

Abstract
Clonostachys rosea is a necrotrophic mycoparasitic fungus, used for biological control of plant pathogenic fungi. A better understanding of the underlying mechanisms resulting in successful biocontrol is important for knowledge‐based improvements of the application and use of biocontrol in agricultural production systems. Transcriptomic analyses revealed that C. rosea responded with both common and specific gene expression during interactions with the fungal prey species Botrytis cinerea and Fusarium graminearum. Genes predicted to encode proteins involved in membrane transport, biosynthesis of secondary metabolites and carbohydrate‐active enzymes were induced during the mycoparasitic attack. Predicted major facilitator superfamily (MFS) transporters constituted 54% of the induced genes, and detailed phylogenetic and evolutionary analyses showed that a majority of these genes belonged to MFS gene families evolving under selection for increased paralog numbers, with predicted functions in drug resistance and transport of carbohydrates and small organic compounds. Sequence analysis of MFS transporters from family 2.A.1.3.65 identified rapidly evolving loop regions forming the entry to the transport tunnel, indicating changes in substrate specificity as a target for selection. Deletion of the MFS transporter gene mfs464 resulted in mutants with increased growth inhibitory activity against F. graminearum, providing evidence for a function in interspecific fungal interactions. In summary, we show that the mycoparasite C. rosea can distinguish between fungal prey species and modulate its transcriptomic responses accordingly. Gene expression data emphasize the importance of secondary metabolites in mycoparasitic interactions.
Keywords: biological control, Clonostachys rosea, gene deletion, membrane transporter, mycoparasitism, transcriptome analysis
1. INTRODUCTION
Chemical pesticides play an important role for maintaining high yields in agricultural productions systems in the world. However, the extensive use of agricultural chemicals has led to concerns about unwanted, negative effects on the environment and human health. One approach to minimize the amount of used pesticides is integrated pest management, which promotes the integrative use of preventive cultural practices, disease‐resistant plant cultivars and mechanical and biological control of pathogen populations. Biological control can also be used in organic agriculture, where chemical pesticides are banned. The use of biological control of plant pathogens by microbial antagonists can therefore be expected to increase, and to become standard procedure, in future agricultural and horticultural production.
Certain species of opportunistic, mycoparasitic fungi from the genera Clonostachys and Trichoderma are currently used as biological control agents to control plant pathogenic fungi. Clonostachys rosea (Link: Fr.) Schroers, Samuels, Seifert & W. Gams, comb. nov. (Schroers, Samuels, Seifert, & Gams, 1999) is an ascomycete fungus that is reported to control diseases caused by a wide range of plant pathogenic fungi, including Alternaria spp. (Jensen, Knudsen, Madsen, & Jensen, 2004), Bipolaris sorokiniana (Jensen, Knudsen, & Jensen, 2002), Botrytis cinerea (Sutton et al., 1997), Fusarium culmorum (Jensen, Knudsen, & Jensen, 2000), Fusarium graminearum (Xue et al., 2009) and Sclerotinia sclerotiorum (Rodriguez, Cabrera, Gozzo, Eberlin, & Godeas, 2011). Several biocontrol mechanisms are reported in C. rosea, including direct parasitism of pathogenic fungi (Li, Huang, Kokko, & Acharya, 2002; Yu & Sutton, 1997), antibiosis (Pachenari & Dix, 1980; Rodriguez et al., 2011), production of fungal cell wall degrading enzymes (Chatterton & Punja, 2009; Mamarabadi, Jensen, & Lubeck, 2008), induction of plant defence reactions (Lahlali & Peng, 2014; Roberti et al., 2008) and plant growth promotion (Roberti et al., 2008). Tolerance towards, and detoxification of, toxic metabolites produced by the fungal prey is also reported to be an important biocontrol trait in C. rosea (Dubey, Jensen, & Karlsson, 2014; Kosawang, Karlsson, Velez et al., 2014).
We recently sequenced the genome of C. rosea strain IK726 and performed a comparative genome analysis with Trichoderma and Fusarium species (Karlsson et al., 2015). Conspicuous features of the C. rosea gene content include a high number of genes encoding ATP‐binding cassette (ABC) and major facilitator superfamily (MFS) membrane transporters, polyketide synthase (PKS) and nonribosomal peptide synthetase (NRPS) secondary metabolite biosynthesis proteins, cytochrome P450 (CYP) and polysaccharide lyase family 1 (PL1) pectin lyases (Karlsson et al., 2015). The selection for high number of genes in these gene families suggests that their gene products may be involved in ecological niche adaptation (Wapinski, Pfeffer, Friedman, & Regev, 2007), such as mycoparasitism. Although C. rosea and Trichoderma spp. are necrotrophic mycoparasites with broad host range, it is not known if, or to what extent, they modulate their responses towards different fungal prey species. A better understanding of the mechanisms that determine the outcome of biocontrol interactions is necessary for science‐based improvements of biocontrol applications in agriculture.
The aim with the current work is to identify C. rosea genes that are specifically induced during interactions with the fungal pathogens B. cinerea or F. graminearum, and genes induced in response to both pathogens, using an RNA‐seq approach. We hypothesize that the transcriptomic response in C. rosea towards the two pathogens differs due to their intrinsic differences in cell wall composition, secondary metabolite spectra, etc. We further hypothesize that there will be an overlap between genes induced during fungal–fungal interactions and genes evolving under diversifying selection (in analogy with effectors in other parasite–host interactions). The results show that C. rosea responds with both common and specific transcriptional changes during interactions with B. cinerea and F. graminearum. Induction of genes putatively encoding drug resistance membrane transporters and proteins involved in biosynthesis of secondary metabolites emphasize the importance of secondary metabolites in mycoparasitic interactions.
2. MATERIALS AND METHODS
2.1. Cultivation conditions
Clonostachys rosea strain IK726 (WT) and mutants derived from it, F. graminearum strain PH‐1 and B. cinerea strain B05.10 were maintained on potato dextrose agar (PDA) medium (Oxoid, Cambridge, UK) at 25°C in darkness unless otherwise specified. Vogel's minimal (VM) medium (Vogel, 1956) with 0.3% glucose was used to grow fungi for transcriptome sequencing, while Czapek dox (CZ) medium (Sigma‐Aldrich, St. Louis, MO, USA) was used to grow fungi for reverse transcription quantitative PCR (RT‐qPCR) gene expression analyses, and phenotypic analyses of mutant strains.
For transcriptome sequencing during interactions, an agar plug of C. rosea mycelium and of the prey fungi F. graminearum or B. cinerea were inoculated at opposite sides of a 9‐cm‐diameter VM agar Petri plate (covered with a nylon membrane for easy harvest) and incubated at 25°C in darkness. Due to different mycelial growth rates, C. rosea was inoculated 5 days prior to the inoculation of F. graminearum or B. cinerea. The growing front (6 mm) of C. rosea mycelium was harvested together with the mycelial front (2 mm) of the prey fungi, 24 hr after hyphal contact between the fungi. Mycelium harvested from C. rosea confronted with itself at the same stage was used as the control treatment. Two biological replicates (different Petri plates) for each treatment were used.
For RT‐qPCR gene expression analysis, zearalenone (ZEA) was obtained from Sigma‐Aldrich and dissolved in methanol, while fungicides were obtained from BASF (Ludwigshafen, Germany [Cantus]), Syngenta (Basel, Switzerland [Amistar and Apron]) and Bayer AG (Leverkusen, Germany [Chipco Green and Teldor]) and dissolved in sterile distilled water. Clonostachys rosea was pregrown for 5 days in 20 ml liquid CZ medium, after which the growth medium was supplemented with ZEA to a final concentration of 10 μg/ml, Apron (mefenoxam) at 2 μg/ml, Amistar (azoxystrobin) at 7.5 μg/ml, Chipco Green (iprodione) at 250 μg/ml or Cantus (boscalid) at 2000 μg/ml. In the control treatments, ZEA and fungicides were replaced with an equal volume of methanol or sterile distilled water, respectively. Fungal mycelia were harvested 2 hr after the addition, washed in distilled water to remove traces of ZEA or fungicides, frozen in liquid nitrogen and stored at −80°C.
2.2. Nucleic acid isolation and manipulation
Genomic DNA was extracted from C. rosea as described previously (Nygren et al., 2008). RNA extraction was performed using the Qiagen RNeasy kit following the manufacturer's protocol (Qiagen, Hilden, Germany). After DNaseI (Fermentas, St. Leon‐Rot, Germany) treatment, five micrograms of total RNA was used for removal of ribosomal RNA using the Ribominus eukaryotic kit for RNA‐seq (Life Technologies, Carlsbad, CA, USA). For each sample, 500 nanograms of ribominus‐depleted RNA was reverse transcribed into cDNA using the SMARTer PCR cDNA synthesis kit following the manufacturer's protocol (Clontech, Mountain View, CA, USA). PCR amplifications were performed on cDNA for 15, 18, 21, 24 and 27 cycles to determine the optimum number of PCR cycles using the conditions: 95°C for 1 min, 95°C for 15 s, 64°C for 30 s and 65°C for 3 min on a GeneAMP PCR system 2700 (Applied Biosystems, Carlsbad, CA, USA). Running the PCR for 18 cycles was found to be optimal based on agarose gel electrophoresis (producing maximum amount of PCR products, but before entering the plateau phase of the reaction), and the PCR product was purified using a PCR purification kit (Fermentas, Waltham, MA, USA), analysed using a 2100 Bioanalyzer Instrument (Agilent Technologies, Santa Clara, CA, USA), quantified using a Qubit fluorometer (Life Technologies) and sent for paired‐end transcriptome sequencing at the SNP&SEQ Technology Platform, Uppsala, Sweden, using Illumina HiSeq2000 equipment. Read lengths were 107 and 144 bp, respectively, and the average insert sizes for the different samples were 305–340 bp.
2.3. Transcriptome data analysis
Quality filtration and adapter removal were performed using Cutadapt ver. 1.2.1 (Martin, 2011), minimum read length was set to 50 bp, and only reads still belonging to a complete pair were kept for further analysis. TopHat ver. 2.0.9 and Cufflinks ver. 2.1.1 packages (Trapnell et al., 2012) were used to map reads to the C. rosea, B. cinerea and F. graminearum reference genomes (Amselem et al., 2011; Cuomo et al., 2007; Karlsson et al., 2015), quantitate and test for differential gene expression. Parameter settings were kept at default, except for intron size adjustment. Fungi generally have short introns; therefore, minimal intron length was set to 5 bp and maximum intron length to 5,000 bp. Reads were analysed as unstranded, and read pair distance was adjusted for each sample. One mismatch was allowed in the analyses. The data were corrected for multiple testing by adjusting p‐values to a false discovery rate (FDR) of .05 using Cuffcompare in Cufflinks. Two of three conditions contained tissue from two species, C. rosea and either of B. cinerea and F. graminearum. Therefore, all reads were aligned to the three reference genomes and only the read pairs with a better alignment to the C. rosea genome than to either of the two other genomes were used in further analyses. To avoid biases in comparisons between conditions, all samples were filtered the same way. Expression levels were determined as reads per kilobase of transcript per million mapped reads.
2.4. Gene expression analysis with RT‐qPCR
RNA extraction was performed using the Qiagen RNeasy kit following the manufacturer's protocol (Qiagen). After DNaseI (Fermentas, St. Leon‐Rot, Germany) treatment, one microgram of total RNA was reverse transcribed in a total volume of 20 μl using Maxima first stand cDNA synthesis kit (Fermentas, St. Leon‐Rot, Germany). Transcript levels were quantified by RT‐qPCR using the SYBR Green PCR Master Mix (Fermentas, St. Leon‐Rot, Germany) and primer pairs listed in Appendix S1, in an iQ5 qPCR System (Bio‐Rad, Hercules, CA, USA) as described previously (Tzelepis, Melin, Jensen, Stenlid, & Karlsson, 2012). Melt curve analysis was performed after the qPCR reactions, to confirm that the signal was the result from a single product amplification. Relative expression levels for target genes in relation to actin (act), shown previously to be constitutively expressed (Kamou et al., 2016; Tzelepis, Dubey, Jensen, & Karlsson, 2015), were calculated from the Ct values by the method (Livak & Schmittgen, 2001). Gene expression analysis was carried out in at least three biological replicates, each based on two technical replicates. Gene expression data were analysed by analysis of variance (ANOVA) using a general linear model approach implemented in Minitab ver. 18 (Minitab Inc., State College, PA, USA). Pairwise comparisons were made using the Fisher method at the 95% significance level.
2.5. MFS transporter gene family evolution
Seven hypocrealean fungi and Neurospora crassa were included in studying the evolutionary history of the MFS gene family. Whole‐genome nucleotide and protein sequences of the mycoparasitic fungi C. rosea (Karlsson et al., 2015), T. atroviride, T. reesei and T. virens (Kubicek et al., 2011; Martinez et al., 2008), and the plant pathogenic fungi F. graminearum, F. solani and F. verticillioides (Coleman et al., 2009; Cuomo et al., 2007; Ma et al., 2010), and the saprotrophic fungus N. crassa (Galagan et al., 2003) were retrieved from the National Center for Biotechnology Information (NCBI). ABC and MFS transporter genes were identified by BlastP analysis in an iterative process described previously (Karlsson et al., 2015), although the predicted C. rosea MFS transporters BN869_T00000646, BN869_T00007234 and BN869_T00002052 were additionally included as templates in the search. MFS transporters were classified into subgroups according to the Transporter Classification Database (TCDB, Saier et al., 2016).
Phylogenetic relationships were taken from Karlsson et al. (2015), while branch lengths were calculated based on a four‐gene alignment including actin, glyceraldehyde 3‐phosphate dehydrogenase, DNA‐directed RNA polymerase II subunit B and translation elongation factor 1 alpha. Coding gene sequences were retrieved from the respective genome sequences. Each gene was aligned individually using Clustal W (Larkin et al., 2007) in MEGA ver. 6 (Tamura, Stecher, Peterson, Filipski, & Kumar, 2013), concatenated and used to calculate branch lengths in MEGA ver. 6. The resulting species phylogeny was calibrated by setting the split between T. reesei and T. virens to 16 million years (de Man et al., 2016).
Major facilitator superfamily gene family evolution analysis was carried out on subgroups that contained ≥2 genes in at least one species and were present in ≥2 species. The program CAFE (Computational Analysis for Gene Family Evolution) ver. 3 (Han, Thomas, Lugo‐Martinez, & Hahn, 2013) was used to test whether gene family sizes were compatible with a stochastic birth‐and‐death model, to estimate gene family size in extinct species and to identify lineages with accelerated rates of gene gain or loss. Mutation rate (λ) was 0.0024 (Karlsson et al., 2015).
2.6. Phylogenetic analyses
Full‐length protein sequences were aligned by MUSCLE (Edgar, 2004), and phylogenetic trees were constructed using neighbour‐joining implemented in MEGA ver. 6. The JTT amino acid substitution model (Jones, Taylor, & Thornton, 1992) was used with uniform rates among sites and pairwise deletion of gaps. Statistical support for branches was assessed by 500 iterations of bootstrap resampling. Conserved protein modules and features were identified using the conserved domain database (CDD) (Marchler‐Bauer et al., 2017) at NCBI.
2.7. Analysis of molecular evolution
Regions of low amino acid conservation in MFS transporter alignments were identified by reverse conservation analysis (RCA), as described by Lee (2008). In short, Rate4Site ver. 2.01 was used to calculate the degree of conservation (S score, high scores correspond to low degree of conservation) for each amino acid position using the empirical Bayesian method (Mayrose, Graur, Ben‐Tal, & Pupko, 2004; Pupko, Bell, Mayrose, Glaser, & Ben‐Tal, 2002). A sliding‐window average (n = 7) of normalized S scores (mean was 0 and standard deviation was 1) was plotted in Excel (Microsoft; W mean score), and significant peaks were defined by values ≥1. Transmembrane helices were predicted using HMMTOP ver. 2 (Tusnady & Simon, 1998, 2001).
2.8. Construction of gene deletion vectors, transformation and mutant validation
Three fragment multisite gateway cloning system (Invitrogen, Carlsbad, CA, USA) was used to construct gene deletion vectors. The ~ 1 kb 5′‐flank and 3′‐flank regions of mfs464, mfs602, fdo1 and cyp1 were amplified from genomic DNA of C. rosea using gene specific primer pairs ups F/ups R and ds F/ds R, respectively, as indicated in Appendix S1. Gateway entry clones of the purified 5′‐flank and 3′‐flank PCR fragments were generated as described by the manufacturer (Invitrogen). The gateway entry clones for the hygromycin (hph) gene, constructed during our previous studies (Dubey, Broberg, Jensen, & Karlsson, 2013; Dubey, Ubhayasekera, Sandgren, Jensen, & Karlsson, 2012; Dubey, Broberg, Sooriyaarachchi et al., 2013), were used. The gateway LR recombination reaction was performed using entry plasmid of respective fragments and destination vector pPm43GW (Karimi, De Meyer, & Hilson, 2005) to generate the deletion vectors. Agrobacterium tumefaciens‐mediated transformation (ATMT) was performed based on a previous protocol for C. rosea (Utermark & Karlovsky, 2008). Putative transformants were selected on a plate containing hygromycin (200 μg/ml). Validation of homologous integration of the deletion cassettes in putative transformants was performed using a PCR screening approach with primer combinations targeting the hph gene (Hyg F/Hyg R) and sequences flanking the deletion cassettes (Appendices S1 and S2) as described previously (Dubey, Jensen, & Karlsson, 2016; Dubey et al., 2014). RT‐PCR analysis was conducted on WT and gene deletion strains using RevertAid premium reverse transcriptase (Fermentas, St. Leon‐Rot, Germany) and primer pairs specific for the respective genes (Appendix S1). The PCR‐positive transformants were tested for mitotic stability, and were purified by two rounds of single spore isolation (Dubey et al., 2014, 2016).
2.9. Phenotypic analyses of gene deletion mutants
For growth rate analysis, a 3‐mm‐diameter agar plug from the growing mycelial front was transferred to solid CZ medium, CZ medium containing the Fusarium mycotoxins ZEA (50 μg/ml) or deoxynivalenol (10 μg/ml); fungicides Cantus (boscalid [succinate dehydrogenase inhibitor] 2000 μg/ml), Amistar (azoxystrobin [electron transport chain inhibitor] 7.5 μg/ml), Chipco green (iprodione [inhibitor of DNA and RNA synthesis, and NADH cytochrome c reductase] 250 μg/ml), Apron (mefenoxam [inhibitor of RNA polymerases] 2 μg/ml) or Teldor (fenhexamid [inhibitor of sterol biosynthesis] 5000 μg/ml); inducers of oxidative stress paraquat (10 mM) or H2O2 (10 mM), cell wall stress SDS (0.05%) or caffeine (0.1%), osmotic stress sodium chloride (0.5 M) or sorbitol (1 M); ions calcium chloride (400 mM), potassium chloride (400 mM), magnesium chloride (400 mM), caesium chloride (20 mM), zinc chloride (20 mM) or lithium chloride (10 mM). In addition, growth rates were analysed under nutrient‐limited conditions at pH 4.0, pH 9.0 or pH 10.0. Limitation for carbon, nitrogen, magnesium, iron or potassium was induced by reducing the concentration of sucrose, sodium nitrate, magnesium sulphate, ferrous sulphate or potassium chloride by 1/10. Colony diameter was measured after 5 days of growth at 25°C in darkness. The concentration of chemicals used for phenotypic analysis in this study was based on our previously published results (Dubey et al., 2014, 2016) with the exception of ions. The appropriate concentration of ions was selected based on screening of C. rosea WT spores and mycelium on CZ plates amended with a series of different concentrations of the respective compounds. For determination of conidial germination, germ tube development and colony formation, WT and deletion strain conidia were inoculated (103 conidia) on solid agar plates using the same concentration of chemical compounds described above with the exception of H2O2 (0.8 mM) and Teldor (fenhexamid 1000 μg/ml). Data for conidial germination and germ tube development were recorded daily using a Leica 165FC microscope (Wetzlar, Germany), while colony diameter was measured 5 days postinoculation. All phenotypic analyses were performed with at least three independent mutants (to avoid phenotypes resulting from ectopic insertions of the deletion cassette), and each based on at least three biological replicates unless otherwise specified.
Antagonistic behaviour of C. rosea WT and deletion strains against F. graminearum or B. cinerea was tested in five biological replicates using an in vitro plate confrontation assay and a culture filtrate assay, as described previously (Dubey et al., 2014, 2016). In addition, a liquid culture confrontation test was performed in five biological replicates, where conidia (105 conidia) from WT and deletion strains were inoculated in 50 ml VM with 0.3% glucose in Erlenmeyer flasks and were allowed to grow for 24 hr at 25°C in darkness. Subsequently, an equal amount of F. graminearum spores were added to the flasks and further incubated for 3 days to allow the antagonistic interaction. All mycelia were harvested, washed with distilled water and used for DNA extraction. The mycelial biomass was estimated by measuring C. rosea and F. graminearum DNA with qPCR using primers specific to β‐tubulin (Dubey et al., 2014; Reischer, Lemmens, Farnleitner, Adler, & Mach, 2004). An in vivo bioassay using a sand seedling test for fusarium foot rot disease on barley was performed in five biological replicates, where each replicate included 15 plants, following the procedure described previously (Dubey et al., 2014, 2016).
3. RESULTS
3.1. Transcriptional response of C. rosea during interactions with B. cinerea and F. graminearum
Dual confrontation assays (C. rosea vs. C. rosea, C. rosea vs. B. cinerea and C. rosea vs. F. graminearum) were monitored using a stereo microscope on a daily basis, and mycelium from the interaction zone was harvested 24 hr after the first hyphal contacts were observed. This occurred 10 days after inoculation of C. rosea for all interactions. Illumina HiSeq paired‐end sequencing of cDNA generated from RNA from the interaction zones resulted in a total of 117 million read pairs, equivalent to 143.5 Gbp of sequence. After removal of low‐quality reads and reads originating from B. cinerea or F. graminearum, 36.8 million read pairs remained that were used for analysis and identification of differentially expressed genes. In total, 41 differentially expressed genes were identified, and functional classification showed that 61% were predicted to encode membrane transporters, 12% were predicted to encode proteins involved in biosynthesis of secondary metabolites, and 7% encoded carbohydrate‐active enzymes (Table 1). Six genes were induced in C. rosea during interactions with both B. cinerea and F. graminearum, while most genes were induced specifically towards either of the two different fungal prey species (Table 1).
Table 1.
Differentially expressed genes in Clonostachys rosea during interactions with Botrytis cinerea or Fusarium graminearum
| Protein ID | Gene name | Putative function | E‐valuea | Expression level in CRb | Expression level in BCb | Expression level in FGb |
|---|---|---|---|---|---|---|
| BN869_T00005393 | Hydantoinase/oxoprolinase | 0 | 2.7 | 16.5 | 16.5 | |
| BN869_T00013543 | 2‐deoxy‐d‐gluconate 3‐dehydrogenase | 1e‐160 | 3.6 | 24.5 | 35.5 | |
| BN869_T00013396 | Uracil permease | 0 | 5.3 | 50.0 | 82.3 | |
| BN869_T00010526 | mfs24 | Maltose permease (MFS) | 0 | 14.4 | 67.2 | 47.2 |
| BN869_T00005531 | mfs166 | Carboxylic acid transporter (MFS) | 0 | 1.0 | 7.1 | 10.2 |
| BN869_T00007347 | mfs104 | Glucose transporter (MFS) | 0 | 29.5 | 77.0 | 76.0 |
| BN869_T00006915 | PL1 pectate lyase | 5e‐165 | 28.4 | 101.9* | 4.0 | |
| BN869_T00008662 | Oxalate decarboxylase | 0 | 0.6 | 20.7* | 0.8 | |
| BN869_T00008498 | Cytochrome P450 | 0 | 1.4 | 43.7* | 3.7 | |
| BN869_T00004216 | Oxalate decarboxylase | 0 | 14.2 | 110.1* | 7.9 | |
| BN869_T00009370 | Cyclopentanone monooxygenase | 0 | 50.8 | 199.7* | 65.1 | |
| BN869_T00006567 | pks9 | Lovastatin diketide synthase | 0 | 0.7 | 2.3* | 0.3 |
| BN869_T00012005 | nps13 | Nonribosomal peptide synthetase | 0 | 1.6 | 3.0* | 0.6 |
| BN869_T00012311 | mfs293 | MFS transporter | 0 | 1.8 | 16.4* | 2.5 |
| BN869_T00009923 | mfs249 | MFS transporter | 2e‐107 | 7.5 | 36.9* | 11.7 |
| BN869_T00007301 | mfs44 | Sugar/quinate transporter (MFS) | 0 | 16.8 | 57.2 | 23.0 |
| BN869_T00006570 | abcC8 | ABC transporter | 0 | 0.7 | 2.7* | 0.4 |
| BN869_T00008321 | GH28 alpha‐L‐rhamnosidase | 8e‐180 | 1.1 | 2.2 | 18.3* | |
| BN869_T00006501 | chiC1 | GH18 killer toxin‐like chitinase | 0 | 1.8 | 1.9 | 90.4* |
| BN869_T00000852 | cyp1 | Isotrichodermin C‐15 hydroxylase | 0 | 2.7 | 0.9 | 35.4* |
| BN869_T00011161 | Norsolorinic acid reductase | 0 | 29.4 | 28.8 | 108.4* | |
| BN869_T00006455 | mfs602 | MFS transporter | 0 | 0.7 | 0.2 | 485.3 |
| BN869_T00007234 | mfs464 | MFS transporter | 0 | 0.9 | 0.9 | 709.9* |
| BN869_T00010393 | mfs49 | Quinate transporter (MFS) | 0 | 1.0 | 1.1 | 23.0* |
| BN869_T00000606 | fdo1 | FAD‐dependent oxidoreductase | 4e‐127 | 1.4 | 1.6 | 121.0* |
| BN869_T00009314 | mfs271 | Allantoate permease (MFS) | 0 | 3.6 | 6.9 | 58.1* |
| BN869_T00002607 | mfs518 | MFS transporter | 0 | 3.8 | 3.6 | 54.0* |
| BN869_T00005826 | Potassium channel subunit beta | 0 | 3.9 | 3.3 | 51.3* | |
| BN869_T00008599 | mfs187 | MFS transporter | 1e‐148 | 8.7 | 9.9 | 37.8* |
| BN869_T00007004 | mfs534 | MFS transporter | 0 | 12.3 | 11.0 | 46.7* |
| BN869_T00009913 | mfs524 | Cycloheximide resistance protein (MFS) | 0 | 12.9 | 6.5 | 75.9* |
| BN869_T00002418 | ptr1 | Peptide transporter | 0 | 27.0 | 32.5 | 103.0* |
| BN869_T00010026 | abcG18 | ABC transporter | 0 | 0.5 | 0.5 | 2.8* |
| BN869_T00011684 | mfs324 | Mononucleotide permease (MFS) | 0 | 1.1 | 1.5 | 12.3* |
| BN869_T00005590 | mfs604 | MFS transporter | 0 | 2.2 | 1.7 | 8.3* |
| BN869_T00006700 | mfs315 | Pantothenate transporter (MFS) | 0 | 69.3 | 73.1 | 232.0* |
| BN869_T00012557 | mfs205 | Riboflavin transporter (MFS) | 9e‐169 | 29.3 | 17.0 | 83.2* |
| BN869_T00008447 | mfs282 | MFS transporter | 0 | 1.6 | 1.4 | 6.0* |
| BN869_T00013156 | mfs291 | Vitamin H transporter (MFS) | 0 | 2.0 | 1.8 | 8.3* |
| BN869_T00000646 | mfs176 | MFS transporter | 4e‐145 | 15.4 | 12.5 | 44.6* |
| BN869_T00011371 | mfs609 | Aminotriazole resistance protein (MFS) | 0 | 45.4 | 43.7 | 109.5* |
ABC, ATP‐binding cassette; FAD, flavin adenine dinucleotide; GH, glycoside hydrolase; MFS, major facilitator superfamily; PL, polysaccharide lyase.
E‐value originates from BlastP analysis of the NCBI nonredundant database, or from the SMART protein analysis tool.
Expression level was determined as reads per kilobase of transcript per million mapped reads values. Treatments are CR = expression level in treatment C. rosea vs. C. rosea, BC = expression level in treatment C. rosea vs. B. cinerea, FG = expression level in treatment C. rosea vs. F. graminearum. BC and FG values indicated in bold are significantly (FDR adjusted p ≤ .05) different from CR values in normal font. BC or FG values marked with an asterisk are significantly (FDR adjusted p ≤ .05) higher in the comparison between BC and FG treatments.
Among the 26 differentially expressed membrane transporter genes, 22 were identified as putative MFS transporters while only two were ABC transporters. The two ABC transporter genes were described previously as abcC8 and abcG18 (Karlsson et al., 2015) and predicted to encode members of the multidrug resistance‐associated protein (MRP, group C) and the pleiotropic drug resistance protein (PDR, group G) ABC transporter groups, respectively. Expression of abcC8 was induced during interaction with B. cinerea, while abcG18 was induced specifically against F. graminearum (Table 1). Three putative MFS transporter genes and one NCS1‐type family uracil transporter (BN869_T00013396) were induced against both pathogens compared with the C. rosea self‐interaction, while 3 and 16 MFS transporter genes were specifically induced against B. cinerea and F. graminearum, respectively. A putative membrane peptide transporter (BN869_T00002418 [ptr1]) was also induced 3.8‐fold against F. graminearum specifically. The highest fold‐change inductions were detected for two predicted MFS transporters (BN869_T00006455 [mfs602] and BN869_T00007234 [mfs464]) that were induced 693‐fold and 789‐fold, respectively, during interaction with F. graminearum (Table 1).
The PKS gene pks9, putatively encoding a reduced polyketide‐type PKS, and the NRPS gene nps13 (Karlsson et al., 2015) were induced during confrontation with B. cinerea in comparison with F. graminearum although expression of nps13 was not significantly different from the C. rosea self‐interaction control treatment. A predicted CYP (BN869_T00000852 [cyp1]) with similarity to an isotrichodermin C‐15 hydroxylase was induced specifically against F. graminearum. The killer toxin‐like chitinase gene chiC1 (Tzelepis et al., 2015) was induced 50‐fold during interactions with F. graminearum, in comparison with the B. cinerea and C. rosea self‐interaction treatments. A predicted FAD‐dependent oxidoreductase (BN869_T00000606 [fdo1]) with similarity to a tetracycline resistance protein was induced 86‐fold against F. graminearum. A putative PL1 pectin lyase gene (BN869_T00006915) was induced during interaction with B. cinerea, but repressed during interaction with F. graminearum (Table 1).
3.2. Validation of differential expression by RT‐qPCR
Validation of RNA‐seq gene expression data with RT‐qPCR was carried out for nine selected genes, including three genes specifically induced during interaction with B. cinerea and six genes specifically induced against F. graminearum. There was a high level of congruence between gene expression patterns measured with the two methods (Table 1, Figure 1). Four minor differences were detected; expression of abcC8 was higher in the C. rosea vs. C. rosea treatment compared with the F. graminearum confrontation using RT‐qPCR but not with RNA‐seq, while expression of ptr1, abcG18 and cyp1 was higher in the F. graminearum confrontation compared with the C. rosea vs. C. rosea treatment using RNA‐seq but not with RT‐qPCR (Figure 1).
Figure 1.
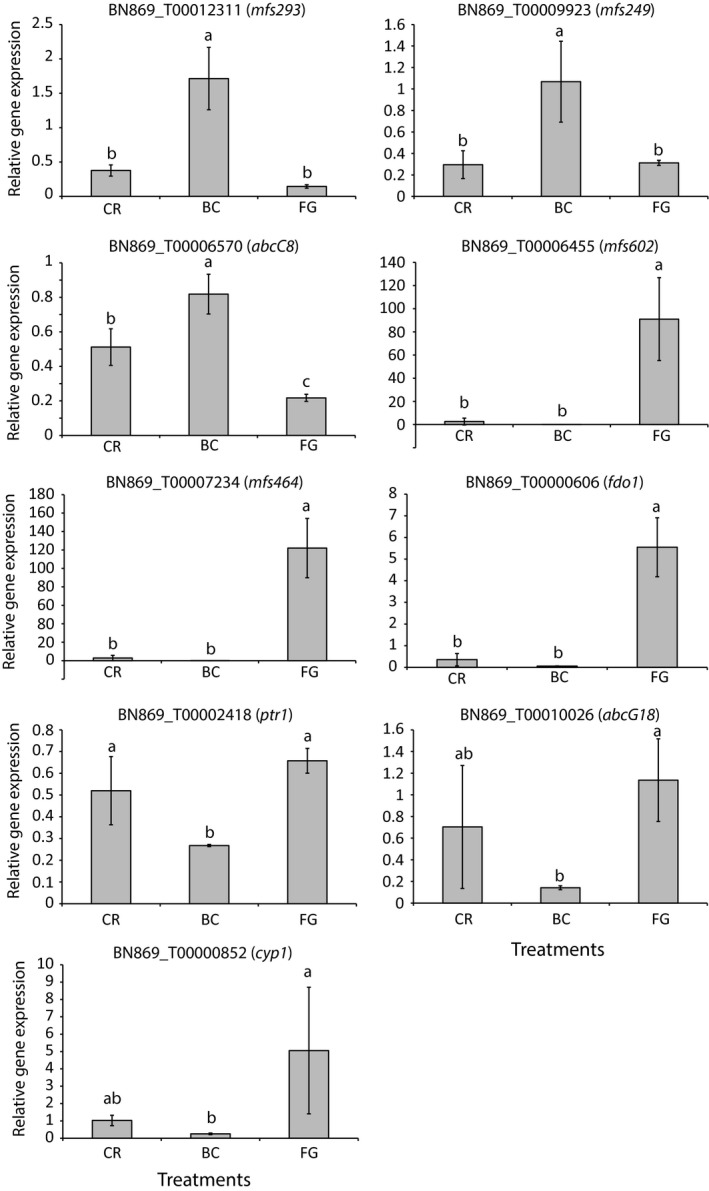
Expression analysis of Clonostachys rosea genes. Gene expression of C. rosea genes was measured with RT‐qPCR during interactions with Botrytis cinerea (BC) or Fusarium graminearum (FG) or during self‐interactions (CR). Relative gene expression was calculated using the method, normalized by actin (act) expression. Error bars represent standard deviation based on three biological replicates. Different letters indicate statistically significant differences (p ≤ .05) based on the Fisher method
3.3. Evolution of the MFS transporter gene family in C. rosea and other hypocrealean fungi
The massive induction of membrane transporter genes in C. rosea during confrontations with fungal prey prompted us to investigate these gene families in more detail. The ABC transporter gene family in C. rosea was previously described in detail (Karlsson et al., 2015), but an iterative Blast approach identified an additional four genes resulting in a total number of 90 ABC transporter genes in the C. rosea genome. Based on a phylogenetic analysis (data not shown), these four additional ABC transporter genes were classified as members of subgroup E (BN869_T00001090) and subgroup F (BN869_T00005647, BN869_T00006720 and BN869_T00007586) according to the Kovalchuk and Driessen (2010) classification system, and named abcE1, abcF3, abcF4 and abcF5, respectively.
Using an iterative Blast approach, we further identified a total of 634 MFS transporter genes in the genome of C. rosea (Appendix S3). The total number of MFS transporter genes in other hypocrealean fungal species ranged from 197 in T. reesei to 587 in F. solani, while N. crassa possessed 132 genes (Appendix S3). All predicted MFS transporters were classified into 124 different subgroups based on sequence similarity and predicted substrate transport preference, according to the TCDB (Saier et al., 2016). The phylogenetic relationship of C. rosea with seven other fungi with varying life strategies (Appendix S4) was used for analysis of MFS transporter gene family expansions and contractions with the program CAFE.
In total, 35 MFS gene families were identified to evolve nonrandomly (p ≤ .05) in at least one species (Appendix S5). In C. rosea specifically, 25 MFS gene families were identified to contain more genes than expected given genetic drift, thereby implying selection to maintain gene duplicates (Table 2). In contrast, no contracted MFS gene families were identified in C. rosea. Based on the predicted functions from TCDB, these 25 MFS gene families were divided into three broad categories; six families involved in drug resistance/secondary metabolite transport, eight families involved in small organic compound transport, and six families involved in carbohydrate transport (Table 2). In addition, one family (MFS 2.A.1.14.38) were predicted to transport sulphur‐compounds, one family (MFS 2.A.1.16.7) were predicted to transport siderophores, while three families lacked any described function. In comparison with the three mycoparasitic Trichoderma species, only two MFS gene families were identified as expanded in both C. rosea and T. atroviride (but not T. virens or T. reesei); MFS 2.A.1.3.65 (multidrug resistance) and MFS 2.A.1.3.68 (acid resistance and pH homoeostasis; Table 2). Three additional MFS gene families were identified as expanded in T. atroviride (MFS 2.A.1.2.6: multidrug resistance, MFS 2.A.1.2.35: caffeine resistance, MFS 2.A.1.3.32: drug resistance) but not in T. virens or T. reesei, while one MFS gene family was expanded in T. virens (MFS 2.A.1.2.79: toxin exporter) but not in T. atroviride or T. reesei (Appendix S5).
Table 2.
Major facilitator superfamily (MFS) gene numbers in groups evolving nonrandomlya in C. rosea
| MFS classificationb | MFS annotationb | CR | FS | FV | FG | TA | TV | TR | NC |
|---|---|---|---|---|---|---|---|---|---|
| MFS 2.A.1.1.7 | Quinate: H+ symporter | 9 | 5 | 8 | 10 | 3 | 4 | 3 | 1 |
| MFS 2.A.1.1.11 | α‐glucoside: H+ symporter | 27 | 31 | 20 | 16 | 9 | 12 | 6 | 1 |
| MFS 2.A.1.1.57 | Monosaccharide:H+ symporter | 8 | 7 | 4 | 1 | 2 | 2 | 1 | 4 |
| MFS 2.A.1.1.68 | Glucose transporter/sensor | 5 | 7 | 5 | 1 | 0 | 0 | 0 | 2 |
| MFS 2.A.1.1.83 | Cellobiose/cellodextrin transporter | 24 | 33 | 24 | 19 | 5 | 8 | 3 | 4 |
| MFS 2.A.1.1.119 | Galacturonic acid uptake porter | 19 | 14 | 14 | 7 | 10 | 6 | 5 | 4 |
| MFS 2.A.1.2.33 | Unknown function | 17 | 25 | 24 | 11 | 6 | 6 | 6 | 3 |
| MFS 2.A.1.2.77 | Drug resistance transporter | 18 | 13 | 10 | 11 | 12 | 8 | 6 | 5 |
| MFS 2.A.1.2.85 | Peroxisomal phenylacetate transporter | 3 | 3 | 1 | 0 | 0 | 1 | 0 | 0 |
| MFS 2.A.1.2.86 | Peroxisomal isopenicillin N importer | 24 | 21 | 16 | 9 | 14 | 13 | 8 | 4 |
| MFS 2.A.1.3.33 | Multidrug resistance porter | 6 | 3 | 1 | 3 | 1 | 2 | 1 | 0 |
| MFS 2.A.1.3.47 | Tri12 trichothecene efflux pump | 6 | 4 | 5 | 2 | 0 | 0 | 0 | 2 |
| MFS 2.A.1.3.65 | Multidrug resistance transporter | 33 | 25 | 21 | 14 | 24 | 19 | 8 | 7 |
| MFS 2.A.1.3.68 | Acid resistance and pH homoeostasis | 4 | 2 | 0 | 0 | 2 | 1 | 0 | 0 |
| MFS 2.A.1.13.4 | Riboflavin uptake transporter | 16 | 16 | 7 | 6 | 11 | 7 | 8 | 1 |
| MFS 2.A.1.13.19 | Unknown function | 20 | 12 | 13 | 9 | 7 | 9 | 6 | 4 |
| MFS 2.A.1.14.3 | Tartrate porter | 20 | 12 | 10 | 8 | 3 | 5 | 2 | 4 |
| MFS 2.A.1.14.4 | Dipeptide porter | 30 | 29 | 16 | 12 | 9 | 11 | 4 | 1 |
| MFS 2.A.1.14.8 | Phthalate porter | 6 | 1 | 3 | 2 | 0 | 0 | 0 | 0 |
| MFS 2.A.1.14.11 | Nicotinate permease | 93 | 51 | 37 | 34 | 22 | 23 | 16 | 13 |
| MFS 2.A.1.14.17 | Pantothenate: H+ symporter | 19 | 18 | 8 | 9 | 2 | 3 | 2 | 2 |
| MFS 2.A.1.14.36 | Thiamine transporter | 14 | 8 | 9 | 7 | 9 | 9 | 6 | 1 |
| MFS 2.A.1.14.37 | Unknown function | 5 | 9 | 2 | 1 | 1 | 0 | 0 | 2 |
| MFS 2.A.1.14.38 | Inorganic sulphur‐compound transporter | 11 | 16 | 13 | 6 | 4 | 4 | 2 | 3 |
| MFS 2.A.1.16.7 | Ferri‐siderophore transporter | 15 | 13 | 7 | 7 | 4 | 6 | 3 | 1 |
Species abbreviations: CR, C. rosea; FG, F. graminearum; FS, F. solani; FV, F. verticillioides; NC, N. crassa; TA, T. atroviride; TR, T. reesei; TV, T. virens.
Gene numbers in bold indicates a significant (p ≤ .05) expansion, while gene numbers in italic indicates a significant (p ≤ .05) contraction of gene family size.
MFS classification and annotation are based on BlastP analysis (E‐value ≤ 1e‐5) of the TCDB transporter classification database.
Nineteen of the 22 differentially expressed C. rosea MFS transporter genes belonged to expanded MFS gene families (Table 2), including five genes in gene family MFS 2.A.1.14.11 (nicotinate permease), three genes in gene family MFS 2.A.1.2.86 (peroxisomal isopenicillin N importer) and two genes each in gene families MFS 2.A.1.1.119 (galacturonic acid uptake) and MFS 2.A.1.14.17 (pantothenate uptake). The highly induced MFS transporter genes mfs602 (BN869_T00006455) and mfs464 (BN869_T00007234) belonged to the expanded MFS gene families 2.A.1.3.65 (multidrug resistance) and 2.A.1.2.33 (unknown function), respectively.
3.4. Phylogenetic relationships of selected MFS transporter gene families
Guided by the high (≥693‐fold) induction of mfs602 and mfs464 during interactions with F. graminearum, but not against B. cinerea, we analysed the phylogenetic relationships of the expanded MFS transporter gene families 2.A.1.3.65 (multidrug resistance) and 2.A.1.2.33 (unknown function). The phylogenetic tree of the MFS 2.A.1.3.65 gene family displayed low resolution among the deeper branches, and incongruence with the species phylogeny in many subgroups (Figure 2), indicative of a birth‐and‐death evolutionary process followed by rapid sequence divergence. The 33 C. rosea paralogous genes were distributed over the phylogeny, rather than being overrepresented in a specific subgroup. The predicted C. rosea MFS602 protein clustered in a well‐supported (99% bootstrap support) subgroup together with single orthologs from Fusarium spp. and Trichoderma spp. (Figure 2). The analysis also showed that C. rosea MFS602 was not orthologous to the previously characterized family 2.A.1.3.65 T. cf. harzianum MFS1 transporter involved in trichodermin secretion and protection against mycotoxins and xenobiotics (Liu, Liu, & Wang, 2012).
Figure 2.
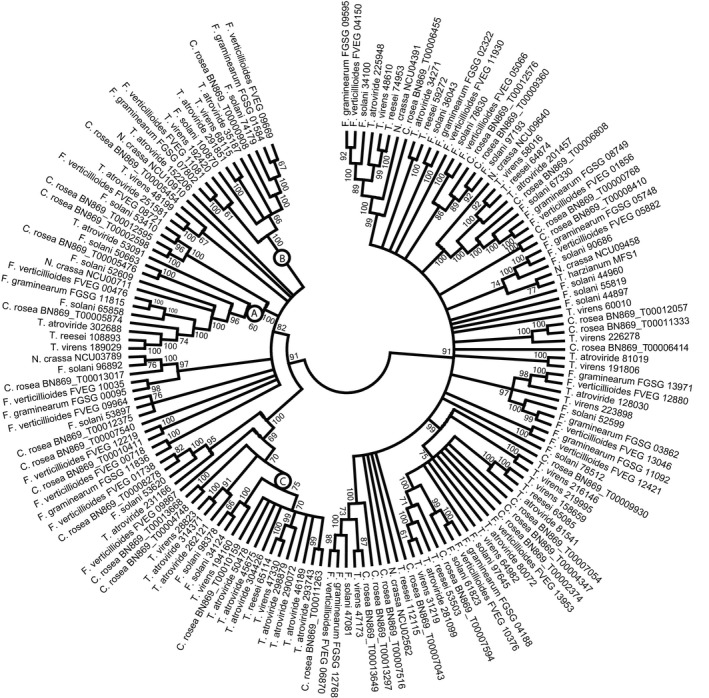
Phylogenetic relationships of MFS transporter family 2.A.1.3.65 among Hypocreales. Predicted amino acid sequences of MFS transporters were aligned by MUSCLE and used to construct a phylogenetic tree using the neighbour‐joining method in the MEGA6 software package. Bootstrap support (≥60%) values from 500 iterations are associated with nodes. Species name and protein ID are given for each MFS transporter
The MFS 2.A.1.3.65 gene family was also expanded in T. atroviride, but not in T. virens or T. reesei. In contrast to C. rosea, the major expansion in T. atroviride was traced to a specific subgroup; seven T. atroviride paralogs clustered together with genes from the other mycoparasites T. virens, T. reesei and C. rosea (Figure 2). Pairwise DNA identity between the seven paralogs was between 46.8% and 59.1%. The seven paralogs were not localized in tandem repeats, but in four different scaffolds in the T. atroviride genome (data not shown). No association with predicted secondary metabolite clusters in T. atroviride was found for any of the seven MFS genes.
The phylogenetic tree of the MFS 2.A.1.2.33 gene family displayed higher resolution of deeper branches compared with the MFS 2.A.1.3.65 family (Figure 3). However, only two cases of orthologs shared between the seven hypocrealean species were identified (C. rosea BN869_T00002983 and BN869_T00000794), indicative of high rates of gene gain and loss. The C. rosea MFS464 transporter was not orthologous to the type transporter of the MFS 2.A.1.2.33 family, the HOL1 transporter from Saccharomyces cerevisiae with unknown function but possibly involved in transport of histidinol and other cations (Wright, Howell, & Gaber, 1996).
Figure 3.
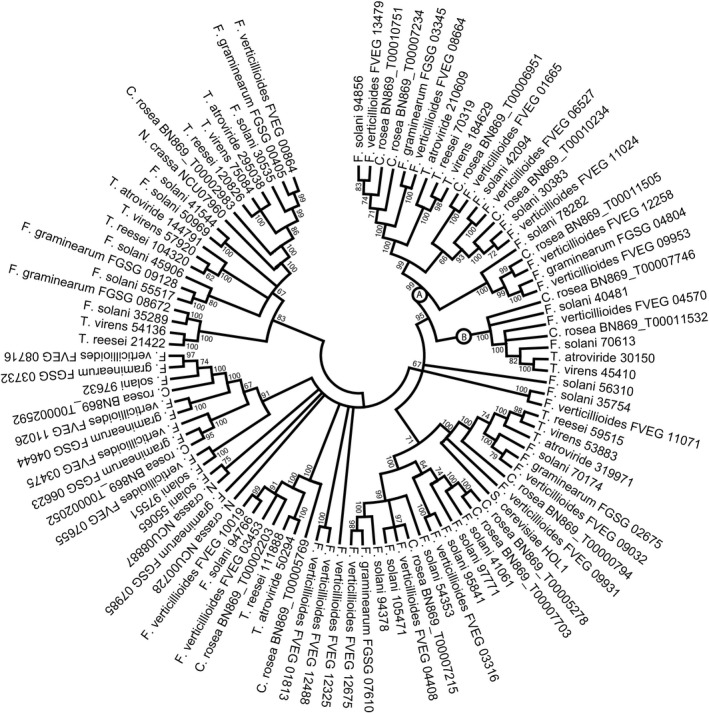
Phylogenetic relationships of MFS transporter family 2.A.1.2.33 among Hypocreales. Predicted amino acid sequences of MFS transporters were aligned by MUSCLE and used to construct a phylogenetic tree using the neighbour‐joining method in the MEGA6 software package. Bootstrap support (≥60%) values from 500 iterations are associated with nodes. Species name and protein ID are given for each MFS transporter
3.5. Structural divergence of MFS 2.A.1.3.65 paralogs
Alignments and RCA analyses were applied to reveal conserved and variable regions between three MFS 2.A.1.3.65 phylogenetic groups with ≥60% bootstrap support (here referred to as clades A, B and C, Figure 2). Ten regions with high amino acid diversity (W ≥ 1) were identified in the MFS 2.A.1.3.65 alignment (Figure 4). Six of these regions displayed signs of functional divergence, defined as high variation (W ≥ 1) in one group in combination with low variation (W < 0.5) in another group and were labelled 1 through 6 (Figure 4).
Figure 4.
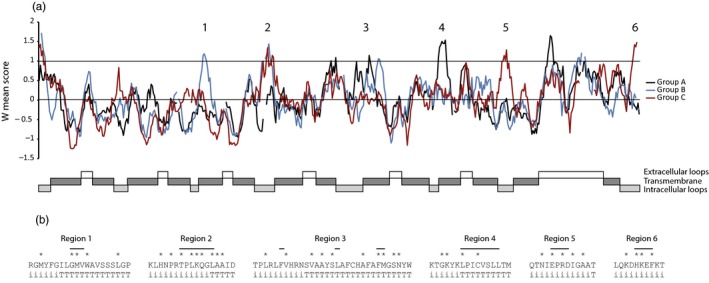
Reverse conservation analysis of family 2.A.1.3.65 MFS transporters. (a) Amino acid conservation was estimated using Rate4Site, based on a MUSCLE alignment of hypocrealean MFS transporters, and plotted as W mean scores (based on S scores for individual positions) in arbitrary units. The black, blue and red lines represent the A, B and C clades indicated in Figure 2, respectively. Regions with signs of functional divergence (W ≥ 1 in one group and W < 0.5 in another group) are indicated. Transmembrane helices and loop structures were predicted with HMMTOP and displayed. (b) Amino acid sequences of regions (indicated by horizontal lines) with signs of functional divergence. Positions with high amino acid diversity (S score ≥1) are indicated by asterisks. Amino acid positions forming parts of transmembrane helices are indicated by T, while positions forming parts of intracellular loops are indicated by i
Five of these regions (1–5) were predicted to be associated with loops that connect transmembrane helices on the inside of the cell and the beginning of the transmembrane helices themselves (Figure 4). Region 6 was predicted to be part of the C‐terminal, internal loop. Hence, all six regions were predicted to form parts of the entry of the transport tunnel, likely influencing substrate specificity.
3.6. Structural divergence of MFS 2.A.1.2.33 paralogs
Four regions with high amino acid diversity (W ≥ 1) were identified by RCA between two MFS 2.A.1.2.33 phylogenetic groups (clades A and B in Figure 3). Only two of these regions displayed signs of functional divergence (W ≥ 1 in group A and W < 0.5 in group B) and were labelled 1 and 2 (Figure 5). Region 1 was located in the end of a large, highly variable internal loop region, while region 2 was located in a C‐terminal internal loop (Figure 5).
Figure 5.
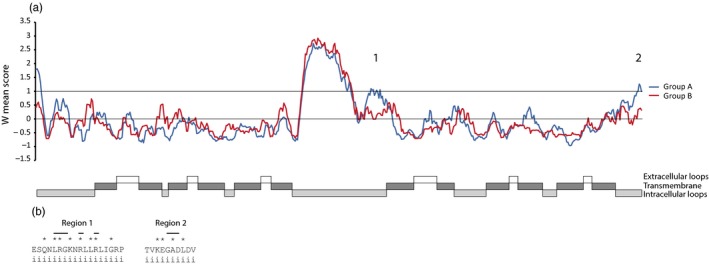
Reverse conservation analysis of family 2.A.1.2.33 MFS transporters. (a) Amino acid conservation was estimated using Rate4Site, based on a MUSCLE alignment of hypocrealean MFS transporters, and plotted as W mean scores (based on S scores for individual positions) in arbitrary units. The blue and red lines represent the A and B clades indicated in Figure 3, respectively. Regions with signs of functional divergence (W ≥ 1 in one group and W < 0.5 in another group) are indicated. Transmembrane helices and loop structures were predicted with HMMTOP and displayed. (b) Amino acid sequences of regions (indicated by horizontal lines) with signs of functional divergence. Positions with high amino acid diversity (S score ≥ 1) are indicated by asterisks. Amino acid positions forming parts of intracellular loops are indicated by i
3.7. Mycotoxin‐ and fungicide‐induced gene expression in C. rosea
As many differentially expressed genes belonged to expanded gene families predicted to be involved in drug resistance/secondary metabolite transport, we analysed gene expression of eight genes used for RNA‐seq validation (Figure 1) in C. rosea during exposure to the Fusarium mycotoxin zearalenone (with antifungal properties [Utermark & Karlovsky, 2007]) and to four fungicides with different mode of action. The ABC transporter gene abcG18 was induced 11,152‐fold after exposure to zearalenone for 2 hr, while the predicted FAD‐dependent oxidoreductase gene fdo1 was induced 2.7‐fold by zearalenone (Table 3). The ABC transporter gene abcC8, the MFS transporter gene mfs602 and fdo1 were induced during exposure to fungicides boscalid and mefenoxam, while mfs602 and fdo1 were additionally induced by iprodione (Table 3).
Table 3.
Expressiona of Clonostachys rosea genes
| Treatment | Genes | |||||||
|---|---|---|---|---|---|---|---|---|
| mfs293 | mfs249 | abcC8 | mfs602 | mfs464 | fdo1 | ptr1 | abcG18 | |
| Zearalenone | 3.8 | ND | 0.8 | 2.4 | D | 2.7* | 18.7 | 11152.3* |
| Zearalenone control | 1.3 | ND | 1.0 | 1.0 | ND | 1.1 | 5.5 | 1.0 |
| Iprodione | ND | ND | 1.0 | 2.0* | ND | 7.4* | 4.3 | ND |
| Boscalid | ND | ND | 3.7* | 4.8* | ND | 4.2* | 1.4 | ND |
| Mefenoxam | ND | ND | 0.4* | 10.9* | ND | 89.3* | 4.6 | D |
| Azoxystrobin | ND | ND | 1.0 | 2.2 | ND | 1.9 | ND | ND |
| Fungicide control | ND | ND | 1.1 | 0.8 | ND | 1.4 | 1.1 | ND |
D, transcripts were detected in ≥3 biological replicates; ND, transcripts were not detected in ≥3 biological replicates.
Relative gene expression in relation with the appropriate control treatment. An asterisk indicates a significant (p ≤ .05) difference compared with the appropriate control treatment as determined by t test.
3.8. Phenotypic analyses of gene deletion strains in C. rosea
Clonostachys rosea gene deletion mutants were generated by replacing mfs602, mfs464, fdo1 and cyp1 with the hygromycin selection cassette (hygB) by ATMT. Successful gene replacements in mitotically stable mutants were confirmed by PCR using primers located within the hygB cassette in combination with primers located upstream and downstream of the constructs (Appendix S2) as described previously (Dubey, Broberg, Jensen et al., 2013; Dubey et al., 2012, 2014, 2016). The expected size of PCR fragments were amplified in selected mfs602, mfs464, fdo1 and cyp1 mutant strains, while no amplification was observed in WT (Appendix S2). Furthermore, RT‐PCR experiments using primers specific to mfs602, mfs464, fdo1 and cyp1 demonstrated the complete loss of the respective transcripts in each mutant (Appendix S2).
When C. rosea WT or the ∆mfs464 mutants were co‐inoculated with F. graminearum in liquid VM with 0.3% glucose, there was a significant (P = .001) 2.1–3.7‐fold reduction in F. graminearum biomass (measured as F. graminearum DNA/C. rosea DNA ratio) after 3 days of interaction (Figure 6). No reductions in F. graminearum biomass were observed for the ∆mfs602, ∆fdo1 or ∆cyp1 mutants. No additional phenotypic effects in any of the four C. rosea gene deletion mutants were observed for any of the studied phenotypes, including tolerance to mycotoxins, fungicides, chemical agents for cell wall, osmotic or oxidative stress, or ion transport/tolerance, nutrient deficiencies, or in vitro dual culture interaction, or biocontrol of fusarium foot rot on barley (data not shown).
Figure 6.
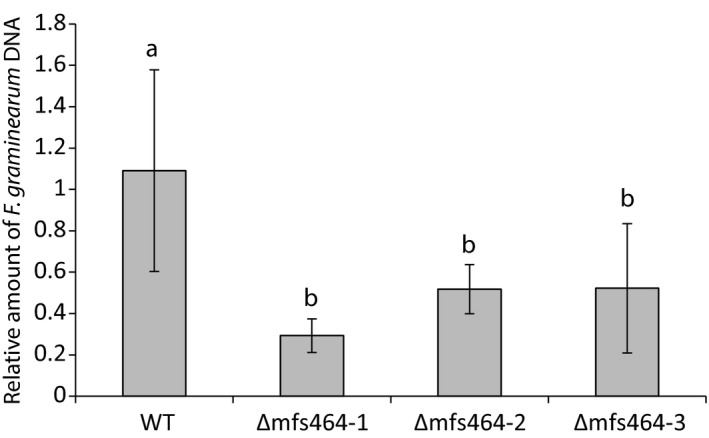
Assay of in vitro antagonism of C. rosea WT and ∆mfs464 mutants. Clonostachys rosea WT and three independent Δmfs464 strains were inoculated in liquid VM media together with F. graminearum. Fungal biomass was estimated after 3 days of interaction by quantifying C. rosea and F. graminearum DNA concentrations using qPCR. Error bars represent standard deviation based on five biological replicates. Different letters indicate statistically significant differences (p ≤ .05) based on the Fisher method
4. DISCUSSION
Necrotrophic mycoparasites such as C. rosea and Trichoderma spp. are assumed to have broad host ranges with little specificity. Evidence to support this view comes from the fact that single species, and sometimes single strains, are efficient biocontrol agents against a range of different plant pathogenic fungi (Jensen et al., 2007), and sometimes even against oomycete and plasmodiophorid pathogens (Lahlali & Peng, 2014; Moller, Jensen, Andersen, Stryhnz, & Hockenhull, 2003). Screening of 75 different species of Trichoderma also showed that all possessed mycoparasitic activity against the plant pathogenic species A. alternata, B. cinerea and S. sclerotiorum (Druzhinina et al., 2011). Therefore, it is intriguing to observe that C. rosea reacts with both common and specific gene expression during interspecific interactions with B. cinerea and F. graminearum, suggesting that C. rosea can distinguish between fungal prey species and modulate its responses accordingly.
A comparative transcriptomic study of T. atroviride, T. reesei and T. virens during interaction with Rhizoctonia solani (Atanasova, Le Crom et al., 2013) revealed both common and specific responses between the three species. The common response included induction of ABC and MFS transporters, proteases and heat shock proteins. Trichoderma atroviride mycoparasitism was characterized by induced genes involved in biosynthesis of secondary metabolites and fungal cell wall degradation, but also small, secreted cysteine‐rich proteins, while the predatory mycoparasitism of T. virens was associated with induction of genes involved in gliotoxin biosynthesis (Atanasova, Le Crom et al., 2013). The weak mycoparasite and wood‐degrading T. reesei induced cellulase and hemicellulase genes.
A comparative genome analysis identified high numbers of genes encoding ABC and MFS transporters, PKS, NRPS, CYP and PL1 genes, and a low number of chitinases, in the C. rosea genome (Karlsson et al., 2015), suggesting that their gene products may be involved in ecological niche adaptation (Wapinski et al., 2007) including biotic interactions, nutrient acquisition and stress modulation. It is therefore notable that members from all of these gene families are also induced during interspecific fungal interactions, indicating that mycoparasitism is the main driver for shaping the genome content in C. rosea. The common response in C. rosea against both fungal preys included sugar and small organic compound transporters that may be involved in nutrient uptake. The specific responses towards the two different fungal preys were dominated by genes predicted to be involved in membrane transport, biosynthesis of secondary metabolites and carbohydrate degradation, which fits well with our current view that degradation of the cell wall of the fungal prey, production of toxic metabolites for poisoning of the fungal prey and the ability to tolerate the counterattack by the fungal prey (by means of toxic metabolites and reactive oxygen species) are key mechanisms for a successful necrotrophic mycoparasite (Druzhinina et al., 2011).
The chiC1 chitinase gene was previously shown to be induced by chitin (Tzelepis et al., 2015) and is predicted to encode a killer toxin‐like chitinase that permeabilize the cell wall of antagonistic species to facilitate entry of toxic metabolites (Karlsson & Stenlid, 2008; Seidl, Huemer, Seiboth, & Kubicek, 2005). This function was previously confirmed for another killer toxin‐like chitinase in C. rosea; deletion of the chiC2 gene resulted in lower growth‐inhibiting activity of culture filtrates against B. cinerea and R. solani, but notably, not against F. graminearum (Tzelepis et al., 2015). This may be significant as the 50‐fold induction of chiC1 against F. graminearum, but not against B. cinerea, may suggest adaptation of killer toxin‐like paralogs towards different fungal preys (driven by differences in cell wall composition, presence of chitinase inhibitors etc.). Differential regulation of killer toxin‐like paralogs was also reported from T. atroviride (Gruber, Vaaje‐Kolstad et al., 2011) and T. virens (Gruber, Kubicek, & Seidl‐Seiboth, 2011), and the saprotrophic Aspergillus nidulans (Tzelepis, Melin, Stenlid, Jensen, & Karlsson, 2014) and N. crassa (Tzelepis et al., 2012). The killer toxin‐like function was also confirmed in a nonmycoparasitic species; deletion of the chiC2‐2 gene in A. nidulans resulted in reduced growth‐inhibiting activity of culture filtrates (Tzelepis et al., 2014).
Clonostachys rosea is known to produce a limited number of secondary metabolites with biotic activity; peptaibols with toxicity against S. sclerotiorum (Rodriguez et al., 2011), polyketides with antibacterial activity (Zhai et al., 2016), glisoprenin inhibitors of appressorium formation (Thines, Eilbert, Anke, & Sterner, 1998) and nematicidal epipolysulfanyldioxopiperazines (Dong, He, Shen, & Zhang, 2005). However, it is not possible at this stage to connect the secondary metabolite biosynthetic genes induced against B. cinerea or F. graminearum with any of these compounds. The pks9 gene induced against B. cinerea is predicted to encode a reduced lovastatin diketide polyketide, and other PKS genes of the same type were induced during mycoparasitism in both T. atroviride (Atanasova, Le Crom et al., 2013) and T. reesei (Atanasova, Knox, Kubicek, Druzhinina, & Baker, 2013). Deletion of the nonreducing type pks4 gene in T. reesei resulted in loss of pigmentation and mutants that were more sensitive to toxic metabolites produced by the fungal prey (Atanasova, Knox, Kubicek et al., 2013). Induction of genes with similarity to isotrichodermin C‐15 hydroxylase (cyp1) and norsolorinic acid reductase in C. rosea during interaction with F. graminearum may indicate production of hitherto unknown secondary metabolites, and similar genes were reported to be induced during mycoparasitism in T. cf. harzianum (Steindorff et al., 2012, 2014; Vieira et al., 2013).
Interpretation of the induction of C. rosea ABC and MFS transporters during interaction with B. cinerea and F. graminearum is made difficult by the fact that both groups of membrane transporters exists as large gene families with a complex phylogenetic substructure that typically reflects their involvement in a wide range of biological functions, including basic metabolism and cellular homoeostasis (Kovalchuk & Driessen, 2010). The ABC transporter gene family in C. rosea has been studied in detail (Karlsson et al., 2015), where ABC transporter group B (multidrug resistance proteins) and G (pleiotropic drug resistance proteins) were shown to evolve under selection for increased gene copy numbers. The abcG18 gene, induced against F. graminearum and during exposure to the Fusarium mycotoxin zearalenone, indeed belongs to group G, suggesting a role of the ABCG18 protein in protecting C. rosea against toxic metabolites produced by fungal prey (at least by Fusarium spp.). Three other ABC transporter genes from group G (abcG5, abcG8 and abcG29) and one gene from group B (abcB26) were also reported to be induced in C. rosea during exposure to ZEA (Karlsson et al., 2015; Kosawang, Karlsson, Jensen, Dilokpimol, & Collinge, 2014), and both ABCG5 and ABCG29 were later shown to be involved in biocontrol of fusarium foot rot disease (Dubey et al., 2014, 2016). The C. rosea abcB1, abcB4, abcB18, abcB20 and abcB26 group B and the abcG8 and abcG25 group G ABC transporter genes were also reported to be induced by exposure to bacterial metabolites (Kamou et al., 2016; Karlsson et al., 2015), suggesting that C. rosea needs to defend itself not only to metabolites from the fungal prey but also to antibiosis from the surrounding microflora in the rhizosphere. The abcC8 gene belongs to the multidrug resistance‐associated proteins, subgroup C‐V (Karlsson et al., 2015) and the induction during interaction with B. cinerea and during exposure to fungicides boscalid and mefenoxam may suggest an involvement in cellular protection against exogenous toxic metabolites and xenobiotics.
The MFS transporter gene family on the other hand is not studied to any detail in C. rosea, or in any other fungus. Our comprehensive evolutionary analysis of the MFS transporter gene family in hypocrealean fungi revealed selection for increased gene copy numbers in families related with drug resistance/secondary metabolite transport, small organic compound transport and carbohydrate transport in C. rosea. MFS transporter genes induced during interactions with B. cinerea and F. graminearum almost exclusively belonged to these expanded MFS families, indicating that efflux‐mediated protection against exogenous or endogenous secondary metabolites and nutrient uptake are important components of the mycoparasitic attack in C. rosea. MFS transporters were reported to be induced in several other mycoparasitic species, including the closely related C. chloroleuca (Moreira, Abreu, Carvalho, Schroers, & Pfenning, 2016; Sun, Sun, & Li, 2015), Trichoderma spp. (Atanasova, Le Crom et al., 2013; Seidl et al., 2009; Steindorff et al., 2014), Tolypocladium ophioglossoides (Quandt, Di, Elser, Jaiswal, & Spatafora, 2016), Escovopsis weberi (de Man et al., 2016), Pythium oligandrum (Horner, Grenville‐Briggs, & Van West, 2012) and Ampelomyces quisqualis (Siozios et al., 2015), but without proper identification of family classification it is difficult to speculate about their exact biological role in these species.
Although gene expression fold‐change induction not necessarily corresponds to the importance of a particular gene product, mfs602 and mfs464 stood out in among the MFS transporters with a ≥693‐fold induction during interaction with F. graminearum, but not during interaction with B. cinerea. This substantial induction, coupled with the fact that both genes belongs to gene families evolving under selection for increased paralog numbers (2.A.1.3.65 and 2.A.1.2.33), indicates that the encoded transporter proteins perform important functions in the mycoparasitic attack of F. graminearum by C. rosea. The phylogenetic structure of both families shows that they evolve by a rapid birth‐and‐death process followed by sequence diversification, indicating that functional diversification between paralogs is the driving force for the observed gene copy number increase, rather than selection for increased protein amount (where sequence conservation between paralogs are expected). MFS602 belong to family 2.A.1.3.65 predicted to be involved in drug resistance, and our analysis of structural divergence showed that loops forming the entry to the transport channel are the targets of the diversification between paralogs, indicating that selection for substrate specificity has been important for the evolution of MFS family 2.A.1.3.65 members in hypocrealean fungi. The MFS 2.A.1.3.65 family also evolve under different evolutionary trajectories in Trichoderma spp., with gene gains in T. atroviride, random genetic drift in T. virens and gene losses in T. reesei, possibly reflecting its current life style transition to a wood‐degrading saprotroph from a mycoparasitic ancestor (Kubicek et al., 2011). The only characterized MFS transporter in Trichoderma spp., MFS1 in T. cf. harzianum, also belongs to family 2.A.1.3.65 and was indeed shown to transport the Trichoderma spp. metabolite trichodermin and consequently affect in vitro antagonism (Liu et al., 2012). MFS1 was also shown to transport xenobiotics such as iprodione and a range of azole‐type fungicides. This dual function fits well with the gene expression profile of mfs602 that was induced during interaction with F. graminearum but also during exposure to the fungicides iprodione, boscalid and fenhexamid. However, deletion of the mfs602 gene in C. rosea did not result in any measurable phenotype. This may not be unexpected, given the large number of similar paralogs present in C. rosea that can mask any potential effect by partially overlapping functions, which is previously reported for other large gene families such as chitinases and hydrophobins (Alcazar‐Fuoli et al., 2011; Mosbach, Leroch, Mendgen, & Hahn, 2011).
The function of MFS464 is more difficult to speculate about, as no member of MFS family 2.A.1.2.33 is characterized to any detail. Even the type transporter of the family, HOL1 from S. cerevisiae, was only shown to transport histidinol after random mutagenesis, which not necessarily provides evidence for its true biological function (Wright et al., 1996). As with family 2.A.1.3.65, the phylogenetic analysis of family 2.A.1.2.33 suggests functional diversification between paralogs, but the structure of these transporters was very different from 2.A.1.3.65 members with one large internal loop. Part of this loop was highly variable in sequence between subgroups, indicating low selective constraints, but another part of this loop displayed a signature of functional diversification. We may speculate that this large, variable internal loop can interact with other proteins, perhaps forming a signalling complex, but this remains to be investigated. Deletion of mfs464 in C. rosea resulted in mutants that displayed increased growth inhibitory activity against F. graminearum when co‐inoculated in liquid medium, providing evidence for a function of MFS464 in interspecific fungal interactions, although the exact nature of this effect remains to be investigated.
In summary, we show that the necrotrophic mycoparasite C. rosea can distinguish between fungal prey species and modulate its transcriptomic responses accordingly. Genes predicted to encode membrane transporters involved in drug resistance/secondary metabolite transport and biosynthesis of PKS and NRPS secondary metabolites forms a large part of the interaction transcriptomes, emphasizing the importance of secondary metabolites in mycoparasitic interactions. The overlap between genes induced during the mycoparasitic attack and genes/gene families evolving under diversifying selection is equivalent to the effector concept in parasite–host interactions where the co‐evolutionary arms race results in rapidly evolving parasite effectors and host receptors. Identification of mechanisms resulting in successful biocontrol of plant pathogenic fungi is important for knowledge‐based improvements of biocontrol efficacy in agricultural production systems.
CONFLICT OF INTEREST
None declared.
DATA ARCHIVING STATEMENT
The transcriptome sequence data generated and analysed in this work are deposited in the short read archive (SRA) at NCBI, with accession numbers SRX3389264 ‐ SRX3389269.
Supporting information
ACKNOWLEDGEMENTS
We acknowledge financial support from the Department of Forest Mycology and Plant Pathology, the Carl Trygger Foundation for Scientific Research (grant no. CTS12:233) and the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (FORMAS, grant nos. 2012‐7768‐22608‐32 and 942‐2015‐1128). Sequencing was performed by the SNP&SEQ Technology Platform in Uppsala. The facility is part of the National Genomics Infrastructure (NGI) Sweden and Science for Life Laboratory. The SNP & SEQ Platform is also supported by the Swedish Research Council and the Knut and Alice Wallenberg Foundation.
Nygren K, Dubey M, Zapparata A, et al. The mycoparasitic fungus Clonostachys rosea responds with both common and specific gene expression during interspecific interactions with fungal prey. Evol Appl. 2018;11:931–949. https://doi.org/10.1111/eva.12609
Kristiina Nygren and Mukesh Dubey should be considered joint first author.
REFERENCES
- Alcazar‐Fuoli, L. , Clavaud, C. , Lamarre, C. , Aimanianda, V. , Seidl‐Seiboth, V. , Mellado, E. , & Latge, J. P. (2011). Functional analysis of the fungal/plant class chitinase family in Aspergillus fumigatus . Fungal Genetics and Biology, 48, 418–429. https://doi.org/10.1016/j.fgb.2010.12.007 [DOI] [PubMed] [Google Scholar]
- Amselem, J. , Cuomo, C. A. , van Kan, J. A. L. , Viaud, M. , Benito, E. P. , Couloux, A. , … Dickman, M. (2011). Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea . PLoS Genetics, 7, e1002230 https://doi.org/10.1371/journal.pgen.1002230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasova, L. , Knox, B. P. , Kubicek, C. P. , Druzhinina, I. S. , & Baker, S. E. (2013). The polyketide synthase gene pks4 of Trichoderma reesei provides pigmentation and stress resistance. Eukaryotic Cell, 12, 1499–1508. https://doi.org/10.1128/Ec.00103-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasova, L. , Le Crom, S. , Gruber, S. , Coulpier, F. , Seidl‐Seiboth, V. , Kubicek, C. P. , & Druzhinina, I. S. (2013). Comparative transcriptomics reveals different strategies of Trichoderma mycoparasitism. BMC Genomics, 14, 121 https://doi.org/10.1186/1471-2164-14-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterton, S. , & Punja, Z. K. (2009). Chitinase and beta‐1,3‐glucanase enzyme production by the mycoparasite Clonostachys rosea f. catenulata against fungal plant pathogens. Canadian Journal of Microbiology, 55, 356–367. https://doi.org/10.1139/W08-156 [DOI] [PubMed] [Google Scholar]
- Coleman, J. J. , Rounsley, S. D. , Rodriguez‐Carres, M. , Kuo, A. , Wasmann, C. C. , Grimwood, J. , … VanEtten, H. D. (2009). The genome of Nectria haematococca: Contribution of supernumerary chromosomes to gene expansion. PLoS Genetics, 5, e1000618 https://doi.org/10.1371/journal.pgen.1000618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuomo, C. A. , Gueldener, U. , Xu, J. R. , Trail, F. , Turgeon, B. G. , Di Pietro, A. , … Kistler, H. C. (2007). The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science, 317, 1400–1402. https://doi.org/10.1126/science.1143708 [DOI] [PubMed] [Google Scholar]
- de Man, T. J. B. , Stajich, J. E. , Kubicek, C. P. , Teiling, C. , Chenthamara, K. , Atanasova, L. , … Gerardo, N. M. (2016). Small genome of the fungus Escovopsis weberi, a specialized disease agent of ant agriculture. Proceedings of the National Academy of Sciences of the United States of America, 113, 3567–3572. https://doi.org/10.1073/pnas.1518501113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, J. Y. , He, H. P. , Shen, Y. M. , & Zhang, K. Q. (2005). Nematicidal epipolysulfanyldioxopiperazines from Gliocladium roseum . Journal of Natural Products, 68, 1510–1513. https://doi.org/10.1021/Np0502241 [DOI] [PubMed] [Google Scholar]
- Druzhinina, I. S. , Seidl‐Seiboth, V. , Herrera‐Estrella, A. , Horwitz, B. A. , Kenerley, C. M. , Monte, E. , … Kubicek, C. P. (2011). Trichoderma: The genomics of opportunistic success. Nature Reviews Microbiology, 9, 749–759. https://doi.org/10.1038/Nrmicro2637 [DOI] [PubMed] [Google Scholar]
- Dubey, M. K. , Broberg, A. , Jensen, D. F. , & Karlsson, M. (2013). Role of the methylcitrate cycle in growth, antagonism and induction of systemic defence responses in the fungal biocontrol agent Trichoderma atroviride . Microbiology, 159, 2492–2500. https://doi.org/10.1099/mic.0.070466-0 [DOI] [PubMed] [Google Scholar]
- Dubey, M. K. , Broberg, A. , Sooriyaarachchi, S. , Ubhayasekera, W. , Jensen, D. F. , & Karlsson, M. (2013). The glyoxylate cycle is involved in pleotropic phenotypes, antagonism and induction of plant defence responses in the fungal biocontrol agent Trichoderma atroviride . Fungal Genetics and Biology, 58–59, 33–41. https://doi.org/10.1016/j.fgb.2013.06.008 [DOI] [PubMed] [Google Scholar]
- Dubey, M. K. , Jensen, D. F. , & Karlsson, M. (2014). An ATP‐binding cassette pleiotropic drug transporter protein is required for xenobiotic tolerance and antagonism in the fungal biocontrol agent Clonostachys rosea . Molecular Plant‐Microbe Interactions, 27, 725–732. https://doi.org/10.1094/MPMI-12-13-0365-R [DOI] [PubMed] [Google Scholar]
- Dubey, M. , Jensen, D. F. , & Karlsson, M. (2016). The ABC transporter ABCG29 is involved in H2O2 tolerance and biocontrol traits in the fungus Clonostachys rosea . Molecular Genetics and Genomics, 291, 677–686. https://doi.org/10.1007/s00438-015-1139-y [DOI] [PubMed] [Google Scholar]
- Dubey, M. K. , Ubhayasekera, W. , Sandgren, M. , Jensen, D. F. , & Karlsson, M. (2012). Disruption of the Eng18B ENGase gene in the fungal biocontrol agent Trichoderma atroviride affetcs growth, conidation and antagonistic ability. PLoS One, 7, e36152 https://doi.org/10.1371/journal.pone.0036152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792–1797. https://doi.org/10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan, J. E. , Calvo, S. E. , Borkovich, K. A. , Selker, E. U. , Read, N. D. , Jaffe, D. , … Birren, B. (2003). The genome sequence of the filamentous fungus Neurospora crassa . Nature, 422, 859–868. https://doi.org/10.1038/nature01554 [DOI] [PubMed] [Google Scholar]
- Gruber, S. , Kubicek, C. P. , & Seidl‐Seiboth, V. (2011). Differential regulation of orthologous chitinase genes in mycoparasitic Trichoderma species. Applied and Environmental Microbiology, 77, 7217–7226. https://doi.org/10.1128/Aem.06027-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber, S. , Vaaje‐Kolstad, G. , Matarese, F. , Lopez‐Mondejar, R. , Kubicek, C. P. , & Seidl‐Seiboth, V. (2011). Analysis of subgroup C of fungal chitinases containing chitin‐binding and LysM modules in the mycoparasite Trichoderma atroviride . Glycobiology, 21, 122–133. https://doi.org/10.1093/glycob/cwq142 [DOI] [PubMed] [Google Scholar]
- Han, M. V. , Thomas, G. W. C. , Lugo‐Martinez, J. , & Hahn, M. W. (2013). Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Molecular Biology and Evolution, 30, 1987–1997. https://doi.org/10.1093/molbev/mst100 [DOI] [PubMed] [Google Scholar]
- Horner, N. R. , Grenville‐Briggs, L. J. , & Van West, P. (2012). The oomycete Pythium oligandrum expresses putative effectors during mycoparasitism of Phytophthora infestans and is amenable to transformation. Fungal Biology, 116, 24–41. https://doi.org/10.1016/j.funbio.2011.09.004 [DOI] [PubMed] [Google Scholar]
- Jensen, B. , Knudsen, I. M. B. , & Jensen, D. F. (2000). Biological seed treatment of cereals with fresh and long‐term stored formulations of Clonostachys rosea: Biocontrol efficacy against Fusarium culmorum . European Journal of Plant Pathology, 106, 233–242. https://doi.org/10.1023/A:1008794626600 [Google Scholar]
- Jensen, B. , Knudsen, I. M. B. , & Jensen, D. F. (2002). Survival of conidia of Clonostachys rosea on stored barley seeds and their biocontrol efficacy against seed‐borne Bipolaris sorokiniana . Biocontrol Science and Technology, 12, 427–441. https://doi.org/10.1080/09583150220146013 [Google Scholar]
- Jensen, D. F. , Knudsen, I. M. B. , Lubeck, M. , Mamarabadi, M. , Hockenhull, J. , & Jensen, B. (2007). Development of a biocontrol agent for plant disease control with special emphasis on the near commercial fungal antagonist Clonostachys rosea strain ‘IK726’. Australasian Plant Pathology, 36, 95–101. https://doi.org/10.1071/Ap07009 [Google Scholar]
- Jensen, B. , Knudsen, I. M. B. , Madsen, M. , & Jensen, D. F. (2004). Biopriming of infected carrot seed with an antagonist, Clonostachys rosea, selected for control of seedborne Alternaria spp. Phytopathology, 94, 551–560. https://doi.org/10.1094/Phyto.2004.94.6.551 [DOI] [PubMed] [Google Scholar]
- Jones, D. T. , Taylor, W. R. , & Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Computer Applications in the Biosciences, 8, 275–282. [DOI] [PubMed] [Google Scholar]
- Kamou, N. N. , Dubey, M. , Tzelepis, G. , Menexes, G. , Papadakis, E. N. , Karlsson, M. , … Jensen, D. F. (2016). Investigating the compatibility of the biocontrol agent Clonostachys rosea IK726 with prodigiosin‐producing Serratia rubidaea S55 and phenazine‐producing Pseudomonas chlororaphis ToZa7. Archives of Microbiology, 198, 369–377. https://doi.org/10.1007/s00203-016-1198-4 [DOI] [PubMed] [Google Scholar]
- Karimi, M. , De Meyer, B. , & Hilson, P. (2005). Modular cloning in plant cells. Trends in Plant Science, 10, 103–105. https://doi.org/10.1016/j.tplants.2005.01.008 [DOI] [PubMed] [Google Scholar]
- Karlsson, M. , Durling, M. B. , Choi, J. , Kosawang, C. , Lackner, G. , Tzelepis, G. D. , … Jensen, D. F. (2015). Insights on the evolution of mycoparasitism from the genome of Clonostachys rosea . Genome Biology and Evolution, 7, 465–480. https://doi.org/10.1093/gbe/evu292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson, M. , & Stenlid, J. (2008). Comparative evolutionary histories of the fungal chitinase gene family reveal non‐random size expansions and contractions due to adaptive natural selection. Evolutionary Bioinformatics Online, 4, 47–60. https://doi.org/10.4137/EBO.S4198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosawang, C. , Karlsson, M. , Jensen, D. F. , Dilokpimol, A. , & Collinge, D. B. (2014). Transcriptomic profiling to identify genes involved in Fusarium mycotoxin deoxynivalenol and zearalenone tolerance in the mycoparasitic fungus Clonostachys rosea . BMC Genomics, 15, 55 https://doi.org/10.1186/1471-2164-15-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosawang, C. , Karlsson, M. , Velez, H. , Rasmussen, P. H. , Collinge, D. B. , Jensen, B. , & Jensen, D. F. (2014). Zearalenone detoxification by zearalenone hydrolase is important for the antagonistic ability of Clonostachys rosea against mycotoxigenic Fusarium graminearum . Fungal Biology, 118, 364–373. https://doi.org/10.1016/j.funbio.2014.01.005 [DOI] [PubMed] [Google Scholar]
- Kovalchuk, A. , & Driessen, A. J. M. (2010). Phylogenetic analysis of fungal ABC transporters. BMC Genomics, 11, 177 https://doi.org/10.1186/1471-2164-11-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek, C. P. , Herrera‐Estrella, A. , Seidl‐Seiboth, V. , Martinez, D. A. , Druzhinina, I. S. , Thon, M. , … Grigoriev, I. V. (2011). Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma . Genome Biology, 12, R40 https://doi.org/10.1186/gb-2011-12-4-r40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahlali, R. , & Peng, G. (2014). Suppression of clubroot by Clonostachys rosea via antibiosis and induced host resistance. Plant Pathology, 63, 447–455. https://doi.org/10.1111/ppa.12112 [Google Scholar]
- Larkin, M. A. , Blackshields, G. , Brown, N. P. , Chenna, R. , McGettigan, P. A. , McWilliam, H. , … Higgins, D. G. (2007). Clustal W and clustal X version 2.0. Bioinformatics, 23, 2947–2948. https://doi.org/10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- Lee, T. (2008). Reverse conservation analysis reveals the specificity determining residues of cytochrome P450 family 2 (CYP 2). Evolutionary Bioinformatics Online, 4, 7–16. https://doi.org/10.4137/EBO.S291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G. Q. , Huang, H. C. , Kokko, E. G. , & Acharya, S. N. (2002). Ultrastructural study of mycoparasitism of Gliocladium roseum on Botrytis cinerea . Botanical Bulletin of Academia Sinica, 43, 211–218. [Google Scholar]
- Liu, M. , Liu, J. , & Wang, W. M. (2012). Isolation and functional analysis of Thmfs1, the first major facilitator superfamily transporter from the biocontrol fungus Trichoderma harzianum . Biotechnology Letters, 34, 1857–1862. https://doi.org/10.1007/s10529-012-0972-x [DOI] [PubMed] [Google Scholar]
- Livak, K. J. , & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2−∆∆Ct method. Methods, 25, 402–408. https://doi.org/10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Ma, L. J. , van der Does, H. C. , Borkovich, K. A. , Coleman, J. J. , Daboussi, M. J. , Di Pietro, A. , … Rep, M. (2010). Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium . Nature, 464, 367–373. https://doi.org/10.1038/Nature08850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamarabadi, M. , Jensen, B. , & Lubeck, M. (2008). Three endochitinase‐encoding genes identified in the biocontrol fungus Clonostachys rosea are differentially expressed. Current Genetics, 54, 57–70. https://doi.org/10.1007/s00294-008-0199-5 [DOI] [PubMed] [Google Scholar]
- Marchler‐Bauer, A. , Bo, Y. , Han, L. Y. , He, J. E. , Lanczycki, C. J. , Lu, S. N. , … Bryant, S. H. (2017). CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Research, 45, D200–D203. https://doi.org/10.1093/nar/gkw1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet Journal, 17, 10–12. https://doi.org/10.14806/ej.17.1.200 [Google Scholar]
- Martinez, D. , Berka, R. M. , Henrissat, B. , Saloheimo, M. , Arvas, M. , Baker, S. E. , … Brettin, T. S. (2008). Genome sequencing and analysis of the biomass‐degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nature Biotechnology, 26, 553–560. https://doi.org/10.1038/Nbt1403 [DOI] [PubMed] [Google Scholar]
- Mayrose, I. , Graur, D. , Ben‐Tal, N. , & Pupko, T. (2004). Comparison of site‐specific rate‐inference methods for protein sequences: Empirical Bayesian methods are superior. Molecular Biology and Evolution, 21, 1781–1791. https://doi.org/10.1093/molbev/msh194 [DOI] [PubMed] [Google Scholar]
- Moller, K. , Jensen, B. , Andersen, H. P. , Stryhnz, H. , & Hockenhull, J. (2003). Biocontrol of Pythium tracheiphilum in Chinese cabbage by Clonostachys rosea under field conditions. Biocontrol Science and Technology, 13, 171–182. https://doi.org/10.1080/958315021000073448 [Google Scholar]
- Moreira, G. M. , Abreu, L. M. , Carvalho, V. G. , Schroers, H. J. , & Pfenning, L. H. (2016). Multilocus phylogeny of Clonostachys subgenus Bionectria from Brazil and description of Clonostachys chloroleuca sp. nov. Mycological Progress, 15, 1031–1039. https://doi.org/10.1007/s11557-016-1224-6 [Google Scholar]
- Mosbach, A. , Leroch, M. , Mendgen, K. W. , & Hahn, M. (2011). Lack of evidence for a role of hydrophobins in conferring surface hydrophobicity to conidia and hyphae of Botrytis cinerea . BMC Microbiology, 11, 10 https://doi.org/10.1186/1471-2180-11-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygren, C. M. R. , Eberhardt, U. , Karlsson, M. , Parrent, J. L. , Lindahl, B. D. , & Taylor, A. F. S. (2008). Growth on nitrate and occurrence of nitrate reductase‐encoding genes in a phylogenetically diverse range of ectomycorrhizal fungi. New Phytologist, 180, 875–889. https://doi.org/10.1111/j.1469-8137.2008.02618.x [DOI] [PubMed] [Google Scholar]
- Pachenari, A. , & Dix, N. J. (1980). Production of toxins and wall degrading enzymes by Gliocladium roseum . Transactions of the British Mycological Society, 74, 561–566. https://doi.org/10.1016/S0007-1536(80)80057-X [Google Scholar]
- Pupko, T. , Bell, R. , Mayrose, I. , Glaser, F. , & Ben‐Tal, N. (2002). Rate4Site: An algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics, 18, S71–S77. https://doi.org/10.1093/bioinformatics/18.suppl_1.S71 [DOI] [PubMed] [Google Scholar]
- Quandt, C. A. , Di, Y. M. , Elser, J. , Jaiswal, P. , & Spatafora, J. W. (2016). Differential expression of genes involved in host recognition, attachment, and degradation in the mycoparasite Tolypocladium ophioglossoides . G3 (Bethesda, Md.), 6, 731–741. https://doi.org/10.1534/g3.116.027045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reischer, G. H. , Lemmens, M. , Farnleitner, A. , Adler, A. , & Mach, R. L. (2004). Quantification of Fusarium graminearum in infected wheat by species specific real‐time PCR applying a TaqMan probe (vol 59, pg 141, 2004). Journal of Microbiological Methods, 59, 437 https://doi.org/10.1016/j.mimet.2004.09.001 [DOI] [PubMed] [Google Scholar]
- Roberti, R. , Veronesi, A. , Cesari, A. , Cascone, A. , Di Berardino, I. , Bertini, L. , & Caruso, C. (2008). Induction of PR proteins and resistance by the biocontrol agent Clonostachys rosea in wheat plants infected with Fusarium culmorum . Plant Science, 175, 339–347. https://doi.org/10.1016/j.plantsci.2008.05.003 [Google Scholar]
- Rodriguez, M. A. , Cabrera, G. , Gozzo, F. C. , Eberlin, M. N. , & Godeas, A. (2011). Clonostachys rosea BAFC3874 as a Sclerotinia sclerotiorum antagonist: Mechanisms involved and potential as a biocontrol agent. Journal of Applied Microbiology, 110, 1177–1186. https://doi.org/10.1111/j.1365-2672.2011.04970.x [DOI] [PubMed] [Google Scholar]
- Saier, M. H. , Reddy, V. S. , Tsu, B. V. , Ahmed, M. S. , Li, C. , & Moreno‐Hagelsieb, G. (2016). The transporter classification database (TCDB): Recent advances. Nucleic Acids Research, 44, D372–D379. https://doi.org/10.1093/nar/gkv1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroers, H. J. , Samuels, G. J. , Seifert, K. A. , & Gams, W. (1999). Classification of the mycoparasite Gliocladium roseum in Clonostachys as C. rosea, its relationship to Bionectria ochroleuca, and notes on other Gliocladium‐like fungi. Mycologia, 91, 365–385. https://doi.org/10.2307/3761383 [Google Scholar]
- Seidl, V. , Huemer, B. , Seiboth, B. , & Kubicek, C. P. (2005). A complete survey of Trichoderma chitinases reveals three distinct subgroups of family 18 chitinases. FEBS Journal, 272, 5923–5939. https://doi.org/10.1111/j.1742-4658.2005.04994.x [DOI] [PubMed] [Google Scholar]
- Seidl, V. , Song, L. F. , Lindquist, E. , Gruber, S. , Koptchinskiy, A. , Zeilinger, S. , … Kubicek, C. P. (2009). Transcriptomic response of the mycoparasitic fungus Trichoderma atroviride to the presence of a fungal prey. BMC Genomics, 10, 567 https://doi.org/10.1186/1471-2164-10-567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siozios, S. , Tosi, L. , Ferrarini, A. , Ferrari, A. , Tononi, P. , Bellin, D. , … Pertot, I. (2015). Transcriptional reprogramming of the mycoparasitic fungus Ampelomyces quisqualis during the powdery mildew host‐induced germination. Phytopathology, 105, 199–209. https://doi.org/10.1094/Phyto-01-14-0013-R [DOI] [PubMed] [Google Scholar]
- Steindorff, A. S. , Ramada, M. H. S. , Coelho, A. S. G. , Miller, R. N. G. , Pappas, G. J. , Ulhoa, C. J. , & Noronha, E. F. (2014). Identification of mycoparasitism‐related genes against the phytopathogen Sclerotinia sclerotiorum through transcriptome and expression profile analysis in Trichoderma harzianum . BMC Genomics, 15, 204 https://doi.org/10.1186/1471-2164-15-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steindorff, A. S. , Silva, R. D. , Coelho, A. S. G. , Nagata, T. , Noronha, E. F. , & Ulhoa, C. J. (2012). Trichoderma harzianum expressed sequence tags for identification of genes with putative roles in mycoparasitism against Fusarium solani . Biological Control, 61, 134–140. https://doi.org/10.1016/j.biocontrol.2012.01.014 [Google Scholar]
- Sun, Z. B. , Sun, M. H. , & Li, S. D. (2015). Identification of mycoparasitism‐related genes in Clonostachys rosea 67‐1 active against Sclerotinia sclerotiorum . Scientific Reports, 5, 18169 https://doi.org/10.1038/srep18169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton, J. C. , Li, D. W. , Peng, G. , Yu, H. , Zhang, P. G. , & Valdebenito Sanhueza, R. M. (1997). Gliocladium roseum – A versatile adversary of Botrytis cinerea in crops. Plant Disease, 81, 316–328. https://doi.org/10.1094/Pdis.1997.81.4.316 [DOI] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. , & Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30, 2725–2729. https://doi.org/10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thines, E. , Eilbert, F. , Anke, H. , & Sterner, O. (1998). Glisoprenins C, D and E, new inhibitors of appressorium formation in Magnaporthe grisea, from cultures of Gliocladium roseum . The Journal of Antibiotics, 51, 117–122. https://doi.org/10.7164/antibiotics.51.117 [DOI] [PubMed] [Google Scholar]
- Trapnell, C. , Roberts, A. , Goff, L. , Pertea, G. , Kim, D. , Kelley, D. R. , … Pachter, L. (2012). Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and Cufflinks. Nature Protocols, 7, 562–578. https://doi.org/10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnady, G. E. , & Simon, I. (1998). Principles governing amino acid composition of integral membrane proteins: Application to topology prediction. Journal of Molecular Biology, 283, 489–506. https://doi.org/10.1006/jmbi.1998.2107 [DOI] [PubMed] [Google Scholar]
- Tusnady, G. E. , & Simon, I. (2001). The HMMTOP transmembrane topology prediction server. Bioinformatics, 17, 849–850. https://doi.org/10.1093/bioinformatics/17.9.849 [DOI] [PubMed] [Google Scholar]
- Tzelepis, G. , Dubey, M. , Jensen, D. F. , & Karlsson, M. (2015). Identifying glycoside hydrolase family 18 genes in the mycoparasitic fungal species Clonostachys rosea . Microbiology, 161, 1407–1419. https://doi.org/10.1099/mic.0.000096 [DOI] [PubMed] [Google Scholar]
- Tzelepis, G. D. , Melin, P. , Jensen, D. F. , Stenlid, J. , & Karlsson, M. (2012). Functional analysis of glycoside hydrolase family 18 and 20 genes in Neurospora crassa . Fungal Genetics and Biology, 49, 717–730. https://doi.org/10.1016/j.fgb.2012.06.013 [DOI] [PubMed] [Google Scholar]
- Tzelepis, G. D. , Melin, P. , Stenlid, J. , Jensen, D. F. , & Karlsson, M. (2014). Functional analysis of the C‐II subgroup killer toxin‐like chitinases in the filamentous ascomycete Aspergillus nidulans . Fungal Genetics and Biology, 64, 58–66. https://doi.org/10.1016/j.fgb.2013.12.009 [DOI] [PubMed] [Google Scholar]
- Utermark, J. , & Karlovsky, P. (2007). Role of zearalenone lactonase in protection of Gliocladium roseum from fungitoxic effects of the mycotoxin zearalenone. Applied and Environmental Microbiology, 73, 637–642. https://doi.org/10.1128/Aem.01440-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utermark, J. , & Karlovsky, P. (2008). Genetic transformation of filamentous fungi by Agrobacterium tumefaciens . Nature Protocols Online. https://doi.org/10.1038/nprot.2008.83 [Google Scholar]
- Vieira, P. M. , Coelho, A. S. G. , Steindorff, A. S. , de Siqueira, S. J. L. , Silva, R. D. , & Ulhoa, C. J. (2013). Identification of differentially expressed genes from Trichoderma harzianum during growth on cell wall of Fusarium solani as a tool for biotechnological application. BMC Genomics, 14, 177 https://doi.org/10.1186/1471-2164-14-177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel, H. J. (1956). A convenient growth medium for Neurospora (medium N). Microbial Genetics Bulletin, 13, 42–43. [Google Scholar]
- Wapinski, I. , Pfeffer, A. , Friedman, N. , & Regev, A. (2007). Natural history and evolutionary principles of gene duplication in fungi. Nature, 449, 54–61. https://doi.org/10.1038/nature06107 [DOI] [PubMed] [Google Scholar]
- Wright, M. B. , Howell, E. A. , & Gaber, R. F. (1996). Amino acid substitutions in membrane‐spanning domains of Hol1, a member of the major facilitator superfamily of transporters, confer nonselective cation uptake in Saccharomyces cerevisiae . Journal of Bacteriology, 178, 7197–7205. https://doi.org/10.1128/jb.178.24.7197-7205.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue, A. G. , Voldeng, H. D. , Savard, M. E. , Fedak, G. , Tian, X. , & Hsiang, T. (2009). Biological control of fusarium head blight of wheat with Clonostachys rosea strain ACM941. Canadian Journal of Plant Pathology, 31, 169–179. https://doi.org/10.1080/07060660909507590 [Google Scholar]
- Yu, H. , & Sutton, J. C. (1997). Morphological development and interactions of Gliocladium roseum and Botrytis cinerea in raspberry. Canadian Journal of Plant Pathology, 19, 237–246. https://doi.org/10.1094/PDIS.1997.81.4.316 [Google Scholar]
- Zhai, M. M. , Qi, F. M. , Li, J. , Jiang, C. X. , Hou, Y. , Shi, Y. P. , … Weu, Q. X. (2016). Isolation of secondary metabolites from the soil‐derived fungus Clonostachys rosea YRS‐06, a biological control agent, and evaluation of antibacterial activity. Journal of Agricultural and Food Chemistry, 64, 2298–2306. https://doi.org/10.1021/acs.jafc.6b00556 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials