

Abstract
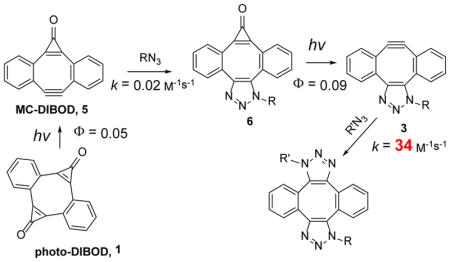
An order of magnitude difference in photo-reactivity between bis- (photo-DIBOD, 1) and mono-cyclopropenone - caged dibenzocyclooctadiynes (MC-DIBOD, 5) allows for selective mono-decarbonylation of 1. Alternatively, 5 is prepared by selective mono-cyclopropanation of dibenzo[a, e]cyclooctadiyne. MC-DIBOD (5) permits efficient sequential SPAAC cross-linking of azide-derivatized substrates. Cycloaddition to 5 converts an azide moiety into a photo-caged form of triazole-fused dibenzo[a, e]cyclooctyne (3). While azide reactivity MC-DIBOD (5) and dibenzo[a, e] cyclooctadiyne (DIBOD, 2) is similar to other cyclooctynes, fusion of triazole to dibenzocyclooctyne system in 3 results in three orders of magnitude enhancement in SPAAC rates. In methanol, 3 reacts with butyl azide at an astonishing rate of 34 M−1s−1, thus representing the most reactive cyclooctyne analog reported so far. MC-DIBOD (5) was utilized in the preparation of mixed bis-triazoles and derivatization of the protein (BSA) with fluorescent dye and polyethylene glycol.
Graphical Abstract
Introduction
Catalyst-free cycloaddition of organic azides across the triple bond in cyclooctynes (SPAAC)1 has become one of the most popular “click-chemistries” employed for the functionalization, cross-linking, and immobilization of various substrates.2 The utility of this technique is hampered by the need of the differential functionaliztion of the substrates involved, one with an azide group, and the second with cyclooctyne moiety. The azide functionalization is well developed for a large variety of substrates, but incorporation of the cyclooctyne fragment is often synthetically challenging and expensive. Cross-linking of two azide-derivatized units by a bis-cyclooctyne linker, therefore, provides an attractive alternative to cyclooctyne-azide coupling.3,4,5 Sondheimer diyne (DIBenzo[a, e]cycloOctaDiyne or DIBOD 2) is the most atom economical double-SPAAC cross-linker, as both strained triple bonds belong to the same eight-membered ring.4 The parent dibenzocyclooctadiyne (2a) has found applications in the macrocyclization of bis-azide functionalized peptides,6 post-synthetic modification of MOF thin films,7 protein derivatization,4a the development of fluorescent dyes,8 and the preparation of tetramers of HIV-related peptides.9 The utility of the DIBOD cross-linkers (2), however, is limited by its short shelf lifetime and rapid decomposition in aqueous solutions (τ½~ 10 min at pH = 7.4).10 To alleviate this shortcoming, we have recently synthesized photochemical precursors to diynes 2a–c, dibenzo[a, e]dicyclopropa[c, g][8]annulene-1,6-dione (photo-DIBOD, 1a–c).10 Both triple bonds in photo-DIBOD (2) are masked as cyclopropenone moieties, which are known to undergo quantitative decarbonylation upon UVA11 or NIR12 photolysis. Photo-DIBODs (1) is a very stable compound, despite the presence of an antiaromatic cyclooctatetraene core, substantial angle strain, and high unsaturation (DU= 15). Irradiation of photo-DIBOD (1) with 350 or 420 nm fluorescent lamps in the presence of two azide-functionalized substrates results in the efficient formation of bis-triazole 4, via the apparent formation of Sondheimer diyne (2) and mono-adduct (3, Scheme 1).10 In addition to enhanced stability of the precursor 1, photochemical generation of reactive diyne 2 allows for the control of the ligation of two azides in space and in time.13
Scheme 1.

Azide cross-linking using bis-caged dibenzo[a, e]cyclooctyne
While DIBOD (2) is an efficient cross-linking reagent, selective conjugation of two different azides is hard to achieve, because the addition of the second equivalent of azide to mono-triazole 3 proceeds much faster than the first “click” step. The actual rates of the addition of organic azides across the triple bond of the didehydro[8]annulene 3 were unknown before the present work (vide infra). To achieve a high degree of selectivity in the sequential conjugation of different azide-tagged substrates, we envisaged a strategy utilizing a mono-cyclopropenone protected analog of Sondheimer diyne (6,7-didehydro-1H-dibenzo[a, e]cyclopropa[c][8]annulene-1-one, MC-DIBOD, 5). The addition of the first azide to MC-DIBOD (5) is followed by the photo-release of second triple bond, making it available for the reaction with the second azide (Scheme 2, isomers of 3 and 4 are omitted for clarity).
Scheme 2.

Selective cross-linking of two azide-derivatized substrates using MC-DIBOD (5)
Our interest in this system was further piqued by the recent report of the preparation of 4,9-dimethoxy-substituted analog of 5 (R1, R3= OCH3, R2= H)14 This compound reacts with azide to give a fluorescent analog of triazole 6 (R1, R3= OCH3, R2= H, Scheme 2), which was found to be photochemically inert. Since it was the first example of a photo-stable and fluorescent cyclopropenone, known to the authors, we have decided to conduct a detailed investigation of the photochemistry and photophysics of this system.
Results and Discussion
To synthesize MC-DIBODs 5a, b we have employed our recently developed methodology of selective mono-cyclopropanation the symmetric diynes. Thus, the treatment of the Sondheimer diyne 2a or 2,9-dibutoxy-substituted DIBOD (2b) with 1 equivalent of trifluoromethyltrimethylsilane15 and sodium iodide in dilute THF, followed by silica gel - promoted hydrolysis afforded the mono-caged diyne in 70% yield (Scheme 3).
Scheme 3.

Synthesis of MC-DIBOD (5)
Interestingly, MC-DIBOD (5a) can be generated photochemically by selective decarbonylation of one of the cyclopropenone moieties in photo-DIBOD (1a). The controlled irradiation of methanol-DCM solution of bis-cyclopropenone 1a with 350 nm fluorescent lamps results in the formation of MC-DIBOD (5a) in 90% preparative yield. The main byproduct of this reaction is DIBOD (2a). MC-DIBOD (5a) can be also converted into diyne 2, but that requires much longer irradiation. This selective sequential decarbonylation of cyclopropenone moieties in photo-DIBOD (1a) is possible due to the significant difference in quantum yields of the photoreaction between cyclopropenones 1a (Φ350 = 0.05) and 5a (Φ350 = 0.006). The loss of one carbonyl group results in the blue shift of characteristic diaryl-substituted cyclopropenone bands from 342 (log ε=4.0) and 361 nm (log ε= 4.1) in 1a to 340 (log ε=3.8) and 353 nm (log ε=3.9) in 5a, accompanied by a weak hypochromic effect (Fig. 1). The position and intensity of two stronger bands at shorter wavelengths (260 and 268 nm, log ε~ 4.8) are not significantly affected by the mono-decarbonylation of 1a.
Figure 1.
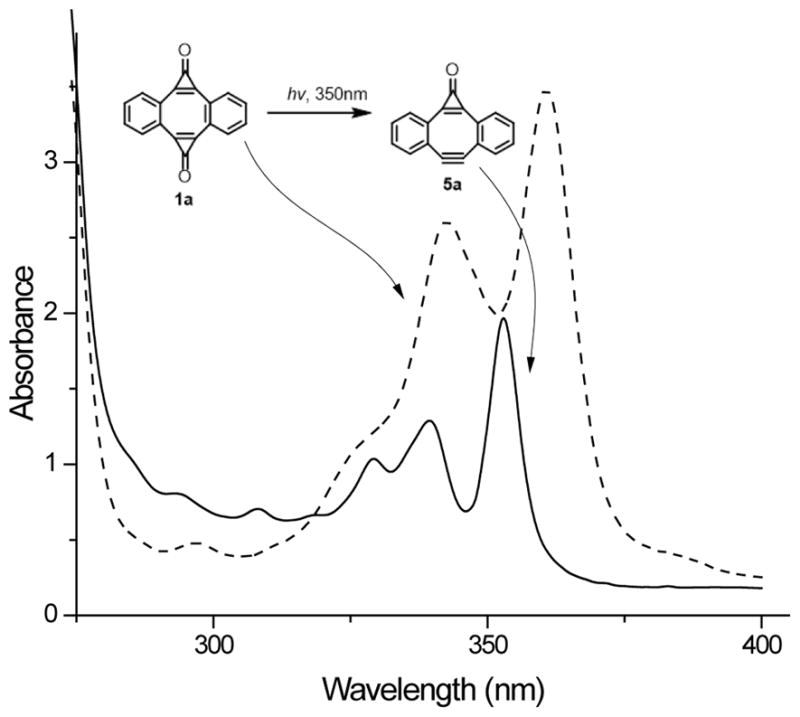
UV Spectra of ca. 0.22 mmol solution of photo-DIBOD (1a, solid line) and MC-DIBOD (5a, dashed line) in MeOH-DCM (9:1).
MC-DIBODs 5a, b, as well as their parent photo-DIBODs 1a, b, show no detectable fluorescence. The reduced quantum yields of photo-decarbonylation of 1 and 5 compared to their saturated analogue 4,9-dialkoxy-6,7-dihydro-1H-dibenzo[a, e]cyclopropa[c] [8]annulene-1-one (photo-DIBO)16 and acyclic diaryl-cyclopropenones,11a as well as their lack of fluorescence, suggest that these rather rigid compounds undergo surprisingly efficient vibrational relaxation.
The reactivity of MC-DIBOD 5 towards organic azides is similar to other carbocyclic dibenzocyclooctynes and this reaction produces a nearly quantitative yield of corresponding triazoles. Thus, 1-(butyl)-dibenzo[3,4:7,8]cyclopropa[5,6]cycloocta[1,2-d]-1,2,3-triazol-8-one (6a-Bu) and two isomers 5,10-dibutoxy-1-(butyl)-dibenzo[3,4:7,8] cyclopropa [5,6]cycloocta[1,2-d]-1,2,3-triazol-8-one (6b-Bu) were isolated in 90–95% yield upon reaction of 5a, b with butyl azide (Scheme 4).
Scheme 4.
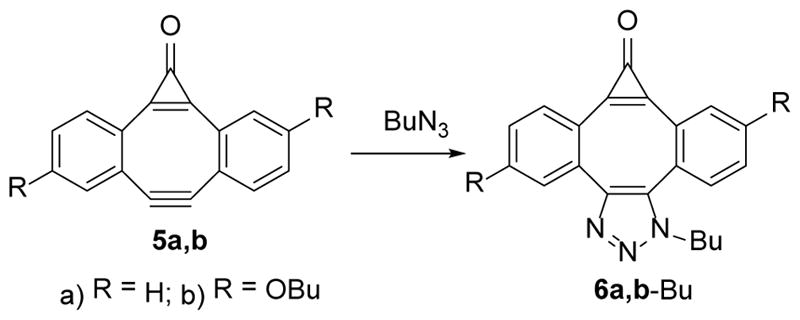
SPAAC of butyl azide across the triple bond of MC-DIBOD (5)
The conversion of MC-DIBOD 5 into triazole 6 is accompanied by the disappearance of 353 nm and 340 nm bands and the formation of the broad band centered at 310 nm (log ε=3.86, Fig. 2). In a sharp contrast to 5,11-dimethoxy-substituted analog of 6a, which was reported to be moderately fluorescent (ΦF= 0.12),14 both the parent triazole 6a-Bu and its 5,10-dibutoxy-substituted analog 6b-Bu show very weak fluorescence. In DCM:MeOH solution fluorescent quantum yields are ΦF= 0.0039 ±0.0006 and ΦF= 0.0076 ±0.0006 correspondingly. The aqueous medium (1:4 MeOH:H2O) somewhat enhances fluorescence of 6a-Bu (ΦF= 0.0119 ±0.0009), but its efficiency still remains very low. Furthermore, irradiation of the triazole 6a-Bu with UVB fluorescent lamp for 5 min results in the smooth loss of carbon monoxide and the formation of triazole-fused didehydro[8]annulene 3a-Bu, which can be followed by the bleaching of 310 nm band (Scheme 5, Fig. 2). Alkyne 3a-Bu could be detected by ESI-MS in dilute solution, but upon concentration, even at low temperature, forms a mixture of dimers and oligomers.
Figure 2.
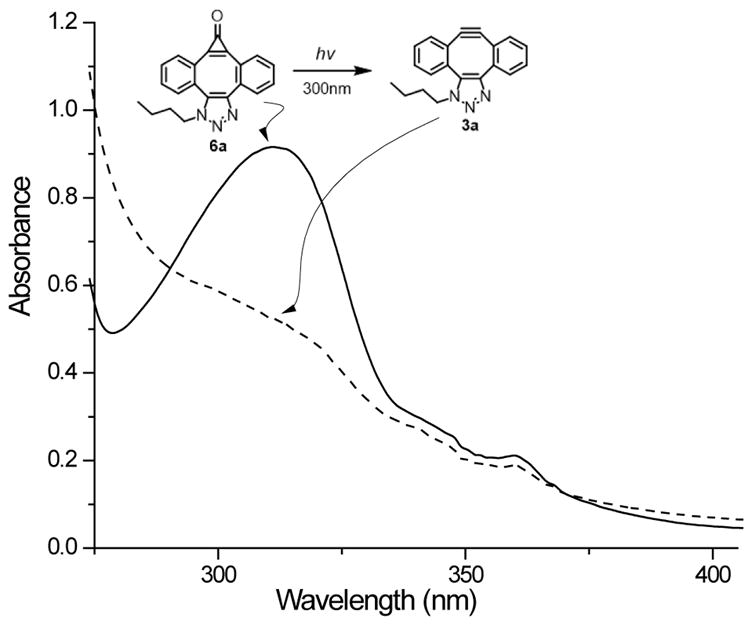
UV Spectra of ca. 0.18 mmol solution of 1-(butyl)-dibenzo[3,4:7,8]cyclopropa[5,6]cycloocta[1,2-d]-1,2,3-triazol-8(1H)-one (6a-Bu, solid line) and mono-triazole (3a-Bu, dashed line) in MeOH-DCM (9:1).
Scheme 5.

Photo-decarbonylation of MC-DIBOD (5) followed by SPAAC of benzyl azide.
Photolysis of 1-(butyl)-dibenzo[3,4:7,8]cyclopropa[5,6]cycloocta[1,2-d]-1,2,3-triazol-8(1H)-one 6a-Bu in the presence of benzyl azide results in the formation of two isomeric of bis-triazole 7 and 8 in 78% isolated yield (Scheme 5).
It is interesting to note that the photo-decarbonylation of triazole 6 is over an order of magnitude more efficient (Φ= 0.09 ± 0.01) than the MC-DIBOD 5. This fortuitous difference in photo-sensitivity between photo-DIBOD 1 (Φ= 0.05), MC-DIBOD 5 (Φ= 0.006), and triazole 6 (Φ= 0.09) allows for selective cross-linking of different azide-derivatized substrates. Thus, one-pot synthesis of mixed bis-triazoles 4 (Scheme 1) was demonstrated using sequential photo-decarbonylation of photo-DIBOD 1a. A solution containing an equimolar mixture of bis-cyclopropenone 1a and butyl azide was irradiated with 350 nm fluorescent lamps for 6 min and incubated for 3 hours. The addition of one equivalent of benzyl azide, followed by 5 min 300 nm photolysis allowed us to isolate bis-triazoles in a good yield 7 and 8 (Scheme 6).
Scheme 6.

Selective one-pot formation of mixed triazoles using photo-DIBOD 1a
Kinetics of SPAAC
Successful preparation of MC-DIBOD 5 gave us access to mono-triazole 3 and allowed for the direct experimental comparison of the azide reactivity of Sondheimer diyne (2), its cyclopropenone- (5) and triazole-fused (3) derivatives. The rate of this 1,3-dipolar cycloaddition (SPAAC) depends on the distortion of the bond angles in the acetylenic fragment,17 and the electronic effects of substituents.18 Since compounds 2, 3, and 5 possess the same dibenzo[a, e]cyclooctatetraene core, the reactivity in the series should follow the apparent strain increase from 3 to 5 to 2. Some experimental evidence, as well as the density functional computation analysis, suggests, however, that the addition of a second equivalent of azide to the DIBOD (2a) proceeds much faster than the first.19 To assess the relative reactivity of strained cycloalkynes 2, 3, and 5 in SPAAC, we have determined the second-order rate constants of the addition of butyl azide. The accurate rate measurements were conducted under pseudo-first order conditions using 20-fold or higher concentration of azide at 25.0 ± 0.1° C in methanol-DCM mixture (1:1).20 The second order rate constants were calculated by the least square analysis of the dependence of the observed rates on the azide concentration (Figure 3). The SPAAC reactivity of the DIBOD (k(BuN3) = (6.65 ± 0.03) ×10−2 M−1s−1 for 2a and k(BuN3) = (6.5 ± 0.02) ×10−2 M−1s−1 for 2b) is surprisingly similar to that of significantly less strained dibenzo[a, e]cyclooctyne (DIBO),16 and agrees well with previous estimates.4 MC-DIBOD (5a), as expected, is 3–4 fold less reactive than 2a (k(BuN3) = (1.66 ± 0.02) ×10−2 M−1s−1, Figure 3). To our great surprise, mono-triazole 3a (R= Bu), generated by photo-decarbonylation of 6a-Bu, is more than 500-fold more reactive than DIBOD 2a (k(BuN3) = (34 ± 1 M−1s−1). This rate represents the fastest ever measured cyclooctyne-azide click reaction in organic solution reported to date, beating the previous record set by ODIBO (1.7 M−1s−1)21 by more than an order of magnitude.
Figure 3.
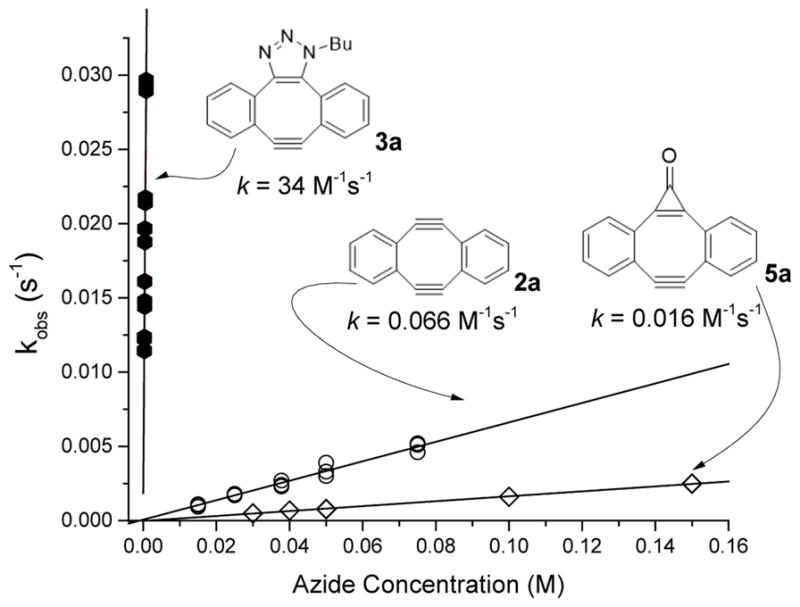
Observed pseudo-first order rate constants of reaction of acetylenes 2a (○), 3a-Bu (●), and 5a (◇) at different concentration of butyl azide in MeOH-DCM (1:1).
To test the suitability of MC-DIBOD (5) for the ligation of biologically relevant azides, we have employed it for the functionalization a model azide-tagged protein, bovine serum albumin (BSA-N3).10 BSA-N3 was treated cyclopropenone 5a to produce MC-DIBOD derivatized protein 9 (Scheme 7). The BSA derivative 9 was irradiated for 8 min with 300 nm fluorescent lamps and incubated with Rhodamine B azide.22 The in-gel fluorescent image of the SDS-PAGE of the resulting protein demonstrates the efficient labeling of azido-BSA with the dye (Lane 1, Fig. 4). An overnight incubation of the triazole 9 with Rhodamine B azide in the dark does not produce labelled BSA 11 (Lane 2, Fig. 4).
Scheme 7.

Cross-coupling of azido-BSA with Rhodamine B azide using MC-DIBOD 5a
Figure 4.
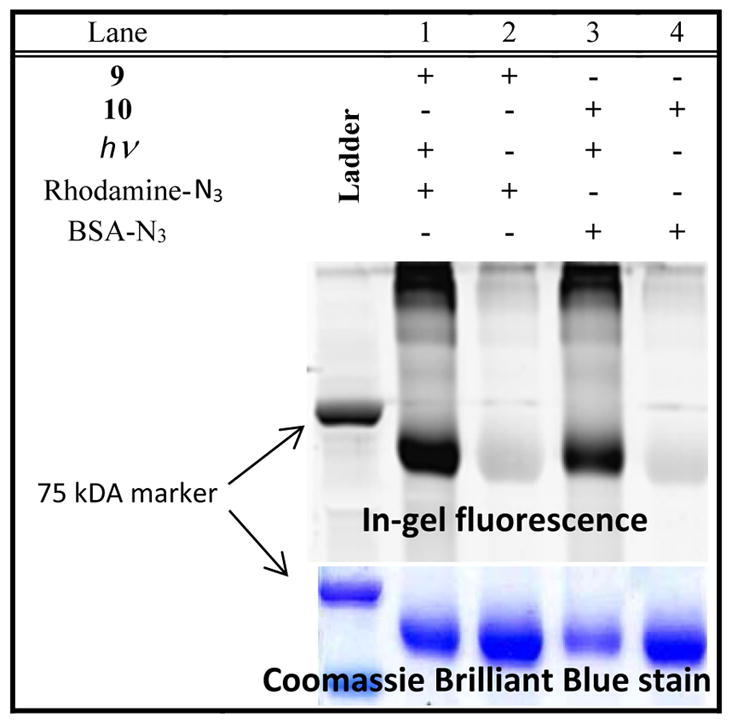
SDS-PAGE analysis of BSA-N3 photo-conjugation with Rhodamine B azide. Lane 1: BSA-N3 treated with MC-DIBOD 5a and irradiated in the presence of Rhodamine B – azide; Lane 2: BSA-N3 treated with 5a and then Rhodamine B – azide in the dark; Lane 3: Rhodamine B – azide reacted with 5a and irradiated in the presence of BSA-N3; Lane 4: Rhodamine B – azide reacted with 5a and incubated with BSA-N3 in the dark.
To illustrate the selectivity of the sequential azide cross-linking, the BSA-Rhodamine B conjugate 11 has been prepared by an alternative procedure. MC-DIBOD – equipped Rhodamine B 10 was prepared in situ by the reaction of Rhodamine B azide with 5a (Scheme 7). Triazole 10 has been added to the solution of BSA-N3 in PBS and irradiated for 8 min. The resulting conjugate 11 shows as a brightly fluorescent band on the gel (Lane 3, Fig. 4). The dark control of the BSA-N3 mixture with 10 produces no fluorescent bands on SDS-PAGE (Lane 4, Fig. 4).
The preparation of mixed triazoles (7, 8, Scheme 5) and cross-conjugation of BSA-N3 with Rhodamine B azide demonstrated the utility of MC-DIBOD 5 for cross-linking of small azides and for the protein functionalization. The applicability of this platform for the cross-linking of larger substrates was demonstrated on the example of BSA-N3 and PEG5000-azide23 cross-linking (Scheme 8). First, PEG-azide was incubated with an equimolar amount of MC-DIBOD 5a for 12 hours at room temperature. The IR spectrum of the product showed disappearance of azide stretching vibrations at 2100 cm−1. Three fold excess of the crude triazole 12 was added to a PBS solution of BSA-N3, irradiated with 300 nm lamps for 8 min, and incubated overnight. The BSA-PEG5000 conjugate was isolated by spin filtration (MWCO 10,000) and purified on a Sephadex column. According to the MALDI-TOF spectrum, the product contains a mixture of the native BSA with proteins bearing one, two, three, and even four PEG fragments (Fig. S3).20
Scheme 8.

PEGylation of BSA
As we reported previously,10 this inhomogeneity of the protein functionalization is due non-selective derivatization of native BSA. The treatment of the BSA-N3 sample used in this experiment with PEG5000-acetylene24 under conventional copper-catalyzed click conditions produced the same mixture of BSA-PEG5000 conjugates (Fig. S5).20 Thus, the photo-derivatization of azido-protein with MC-DIBOD derivative 12 occurs only at azide groups and is at least as efficient as CuAAC coupling. The MALDI-TOF spectrum of the protein isolated after the incubation of BSA-N3, with triazole 12 in the dark overnight contains only the single mass peak of a native protein (Fig S4). 20
Conclusions
An order of magnitude difference in the photo-sensitivity between bis-cyclopropenone derivative of dibenzo[a, e]cyclooctatetraene 1 and mono-cyclopropenone 5 allows for the selective mono-decarbonylation of photo-DIBOD (1, Scheme 9). MC-DIBOD (5) was also synthesized by selective mono-cyclopropanation of dibenzocyclooctadiyne (2). The addition of an azide across the triple bond of MC-DIBOD (5) to give triazole 6 restores the photo-reactivity and permits the efficient generation of triazole-fused didehydrodibenzo[a, e][8]annulene 3 (the mono-adduct of azide to DIBOD 2, Scheme 9). The alkyne 3 displays extraordinary reactivity towards azides: the rate of addition of butyl azide in methanol (34 M−1s−1) represents the fastest ever reported SPAAC reaction of cyclooctynes in organic solvents.
Scheme 9.

This fortuitous combination of photo-sensitivity and azide reactivity of dibenzo[a, e]cyclooctatetraene derivatives 1 – 6, makes MC-DIBOD 5 a very convenient platform for selective sequential cross-linking of azide-tagged substrates. We have illustrated the utility of the cross-linker 5 by one-pot preparation of mixed bis-triazoles, cross-conjugation of azido-derivatized protein and azido-terminated polymer, and by the efficient labelling of protein with fluorescent dye.
Experimental
General Methods
Tetrahydrofuran was freshly distilled from sodium benzophenone ketyl prior to use. Dichloromethane was freshly distilled from CaH2 prior to use. Solutions were prepared using HPLC grade water and methanol. Flash chromatography was performed using 40–63 μm silica gel. Photolyses were conducted using a photoreactor equipped with sixteen 4W fluorescent lamps with emission at 300 or 350 nm. The quantum yields of photolysis were measured against the 4-nitroveratrole actinometer.25 NMR spectra were recorded using 400 MHz spectrometer in deuterochloroform and referenced to TMS unless otherwise noted. High resolution mass spectra were obtained using electron spray ionization and orbitrap mass analyzer.
Materials
All reagents were purchased from commercial sources and used as received, unless otherwise noted. Rhodamine-B azide,22 BSA-N3,10 PEG5000 azide,10 DIBOD (2a, b),10 and photo-DIBOD (1a, b)10 were prepared following the procedures reported previously.
6,7-Dehydrodibenzo[a, e]cyclopropa[c][8]annulen-1-one (5a)
TMSCF3 (0.191 mL, 1.29 mmol) was added to a solution of Sondheimer diyne (0.258 g, 1.29 mmol) and NaI (0.232 g, 1.55°C mmol) in THF (24.3 mL) in a pressure vessel. The reaction mixture was heated at 110 for 2 h, cooled to r.t., and quenched by adding a saturated bicarbonate solution (20 mL), diluted in DCM (200 mL) and extracted with deionized water and brine. The organic phase was dried over anhydrous K2CO3, concentrated, and loaded onto a silica column (10% DCM/hexanes to 1% MeOH/DCM). The difluorocyclopropene immediately hydrolyzed upon chromatography to give 0.195 g (67%) of the title compound as a bright yellow solid (m.p. 204°C decomp.). 1H NMR: 7.55 (d, J=7.4 Hz, 2H), 7.23 (m, 4H), 6.91 (d, J=8.3Hz, 2H) 13C NMR: 154.3, 150.7, 135.2, 133.7, 132.1, 130.1, 127.2, 125.2, 106.8. IR: 1845cm−1 (C=O). HRMS m/z: (M+H+) Calcd. for C17H8O: 229.0648, found: 229.0649.
Preparation of 5a from photo-DIBOD 1a
A solution of 1a (25 mg, 0.098 mmol) in 440 mL of DCM: MeOH mixture (1: 9) was irradiated using 350 nm lamps. The consumption of starting material was followed by the disappearance of the band at 363 nm (ca. 4 min of exposure). The solvent was evaporated in vacuum and the crude mixture was purified via flash chromatography (0.5 – 2% DCM/MeOH) to give 20 mg (90%) of the desired product as a yellow solid.
Preparation of 5b from photo-DIBOD 1b
A solution of bis-butoxy photo-DIBOD 1b (39 mg, 0.098 mmol) in 0.5 L of DCM: MeOH mixture (1:9) was irradiated using 350 nm lamps following the disappearance of λmax at 370 nm (ca. 5 min of irradiation). The solution was evaporated in vacuum and the crude mixture was purified via flash chromatography (0.5–2% DCM/MeOH) to give 34 mg (73%) of the desired product as a yellow solid. 1H-NMR: 7.45 (d, J = 8.5 Hz, 1H), 7.08 (d, J = 2.6 Hz, 1H), 6.81 (d, J = 8.4 Hz, 1H), 6.68 (dd, J = 8.5, 2.6 Hz, 1H), 6.58 (dd, J = 8.5, 2.6 Hz, 1H), 6.40 (s, 1H), 3.95 (q, J = 6.6Hz, 4H), 1.75 (m, 4H), 1.48 (m, 4H), .99 (t, J = 7.4 Hz, 6H). 13C-NMR: 163.5, 160.6, 154.1, 151.2, 145.8, 137.1, 133.8, 128.4, 127.9, 124.2, 121.2, 118.5, 116.0, 114.5, 113.4, 107.9, 104.7, 68.4, 68.2, 31.1, 31.0, 19.1, 13,8, 13.8. HRMS m/z: (M+H+) Calcd. for C25H25O3: 373.1798, found: 373.1795.
1-butyldibenzo[3,4;7,8]cycloocta[1,2-d][1,2,3]triazol-8-one (6a)
Butyl azide (43 mg, 0.44 mmol) was added to a solution of cyclopropenone 5a (50 mg, 0.22 mmol), in 20 mL of DCM/MeOH mixture (1:4). The mixture was stirred at r.t. overnight, concentrated, and purified via flash chromatography (0.5% MeOH/DCM) to give 66 mg (92%) of the title compound as an off-white solid (m.p. 153–156°C). 1H NMR: 7.90 (d, J=7.7Hz, 1H), 7.73 (d, J=7.6Hz, 1H), 7.65–7.53 (m, 4H), 7.46 (t, J=7.6 Hz, 1H) 7.39 (d, J=7.6Hz, 1H), 4.35 (m 1H), 4.18 (m, 1H), 1.69 (m, 2H), 1.21 (m, 2H), .81 (m, 3H). 13C NMR: 157.7, 154.2, 151.9, 142.9, 133.8, 133.1, 132.7, 132.2, 132.1, 131.7, 131.5, 131.3, 130.5, 129.3, 128.9, 126.9, 124.4, 49.2, 32.0, 19.6, 13.3. IR: 1852 cm−1 (C=O). HRMS m/z: (M+H+) Calcd. for C21H17N3O: 328.1444, found: 328.1444.
6,11-dibutoxy-1-butyldibenzo[3,4:7,8]cyclopropa[5,6]cycloocta[1,2-d][1,2,3]triazol-8-one (6b) and 5,10-dibutoxy-1-butyldibenzo[3,4:7,8]cyclopropa[5,6]cycloocta[1,2-d][1,2,3]triazol-8-one (6b′)
Butyl azide (11 mg, 0.11 mmol) was added to a solution of the mono-cyclopropenone 5b (34 mg, 0.091 mmol) in 1mL of MeOH/DCM (1:1). The mixture was stirred for 12 h, evaporated, purified via flash chromatography (1% MeOH/DCM), evaporated, and recrystallized from DCM/hexanes to give 0.025 g (58%) of the desired product as a yellow solid (mixture of isomers). 1H and 13C NMR spectra are provide in supporting information file(S12-S-13).20 HRMS m/z: (M+H+) Calcd. for C29H34N3O3: 472.2595, found: 472.2593.
1-benzyl-8-butyldibenzo[3,4;7,8]cycloocta[1,2-d:5,6-d′]bis([1,2,3]triazole) (7) and 1-benzyl-10-butyldibenzo[3,4;7,8]cycloocta[1,2-d:5,6-d′]bis([1,2,3]triazole) (8)
A solution of the cyclopropenone 6a-Bu (30 mg, 0.092 mmol) in 0.5 L of DCM/MeOH mixture (1:1), was irradiated for 8 min at 300 nm. After irradiation, benzyl azide (610 mg, 4.6 mmol) was added. The mixture was stirred at r.t. overnight, concentrated, and purified via flash chromatography (0.3% MeOH/DCM) to give 31 mg (78%) of a mixture of the bis-triazoles as viscous oil. Triazoles could be partially separated for NMR characterization: 1H NMR: 7.75 (d, J=6.7Hz, 1H), 7.68 (d, J=7.7Hz, 1H), 7.51 (m, 4H), 7.39 (t, J=8.2Hz, 1H) 7.26 (m, 3H), 7.10 (d, J=7.2Hz, 2H), 7.01 (m, 2H), 5.50–5.33 (dd, J=90.8, 15.3Hz, 1H) 4.32 (m 1H), 4.18 (m, 1H), 1.67 (m, 2H), 1.04 (m, 2H), .74 (m, 3H).13C NMR: 145.2, 144.8, 135.1, 135.0, 134.4, 132.9, 132.7, 131.4, 131.3, 130.2, 130.0, 129.9, 129.4, 128.9, 128.8, 128.6, 128.3, 127.1, 127.0, 126.4, 52.2, 48.4, 31.8, 19.4, 13.2. HRMS m/z: (M+H+) Calcd. for C27H25N6: 433.2135, found: 433.2130.
Preparation of bis-triazoles 7 and 8 from 1a
A solution of DIBOD 1a (25 mg, 0.098 mmol) in 0.5 L of DCM/MeOH (1:10) was irradiated with 350 nm lamps, following the disappearance of absorption peak at 363 nm (ca. 6 minutes). Butyl azide (19 mg, 0.195 mmol) was added to the photolysate and the reaction mixture was stirred for 3 h at r.t. Benzyl azide (1.8 mL, 0.98 mmol) was added to the mixture, irradiated with 300 nm lamps for 5 minutes, stirred for 1 h, and solvents were removed in vacuum. The crude material was purified via flash chromatography (DCM to 3%MeOH/DCM) to give 0.020 g (47%) the mixture of 7 and 8 as a tan oil.
N-(6-(diethylamino)-9-(2-((3-(8-oxodibenzo[3,4:7,8]cyclopropa[5,6]cycloocta[1,2-d][1,2,3]triazol1(8H)-yl)propoxy)carbonyl)phenyl)-3H-xanthen-3-ylidene)-N-ethylethaneaminium chloride 10
MC-DIBOD 5a (0.028 g, 0.123 mmol) was added to a solution of rhodamine azide (50 mg, 0.094 mmol) in 5mL of MeOH/DCM (9:1). The mixture was stirred overnight, evaporated and purified via flash chromatography (1% MeOH/DCM) to give 65 mg (91%) of the desired compound as a violet, amorphous solid. 1H-NMR 8.03–8.02 (d, J = 7.7 Hz, 1H), 7.88 (d, J = 7.7Hz, 1H), 7.82–7.76 (m, 2H), 7.69–7.57 (m, 6H), 7.52–7.49 (t, J = 8.4 Hz, 2H), 7.10–7.05 (m, 2H), 7.02–6.99 (dd, J = 9.6, 2.3 Hz, 1H), 6.93–6.91 (dd, J = 9.6, 2.3 Hz, 1H), 6.81–6.79 (m, 2H), 4.46–4.41 (m, 1H), 4.31–4.28 (m, 1H), 4.05–3.94 (m, 2H), 3.64 (m, 8H), 1.95 (m, 2H), 1.33 (m, 12H). 13C-NMR; 12.6, 28.7, 46.1, 46.1, 46.4, 62.1, 96.2, 113.38, 113.41, 113.6, 114.0, 114.3, 114.5, 124.3, 126.4, 128.2, 129.3, 129.4, 130.2, 130.4, 130.8, 131.2, 131.3, 131.6, 132.1, 131.2, 132.9, 133.1, 133.2, 133.2, 133.6, 133.7, 142.4, 142.9, 151.8, 152.1, 154.3, 155.5, 155.6, 157.4, 157.6, 157.7, 158.6, 164.5. HRMS m/z: (M+) Calcd. for C48H44N5O4754.3388 Found: 754.3391.
Kinetics
The accurate rate measurements of butyl azide addition to alkynes 2a, 3a, and 5a were performed in MeOH-DCM (1:1) solution at 25.0±0.1°C under pseudo-first order conditions, using 20 fold or higher excess of azide. The consumption of alkynes was followed by the decay of the absorbance band of the starting material: 287 nm for 2a, 266 nm for 3a, and 350 nm for 5a. The experimental data fits the single exponential equation well (Figure S1 and S2).20 Linear dependence of the observed pseudo-first order rate constants on azide concentration (Table S1–S3) was analyzed by the least squares method to obtain the bimolecular rate constant (Inserts in Figures S1 and S2).20
Photo-conjugation of BSA-N3 with Rhodamine B azide
Procedure 1
Rhodamine B azide (95 mg, 0.180 mmol) was added to a solution of MC-DIBOD 5a (41 mg, 0.18 mmol) in 8 mL of DCM/MeOH (1:3) mixture, stirred overnight at r.t., and solvents evaporated in a vacuum. The crude solid was used in the subsequent step without further purification. A solution of the triazole 10 (2 mg, 3.90 μmol) in MeOH (241 μL) was added to a solution of BSA-N3 (17.5 mg, 0.265 μmol), in PBS (10 mL). The solution was irradiated for 8 min at 300 nm and stirred overnight in the dark. The resulting protein was concentrated by spin filtration (MWCO 10,000), purified on a PD-10 Sephadex column, and lyophilized. The BSA-Rhodamine conjugate, as well as a dark control, were resolved on 12% SDS-PAGE gel and visualized through in-gel fluorescence using a GE Typhoon scanner with excitation wavelength fixed at 532 nm and emission wavelength fixed at 580 nm.
Procedure 2
A solution of MC-DIBOD 5a (5 mg, 0.022 mmol) in MeOH (2 mL) was added to a solution of BSA-N3 (145 mg, 2.19 μmol) in PBS (10 mL), the reaction mixture was stirred overnight at r.t., concentrated by spin filtration (MWCO 10,000), purified on a PD-10 Sephadex column, and lyophilized. A portion of the resulting triazole 9 (25 mg, 0.379 μmol) was reconstituted in PBS (2.5 mL), and Rhodamine B azide (2 mg, 3.79 μmol) was added to it. The solution was irradiated for 8 min at 300 nm and stirred overnight in the dark. The resulting protein was concentrated by spin filtration (MWCO 10,000), purified on a PD-10 Sephadex column, and lyophilized. The BSA-Rhodamine conjugate, as well as a dark control, was resolved on 12% SDS-PAGE gel and visualized through in-gel fluorescence using a GE Typhoon scanner with excitation wavelength fixed at 532 nm and emission wavelength fixed at 580 nm.
Photo-crosslinking of BSA-N3 and PEG5000-N3
PEG5000-N3 (0.500g, 0.100 mmol) was added to a solution of 5a (23 mg, 0.10 mmol) in MeOH, the mixture was stirred at r.t. for 12 h, and concentrated to give 519 mg (99%) of the crude product as a white solid. Evaluation of the IR spectrum of the product revealed the disappearance of (N=N=N) stretch at 2100 cm−1. The crude solid was used in the subsequent step without further purification. A solution of the PEG-triazole 12 (5 mg, 0.956 μmol) in MeOH (1 mL) was added slowly to a solution of BSA-N3 (21 mg, 0.319 μmol) in PBS buffer (2 mL). The mixture was irradiated at 300 nm for 8 min and incubated overnight. The resulting protein was concentrated by spin filtration (MWCO 10,000), purified on a PD-10 Sephadex column, and lyophilized. The product was characterized by MALDI-TOF analysis and compared with a dark control (same treatment without irradiation). The MALDI-TOF analysis of the BSA-PEG5000 conjugate revealed multi-functionalization of BSA (Figure S3). Incubation of the mixture containing the triazole 12, BSA-N3, and PEG5000-azide in PBS overnight, didn’t produce any BSA-PEG5000 conjugate (Figure S4).20
Copper-catalyzed click (CuAAC) conjugation of BSA-N3 and propargyl-PEG5000
A solution BSA-N3 (13.2 mg, 0.20 μmol), propargyl-PEG5000 (10 mg, 2.0 μmol), ascorbic acid (7.0 μg, 0.040 μmol) and CuSO4 (3.2 μg, 0.020 μmol) in PBS (7 mM, 6 mL) was stirred for 12 h, concentrated to 1.5 mL by spin filtration (MWCO 10,000), purified over a PD-10 column, and lyophilized. The MALDI-TOF analysis of the protein shows that it contains a mixture of unmodified BSA and BSA molecules derivatized with one, two, three, and four PEG5000 fragments (Fig. S5).20 The ratio of these proteins (60: 23: 11) was almost identical to MC-DIBOD 5a pegylation experiment.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Dr. Y. G. Zheng and Mr. Z. Han’s help in the analysis of dye-labeled BSA. This project was supported by grants from the National Science Foundation (CHE-1565646 and CHE-1213789).
Footnotes
Supporting Information Available: Kinetic and MALDI-TOF data and NMR spectra of newly synthesized compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Baskin J, Prescher J, Laughlin S, Agard N, Chang P, Miller I, Lo A, Codelli J, Bertozzi CR. Proc Natl Acad Sci USA. 2007;104:16793. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Some recent reviews: Delaittre G, Guimard NK, Barner-Kowollik C. Acc Chem Res. 2015;48:1296. doi: 10.1021/acs.accounts.5b00075.Shieh P, Bertozzi CR. Org Biomol Chem. 2014;12:9307. doi: 10.1039/c4ob01632g.Tang W, Becker ML. Chem Soc Rev. 2014;43:7013. doi: 10.1039/c4cs00139g.
- 3.Arumugam S, Popik VV. J Org Chem. 2014;79:2702. doi: 10.1021/jo500143v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Kii L, Shiraishi A, Hiramatsu T, Matsushita T, Uekusa H, Yoshida S, Yammamoto M, Kudo A, Hagiwara M, Hosoya T. Org Biomol Chem. 2010;8:405. doi: 10.1039/c0ob00003e. [DOI] [PubMed] [Google Scholar]; (b) Xu F, Peng L, Shinohara K, Morita T, Yoshida S, Hosoya T, Orita A, Otera JJ. Org Chem. 2014;79:11592. doi: 10.1021/jo502248p. [DOI] [PubMed] [Google Scholar]; (c) Yoshida S, Shiraishi A, Kanno K, Matsushita T, Johmoto K, Uekusa H, Hosoya T. Sci Rep. 2011;1:8. doi: 10.1038/srep00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Delft P, de Witte W, Meeuwenoord NJ, van der Heden van Noort G, Versluis F, Olsthoorn RCL, Overkleeft HS, van der Marel GA, Filippov DV. Eur J Org Chem. 2014;34:7566. [Google Scholar]
- 6.Lau YH, Wu Y, Rossmann M, Tan BX, de Andrade P, Tan YS, Verma C, McKenzie GJ, Venkitaraman AR, Hyvonen M, Spring DR. Angew Chem Int Ed. 2015;54:15410. doi: 10.1002/anie.201508416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z, Liu J, Arslan HK, Grosjean S, Hagendorn T, Gliemann H, Brase S, Woll C. Langmuir. 2013;29:15958. doi: 10.1021/la403854w. [DOI] [PubMed] [Google Scholar]
- 8.Horner A, Volz D, Hagendorn T, Furniss D, Greb L, Ronicke F, Nieger M, Schepers U, Brase S. RSC Adv. 2014;4:11528. [Google Scholar]
- 9.Hashimoto C, Nomura W, Narumi T, Fujino M, Nakahara T, Yamamoto N, Murakami T, Tamamura H. Bioorg Med Chem. 2013;21:6878. doi: 10.1016/j.bmc.2013.09.037. [DOI] [PubMed] [Google Scholar]
- 10.Sutton AD, Yu SH, Steet R, Popik VV. Chem Commun. 2016;52:553. doi: 10.1039/c5cc08106h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Poloukhtine A, Popik VV. J Org Chem. 2003;68:7833. doi: 10.1021/jo034869m. [DOI] [PubMed] [Google Scholar]; (b) Poloukhtine A, Popik VV. J Phys Chem A. 2006;110:1749. doi: 10.1021/jp0563641. [DOI] [PubMed] [Google Scholar]
- 12.Urdabaev NK, Poloukhtine A, Popik VV. Chem Comm. 2006:454. doi: 10.1039/b513248g. [DOI] [PubMed] [Google Scholar]
- 13.Recent review on photo-click chemistry: Tasdelen MA, Yagci Y. Angew Chem Int Ed. 2013;52:5930. doi: 10.1002/anie.201208741.
- 14.Friscourt F, Fahrni CJ, Boons GJ. J Am Chem Soc. 2012;134:18809. doi: 10.1021/ja309000s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Wang F, Luo T, Hu J, Wang Y, Krishnan H, Jog P, Ganesh S, Prakash G, Olah G. Angew Chem Int Ed. 2011;50:7153. doi: 10.1002/anie.201101691. [DOI] [PubMed] [Google Scholar]; (b) Wang F, Zhang W, Zhu J, Li H, Huang KW, Hu J. Chem Comm. 2011;47:2411. doi: 10.1039/c0cc04548a. [DOI] [PubMed] [Google Scholar]
- 16.Poloukhtine AA, Mbua NE, Wolfert MA, Boons GJ, Popik VV. J Am Chem Soc. 2009;131:15769. doi: 10.1021/ja9054096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Ess DH, Jones GO, Houk KN. Organic Letters. 2008;10:1633. doi: 10.1021/ol8003657. [DOI] [PubMed] [Google Scholar]; (b) Gordon CG, Mackey JL, Jewett JC, Sletten EM, Houk KN, Bertozzi CR. J Am Chem Soc. 2012;134:9199. doi: 10.1021/ja3000936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Gold B, Dudley GB, Alabugin IV. J Am Chem Soc. 2012;135:1558. doi: 10.1021/ja3114196. [DOI] [PubMed] [Google Scholar]; (b) Gold B, Batsomboon P, Dudley GB, Alabugin IV. J Org Chem. 2014;79:6221. doi: 10.1021/jo500958n. [DOI] [PubMed] [Google Scholar]
- 19.Kii L, Shiraishi A, Hiramatsu T, Matsushita T, Uekusa H, Yoshida S, Yammamoto M, Kudo A, Hagiwara M, Hosoya T. Org Biomol Chem. 2010;8:405. doi: 10.1039/c0ob00003e. [DOI] [PubMed] [Google Scholar]
- 20.Supporting Information
- 21.McNitt CD, Popik VV. Org Biomol Chem. 2012;10:8200. doi: 10.1039/c2ob26581h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marculesca C, Kossen H, Morgan R, Mayer P, Fletcher S, Tolner B, Chester K, Jones L, Baker J. Chem Comm. 2014;50:7139. doi: 10.1039/c4cc02107j. [DOI] [PubMed] [Google Scholar]
- 23.Roeder R, Rungta P, Tsyalkovvsky V, Bandera Y, Foulger S. Soft Matter. 2012;8:5493. [Google Scholar]
- 24.Gauthier M, Klok H. Chem Comm. 2008;23:2591. doi: 10.1039/b719689j. [DOI] [PubMed] [Google Scholar]
- 25.Pavilockova L, Kuzmic P, Soucek M. Coll Czech Chem Comm. 1986;51:368. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.