJ, Scanlon MJ. Characterization of the drug binding specificity of rat liver fatty acid binding protein. J Med Chem. 2008;51:3755–3764. doi: 10.1021/jm701192w. [DOI] [PubMed] [Google Scholar]
- 28.Chi YM, Nakamura M, Zhao XY, Yoshizawa T, Yan WM, Hashimoto F, Kinjo J, Nohara T, Sakurada S. Anti-inflammatory Activity of 4,4′-Dihydroxy-a-truxillic Acid. Biol Pharm Bull. 2006;29:489–493. doi: 10.1248/bpb.29.489. [DOI] [PubMed] [Google Scholar]
- 29.Chi YM, Nakamura M, Zhao XY, Yoshizawa T, Yan WM, Hashimoto F, Kinjo J, Nohara T, Sakurada S. Antinociceptive Activities of a-Truxillic Acid and b-Truxinic Acid Derivatives. Biol Pharm Bull. 2006;29:580–584. doi: 10.1248/bpb.29.580. [DOI] [PubMed] [Google Scholar]
- 30.Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008;7:489–503. doi: 10.1038/nrd2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lan H, Cheng CC, Kowalski TJ, Pang L, Shan L, Chuang CC, Jackson J, Rojas-Triana A, Bober L, Liu L, Voigt J, Orth P, Yang X, Shipps GW, Jr, Hedrick JA. Small-molecule inhibitors of FABP4/5 ameliorate dyslipidemia but not insulin resistance in mice with diet-induced obesity. J Lipid Res. 2011;52:646–656. doi: 10.1194/jlr.M012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaczocha M, Glaser ST, Maher T, Clavin B, Hamilton J, O’Rourke J, Rebecchi M, Puopolo M, Owada Y, Thanos PK. Fatty acid binding protein deletion suppresses inflammatory pain through endocannabinoid/N-acylethanolamine-dependent mechanisms. Mol Pain. 2015;11:52. doi: 10.1186/s12990-015-0056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 34.Hui X, Li H, Zhou Z, Lam KS, Xiao Y, Wu D, Ding K, Wang Y, Vanhoutte PM, Xu A. Adipocyte fatty acid-binding protein modulates inflammatory responses in macrophages through a positive feedback loop involving c-Jun NH2-terminal kinases and activator protein-1. J Biol Chem. 2010;285:10273–10280. doi: 10.1074/jbc.M109.097907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bogdan D, Falcone J, Kanjiya MP, Park SH, Carbonetti G, Studholme K, Gomez M, Lu Y, Elmes MW, Smietalo N, Yan S, Ojima I, Puopolo M, Kaczocha M. Fatty acid binding protein 5 controls microsomal prostaglandin E synthase 1 (mPGES-1) induction during inflammation. J Biol Chem. 2018;293:5295–306. doi: 10.1074/jbc.RA118.001593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peng X, Studholme K, Kanjiya MP, Luk J, Bogdan D, Elmes MW, Carbonetti G, Tong S, Gary Teng YH, Rizzo RC, Li H, Deutsch DG, Ojima I, Rebecchi MJ, Puopolo M, Kaczocha M. Fatty-acid-binding protein inhibition produces analgesic effects through peripheral and central mechanisms. Mol Pain. 2017;13 doi: 10.1177/1744806917697007. 1744806917697007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sterling T, Irwin JJ. ZINC 15–Ligand Discovery for Everyone. J Chem Inf Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O. Diffraction, P. CrysAlis, Oxford Diffraction Ltd, Yarnton, England, (2009).
- 39.Farrugia LJ. WinGX and ORTEP for Windows: an update. J Appl Crystallogr. 2012;45:849–854. [Google Scholar]
- 40.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JA, Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J Appl Crystallogr. 2009;42:339–341. [Google Scholar]
- 41.Gruene T, Hahn HW, Luebben AV, Meilleur F, Sheldrick GM. Refinement of macromolecular structures against neutron data with SHELXL2013. J Appl Crystallogr. 2014;47:462–466. doi: 10.1107/S1600576713027659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu SX, Jin HZ, Shan L, Zeng HW, Chen BY, Sun QY, Zhang WD. Inhibitory effect of 4, 4′-dihydroxy-α-truxillic acid derivatives on NO production in lipopolysaccharide-induced RAW 264.7 macrophages and exploration of structure–activity relationships. Bioorg Med Chem Lett. 2013;23:2207–2211. doi: 10.1016/j.bmcl.2013.01.091. [DOI] [PubMed] [Google Scholar]
- 43.Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J Cheminform. 2012;4:17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J Comput Chem. 1996;17:490–519. [Google Scholar]
- 45.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 46.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris GM, Huey R, Olson AJ. Using AutoDock for ligand-receptor docking. Curr Protoc Bioinformatics. 2008:14. doi: 10.1002/0471250953.bi0814s24. Chapter 8. Unit 8. [DOI] [PubMed] [Google Scholar]
- 48.Thumser AE, Evans C, Worrall AF, Wilton DC. Effect on ligand binding of arginine mutations in recombinant rat liver fatty acid-binding protein. Biochem J. 1994;297(Pt 1):103–107. doi: 10.1042/bj2970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.