

Abstract
Antigen-presenting cells (APCs) sense microorganisms via pathogen-associated molecular patterns (PAMPs) by both extra- and intracellular Toll-like Receptors (TLRs), initiating immune responses against invading pathogens. Bacterial PAMPs include extracellular lipopolysaccharides and intracellular unmethylated CpG-rich oligodeoxynucleotides (CpG). We hypothesized that a biomimetic approach involving antigen-loaded nanoparticles (NP) displaying Monophosphoryl Lipid A (MPLA) and encapsulating CpG may function as an effective “artificial bacterial” biomimetic vaccine platform. This hypothesis was tested in vitro and in vivo using NP assembled from biodegradable poly(lactic-co-glycolic acid) (PLGA) polymer, surface-modified with MPLA, and loaded with CpG and model antigen Ovalbumin (OVA). First, CpG potency, characterized by cytokine profiles, titers, and antigen-specific T cell responses, was enhanced when CpG was encapsulated in NP compared to equivalent concentrations of surface-presented CpG, highlighting the importance of biomimetic presentation of PAMPs. Second, NP synergized surface-bound MPLA with encapsulated CpG in vitro and in vivo, inducing greater pro-inflammatory, antigen-specific T helper 1 (Th1)-skewed cellular and antibody-mediated responses compared to single PAMPs or soluble PAMP combinations. Importantly, NP co-presentation of CpG and MPLA was critical for CD8+ T cell responses, as vaccination with a mixture of NP presenting either CpG or MPLA failed to induce cellular immunity. This work demonstrates a rational methodology for combining TLR ligands in a context-dependent manner for synergistic nanoparticulate vaccines.
Keywords: Nanoparticle, vaccine, PAMP, TLR, biomimetic, cellular immunity
Introduction
Despite incomplete mechanistic understanding, one of the most effective vaccines ever formulated is the live attenuated yellow fever vaccine 17D (YF-17D), inoculated in 400 million people for over 65 years. Its efficacy likely results from robust activation of Dendritic Cell (DC) subsets through Toll-like Receptors (TLRs), specifically TLR2, TLR7, and TLR9 to increase secretion of pro-inflammatory cytokines IL-12p40, IL-6, and IFN-α [1]. TLRs link innate and adaptive immunity by both inducing Antigen-Presenting Cell (APC) maturation for T cell activation and by attenuating suppressor functions of CD4+CD25+ regulatory T cells [2]. The TLR-triggering mechanism underlying this vaccine’s polyvalent immune response motivated the design of newer strategies using multiple Pathogen-Associated Molecular Patterns (PAMPs) for combinatorial TLR stimulation to overcome the failures of several commercially available vaccines to induce cellular immunity [3, 4]. For example, in both human and mouse DCs, activation of TLR3 and TLR4 together with TLR7, TLR8, and TLR9 improved and sustained T helper type 1 (Th1) responses, demonstrated by enhanced IL-12 and IL-23 [5]. Recent work has shown that mice immunized with multiple TLR ligands were protected against lethal avian and swine influenza strains, and immunized rhesus macaques were protected against H1N1 influenza [6]. Further investigations demonstrated synergistic effects of ligands for TLR4, TLR7/8, and TLR9 in rhesus macaques, showing distinct activation patterns of local and systemic innate immunity [7].
A critical component in vaccine design is the delivery vehicle, the properties of which may influence the effects of delivered antigen and adjuvants [8]. Nanoparticles (NP) encapsulating antigen(s) are promising vaccines for numerous reasons, including clinical relevance [9], flexible and reproducible control of antigen and adjuvant configurations, and APC targeting [10, 11]. Because NP-based vaccines are modular systems comprised of at least three key components: a carrier, antigen(s), and adjuvant(s), these variables, which are often well-characterized, can be reliably selected for or arranged in different ways to allow optimal immune responses to a specific pathogen. Variables such as the material and size of the core matrix, surface attachment of ligands that target DCs or epithelial cells, or ligands that protect the carrier during trafficking or facilitate drainage to lymphatics, can increase the overall efficacy of the vaccine response with lower overall quantities of antigens and adjuvants [12]. The distinguishing feature is the flexibility of these systems, such that NP can address critical issues to optimize the vaccine response, including targeting different DC subsets for tailored priming for antigen presentation [10], and delivery of complex mixtures of antigens to induce reactivity against multiple viral and tumor epitopes. Recently, NP encapsulating a pool of peptides derived from viruses and cancer oncogenes enabled antigen recognition by different HLA haplotypes and stimulation of antigen-specific immunity in response to multiple peptides [13]. NP enable the assembly of different combinations of recognition and antigen components to affect a broad-spectrum CD4 and CD8 vaccine response [14] [15]. APCs efficiently internalize NP, enabling synchronous high intracellular concentrations of antigen and adjuvant [16, 17]. Furthermore, particulate encapsulation of vaccine components protects from non-specific immune activation and proteasomal degradation and maintains proximity of antigens and adjuvants; well-designed NP vaccines facilitate antigen-cross presentation to CD4+ and CD8+ T cells while stimulating antibody-mediated immunity [14, 15].
An advantage of NP-based vaccine systems is the ability to assemble different combinations of PAMPs and antigens for optimal vaccine responses [13, 18]. Recent work has established the importance of targeting TLRs through PAMPs on the surfaces or interiors of NP [19]. Further work has shown the importance of combining TLR ligands to enhance vaccine responses [20], inducing immunity against diseases such as influenza [21], cancer [22], hepatitis B [23], and West Nile Encephalitis [24].
In this work, we sought to construct a versatile, pathogen-mimicking NP vaccine system using a logical approach that can be easily adapted to the requirements of a particular vaccine. This platform displays LPS-derived Monophosphoryl Lipid A (MPLA), a TLR4 agonist and clinically approved Th1-skewing adjuvant that is 100–10,000 times less toxic than LPS [25–27] on NP constructed from biocompatible polyester poly(lactic-co-glycolic acid) (PLGA). The cores of NP were loaded with model antigen Ovalbumin (OVA) and another adjuvant, synthetic oligodeoxynucleotides rich in unmethylated CpG motifs (CpG) that ligate endosomal TLR9 [28], facilitating targeted release of antigen and CpG into intracellular compartments. Based on demonstrations of the necessity of adjuvant and antigen to be presented within the same particle for cross-priming and in vivo CD8+ responses, NP contained both antigen and adjuvants [29, 30].
Additionally, we sought to understand the influence of combinations and context of PAMP presentation, both in terms of spatial location and proximity, on the in vitro and in vivo antigen-specific immune response. We hypothesized that arranging PAMPs on or inside NP, which are in the size range of viruses and bacteria, created biomimetic platforms for efficient interactions with APCs that would outperform suspensions of equivalent doses of antigen and adjuvants. To verify, we compared NP to soluble antigen/adjuvant formulations to determine if maximal vaccine efficacy required a physiologically relevant arrangement of PAMPs, or if PAMP combinations would suffice. Furthermore, we compared the vaccine efficacy of artificial bacterial biomimetic NP, antigen-loaded NP containing both MPLA and CpG, tethering adjuvants, against a mixture of NP presenting either MPLA or CpG, in which each adjuvant remained in close proximity to antigen but could be distant from one another. Realizing that subsets of APCs express different Pattern-Recognition Receptors (PRRs) and perform complementary functions [31], we hypothesized that bundling PAMPs with antigen would maximize antigen-specific immune activation. Our approach is biomimetic in that vaccine NP approximate bacterial pathogens with protein antigen and DNA motifs inside and MPLA, a cell wall component, on the surface (Fig. 1A). We show that spatial and combinatorial presentation of PAMPs in or on NP influences the magnitude and direction of vaccine responses; artificial bacterial NP create a synergistic antibody-mediated and cellular antigen response in vitro and in vivo.
Figure 1.
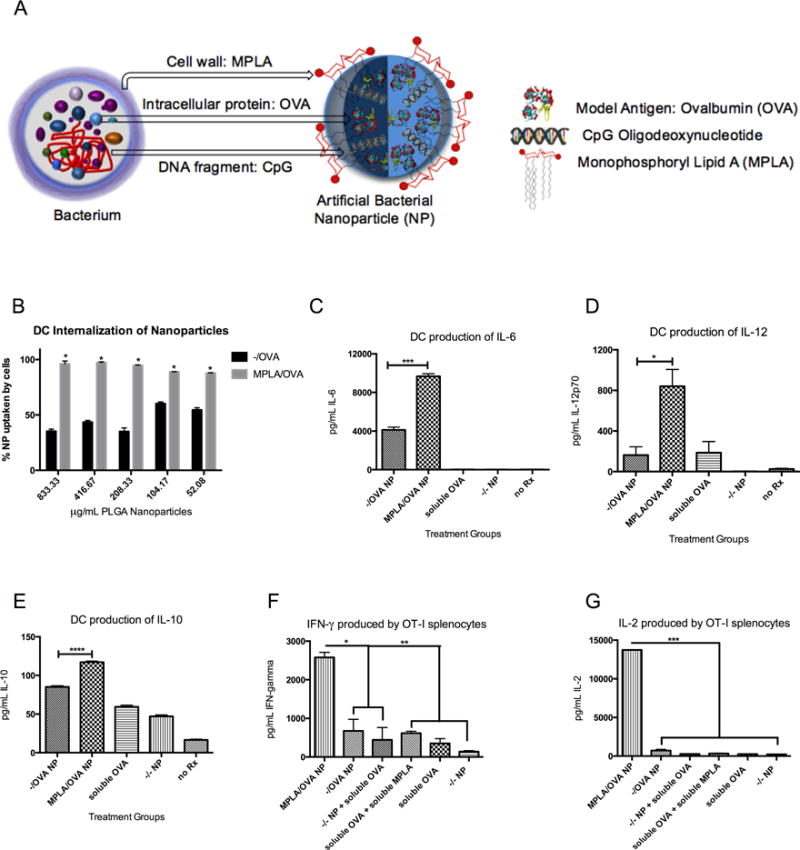
MPLA on the surface of NP, mimicking bacterial cell walls (A), increases BMDC uptake, pro-inflammatory cytokine production, and antigen-specific T cell proliferation. (B) BMDCs incubated with titrated PLGA NP internalize MPLA-coated particles more avidly than blank NP (*p<.0001 for all concentrations) (C) After 24h with NP, BMDCs produce a higher amount of pro-inflammatory cytokines IL-6 (***p=.0001), (D) IL-12 (*p=.0217), and (E) IL-10 (****p<.0001) in response to MPLA/OVA vs. −/OVA NP. (F) Co-incubating OVA-specific OT-I splenocytes with NP-treated BMDCs results in greater production of proliferative cytokines IFN-gamma (*p<.05, **p<.01) (G) and IL-2 (****p<.0001) after 5 days. Results are representative of three independent experiments. Error bars represent Standard Experimental Error (SEM).
Materials and Methods
Materials
Fully phosphorothioated Type B CpG 1826 ODN were synthesized by Yale Keck Facility (sequence: 5’ TCC ATG ACG TTC CTG ACG TT) and conjugated to biotin. Poly(vinyl alcohol) (PVA), palmitic acid N-hydroxy-succinimide ester, Atto 565-biotin, Coumarin 6, Ovalbumin, and chloroform were purchased from Sigma-Aldrich. Synthetic MPLA was purchased from Invivogen, avidin from Invitrogen, and research-grade PLGA (50:50, iv.55-.75 dL/g) from Durect.
NP Synthesis and Characterization
PLGA NP were synthesized using a water/oil/water (w/o/w) double emulsion technique previously described [24, 32]. Briefly, polymer was dissolved in chloroform at 50 mg/mL, OVA (100 mg/mL in PBS) added dropwise to polymer under vortex, then subjected to 30 second sonication using a Tekmar Sonic Distributor at 38% amplitude. Polymer/OVA was added dropwise to an aqueous solution of PVA (sometimes containing avidin-palmitate or MPLA) under vortex before further sonication then stirring in 0.2% PVA for NP hardening and solvent evaporation. NP were collected and rinsed by centrifugation at 12,000 rpm, flash frozen, lyophilized, and stored at −20°C. Immediately before experiments, avidin-coated NP were suspended in PBS and incubated with biotinylated CpG to surface-attach the ODN.
Scanning Electron Microscopy was used to characterize NP morphology, and hydrodynamic diameters were measured using Dynamic Light Scattering (DLS). For DLS and zeta potential measurements, employing a Malvern Zetasizer, NP were suspended in deionized water at pH 6.5 and titrated from 100 μg/mL to 6.25 μg/mL, and average values taken across titrations. Dissolved NP were assayed for loading of OVA by Micro BCA protein assay and CpG by PicoGreen DNA Detection Assay. Blank, empty NP were used as negative controls.
A dye conjugation assay was developed to provide evidence that acyl chains of MPLA partition into PLGA during NP synthesis, resulting in NP surface-presenting bioactive MPLA carbohydrate structures. MPLA was reacted with N,N-Carbonyl Diimidazole (CDI) to convert the free hydroxyl group on MPLA to an imidazole carbamate reactive intermediate (termed ‘MPLA-imidazole carbamate) (Supplementary Figure 1A). NP were formulated, as previously described, using either MPLA-imidazole carbamate (resulting from CDI reaction) or non-reacted MPLA (with intact hydroxyl group). NP were collected, rinsed twice, and incubated with rhodamine that had been previously conjugated to polyamidoamine (PAMAM) dendrimer (termed ‘rhodamine’) to introduce functional amine groups, in aqueous buffer overnight, to allow stable urethane (N-alkyl carbamate) linkages to form between amine and imidazole groups [33] (Supplementary Figure 1B). If MPLA were on the outside of NP, imidazole will be exposed and rhodamine conjugated. Else, no differences between adsorbed dye on surfaces of control NP (MPLA and blank NP) would be observed.
After reaction, NP were collected, rinsed twice, and fluorescence quantified using both a spectrophotometer and Bruker Image Stream X with excitation/emission = 550/670 nm. Fluorescence was evaluated using a standard curve of dye and subtracting nonspecific dye conjugation and adsorption from control NP (MPLA and blank NP).
In vitro BMDC internalization, activation and coculture experiments
Bone-marrow-derived dendritic cells (BMDCs) were cultured from marrow of C57BL/6 mice, plated in 96 well plates, and titrated with NP as previously described [12]. After 24 hours, supernatants were collected for analysis, NP were washed out, and OT-I splenocytes added to BMDCs at a 2:1 ratio. After 3 or 5 days, plates were centrifuged to pellet cells, and supernatants saved for cytokine analysis by ELISA according to manufacturer instructions.
For internalization studies, NP were made with Coumarin 6 or Atto 565-biotin so that groups were commensurately fluorescent as measured by flow cytometry. BMDCs were incubated with NP at 37°C for 24 hours, washed, and analyzed for the percentage of fluorescent-positive CD11c+ cells using a BD Biosciences LSR II Flow Cytometer.
Vaccination experiments
C57BL/6 mice were injected subcutaneously with treatments normalized to 50 μg OVA per mouse, and boosted with equivalent doses at day 14. Blood was collected retro-orbitally 7, 14, and 21 days post-prime. After 21 days, mice were sacrificed and spleens harvested for antigen-specific lymphocyte phenotyping, staining with fluorescent antibodies against CD4, CD8, CD44, CD127, and KLRG1 (eBioscience), and ex vivo antigen stimulation.
Serum was analyzed for cytokines and anti-OVA isotype antibodies by ELISA as previously described [34]. Titer was calculated as the inverse dilution at which the absorbance equaled that of the control (PBS-injected mice) plus 2 SD. Linear regression of dilution versus antibody curves was calculated after a log transformation of values.
TLR Trigger Screening Experiments
Reagents were screened using a customized PRR Ligand Screening Service offered by Invivogen (San Diego, CA) (www.invivogen.com/custom-tlr-screening), with human and mouse TLR stimulation quantified by NF-κB activation, which induces a secreted embryonic alkaline phosphatase (SEAP) reporter transfected in HEK293 cells expressing a given TLR. Reagents were tested in triplicate compared to control ligands. In a 96-well plate (200 μL total volume) containing the appropriate cells (50,000–75,000 cells/well), 20 μL of the test article or the positive control ligand was added. The media added to the wells detects NF-κB induced SEAP expression. Optical density (OD) was read at 650 nm after 16–24 hours on a Molecular Devices SpectraMax 340PC absorbance detector.
Statistical analysis
GraphPad Prism (version 6) was used for statistics. Two-tailed t-tests and ANOVA with Bonferonni’s post-test were performed. p-value of 0.05 or less was considered statistically significant.
Study approval
All animal studies were approved by Yale University Institutional Animal Care and Use Committee.
Results
Nanoparticle fabrication and characterization
Previously, we have shown that fatty acid chains embed in the PLGA polymer matrix during emulsion formulation [32, 35], which afforded a strategy for surface attachment of MPLA; the fatty acid portion anchored in the matrix while the hydrophilic phosphoryl head remained surface-exposed without compromising ligand binding to extracellular TLR4 [32]. A dye conjugation assay confirmed surface-presentation of MPLA, and its concentration on NP was determined to be 3.80 +/− 0.23 μg MPLA per mg NP (Supplementary Figure 1).
Because CpG ODN lack a fatty acid and the entire molecule ligates TLR9, NP were formulated with avidin on the surface for further modification with biotinylated CpG. This strategy enabled maximal exposure and prolonged presentation of CpG or MPLA over several weeks without hindering antigen release from polymer matrix [34].
Using this method, we constructed a set of NP configurations shown in Table 1 with similar properties, including OVA loading (average loading = 53 μg OVA per mg of NP), hydrodynamic diameter, and surface charge.
Table 1.
Schematic representations of biomimetic NP, which exhibited similar physical properties of antigen loading, hydrodynamic diameter, and zeta potential in water.
| Nomenclature | −/− | −/OVA | −/OVA+CpG | MPLA/OVA | CpG/OVA | MPLA/OVA+CpG |
|---|---|---|---|---|---|---|
| Schematic |
|

|

|
|

|
|
| Description | Blank surface, empty interior | Blank surface, OVA-loaded | Blank surface, OVA and CpG Inside | MPLA on surface, OVA inside | CpG on surface, OVA inside | MPLA on surface, OVA and CpG inside |
| Antigen Loading (μg/mg NP) | n/a | 54.0 | 60.2 | 48.0 | 60.5 | 41.0 |
| Diameter (nm) | 216.0 +/− 82.1 | 238.8 +/− 94.3 | 231.5+/− 99.1 | 263.7+/− 111.6 | 210.8+/− 103.7 | 298.0 +/− 114.2 |
| Zeta potential (mV) | −16.0 +/− 4.9 | −11.1 +/− 5.3 | −14.4 +/− 4.6 | −17.3+/− 5.7 | −14.5 +/− 5.9 | −16.3+/− 4.2 |
MPLA-coated NP enter and activate DCs
Because MPLA engages TLR4 on APC surfaces, we conjectured that MPLA-modified, antigen-loaded NP (termed ‘MPLA/OVA NP’) would mimic bacterial cell walls and increase phago- and endocytosis by bone marrow-derived dendritic cells (BMDCs) and expression of costimulatory molecules and cytokines. Using fluorescently labeled NP, we show that independent of concentration, MPLA-decorated NP were internalized significantly more in BMDCs compared to similarly sized NP with no surface modification (Fig 1B). Furthermore, modification of OVA-loaded NP with MPLA increased the production of pro-inflammatory cytokines IL-6 and IL-12 (Fig 1C, 1D) as well as anti-inflammatory IL-10 (1E). OT-I splenocytes, transgenic CD8+ T cells specific for MHC-Class-I-restricted OVA epitope (SIINFEKL), secreted more proliferative cytokines IFN-γ and IL-2 (Fig 1F and G) when cocultured with BMDCs incubated with MPLA/OVA NP for 24 hours than with antigen-matched controls. These in vitro results indicate that adjuvanted NP mediated antigen cross-presentation.
CpG is more potent when presented in NP interiors than surfaces
Since CpG binds an intracellular receptor, we wondered if the context of CpG presentation (dose-matched NP surface presentation versus encapsulation) affects its potency as a TLR9 ligand, theorizing that encapsulated CpG would be most effective at ligating TLR9. To investigate, OVA-loaded NP were surface-modified with CpG (CpG/OVA NP) and compared to NP encapsulating the same amount of OVA and CpG (−/OVA+CpG NP). We observed a concentration-dependent enhancement in potency of encapsulated CpG in terms of BMDC activation, antigen presentation, and subsequent antigen-specific T cell proliferation and production of proliferative cytokines (Figure 2). DC secretion of IL-6 and IL-12 was increased when CpG was inside NP compared to on surfaces (Fig. 2A, B), an effect that was significant at low, but not high, CpG concentrations, suggesting an adjuvant availability mechanism. It is possible that surface-bound CpG is restricted in motion and solubility, limiting receptor engagement. Co-incubating OT-I splenocytes led to similar results in proliferative cytokine production of IL-2 and IFN-γ, such that more vigorous T cell activation resulted from encapsulated CpG compared to surface modification (Fig 2C, D).
Figure 2.
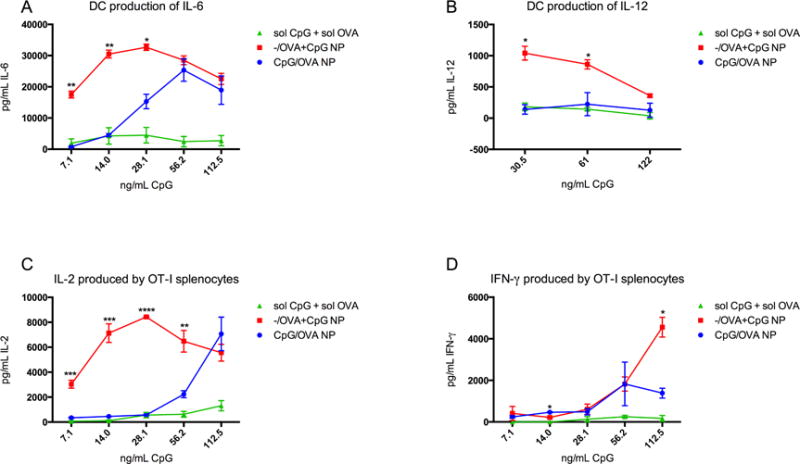
Encapsulating CpG in NP enhances in vitro adjuvant properties. BMDCs produced larger quantities of pro-inflammatory cytokines IL-6 (A) and IL-12 (B) in response to CpG inside NP compared to soluble CpG and OVA. At low CpG concentrations, the potency of encapsulated adjuvant is significantly higher (**p<.005) than surface-associated or soluble. At higher CpG concentrations, the encapsulation-enhancement is abrogated. Co-incubated OT-I splenocytes also showed differential response to BMDCs primed with soluble CpG and OVA, CpG/OVA NP, or −/OVA+CpG NP. Proliferative cytokine IL-2 from OT-I cells is markedly increased when antigen and adjuvant are delivered in particulate form versus soluble (C), and at low CpG concentrations, the IL-2 response is amplified by CpG encapsulation but not high CpG concentrations. Interestingly, while OT-I IFN-γ response is influenced by nanoparticulate CpG localization (D), the enhanced potency of encapsulated CpG is observed at high CpG concentrations instead of low (*p<.05) Results represent 3 independent experiments.
Artificial Bacterial NP drive the most robust BMDC activation profile
Having determined the optimal context for CpG delivery and efficacy of surface-bound MPLA, we formulated “artificial bacterial” biomimetic NP with MPLA on the surface and encapsulating CpG and OVA (termed ‘MPLA/OVA+CpG’) to investigate possible PAMP synergy and the antigen-specific vaccine response. Artificial bacterial NP were more pro-inflammatory in vitro than NP with either CpG or MPLA (Figure 3). When incubated with BMDCs, MPLA/OVA+CpG NP elicited higher IL-6 and IL-12 than NP presenting either PAMP (Fig 3A, B). Interestingly, artificial bacterial NP incubation led to lower IL-10 production (Fig 3C), suggesting that dual activation of TLR4 and TLR9 may attenuate anti-inflammatory responses. Furthermore, MPLA/OVA+CpG NP increased antigen cross-presentation (Fig. 3D). To determine if these differences resulted from differential NP uptake, DCs were incubated with fluorescent NP with MPLA, CpG, or nothing on surfaces (blank NP). While surface modification with MPLA enhanced internalization (Fig 1A), no significant differences were observed in the uptake of CpG-modified or blank NP. In the presence of an equimolar mixture of NP with MPLA or CpG on surfaces, APCs preferentially internalized MPLA-decorated particles over CpG (%cells MPLA/OVA+), revealing that increased uptake of MPLA-decorated NP is a possible mechanism for the observed vaccine potency (Fig 3F). As NP concentration decreased, the number of DCs internalizing both MPLA/OVA and CpG/OVA NP increased (%cells double positive), suggesting that DCs saturate uptake capacity with MPLA-decorated NP before taking in CpG/OVA NP (Fig 3F). DC targeting is especially important since direct TLR stimulation leads to appropriate adaptive responses as opposed to DCs matured by indirect inflammatory mediators [36]. T cell proliferation and cytokine production from DC exposure to MPLA/OVA+CpG NP versus MPLA/OVA NP corroborated that enhanced NP uptake as well as TLR9 activation by encapsulated CpG are important drivers of immune activation.
Figure 3.
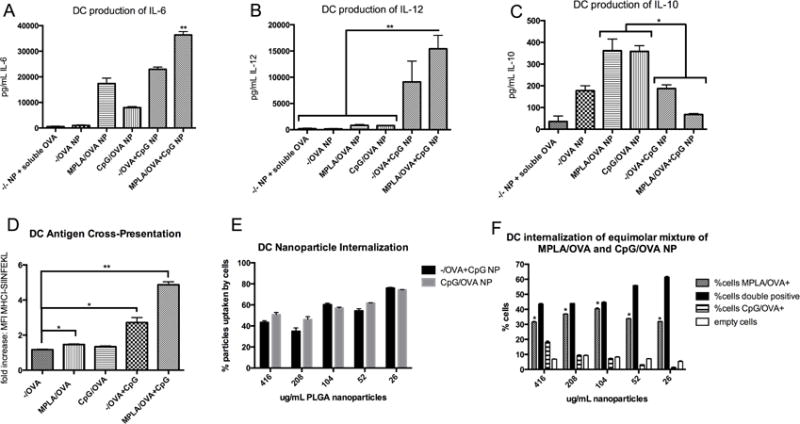
Artificial Bacterial NP synergize MPLA and CpG to activate APCs. Incubating MPLA/OVA+CpG NP with BMDCs induces pro-inflammatory cytokines IL-6 (**p<.01 versus all other treatments) (A) and IL-12 (B) while decreasing anti-inflammatory cytokine IL-10 (*p<.05) (C). Flow cytometry reveals that antigen delivered by MPLA/OVA+CpG NP significantly increases MHC Class I presentation (D); to determine if cross-presentation results from NP internalization differences, BMDCs were incubated with fluorescent NP with CpG, MPLA, or nothing on surfaces. CpG-modification does not increase intracellular concentration (E), and when incubated with an equimolar ratio of MPLA/OVA and CpG/OVA NP, BMDCs preferentially internalize MPLA/OVA NP (F). At high NP concentrations, approximately 30% of BMDCs contain only MPLA/OVA NP and 40% contain both types of NP (double positive), and at lower NP concentrations, the proportion of BMDCs with both MPLA/OVA and CpG/OVA NP inside increases while the proportion of cells containing only CpG/OVA NP decreases, indicating a concentration-independent enhanced uptake of MPLA/OVA NP. These results were replicated in 3 independent experiments.
Artificial Bacterial NP elicit a Th1-skewed antibody-mediated response
To evaluate antibody-mediated and cellular responses, C57BL/6 mice were subcutaneously inoculated with 50 μg OVA per mouse, either as a soluble protein with adjuvants MPLA and CpG, or in various NP configurations, and boosted after 14 days with identical doses [32]. Control groups were vaccinated with alum and OVA or sham injected with saline (PBS). To confirm the utility of tethering PAMPs with OVA on or in NP, one group received a cocktail of OVA-loaded NP that either surface-presented MPLA or encapsulated CpG (MPLA/OVA & −/OVA+CpG) to compare with artificial bacterial NP, MPLA/OVA+CpG NP, in which PAMPs are spatially restricted. All vaccinated groups exhibited OVA-specific titers within 7 days post-vaccination (compared to PBS-injected animals). After 21 days, vaccination with MPLA/OVA+CpG NP, −/OVA+CpG NP, and MPLA/OVA & −/OVA+CpG NP resulted in significantly higher total anti-OVA IgG titers than mice vaccinated with −/OVA NP and soluble dose-matched OVA, CpG, and MPLA, suggesting that encapsulated CpG may be the most influential driver of total antigen-specific IgG. This result is in agreement with previous reports of TLR stimulation increasing the magnitude of antibody responses [6, 37]. Th1-associated isotype IgG2b titers were over five-fold greater in mice vaccinated with artificial bacterial NP than soluble antigen/adjuvant mixture and 6-fold greater than mice dosed with −/OVA NP (Fig 4B). Interestingly, after 7 days, IgG2b titers of mice vaccinated with −/OVA+CpG NP were twice as high (statistically significant) as those vaccinated with CpG/OVA NP, suggesting an early Th1 response elicited by encapsulated CpG. After 21 days, mice vaccinated with NP encapsulating CpG showed the highest levels of anti-OVA total IgG, nearly three times greater than titers elicited by −/OVA NP vaccination (Fig 4C). IgG2b titers and IgG1 titers were similarly highest in artificial bacterial NP groups. Furthermore, TNF-α, a pleiotropic cytokine that may represent non-specific inflammation and is thus a poor prognostic, was elevated in the serum of mice vaccinated with alum, CpG/OVA NP, and soluble controls; these elevated levels were not observed after vaccination with NP encapsulating CpG or bundling TLR ligands (Fig 4D).
Figure 4.
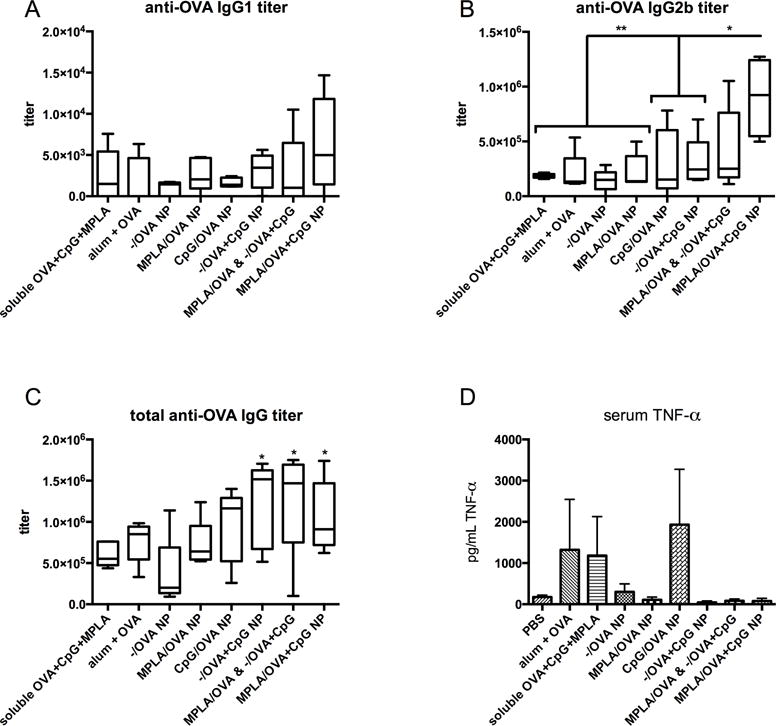
OVA-specific titers show a Th1-skewed antibody-mediated response.
Mice were vaccinated with treatment groups, including NP and soluble controls, normalized to OVA content. A mixture of NP, each containing antigen and presenting either MPLA or CpG, was administered to compare with MPLA/OVA+CpG NP to determine if PAMPs must be tethered for optimal responses. 21 days post-prime, mice inoculated with MPLA/OVA+CpG NP had the highest IgG1 titers (A) and significantly increased IgG2b (B) and total IgG titers (C) compared to NP with no adjuvant (−/OVA NP). The five-fold increase in IgG2b titers resulting from artificial bacteria NP versus soluble OVA, MPLA, and CpG suggests a NP-mediated Th1-skewed antibody-mediated response as IgG1 titers were lower. Interestingly, −/CpG+OVA NP groups also showed significantly higher anti-OVA total IgG compared to −/OVA-vaccinated animals, a result not recapitulated by CpG/OVA NP (C). A marker of nonspecific inflammation, elevated serum TNF-α levels over saline-injected mice was detected by ELISA (D). Serum TNF-α levels are highest in mice treated with exposed CpG (soluble or on NP surface) or alum and low in serum of mice vaccinated with CpG inside NP (MPLA/OVA+CpG and −/OVA+CpG).
Artificial Bacterial NP induce cellular immunity
To analyze the cellular response, splenocytes were harvested 21 days after vaccination, phenotyped by flow cytometry, or stimulated ex vivo with OVA. Activated, antigen-specific CD8+ T cells were quantified by staining splenocytes with a fluorophore-conjugated mouse tetramer H2k-MHC-I SIINFEKL. Consistent with antibody titer results, a greater frequency of antigen-specific T cells was observed in mice vaccinated with MPLA/OVA+CpG NP (Fig. 5A). Notably, vaccination with the cocktail of NP presenting individual PAMPs with OVA failed to elicit cellular immunity (Fig 5A), confirming the necessity of a single NP presenting both PAMPs in a manner that mimics bacteria in terms of components and spatial presentation. Antigen-specific CD8+ splenocytes were effector-memory phenotype characterized by CD127 low, CD62L low, CD44 high and KLRG1 high markers (Figure 5B). Upon restimulation with soluble antigen, splenocytes from all NP-vaccinated groups proliferated, in contrast to splenocytes from control animals (Figure 5C). Interestingly, after OVA stimulation, the amount of Th2 cytokine IL-5 was significantly decreased in some NP-vaccinated groups, most significantly MPLA/OVA, compared to PBS mice, an effect not seen with alum vaccination (Fig 5D).
Figure 5.
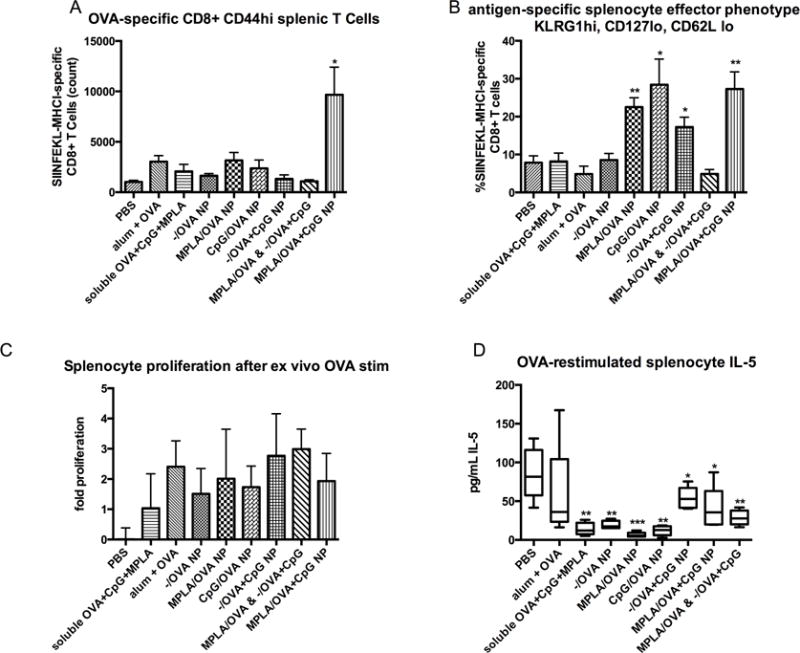
Artificial bacterial NP vaccines induce cellular immunity. 21 days after vaccination, splenocytes were harvested and either restimulated with soluble OVA or stained with OVA-specific T Cell Receptor (TCR) pentamer and antibodies against CD4, CD8, KLRG1, CD44, CD127 and CD62L for flow cytometry analysis. CD8 and tetramer positive staining shows a significantly higher number of antigen-specific, activated CD8+ T cells in spleens of mice inoculated with artificial bacteria NP (A) (p<.05 vs. PBS). Significant cellular immunity was not generated by vaccination with a mixture of OVA-loaded NP presenting either MPLA or CpG, confirming the necessity of spatially restricting the combination of PAMPs. Gating on this population reveals that antigen-specific T cells are activated effector phenotype characterized by high levels of KLRG1 and CD44, and low levels of CD127 and CD62L (B). This activated phenotype is most substantial in PAMP-modified NP versus −/OVA NP (*p<.05 **p<.001 vs. PBS). OVA-pulsed splenocytes of vaccinated mice proliferate ex vivo (C) and produce Th2 cytokine IL-5; vaccinated groups produce less IL-5 than splenocytes from control mice (D), suggesting Th1 skewing.
Artificial Bacterial NP stimulate multiple mouse and human TLRs
To quantify the synergy of TLR ligands presented in a physiologically relevant manner, screening experiments were performed by incubating reagents with human and mouse TLR-transfected HEK293 reporter cells. TLR triggering was determined by statistical significance of NF-kB activation over negative control ligands (indicated by red bars), and to truly compare the approximation of bacteria by NP, OVA-expressing heat-inactivated E.coli was screened along with high-dose TLR ligands as positive controls. Soluble MPLA and pyrogen-free OVA were assayed as further controls.
NP-mediated synergy of MPLA and CpG was observed for both mouse and human TLR2, in which artificial bacterial NP, as well as E.coli, induced significant activation over soluble MPLA and antigen (Fig 6A, B). As expected, MPLA, E.coli, and MPLA/OVA+CpG NP induced high levels of mouse TLR4 activation, while CpG added to human TLR4 activation (Fig 6C, D). Interestingly MPLA/OVA+CpG NP, along with E.coli, induced statistically significant activation of both mouse and human TLR5 versus negative controls and soluble MPLA (Fig 6E, F). CpG bundled in artificial bacterial NP was the most potent mouse TLR9 stimulant (greater than E.coli) (Fig 6G), a species-specific effect confirmed by lack of activation of human TLR9 (Fig 6H). Taken together, these results suggest that artificial bacterial NP, similar to E.coli, induce immunity by activation of TLRs beyond those associated with comprising adjuvants MPLA and CpG in both mouse and human systems.
Figure 6.
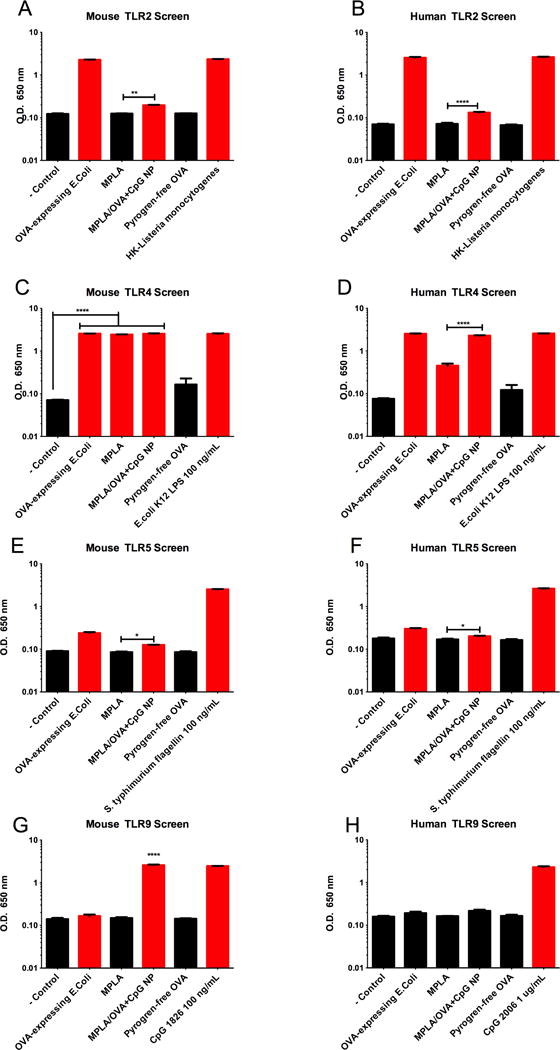
Artificial Bacterial NP trigger multiple mouse and human TLRs. Artificial bacteria NP, sterile antigen OVA, MPLA, and heat-inactivated OVA-expressing whole E.Coli were screened for TLR activation using TLR-transfected HEK293 reporter cell lines; red bars indicate significance over negative control ligands (A-H). Artificial bacteria NP containing ligands for mouse TLR4 and TLR9 significantly trigger mouse TLR2 (A), TLR4 (C), TLR5 (E), and TLR9 (G). MPLA/OVA+CpG also significantly stimulate human TLR2 (B), TLR3, TLR4 (D), TLR5 (F), TLR7, and TLR8 (data not shown/supplementary). Interestingly, artificial bacteria NP were more potent TLR triggers than OVA-expressing E.coli for mouse TLR5 (E) and human TLR5 (F) and mouse TLR9 (G).
Discussion
In this study, we aimed to recapitulate bacterial pathogens with antigen-encapsulating NP logically presenting two TLR ligands. Our hypothesis that combinatory TLR ligands can enhance vaccine potency was motivated by synergies shown between TLR2 and TLR4 [38], TLR3 and TLR7, and TLR1 and TLR2 [39]. Though an elegant study using emulsions containing MPLA and CpG showed synergy between these ligands for Leishmaniasis immunotherapy [40], solid biodegradable NP comprised of spatially appropriate ligands for TLR4 and TLR9 have not been demonstrated as versatile vaccine platforms. We deliberately chose clinically relevant adjuvants with complementary functions, designing vaccine vehicles to maximize Th1 responses while minimizing toxicity. For example, high serum TNF-α, a metric of non-specific inflammation, is a clinical roadblock for CpG adjuvants [41, 42]. We observed high serum TNF-α levels in mice vaccinated with alum, soluble controls, or NP surface presenting CpG, but low levels of TNF in groups vaccinated with NP-encapsulated CpG; therefore, the proposed NP vaccine platform may alleviate adjuvant-mediated systemic toxicity.
Delivering ligands to TLR4 and TLR9 is challenging because of receptor localization outside and inside cells. As such, we conjectured that a NP platform presenting both agonists in a physiologically-relevant context may be an optimal way to investigate PAMP synergy for vaccination. Manipulating formulation conditions enables NP to deliver one or more antigens (including proteins, peptides, and nucleic acids) and diverse adjuvants, and the inherent targeting of APCs makes for efficient vaccines that leverage innate immune stimulation to build adaptive immune responses. While hurdles remain between preclinical development and clinical use of nanoparticulate vaccines, we are encouraged by the successes of companies like Selecta Biosciences, which has a biodegradable polymer-based NP vaccine in Phase I clinical trials, and Novavax, which has completed several clinical trials evaluating their Virus-Like Particle (VLP) technology against Respiratory Syncytial Virus (RSV), pandemic flu, and Ebola. We highlight these companies as relevant to the present work because their NP-based vaccines are modular, synthetic platforms that are similar to what we describe here. Thus, the advancement of these technologies offer the potential for engineering qualitative and quantitative elements for optimizing antigen and adjuvant delivery to APCs and targeting delivered agents to specific intracellular compartments. Despite the clear advantages offered by NP systems for vaccination, such as usage of lower amounts of antigens and adjuvants and improved antigen-specific cellular and antibody responses, compared to current technologies such as alum, the translational potential of NP platforms as vaccines is yet to be fully realized.
Perhaps one reason for the lag time between preclinical development and clinical use of NP vaccines is that reliable, cost-effective, large-scale manufacturing of NP is still in its infancy. In terms of Good Manufacturing Practice (GMP) operations, there are no precedents on a massive scale of such complex systems involving at least three different components: an antigen, adjuvant, and vehicle that fit with regulatory guidelines defining parameters such as number of ligands on NP surfaces, amount of encapsulated components, and NP size and charge, among other important formulation metrics. While economies of scale may pave the way for manufacturing on a large scale, the most significant hurdle for translation is likely cultural acceptability for prophylactic nanotechnology; notably, while acutely sick patients who have limited therapeutic options may be open to NP treatments, which can offer renewed hope against their diseases, healthy people may find the use of NP as a prophylactic an unwarranted risk. While a full discussion of these topics is beyond the scope of this paper, we make these points because end user considerations affect the translational potential and adoption of any new technology. Thus, the technical components involving clinical efficacy are but a fraction of the issues hindering the translational potential of NP-based vaccines. Affordability, accessibility, and acceptance for such newer technologies are key factors that will motivate financing of clinical trials and manufacturing for widespread use.
Immobilizing PAMPs on or in NP removes the influence of dynamic, spontaneous formation of supramolecular structures that affect biological activity. For instance, LPS in cubic or hexagonal structures activates TLR4 more potently than lamellar aggregate structures formed by cylindrical LPS molecules [43]. Similarly, the sequence-specific effects of different types of CpG can change if CpG forms micelles or is encapsulated in liposomes [44–46]; NP-encapsulation of linear Type B CpG may create structures similar to the self-complementary palindromes that naturally form from other types of CpG. Because MPLA has a low critical micelle concentration (CMC), we theorized that soluble mixtures of OVA, CpG and MPLA could form micelles with differential localization of antigen and CpG. Given this possibility, we chose to immobilize PAMPs using solid NP to reduce the degrees of freedom while investigating PAMP synergy. While a logical extension of this work might test NP encapsulating MPLA, the physiochemical properties of the polymer and PAMP preclude its entrapment into stable NP. As MPLA is an amphiphilic molecule with a hydrophilic phosphatidic head and hydrophobic fatty acid tail, MPLA partitions to NP surfaces, with the phosphatidic head group exposed to the aqueous phase and the fatty acid group embedded in the polymer phase, during the PLGA NP synthesis procedure used in this study. This phenomenon recapitulates our previous work with avidin-fatty acid conjugates for NP surface modification and display of biotinylated moieties [16, 35, 47–50]. Conjugating MPLA to PLGA polymer leads to unstable aggregates and would block the binding site of MPLA to TLR4.
CpG and MPLA, alone or in combination with polymer, may initiate responses beyond TLR stimulation. Recently, two groups demonstrated that intracellular LPS is recognized by the non-canonical inflammasome, caspase-11, in mouse macrophages [51] [52], which can respond to cytosolic MPLA independent of TLR4 activation. Thus, coating NP with MPLA may activate APCs with more potency than soluble because of greater intracellular concentration to activate caspase-11. CpG encapsulated in NP leads to TLR9 ligation while bypassing surface receptors DEC-205, which can drive an anti-inflammatory response depending on ligand density [16], and receptor for advanced glycation end-products (RAGE) [53], which may modulate the immune response to CpG, NP, and antigen [54].
There are many potential reasons that CpG encapsulated in NP is more potent than its surface-presentation. Entrapment of CpG within the PLGA matrix may confer advantageous TLR9 ligand protection during NP endocytosis by APCs. Additionally, prolonged release of CpG from NP may enable continuous ligation of TLR9, whereas surface presented CpG would immediately engage TLR9; this adjuvant persistence may be important for durable immune responses [55]. If TLR9 clusters in endosomes after initial engagement, a more efficient ligation of CpG and TLR9 would occur when CpG is inside NP. Surface tethering of CpG ODNs may compromise structural integrity and restrict the degree of ligand motion, in contrast to CpG liberated from within NP.
Conclusions
Recently, it has been proposed that DC function is not predetermined by lineage alone but may also be influenced by the variety of PAMPs that stimulate those DCs [56]. As such, the artificial bacterial vaccine platform induces a broad DC functional response by exposing the same cell to antigen and combinations of PAMPs. Cellular immunity resulted only from NP presenting both TLR ligands, confirming the importance of spatial orientation of PAMPs reminiscent of viruses or bacteria. This finding is supported by recent work using NP to tether antigen and CpG, showing functional memory CD8+ responses only when antigen and PAMP were colocalized [57]. We present a proof-of-concept with two different PAMPs incorporated in and on a NP; it is possible to engineer these systems to present multiple PAMP adjuvants. This strategy of multivalent delivery has been demonstrated with delivery of multiple antigens for induction of a polyvalent vaccine response [13].
Our data suggests that structural and compositional elements of pathogens, including viruses and bacteria, can be recapitulated in biodegradable polymeric NP that have an established safety record. This work highlights the importance of synchronous delivery of antigen and physiologically arranged TLR ligands to the same cell. This formulation of clinically relevant adjuvants and polymers [58] demonstrates a rational, modular, and translational approach to improved vaccine design.
Supplementary Material
Acknowledgments
We would like to thank Drs. Kim Bottomly and Stacey Hero for intellectual input. OVA-expressing heat-inactivated E.Coli was a kind gift from Dr. Alexander Grishin. ALS, MJC, and TMF designed research; ALS performed experiments and analyzed data, and TMF, MJC, and ALS discussed results and wrote conclusions. This work was supported in part by an NIH grant R01AI093775.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflict of interest.
References
- 1.Querec T, Bennouna S, Alkan S, Laouar Y, Gorden K, Flavell R, et al. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J Exp Med. 2006;203:413–24. doi: 10.1084/jem.20051720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 3.Todoroff J, Lemaire MM, Fillee C, Jurion F, Renauld JC, Huygen K, et al. Mucosal and systemic immune responses to Mycobacterium tuberculosis antigen 85A following its co-delivery with CpG, MPLA or LTB to the lungs in mice. PloS one. 2013;8:e63344. doi: 10.1371/journal.pone.0063344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthews K, Chung NP, Klasse PJ, Moore JP, Sanders RW. Potent induction of antibody-secreting B cells by human dermal-derived CD14+ dendritic cells triggered by dual TLR ligation. Journal of immunology. 2012;189:5729–44. doi: 10.4049/jimmunol.1200601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nature immunology. 2005;6:769–76. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, et al. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470:543–7. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwissa M, Nakaya HI, Oluoch H, Pulendran B. Distinct TLR adjuvants differentially stimulate systemic and local innate immune responses in nonhuman primates. Blood. 2012;119:2044–55. doi: 10.1182/blood-2011-10-388579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, Rothstein SN, Little SR, Edenborn HM, Meyer TY. The effect of monomer order on the hydrolysis of biodegradable poly(lactic-co-glycolic acid) repeating sequence copolymers. Journal of the American Chemical Society. 2012;134:16352–9. doi: 10.1021/ja306866w. [DOI] [PubMed] [Google Scholar]
- 9.Cancer NAfNi, editor. Nanotechnology in Clinical Trials. National Cancer Institute; [Google Scholar]
- 10.Moon JJ, Suh H, Li AV, Ockenhouse CF, Yadava A, Irvine DJ. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1080–5. doi: 10.1073/pnas.1112648109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irvine DJ, Swartz MA, Szeto GL. Engineering synthetic vaccines using cues from natural immunity. Nature materials. 2013;12:978–90. doi: 10.1038/nmat3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, et al. Combination delivery of TGF-beta inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nature materials. 2012;11:895–905. doi: 10.1038/nmat3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sehgal K, Ragheb R, Fahmy TM, Dhodapkar MV, Dhodapkar KM. Nanoparticle-Mediated Combinatorial Targeting of Multiple Human Dendritic Cell (DC) Subsets Leads to Enhanced T Cell Activation via IL-15-Dependent DC Crosstalk. Journal of immunology. 2014;193:2297–305. doi: 10.4049/jimmunol.1400489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarti F, Perera G, Hintzen F, Kotti K, Karageorgiou V, Kammona O, et al. In vivo evidence of oral vaccination with PLGA nanoparticles containing the immunostimulant monophosphoryl lipid A. Biomaterials. 2011;32:4052–7. doi: 10.1016/j.biomaterials.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 15.Bershteyn A, Hanson MC, Crespo MP, Moon JJ, Li AV, Suh H, et al. Robust IgG responses to nanograms of antigen using a biomimetic lipid-coated particle vaccine. Journal of controlled release : official journal of the Controlled Release Society. 2012;157:354–65. doi: 10.1016/j.jconrel.2011.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bandyopadhyay A, Fine RL, Demento S, Bockenstedt LK, Fahmy TM. The impact of nanoparticle ligand density on dendritic-cell targeted vaccines. Biomaterials. 2011;32:3094–105. doi: 10.1016/j.biomaterials.2010.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elamanchili P, Diwan M, Cao M, Samuel J. Characterization of poly(D,L-lactic-co-glycolic acid) based nanoparticulate system for enhanced delivery of antigens to dendritic cells. Vaccine. 2004;22:2406–12. doi: 10.1016/j.vaccine.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 18.Hamdy S, Haddadi A, Somayaji V, Ruan D, Samuel J. Pharmaceutical analysis of synthetic lipid A-based vaccine adjuvants in poly (D,L-lactic-co-glycolic acid) nanoparticle formulations. Journal of pharmaceutical and biomedical analysis. 2007;44:914–23. doi: 10.1016/j.jpba.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 19.Demento SL, Siefert AL, Bandyopadhyay A, Sharp FA, Fahmy TM. Pathogen-associated molecular patterns on biomaterials: a paradigm for engineering new vaccines. Trends in Biotechnology. 2011;29:294–306. doi: 10.1016/j.tibtech.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lynn GM, Laga R, Darrah PA, Ishizuka AS, Balaci AJ, Dulcey AE, et al. In vivo characterization of the physicochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat Biotechnol. 2015;33:1201–10. doi: 10.1038/nbt.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nembrini CSA, Dane KY, Ballester M, van der Vlies A, Marsland BJ, Swartz MA, Hubbell JA. Nanoparticle conjugation of antigen enhances cytotoxic T-cell responses in pulmonary vaccination. Proc Natl Acad Sci USA. 2011;108:989–97. doi: 10.1073/pnas.1104264108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamdy S, Molavi O, Ma Z, Haddadi A, Alshamsan A, Gobti Z, et al. Co-delivery of cancer-associated antigen and Toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity. Vaccine. 2008;26:5046–57. doi: 10.1016/j.vaccine.2008.07.035. [DOI] [PubMed] [Google Scholar]
- 23.Chong CS, Cao M, Wong WW, Fischer KP, Addison WR, Kwon GS, et al. Enhancement of T helper type 1 immune responses against hepatitis B virus core antigen by PLGA nanoparticle vaccine delivery. Journal of controlled release : official journal of the Controlled Release Society. 2005;102:85–99. doi: 10.1016/j.jconrel.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 24.Demento SL, Bonafe N, Cui W, Kaech SM, Caplan MJ, Fikrig E, et al. TLR9-targeted biodegradable nanoparticles as immunization vectors protect against West Nile encephalitis. Journal of immunology. 2010;185:2989–97. doi: 10.4049/jimmunol.1000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mata-Haro VCC, Martin M, Chilton PM, Casella CR, Mitchell TC. The Vaccine Adjuvant Monophosphoryl Lipid A as a TRIF-Biased Agonist of TLR4. Science. 2007;316:1628–32. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 26.Salkowski CADG, Vogel SN. Lipopolysaccharide and monophosphoryl lipid A differentially regulate interleukin-12, gamma interferon, and interleukin-10 mRNA production in murine macrophages. Infection and Immunity. 1997;65:3239–47. doi: 10.1128/iai.65.8.3239-3247.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ismaili JRJ, Aksoy E, Vekemans J, Vincart B, Amraoui Z, van Laethem F, Goldman M, Dubois PM. Monophosphoryl Lipid A Activates Both Human Dendritic Cells and T Cells. Journal of immunology. 2002;168:926–32. doi: 10.4049/jimmunol.168.2.926. [DOI] [PubMed] [Google Scholar]
- 28.Hemmi HTO, Kawal T, Kalsho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–5. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 29.Kaiser-Schulz G, Heit A, Quintanilla-Martinez L, Hammerschmidt F, Hess S, Jennen L, et al. Polylactide-Coglycolide Microspheres CoEncapsulating Recombinant Tandem Prion Protein with CpG-Oligonucleotide Break Self-Tolerance to Prion Protein in Wild-Type Mice and Induce CD4 and CD8 T Cell Responses. Journal of immunology. 2007;179:2797–807. doi: 10.4049/jimmunol.179.5.2797. [DOI] [PubMed] [Google Scholar]
- 30.Schlosser E, Mueller M, Fischer S, Basta S, Busch DH, Gander B, et al. TLR ligands and antigen need to be coencapsulated into the same biodegradable microsphere for the generation of potent cytotoxic T lymphocyte responses. Vaccine. 2008;26:1626–37. doi: 10.1016/j.vaccine.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 31.Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315:107–11. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 32.Demento SL, Eisenbarth SC, Foellmer HG, Platt C, Caplan MJ, Mark Saltzman W, et al. Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine. 2009;27:3013–21. doi: 10.1016/j.vaccine.2009.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hermanson G. Bioconjugate Techniques. Second. Elsevier, Inc.; 2008. [Google Scholar]
- 34.Demento SL, Cui W, Criscione JM, Stern E, Tulipan J, Kaech SM, et al. Role of sustained antigen release from nanoparticle vaccines in shaping the T cell memory phenotype. Biomaterials. 2012;33:4957–64. doi: 10.1016/j.biomaterials.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park J, Mattessich T, Jay SM, Agawu A, Saltzman WM, Fahmy TM. Enhancement of surface ligand display on PLGA nanoparticles with amphiphilic ligand conjugates. J Control Release. 2011;156:109–15. doi: 10.1016/j.jconrel.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sporri R, Reis e Sousa C. Inflammatory mediators are insufficient for full dendritic cell activation and promote expansion of CD4+ T cell populations lacking helper function. Nature immunology. 2005;6:163–70. doi: 10.1038/ni1162. [DOI] [PubMed] [Google Scholar]
- 37.Wille-Reece U, Flynn BJ, Loré K, Koup RA, Miles AP, Saul A, et al. Toll-like receptor agonists influence the magnitude and quality of memory T cell responses after prime-boost immunization in nonhuman primates. The Journal of Experimental Medicine. 2006;203:1249–58. doi: 10.1084/jem.20052433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Warger T, Hilf N, Rechtsteiner G, Haselmayer P, Carrick DM, Jonuleit H, et al. Interaction of TLR2 and TLR4 ligands with the N-terminal domain of Gp96 amplifies innate and adaptive immune responses. The Journal of biological chemistry. 2006;281:22545–53. doi: 10.1074/jbc.M502900200. [DOI] [PubMed] [Google Scholar]
- 39.van Duin D, Medzhitov R, Shaw AC. Triggering TLR signaling in vaccination. Trends in immunology. 2006;27:49–55. doi: 10.1016/j.it.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 40.Raman VS, Bhatia A, Picone A, Whittle J, Bailor HR, O’Donnell J, et al. Applying TLR synergy in immunotherapy: implications in cutaneous leishmaniasis. Journal of immunology. 2010;185:1701–10. doi: 10.4049/jimmunol.1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, et al. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507:519–22. doi: 10.1038/nature12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bourquin CAD, Zwiorek K, Lanz A-L, Fuchs S, Weigel S, Wurzenberger C, von der Borch P, Golic M, Moder S, Winter G, Coester C, Endres S. Targeting CpG Oligonucleotides to the Lymph Node by Nanoparticles Elicits Efficient Antitumoral Immunity. Journal of immunology. 2008;181:2990–8. doi: 10.4049/jimmunol.181.5.2990. [DOI] [PubMed] [Google Scholar]
- 43.Maeshima N, Fernandez RC. Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Frontiers in cellular and infection microbiology. 2013;3:3. doi: 10.3389/fcimb.2013.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jurk M, Kritzler A, Debelak H, Vollmer J, Krieg AM, Uhlmann E. Structure-activity relationship studies on the immune stimulatory effects of base-modified CpG toll-like receptor 9 agonists. ChemMedChem. 2006;1:1007–14. doi: 10.1002/cmdc.200600064. [DOI] [PubMed] [Google Scholar]
- 45.Vollmer J, Krieg AM. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Advanced drug delivery reviews. 2009;61:195–204. doi: 10.1016/j.addr.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 46.Vollmer J, Weeratna R, Payette P, Jurk M, Schetter C, Laucht M, et al. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. European journal of immunology. 2004;34:251–62. doi: 10.1002/eji.200324032. [DOI] [PubMed] [Google Scholar]
- 47.Park J, Fong PM, Lu J, Russell KS, Booth CJ, Saltzman WM, et al. PEGylated PLGA nanoparticles for the improved delivery of doxorubicin. Nanomedicine : nanotechnology, biology, and medicine. 2009;5:410–8. doi: 10.1016/j.nano.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fahmy TM, Samstein RM, Harness CC, Mark Saltzman W. Surface modification of biodegradable polyesters with fatty acid conjugates for improved drug targeting. Biomaterials. 2005;26:5727–36. doi: 10.1016/j.biomaterials.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 49.Demento SL, Bonafé N, Cui W, Kaech SM, Caplan MJ, Fikrig E, et al. TLR9-targeted biodegradable nanoparticles as immunization vectors protect against West Nile encephalitis. The Journal of Immunology. 2010;185:2989–97. doi: 10.4049/jimmunol.1000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Demento SL, Eisenbarth SC, Foellmer HG, Platt C, Caplan MJ, Saltzman WM, et al. Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine. 2009;27:3013–21. doi: 10.1016/j.vaccine.2009.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nobuhiko Kayagaki MTW, Stowe Irma B, Ramani Sree Ranjani, Gonzalez Lino C, Akashi-Takamura Sachiko, Miyake Kensuke, Zhang Juan, Lee Wyne P, Muszynski Artur, Forsberg Lennart S, Carlson Russell W, Dixit Vishva M. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science. 2013;341:1246–9. doi: 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 52.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–3. doi: 10.1126/science.1240988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lahoud MHAF, Zhang J-G, Meuter S, Policheni AN, Kitsoulis S, Lee C-N, O’Keeffe M, Sullivan LC, Brooks AG, Berry R, Rossjohn J, Mintern JD, Vega-Ramos J, Villadangos JA, Nicola NA, Nussenzweig MC, Stacey KJ, Shortman K, Heath WR, Caminschi I. DEC-205 is a cell surface receptor for CpG oligonucleotides. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:16270–5. doi: 10.1073/pnas.1208796109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sirois CM, Jin T, Miller AL, Bertheloot D, Nakamura H, Horvath GL, et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J Exp Med. 2013;210:2447–63. doi: 10.1084/jem.20120201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kohn JSN, Albert EC, Murphy JC, Langer R, Fox JG. Single-step immunization using a controlled release, biodegradable polymer with sustained adjuvant activity. J Immunological Methods. 1986;95 doi: 10.1016/0022-1759(86)90314-5. [DOI] [PubMed] [Google Scholar]
- 56.Desch AN, Gibbings SL, Clambey ET, Janssen WJ, Slansky JE, Kedl RM, et al. Dendritic cell subsets require cis-activation for cytotoxic CD8 T-cell induction. Nature communications. 2014;5:4674. doi: 10.1038/ncomms5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Titta A, Ballester M, Julier Z, Nembrini C, Jeanbart L, van der Vlies AJ, et al. Nanoparticle conjugation of CpG enhances adjuvancy for cellular immunity and memory recall at low dose. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:19902–7. doi: 10.1073/pnas.1313152110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morris K. BIND Therapeutics Press Release. Cambridge, Mass: 2013. BIND Therapeutics Presents Positive Clinical Data at the AACR 2013 Annual Meeting for Lead Accurin Candidate, BIND-014, in Cancer Patients. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.