

SUMMARY
S-nitrosylation, the oxidative modification of Cys residues by nitric oxide (NO) to form S-nitrosothiols (SNOs), modifies all main classes of proteins and provides a fundamental redox-based cellular signaling mechanism. However, in contrast to other post-translational protein modifications, S-nitrosylation is generally considered to be non-enzymatic, involving multiple chemical routes. We report here that endogenous protein S-nitrosylation in the model organism E. coli depends principally upon the enzymatic activity of the hybrid-cluster protein Hcp, employing NO produced by nitrate reductase. Anaerobiosis on nitrate induces both Hcp and nitrate reductase, thereby resulting in the S-nitrosylation-dependent assembly of a large interactome including enzymes that generate NO (NO synthase), synthesize SNO-proteins (SNO synthase), and propagate SNO-based signaling (trans-nitrosylases) to regulate cell motility and metabolism. Thus, protein S-nitrosylation by NO in E. coli is essentially enzymatic, and the potential generality of the multiplex enzymatic mechanism we describe may support a re-conceptualization of NO-based cellular signaling.
eTOC BLURB
Protein S-nitrosylation by nitric oxide is generally thought to be non-enzymatic. Seth et al. demonstrate that endogenous S-nitrosylation in E. coli depends upon enzymatic activity of the hybrid cluster protein Hcp and involves assembly of an Hcp interactome including enzymes that generate NO, synthesize SNO-proteins, and propagate SNO-based signaling.
INTRODUCTION
Proteins are subject to a panoply of function-regulating post-translational modifications which, as a general rule, are mediated by dedicated enzymatic mechanisms. Nitric oxide (NO)-based post-translational modification by S-nitrosylation, which regulates proteins in most or all functional classes across phylogeny and conveys the large part of the ubiquitous cellular influence of NO (Hess et al., 2005), tests that rule. Although S-nitrosylated proteins (protein S-nitrosothiols, SNOs) may trans-nitrosylate additional proteins with which they interact (Jia et al., 2014; Kornberg et al., 2010; Mitchell and Marletta, 2005; Nakamura et al., 2010; Pawloski et al., 2001; Seth and Stamler, 2011; Wu et al., 2011), providing the basis of an enzymatic mechanism, NO biology is shaped by the tenet that protein SNOs are initially formed non-enzymatically. Theoretical analyses of the mechanism of SNO formation from NO, which entails a one-electron oxidation, have invoked multiple chemical routes in the cellular milieu (e.g. formation of NO+ donating intermediates such as N2O3) (Gould et al., 2013; Hess et al., 2005; Koppenol, 2012; Lancaster, 2017). However, this chemistry in vivo is subject to kinetic constraints and is highly dependent on cellular conditions, which does not reconcile well with a pervasive, regulatory signaling process. Thus, the fundamental question of how protein SNOs are formed de novo from NO, a seminal step in initiating S-nitrosylation-based signaling, has remained unresolved (Lancaster, 2017).
In facultative anaerobic bacteria, including E. coli, anaerobic respiration can be sustained with either fumarate or nitrate as an electron acceptor, but nitrate is preferred (Unden and Bongaerts, 1997). During anaerobic respiration on nitrate (ARN), the nitrate reductase NarGHI is induced (Showe and DeMoss, 1968), and NarGHI serves as a source of NO (Ji and Hollocher, 1988; Ralt et al., 1988) that mediates the S-nitrosylation of multiple proteins (Seth et al., 2012). These include the transcription factor OxyR, and S-nitrosylation of OxyR induces expression of a specialized regulon (i.e. distinct from the regulon induced by oxidation of OxyR under aerobic conditions) (Seth et al., 2012), whose components in turn regulate levels of protein S-nitrosylation (Seth et al., 2012).
Based on these findings, we developed a model system of endogenous (nitrate reductase–dependent) S-nitrosylation in E. coli to screen systematically for candidate enzymatic protein S-nitrosylases. We show that the hybrid-cluster protein Hcp, a component of the SNO-OxyR regulon (Seth et al., 2012), plays an essential role in S-nitrosylation by endogenously generated NO of most cellular SNO-proteins. We find that the S-nitrosylase activity of Hcp is dependent upon its iron-containing cluster(s) and, in vivo, entails its participation in a large multi-protein complex that contains an enzymatic source of NO and multiple substrates for S-nitrosylation, including proteins that propagate signaling by trans-nitrosylation. Thus, protein S-nitrosylation by NO in E. coli is essentially enzymatic, involving activities of three classes of enzymes that coordinately generate NO, convert NO to SNO, and propagate SNO-based signaling. The likely generality of the multiplex mechanism we describe points to a new paradigm in NO biology. Our findings also show that protein S-nitrosylation mediated by this mechanism subserves the adaptation of E. coli to anaerobic existence and plays a major role in resistance to exogenous nitrosative stress.
RESULTS
Hcp is a SNO Synthase and Protein S-nitrosylase
To screen for potential S-nitrosylating activities, we initially assayed by photolysis-chemiluminescence (Stamler et al., 1992) total protein-bound NO (which includes SNO and transition metal-bound NO) during ARN in E. coli in which each of the 74 genes induced during ARN in an OxyR-dependent fashion (Seth et al., 2012) was individually knocked out. Whereas rate of growth and nitrite production (indicative of nitrate reductase-mediated NO generation) did not differ appreciably between the strains (Table S1), they differed widely with respect to magnitude of protein nitrosylation (Figure 1A and Table S1). Thus, evidently, cellular NO levels are not the primary determinant of the amount of S-nitrosylation. The most profound loss of protein nitrosylation was seen with knock-out of hcp (Δhcp) (Figure 1A), which encodes the hybrid cluster protein Hcp, which in E. coli contains a [2Fe-2S] cluster as well as a [4Fe-2S-2O] “hybrid” cluster (van den Berg et al., 2000). Hcp expression is induced both by exogenous NO/SNO donors and during ARN (Filenko et al., 2005; Seth et al., 2012) (verified by qPCR; Figure S1A). A role for Hcp in regulating S-nitrosylation specifically was demonstrated by the finding that, during ARN, Δhcp greatly diminished cellular SNO-protein levels, whereas Hcp overexpression significantly enhanced SNO-protein levels, across many if not most SNO-proteins as revealed by the biotin-switch method (Jaffrey et al., 2001) applied to whole-cell lysates (Figure 1B). Mass spectrometric analysis employing the SNO-RAC method (Forrester et al., 2009) in combination with differential iTRAQ labeling revealed that deletion of Hcp diminished S-nitrosylation of 48/58 SNO-proteins identified in wild-type (WT) cells during ARN (Figure S1B; Tables 1 and S2).
Figure 1. Hcp is a Protein S-nitrosylase.

(A) During ARN, individual genes in the OxyR regulon differentially regulate intracellular protein nitrosylation levels. The greatest decrease in protein nitrosylation resulted from deletion of hcp as measured by photolysis/chemiluminescence (n=3–21, ±SEM). Dashed line=nitrosylation level in WT.
(B) Hcp mediates S-nitrosylation of the majority of SNO-proteins during ARN (biotin switch). HcpF=overexpressed FLAG-tagged Hcp. WT/HcpF=HcpF in WT background. Values are normalized with respect to WT during ARN; N=nitrate, F=fumarate (n=3, ±SEM). Dashed lines indicate lanes removed for clarity.
(C) Increased intracellular NO levels (resulting from knockout of NO metabolizing enzymes) do not restore protein S-nitrosylation during ARN in the absence of Hcp. SNO content of lysates was determined by Hg2+-coupled photolysis-chemiluminescence (n=6, ±SEM). * p < 0.05 versus WT by Student’s t-test.
(D) LpdA is S-nitrosylated in vitro by Hcp. LpdA (1.6 µM) was treated anaerobically with the NO donor DEANO (50 µM; 15 min) and Hcp (1.2 µM) where indicated, and S-nitrosylation was assessed by SNO-RAC. Values are normalized with respect to NO+Hcp, n=2–4±SEM.
(E) GAPDH is S-nitrosylated in vitro by Hcp. GAPDH (12.8 µM) was treated as in (D), n=2–4±SEM.
(F) S-nitrosylation of OxyR in vivo during ARN (6 hr) is largely dependent upon Hcp (SNO-RAC). n=4, ± SEM, *, differs from WT by ANOVA (p < 0.05).
(G) OxyR is S-nitrosylated by Hcp in vitro. OxyR (1.6 µM) was treated as in (D) and (E) above, and S-nitrosylation was detected by SNO-RAC (n=3, ±SEM).
(H) Hcp interacts with OxyR in an NO-dependent manner. His-tagged Hcp (HcpHis6) (20 µg) was added to lysates from Δhcp/OxyRF (cells grown anaerobically on nitrate or fumarate). Samples were treated with DEANO (100 µM) where indicated. Following chemical crosslinking, OxyRF was immunoprecipitated and Hcp was detected by western blotting (OxyRF=overexpressed FLAG-tagged OxyR) (n=3, ±SEM).
Table 1. Hcp-dependent S-nitrosylation of Proteins during ARN.
Proteins that are S-nitrosylated during ARN in WT were identified as proteins that were pulled down by SNO-RAC ≥1.5 fold on nitrate versus fumarate. Hcp-dependent SNO-proteins were identified as those S-nitrosylated ≥1.2 fold on WT versus Δhcp. Included proteins were identified as Hcp-dependent in at least two of four independent experiments. Proteins present in the Hcp interactome (Table S3) are identified with Y.
| Protein | Uniprot | WT Fold- change (N/F) |
Δhcp Fold- change (N/F) |
WT/ Δhcp |
Interact with Hcp |
|---|---|---|---|---|---|
| SerA D-3phosphoglycerate dehydrogenase | P0A9T0 | 4.7 | 1.1 | 4.3 | |
| FdnG Formate dehydrogenase, major subunit | P24183 | 11.2 | 2.7 | 4.1 | Y |
| GlyA Serine hydroxymethyltransferase | P0A825 | 3.8 | 0.9 | 4.2 | |
| EfG Elongation factor G | P0A6M8 | 5.8 | 1.5 | 3.9 | |
| AceE Pyruvate dehydrogenase E1 component | P0AFG8 | 5.2 | 1.4 | 3.7 | Y |
| LeuC 3-isopropylmalate dehydratase large subunit | P0A6A6 | 5.0 | 1.4 | 3.6 | Y |
| GltB Glutamate synthase [NADPH] large chain | P09831 | 4.5 | 1.4 | 3.2 | |
| AdhE Aldehyde-alcohol dehydrogenase | P0A9Q7 | 6.6 | 2.4 | 2.8 | |
| PykA Pyruvate kinase II | P21599 | 3.9 | 1.5 | 2.6 | |
| FdnH Formate dehydrogenase, iron-sulfur subunit | P0AAJ3 | 7.0 | 2.7 | 2.6 | Y |
| NarG Respiratory nitrate reductase 1 alpha chain | P09152 | 7.3 | 2.8 | 2.6 | Y |
| MetE 5-methyltetrahydropteroyltriglutamate-homocysteine | P25665 | 4.0 | 1.7 | 2.4 | Y |
| RpoC DNA-directed RNA polymerase subunit beta' | P0A8T7 | 4.4 | 1.9 | 2.3 | |
| CodA Cytosine deaminase | P25524 | 4.1 | 1.9 | 2.2 | |
| SbcB Exodeoxyribonuclease I | P04995 | 3.8 | 1.8 | 2.1 | |
| GapA Glyceraldehyde-3-phosphate dehydrogenase A | P0A9B2 | 3.9 | 1.9 | 2.1 | Y |
| DnaK Chaperone protein DnaK | P0A6Y8 | 3.2 | 1.8 | 1.8 | Y |
| PflB Formate acetyltransferase 1 | P09373 | 5.4 | 0.6 | 9.0 | |
| TufA Elongation factor Tu 1 | P0CE47 | 6.1 | 1.3 | 4.7 | Y |
| OppA Periplasmic oligopeptide-binding protein | P23843 | 5.3 | 1.2 | 4.4 | |
| Rho Transcription termination factor | P0AG30 | 5.2 | 1.3 | 4.0 | Y |
| ThrA Bifunctional aspartokinase/homoserine | P00561 | 6.1 | 1.6 | 3.8 | |
| Gnd 6-phosphogluconate dehydrogenase | P00350 | 4.9 | 1.3 | 3.8 | |
| AsnS Asparagine--tRNA ligase | P0A8M0 | 4.0 | 1.4 | 2.9 | |
| AtpA ATP synthase subunit alpha | P0ABB0 | 3.4 | 1.3 | 2.6 | |
| Asd Aspartate-semialdehyde dehydrogenase | P0A9Q9 | 5.7 | 2.6 | 2.2 | |
| NarI Respiratory nitrate reductase 1 gamma chain | P11350 | 8.3 | 3.8 | 2.2 | |
| Zwf Glucose-6-phosphate 1-dehydrogenase | P0AC53 | 3.7 | 1.8 | 2.1 | |
| AcnB Aconitate hydratase 2 | P36683 | 3.8 | 1.8 | 2.1 | Y |
| TrxA Thioredoxin-1 | P0AA25 | 4.6 | 2.3 | 2.0 | |
| Eno Enolase | P0A6P9 | 2.1 | 1.2 | 1.8 | |
| NarH Respiratory nitrate reductase 1 beta chain | P11349 | 7.1 | 3.9 | 1.8 | Y |
| NarZ Respiratory nitrate reductase 2 alpha chain | P19319 | 4.4 | 2.5 | 1.8 | |
| SerS Serine--tRNA ligase | P0A8L1 | 2.7 | 1.7 | 1.6 | |
| NarY Respiratory nitrate reductase 2 beta chain | P19318 | 3.9 | 2.5 | 1.6 | |
| PtsG PTS system glucose-specific EIICB component | P69786 | 2.7 | 1.7 | 1.6 | |
| NapA Periplasmic nitrate reductase | P33937 | 5.0 | 3.2 | 1.6 | |
| PyrG CTP synthase | P0A7E5 | 3.3 | 2.1 | 1.6 | Y |
| GuaA GMP synthase [glutamine-hydrolyzing] | P04079 | 2.1 | 1.5 | 1.4 | Y |
| PepQ Xaa-Pro dipeptidase | P21165 | 3.0 | 2.1 | 1.4 | |
| LpdA Dihydrolipoyl dehydrogenase | P0A9P0 | 2.5 | 1.7 | 1.5 | Y |
| FdoG Formate dehydrogenase-O major subunit | P32176 | 2.9 | 2.1 | 1.4 | |
| GuaB Inosine-5'-monophosphate dehydrogenase | P0ADG | 2.3 | 1.6 | 1.4 | |
| RpsA 30S ribosomal protein S1 | P0AG67 | 2.6 | 1.9 | 1.4 | |
| HybC Hydrogenase-2 large chain | P0ACE0 | 2.4 | 1.9 | 1.3 | |
| IlvD Dihydroxy-acid dehydratase | P05791 | 4.0 | 3.3 | 1.2 | |
| NirB Nitrite reductase (NADH) large subunit | P08201 | 3.1 | 2.6 | 1.2 | Y |
| Hcr NADH oxidoreductase HCR | P75824 | 3.3 | 2.7 | 1.2 | Y |
To support the conclusion that diminished cellular protein-SNO levels resulting from deletion of Hcp reflects primarily a role for Hcp as a protein S-nitrosylase, rather than the operation of alternative mechanisms such as compensatory diminishment of cellular NO levels, we examined the effects on protein S-nitrosylation during ARN of combined mutation of Hcp and of NorV, the major NO-consuming reductase under anaerobiosis (Gomes et al., 2002), as well as of the flavoprotein hmp, which encodes a second significant source of NO reductase activity (Hausladen et al., 1998; Membrillo-Hernandez et al., 1999). Steady-state SNO-protein levels were greatly diminished in the absence of Hcp (and were not significantly different from WT in the absence of NorV and/or Hmp) (Figure 1C). Notably, SNO-protein levels remained significantly diminished in the combined absence of Hcp and NorV and/or Hmp (Figure 1C), where endogenous NO levels would be enhanced (Gomes et al., 2002; Hausladen et al., 1998). These findings are consistent with the conclusion that diminished S-nitrosylation in the absence of Hcp is unlikely to result from compensatory alterations in NO abundance, and that the S-nitrosylase activity of Hcp is a principal determinant of cellular protein S-nitrosylation in E. coli during ARN.
To verify directly the function of Hcp as a protein S-nitrosylase, we employed an in vitro assay comprising recombinant Hcp in combination with individual Hcp-dependent SNO-proteins identified during ARN in vivo (Table 1); diethylamine NONOate (DEANO) was included as a source of NO. Assays were carried out under anaerobic conditions, precluding auto-oxidative (O2-dependent) production of nitrosating (NO+-donating) intermediates. Remarkably, when representative substrates of S-nitrosylation during ARN (LpdA and GAPDH; Table 1) were incorporated in the assay, substrate S-nitrosylation (as assessed by SNO-RAC) was found to be dependent on the presence of both Hcp and NO, i.e., NO-mediated S-nitrosylation was negligible in the absence of Hcp but greatly enhanced in its presence (Figures 1D and 1E). Thus, Hcp can S-nitrosylate multiple substrates, and NO itself was ineffective in the absence of Hcp.
Because S-nitrosylation of OxyR mediates the induction of Hcp during ARN (Seth et al., 2012), we examined the role of Hcp in OxyR S-nitrosylation. Although OxyR was not initially identified by mass spectrometry as a substrate of Hcp (Table 1), consistent with low abundance, in vivo assays employing Δhcp demonstrated that S-nitrosylation of OxyR during ARN is in fact Hcp-dependent (Figure 1F) and assays in vitro verified that S-nitrosylation of OxyR by NO (Figure 1G) requires Hcp. Further, OxyR interacted with Hcp in vitro (Figure 1H). Thus, in vivo S-nitrosylation of OxyR by Hcp, which activates OxyR-mediated transcription (Seth et al., 2012), in turn up-regulates Hcp. Accordingly, for cellular S-nitrosylation during ARN, Hcp is an essential component of the regulon induced by OxyR S-nitrosylation.
Hcp Iron Clusters and Cysteines Play an Essential Role in Hcp-Mediated S-nitrosylation
We extended our in vitro analyses with purified recombinant Hcp to gain insight into the enzymatic mechanism of S-nitrosylation. Formally, S-nitrosylation by NO of Cys thiolate (S−) to generate an SNO requires a one-electron oxidation e.g., of NO to NO+. The redox requirements of this process can be met through the intermediate formation of a transition metal nitrosyl, e.g. following charge-transfer (Feδ−-NOδ+) (Gow and Stamler, 1998; Lancaster, 2017; Luchsinger et al., 2003). Following co-incubation of Hcp and DEANO, analysis by Hg2+-coupled photolysis-chemiluminescence revealed that an iron-nitrosyl complex was formed, and that SNO was present within Hcp in equivalent amounts (~ 0.4 FeNO/Hcp and 0.4 SNO/Hcp) (Figure 2A), indicating that Hcp converts NO to SNO. The Fe content of recombinant Hcp as measured by Ferene S assay was 6.1±0.1 Fe/Hcp molecule. Treatment with 50 µM or 200 µM of the transition metal chelator 2’, 2’-bipyridyl decreased Fe content respectively to 3.6±0.08 Fe/Hcp (~ 40% decrease) and 2.6±0.4 Fe/Hcp (~60% decrease). Hcp-dependent S-nitrosylation of OxyR in vitro was diminished by chelator treatment in close proportion to the loss of Fe (Figure 2B), confirming a role for cluster-coordinated iron in the S-nitrosylase activity of Hcp. Further, iron chelation proportionally diminished the production of FeNO and SNO in Hcp in isolation (in the absence of additional substrates) (Figure 2C). Collectively, these findings are consistent with the formation of SNO-Hcp through auto-S-nitrosylation that couples formation of FeNO and SNO. SNO in Hcp may in principle be formed on one or more of the six free Cys or the eight Cys involved in cluster co-ordination (van den Berg et al., 2000), although the stoichiometry of SNO-Hcp formation resulting from exposure to DEANO (~ 0.5 SNO/Hcp) points to a single site.
Figure 2. Metal Clusters and Cysteines Play an Essential Role in Hcp-mediated S-nitrosylation.

(A) Following anaerobic treatment of purified recombinant Hcp (1 µM) with the NO donor PROLINONOate (PROLINO) (20 µM), Hcp-bound NO is present as both FeNO and SNO in approximately equal proportions. Protein-bound NO was measured by Hg2+-coupled photolysis-chemiluminescence (n=5, ±SEM).
(B) Treatment of Hcp with the iron chelator 2’, 2’ bipyridyl (30 min) diminished in vitro S-nitrosylation of OxyR by DEANO, assessed as in Figure 1G; n=3, ±SEM. *p < 0.05 with respect to Hcp + NO by ANOVA.
(C) Iron-sulfur clusters mediate Hcp auto-S-nitrosylation. Hcp was treated with 2’, 2’ bipyridyl chelator as in (B) followed by PROLINO (20 µM) and protein-bound NO was measured by Hg2+-coupled photolysis-chemiluminescence (UT=untreated; Chel=chelator, n=3, ±SEM).
(D) Non-cluster coordinating cysteines in Hcp are not stably S-nitrosylated. Following anaerobic treatment of purified recombinant WT and mutant Hcp (1 µM) with PROLINO (20 µM), protein-bound SNO was measured by Hg2+-coupled photolysis-chemiluminescence (n=3, ±SEM).
(E) Maintained S-nitrosylation by Hcp requires a redox cycle. OxyR (1.2 µM) was treated anaerobically with DEANO (100 µM) in the presence or absence of Hcp (100 nM), the Hcp reductase Hcr (100 nM) and the electron acceptor NAD+ (50 µM). S-nitrosylation of OxyR was assessed by SNO-RAC. Data are presented as SNO-OxyR normalized with respect to total OxyR (n=3, ±SEM).
To refine this analysis, we employed mutation of individual free Cys within Hcp, and assessed the effects on S-nitrosylation as assessed by Hg2+-coupled chemiluminescence. We found that S-nitrosylation of Hcp by DEANO in vitro was unaffected by mutation of any of the six free Cys within Hcp, i.e., Cys not involved in cluster coordination (Figure 2D). We therefore concluded that auto-S-nitrosylation targets a Cys(s) otherwise serving in Fe coordination. Identification by mutational analysis of coordinating Cys targeted by S-nitrosylation was precluded, because Hcp in which coordinating Cys were mutated was not recovered from our expression system presumably due to misfolding that led to sequestration and/or degradation. Taken together, these data are consistent with a mechanism whereby Hcp converts NO into SNO (Hcp-FeNO→Hcp-SNO) within Hcp Fe-clusters followed by trans-S-nitrosylation of additional substrates.
S-nitrosylation by Hcp, which constitutes an oxidative modification, would need to be redox coupled to maintain an enzymatic cycle. The operon encoding Hcp also encodes a cognate reductase, Hcp reductase (Hcr), which uses NADH as co-substrate (van den Berg et al., 2000). Rapid S-nitrosylation of OxyR in vitro by Hcp in limiting concentrations is followed by gradual, spontaneous de-nitrosylation, consistent with limited turnover of NO on Hcp. However, inclusion of Hcr and NAD+ as electron acceptor resulted in sustained S-nitrosylation of OxyR (Figure 2E). Thus, the redox requirements for converting NO to SNO by Hcp can be met readily through a physiological redox cycle. However, our findings do not preclude alternative electron acceptors in vivo that could sustain the enzymatic cycle.
NO-Dependent Formation of an Intramolecular Disulfide within Hcp, Hcp Dimerization and Assembly of a Multi-Protein Complex
NO group transfer by Hcp would require close proximity with substrates. Our finding that S-nitrosylation of dozens of proteins is dependent upon Hcp in situ (Table 1) raises the possibility of a multi-protein complex. The possibility of an Hcp interactome was explored by immunoprecipitation of FLAG-Hcp from extracts of E. coli during ARN or anaerobic exposure to DEANO (as well as control E. coli grown anaerobically on fumarate), followed by mass spectrometric analysis (LC/MS-MS). The interactomes following exposure to endogenous NO (ARN) (Table S3) or DEANO (on fumarate) (Table S4) comprised >100 proteins and 30 proteins respectively. Although the cellular substrate is known to differ greatly between ARN and anaerobic respiration on fumarate, 16/30 DEANO-induced interactome components were also identified as components of the Hcp interactome induced during ARN, indicating substantial NO dependence under both conditions. Notably, the Hcp interactome includes the endogenous source of NO during ARN (NarGHI) and multiple central metabolic enzymes (LpdA, PDH and PflB), as well as homologs of proteins previously characterized in mammalian cells as trans-nitrosylases (GAPDH and thioredoxin (TrxA)) (Kornberg et al., 2010; Mitchell and Marletta, 2005) (Tables S3 and S4). Furthermore, during ARN, S-nitrosylation of 17/113 Hcp interactome components was Hcp dependent (Table 1). Thus, NO induces the formation of a large signaling assembly, presumably comprising components interacting both directly and indirectly with Hcp, which includes machinery and substrates of S-nitrosylation (Figure 3A).
Figure 3. NO-dependent Formation Within Hcp of an Intramolecular Disulfide, Dimerization and Assembly of a Multi-protein Complex Competent for S-nitrosylation.

(A) Hcp-dependent S-nitrosylation during ARN generates a subset of SNO-proteins that are constituents of the Hcp interactome (representing substrates likely S-nitrosylated directly by Hcp), and a larger set of SNO-proteins that are S-nitrosylated through a trans-nitrosylation cascade. The overlap between the Hcp-dependent SNOome and the Hcp interactome is illustrated by a Venn diagram, in which numbers represent individual substrates as identified by mass spectrometry.
(B) NO-dependent intramolecular disulfide formation in Hcp. Hcp (1.8 µM) was treated anaerobically with DEANO (200 µM) followed by SDS-PAGE and Coomassie staining.
(C) Dimerization of Hcp leads to interactome formation. WT E. coli overexpressing FLAG-tagged Hcp (WT/HcpF) were grown anaerobically either in nitrate (10 mM, 6 hr), or in fumarate followed by treatment with DEANO (100 µM). Lysates were analyzed by non-reducing and reducing SDS-PAGE, and Hcp was visualized by western blotting with anti-FLAG anti-body. Representative gel, n=3. At right, longer exposure of nitrate lane.
(D) NO-dependent formation of an intramolecular disulfide in Hcp between Cys102 and Cys155. WT/HcpF were grown as in (C). HcpF was immunoprecipitated followed by non-reducing SDS-PAGE. A single disulfide bond, between Cys102–155, was identified in dimerized but not monomeric Hcp by mass spectrometry (see also Figure S2E). Representative gel, n=3 (UT=untreated; conditions as in 3C).
(E) Cys102 and Cys155 are essential for Hcp dimerization and complex formation. WT/HcpF or WT/HcpF C102/155S were grown anaerobically in the presence of either nitrate (N) or fumarate (F) (10 mM). Lysates were analyzed by non-reducing and reducing SDS-PAGE. Hcp was visualized by western blotting with anti-FLAG antibody (n=2).
(F) Alkylation of free thiols in Hcp decreases S-nitrosylation of OxyR. Hcp was treated anaerobically with iodoacetamide (IAA) (30 min, 37°C), (followed by removal of IAA=Hcp-IAA) and in vitro S-nitrosylation of OxyR was assessed as in Figure 1G (n=3, ±SEM); * differs from Hcp + DEANO treatment by ANOVA, p < 0.05.
(G) Cys102/Cys155 are essential for S-nitrosylation by Hcp. In vitro S-nitrosylation of OxyR was carried out as in (1G). n=3, ±SEM; * differs from Hcp + DEANO treatment by ANOVA, p < 0.05. A dashed line indicates an in-gel cut for clarity.
(H) Cys102/Cys155 are essential for S-nitrosylation by Hcp. WT, Δhcp, Δhcp/HcpF, Δhcp/HcpF C102/155S were grown anaerobically in LB medium in the presence of nitrate (10 mM, 2.5 hr). Lysates were subjected to SNO-RAC and SNO proteins were visualized by SDS-PAGE followed by Coomassie staining.
(I) Hcp undergoes an NO-dependent conformational change. CD spectra of recombinant Hcp with and without DEANO treatment (Representative spectra, n=3).
(J) The NO-induced conformational change is not altered by C102/155S mutation. CD spectra of WT and C102/155S Hcp with and without DEANO treatment. (Representative spectra, n=3).
(K) NO-dependent conformational change in Hcp is abrogated by ascorbate. CD spectra of recombinant Hcp with and without DEANO treatment in the presence or absence of ascorbate (Representative spectra, n=3).
Formation of an NO-dependent Hcp interactome might be expected to reflect an NO-dependent change in Hcp conformation. We observed that exposure of Hcp in vitro to NONOate-derived NO (Figure 3B) or to the S-nitrosylating agent S-nitrosocysteine (CysNO) (Figure S2A) resulted in a decrease in mr (apparent molecular weight) following SDS-PAGE, pointing to intramolecular disulfide formation (Silva and Cidlowski, 1989) as confirmed by reversal of the decrease in mr by thiol reductant (Figures 3B and S2A). These results indicate that SNO may serve as a precursor to disulfide (Arnelle and Stamler, 1995), and we found that the decrease in the mr of Hcp induced by treatment with NO is diminished by inclusion in the reaction mixture of ascorbate, a selective reductant of SNO (Forrester et al., 2007) (Figure S2B). The NO-induced decrease in mr is also eliminated by inclusion in the reaction mixture of glutathione, which would serve to reduce both SNO and disulfide (Figure S2B). Note that mutation of free Cys had no effect on Hcp-SNO levels at steady state (Figure 2D), consistent with a transient, disulfide-promoting S-nitrosylation (involving free Cys) and a stable S-nitrosylation involving coordinating Cys.
We considered the possibility that, in addition to the NAD+-dependent role of Hcr in redox cycling of coordinated iron and thus in auto-S-nitrosylation of Hcp (Figure 2E), NO-dependent formation of an internal disulfide within Hcp might also be regulated by Hcr, which would require NADH as co-factor. We found that inclusion of Hcr and NADH diminishes the NO-induced decrease in the mr of Hcp (Figure S2C). Thus, Hcr may regulate disulfide formation within Hcp that is required for assembly of the Hcp interactome and thereby propagation of S-nitrosylation-based signaling. The role of Hcr would thereby be influenced by cellular redox status (NAD+ versus NADH).
We found that, in cells treated with NO or during ARN, Hcp formed a homodimer (~120 kDa) in association with interactome assembly (Figure 3C); Hcp protein complexes formed within 30 min of ARN (Figure S2D). We identified a disulfide by LC-MS/MS (Cys102/155) following either ARN or exposure to DEANO within dimeric but not monomeric Hcp (Figures 3D and S2E). We determined that both dimer and interactome were disassembled under reducing conditions (Figures 3C, S2D and S2F) (and that no intermolecular disulfides were observed between Hcp and other proteins by mass spectrometry). Further, Hcp with Cys102-155Ser mutation did not form dimers or associate with other proteins in vivo (Figure 3E), and we observed that either prior alkylation of Hcp with iodoacetamide, which would block free but not stably coordinating thiols (Figure 3F), or mutation of Cys102/155 eliminated S-nitrosylation by Hcp of OxyR in vitro (Figure 3G). In addition, mutation of Cys102/155 compromised S-nitrosylation by Hcp of cellular proteins in vivo (Figures 3H and S2G). Specifically, whereas levels of S-nitrosylation during ARN were similar to WT E. coli when WT Hcp was overexpressed in Δhcp, S-nitrosylation was not rescued by overexpression of C102/155S Hcp in Δhcp (Figures 3H and S2G). Thus, S-nitrosylation of Hcp results in the formation of an intramolecular disulfide, which leads to conformational change, dimerization and interactome formation that is required for S-nitrosylation of interactome constituents.
To further assess NO-dependent conformational change in Hcp that would be implicated in dimerization and complex formation, we employed mutational analysis and circular dichroism spectroscopy (CD). As described above, exposure of purified recombinant Hcp to NO derived from DEANO results in the formation of SNO, FeNO and an internal disulfide. Treatment of WT Hcp with DEANO led to a decrease in CD spectral intensity over the range of 200 nm – 220 nm (Figure 3I), and treatment of C102/155S mutant Hcp resulted in a similar change in spectral intensity (Figure 3J), indicating that the conformational change observed reflected a stable NO-dependent modification independent of disulfide formation. We then examined the results of DEANO treatment in the presence of ascorbate, which prevents the stable formation of Hcp-SNO (as in Figure S2B). Notably, DEANO did not elicit a spectral change in the presence of ascorbate (Figure 3K) (it should be noted that ascorbate treatment alone altered the CD spectrum, likely reflecting conformational changes resulting from changes in oxidation state of Hcp’s clusters (Macedo et al., 2002)). These results are consistent with a conformational change that is associated with SNO formation, which might promote disulfide formation and thereby dimerization and complex assembly.
Trans-nitrosylases Propagate Hcp-dependent S-nitrosylation
As noted above, the Hcp interactome includes homologs of proteins that are well established as mammalian trans-nitrosylases including GAPDH and TrxA (Kornberg et al., 2010; Mitchell and Marletta, 2005), which we identified as Hcp-dependent SNO-proteins in vivo (Table 1) and which we verified as substrates for S-nitrosylation by Hcp in vitro (Figures 1E and S3A). Inclusion of the trans-nitrosylases GAPDH and TrxA in the SNO-induced Hcp interactome suggests that S-nitrosylation by Hcp of interactome members will support a signaling cascade that propagates widely via S-nitrosylation, accounting for the Hcp-dependence of S-nitrosylation of the majority of SNO-proteins under ARN. Indeed, whereas S-nitrosylation of 17/113 components of the Hcp interactome during ARN was Hcp-dependent (Table S3), a total of 31 additional proteins were S-nitrosylated in an Hcp-dependent fashion during ARN (Figure 3A and Table 1).
This model, in which trans-nitrosylases propagate Hcp-dependent S-nitrosylation, is further supported by the finding that knockout of GAPDH (ΔgapA) resulted in diminished S-nitrosylation of multiple substrates during ARN (Figure 4A). Analysis by iTRAQ-coupled mass spectrometry revealed that, among the GAPDH-dependent SNO-proteins (Table 2), CodA and NapA were also S-nitrosylated in an Hcp-dependent manner (Table 1), and trans-nitrosylation of CodA and NapA by SNO-GAPDH was verified in vitro (Figures 4B and 4C). Further, remarkably, GAPDH interacted with CodA and NapA only in the presence of NO (Figures 4D and 4E). When equimolar concentrations of OxyR (a SNO-substrate of Hcp), and CodA (a SNO-substrate of GAPDH) were S-nitrosylated in vitro in the presence of Hcp and NO, the proportion of OxyR S-nitrosylated was significantly higher versus CodA (Figure S3B). This finding verifies the specificity of the in vitro assay for direct S-nitrosylation by Hcp and supports the model under which Hcp initially auto-S-nitrosylates, followed by trans-nitrosylation of GAPDH, which in turn binds and trans-nitrosylates CodA and NapA, providing a first demonstration of a trans-nitrosylation cascade.
Figure 4. Trans-nitrosylases Propagate Hcp-dependent S-nitrosylation.

(A) GAPDH-dependent endogenous S-nitrosylation. WT and ΔgapA E. coli were grown anaerobically in either fumarate or nitrate (10 mM, 6 hr) and lysates were subjected to SNO-RAC. SNO-proteins were visualized by Coomassie staining following SDS-PAGE. Representative gel, n=3. Arrows indicate multiple GAPDH-dependent SNO-proteins.
(B) GAPDH trans-nitrosylates CodA in vitro. His-tagged SNO-GAPDH (10 µM) was incubated with His-tagged CodA (2.4 µM) (30 min, 37°C). Reaction mixtures were subjected to SNO-RAC and SNO-proteins were visualized by western blotting with an anti-His antibody, n=3, ±SEM.
(C) GAPDH trans-nitrosylates NapA in vitro. His-tagged SNO-GAPDH was used to transnitrosylate His-tagged NapA as in (B), n=3, ±SEM.
(D) GAPDH interacts directly with CodA in an NO-dependent manner. His-tagged GAPDH (10 µM) was incubated with His-tagged CodA (2.4 µM) in the presence or absence of CysNO (1 mM, 30 min). GAPDH was immunoprecipitated with anti-GAPDH antibody and co-immunoprecipitated CodA was visualized with an anti-His antibody, n=3, ±SEM.
(E) GAPDH interacts directly with NapA in an NO-dependent manner. Co-immunoprecipitation of GAPDH with NapA was done as in (D), n=3, ±SEM.
Table 2. GAPDH-dependent S-nitrosylation of Proteins.
Proteins that are S-nitrosylated by GAPDH during ARN were identified as proteins that were both S-nitrosylated in WT by ≥3.0 fold on nitrate versus fumarate and ≥1.5 fold in WT versus ΔgapA. Proteins listed were identified as GAPDH-dependent in at least two of three independent experiments.
| Protein | Uniprot | WT Fold- change (N/F) |
ΔgapA Fold- change (N/F) |
WT/ ΔgapA |
|---|---|---|---|---|
| IadA Isoaspartyl dipeptidase | P39377 | 4.6 | 1.3 | 3.5 |
| CodA Cytosine deaminase | P25524 | 6.9 | 2.5 | 2.8 |
| NapA Periplasmic nitrate reductase | P33937 | 6.3 | 2.6 | 2.4 |
| FdnG Formate dehydrogenase, major subunit | P24183 | 8.2 | 3.9 | 2.1 |
| FlgI Flagellar P-ring protein | P0A6S3 | 4.0 | 2.3 | 1.7 |
| NarG Respiratory nitrate reductase 1 alpha chain | P09152 | 6.4 | 3.8 | 1.7 |
| PyrB Aspartate carbamoyltransferase | P0A786 | 3.2 | 2.1 | 1.5 |
Hcp-Mediated S-nitrosylation Regulates Cellular Metabolism
We validated Hcp-dependent S-nitrosylation in vivo for representative SNO-proteins in the Hcp interactome (Table 1), including dihydrolipoyl dehydrogenase (LpdA) (a component of the multi-enzyme pyruvate dehydrogenase (PDH) complex and a substrate for Hcp-mediated S-nitrosylation in vitro (Figure 1D)) and NarH, and examined its functional consequences for LpdA, AceE (PDH E1 component), pyruvate formate lyase (PflB) and NarH.
LpdA was S-nitrosylated endogenously in an Hcp-dependent manner during ARN (Figure S4A) as well as in vitro (Figure 1D). LpdA is known to be inhibited by S-nitrosylation (Richardson et al., 2011; Yan et al., 2012), and LpdA activity was inhibited under conditions of Hcp-dependent S-nitrosylation in vitro (Figure 5A). In vivo, inhibition of LpdA would lead to the accumulation of dihydrolipoamide, which can act as a source of reducing equivalents (Feeney et al., 2011) and thereby potentially prevent accumulation of excessive NO (nitrosative stress) during ARN (Stoyanovsky et al., 2005).
Figure 5. Hcp-Mediated S-Nitrosylation Regulates Cellular Metabolism and is Essential for Protection Against Nitrosative Stress and for Motility During Anaerobic Respiration on Nitrate.

(A) LpdA enzymatic activity is inhibited in vitro by Hcp-dependent S-nitrosylation. Following Hcp-dependent S-nitrosylation in vitro (as in Figure 1D), LpdA activity was determined using dihydrolipoamide to lipoamide conversion by monitoring the conversion of NAD+ to NADH, at 340 nm (n=3, ±SEM), * p < 0.05 Hcp + NO versus Hcp; Hcp + NO versus NO; Hcp + NO versus UT (untreated).
(B) Effect of Hcp knockout on acetyl-CoA levels. Acetyl-CoA was measured in WT and Δhcp cells grown anaerobically on either fumarate or nitrate (10 mM, 6 hr) (n=3 ±SEM).
(C) Hcp inhibits nitrate reductase activity. WT and Δhcp E. coli were grown for 4 hr on nitrate anaerobically to induce nitrate reductase activity, washed and re-exposed to nitrate (5 mM). Nitrite formed was measured with the Greiss assay (n=4, ±SEM), * p < 0.05 WT versus Δhcp.
(D) ARN confers Hcp-dependent protection from exogenous nitrosative stress. E. coli cultures were started in minimal medium including 10 mM fumarate or 10 mM nitrate at A600 of 0.1. Where indicated sodium nitroprusside (NP; 2 mM) was added. Growth was followed spectrophotometrically at A600 (n=3, ±SEM). * p < 0.05 WT vs Δhcp.
(E) Hcp is essential for E. coli motility during ARN. WT and Δhcp were introduced onto soft agar plates that were supplemented with either fumarate or nitrate (20 mM). Following growth at 37°C for 18 hrs under anaerobic conditions the plates were photographed under white light. Representative results, n=5.
(F) Hcp is essential for E. coli motility during ARN. Quantification of E. coli motility following assay described in Figure 5E (n=5, ±SEM). * p < 0.05 WT (nitrate) vs Δhcp (nitrate).
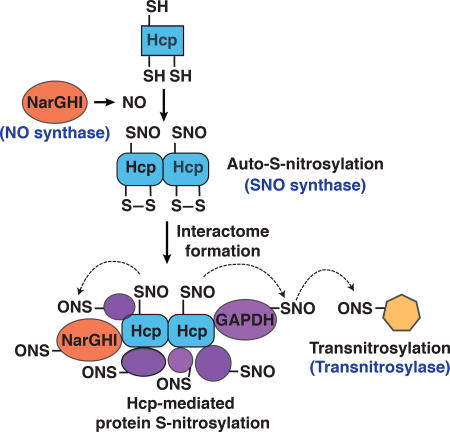
(G) A schematic summary of the Hcp-based enzymatic machinery mediating de novo protein S-nitrosylation and S-nitrosylation signaling cascades in E. coli. NarGHI generates NO (NO synthase activity); Hcp converts NO to SNO (SNO-synthase) and trans-nitrosylates binding partners within the Hcp interactome (S-nitrosylase activity), including additional trans-nitrosylases (e.g. SNO-GAPDH) that further propagate SNO-based signals. The Cys-coordinated iron cluster that mediates auto-S-nitrosylation is depicted schematically in the second section. The Hcp dependent interactome comprises over 100 proteins including enzymes (e.g. NarGHI, LpdA) and transcription factors (OxyR) regulating cell growth and metabolism, and conferring protection against nitrosative stress.
LpdA is a component of the multi-enzyme PDH complex that includes AceE, also identified as a SNO-protein within the Hcp interactome (Table 1). Both PDH and pyruvate formate lyase (PflB) generate acetyl CoA during anaerobic growth. PflB was also S-nitrosylated in an Hcp-dependent manner during ARN (Table 1). We compared intracellular levels of acetyl CoA in WT and Δhcp cells that were grown anaerobically on either fumarate or nitrate, and found that acetyl CoA levels were elevated during ARN versus fumarate in WT cells and that this effect was markedly diminished in Δhcp cells (Figure 5B). Acetyl CoA metabolism is thus aberrant in Δhcp cells. Note that there are multiple potential loci of regulation of acetyl-CoA metabolism by Hcp-mediated S-nitrosylation.
A recent study identified the major respiratory enzyme NarH as subject to S-nitrosylation at Cys184 (Wojdyla et al., 2015). We found that NarH was S-nitrosylated endogenously at Cys184 during ARN (Figure S4B) and that S-nitrosylation was decreased in Δhcp cells (Figure S4B). NarGHI is the major enzyme that converts nitrate to nitrite and is also the primary source of NO production during ARN (Ji and Hollocher, 1988; Ralt et al., 1988; Seth et al., 2012). Nitrate reductase activity was higher in Δhcp cells than in WT cells (Figure 5C) indicating that S-nitrosylation by Hcp inhibits NarGHI in vivo. NO generated during ARN is self-limiting and does not exceed micromolar concentrations (Ji and Hollocher, 1988) (even in the presence of millimolar nitrate and nitrite), avoiding toxicity of NO, and this effect may reflect inhibition of NarGHI by S-nitrosylation. Taken together, our findings indicate that Hcp-dependent S-nitrosylation of metabolic enzymes broadly regulates cellular metabolism during ARN.
Hcp-dependent S-nitrosylation is Essential for Protection Against Nitrosative Stress and for Motility During ARN
We showed previously that deletion of Hcp (or of its transcriptional activator OxyR) renders E. coli more susceptible to exogenous nitrosative stress exerted either by application of S-nitrosoglutathione or by NO generated by activated macrophages (Seth et al., 2012). It has recently been reported that Hcp can function as an NO reductase, operating specifically to eliminate low (nanomolar) levels of NO (Wang et al., 2016). In this scenario, enhanced abundance of NO might result in cellular injury through excessive S-nitrosylation (nitrosative stress) (Fang et al., 2016; Hausladen et al., 1996). However, our finding that deletion of Hcp greatly suppresses rather than enhances protein S-nitrosylation strongly suggests that it is the S-nitrosylase rather than the NO reductase function of Hcp that contributes to resistance to nitrosative stress.
We provided further support for this conclusion by examining the effects on nitrate-or fumarate-grown WT and Δhcp cells of exogenous nitrosative stress induced by levels of NO that far exceed those where Hcp’s NO reductase activity is operative (nanomolar) (Wang et al., 2016) and by using an NO donor that can directly S-nitrosylate proteins (sodium nitroprusside, NP) without a general requirement of Hcp (Grossi and D'Angelo, 2005). On fumarate, both WT and Δhcp are inhibited to the same extent by 2 mM NP (Figure 5D). However, on nitrate (where hcp is induced), growth of the Δhcp strain is inhibited to a significantly greater extent than WT (Figure 5D). Moreover, on nitrate, levels of SNO actually decline following NP exposure in the WT but increase in Δhcp, consistent with induction of a protective regulon (including hcp) during ARN (Figure S5A). Thus, growth (which is inhibited by NO) does not simply reflect SNO levels, but rather the targets of S-nitrosylation. That is, Hcp-dependent S-nitrosylation is protective whereas non-specific S-nitrosylation is inhibitory. Further, we examined the effects on growth during ARN of single or combined deletion of Hcp and NorV (the major NO reductase). Whereas all mutants grew at rates comparable to WT on fumarate (where NarGHI is not induced and NO is not produced) (Figure S5B), the growth of mutants that lacked both hcp and norV was inhibited during ARN (Figure S5B). This finding is consistent with the conclusion that endogenous nitrosative stress (as assessed by growth rate) conveyed by NO production during ARN is ameliorated by the activity of dedicated NO reductases (NorV) such that the protective effect of Hcp is revealed only in their absence. Taken together, our results indicate that protein S-nitrosylation mediated by Hcp supports growth under conditions of nitrosative stress, consistent with our previous findings that deletion of hcp results in enhanced suppression of growth of E. coli by S-nitrosoglutathione and by activated macrophages (Seth et al., 2012). We speculate that the NO reductase activity of Hcp, which only operates at very low NO concentrations (Wang et al., 2016), may serve principally to regulate the local concentration of NO within the Hcp interactome to ensure effective S-nitrosylation.
The targeting by Hcp-mediated S-nitrosylation of manifold targets including multiple components of cellular metabolism (Figures 5A–5C) predicts that Hcp-mediated S-nitrosylation would play appreciable functional roles in E. coli during ARN. As evidence for functionality that is impacted by Hcp-mediated protein S-nitrosylation, we found that deletion of Hcp profoundly inhibited “swimming” motility during ARN, as assessed by assay of spreading, whereas the absence of Hcp had no effect on motility during anaerobic growth on fumarate (Figures 5E and 5F). It is therefore of interest that flagellar protein FlgI is among the seven proteins identified as subject to trans-nitrosylation by GAPDH during ARN (Table 2). Thus, protein S-nitrosylation by Hcp plays an essential role in at least some important aspects of E. coli functionality during ARN, as it does in protection against nitrosative stress (vida supra).
DISCUSSION
A Multiplex Enzymatic Machinery for Cellular Protein S-nitrosylation in E. coli
How SNO-proteins are generated de novo has remained a major unanswered question in the field of NO biology. Under the prevailing theoretical framework, de novo SNO formation is mediated through multiple, non-enzymatic channels that are mechanistically dependent on cellular conditions, and are therefore poorly suited to serve as the basis of the pervasive cellular signaling that S-nitrosylation provides (Hess et al., 2005). Thus, our findings of a critical enzyme responsible for cellular protein S-nitrosylation and of its functional connectivity to additional classes of enzymes, operating coordinately to propagate NO-based signaling, potentially changes thinking in the field.
The multiplex enzymatic machinery for S-nitrosylation (and NO-based cellular signaling more generally) that we have described here (summarized in Figure 5G) is analogous to ubiquitinylation (Chaugule and Walden, 2016), where a small number (2 known) of E1 ubiquitin activators bring into play the ubiquitin moiety (by analogy to the three mammalian NO synthases and bacterial and plant nitrate reductases, which may be viewed as putting NO in play). Ubiquitin is then transferred to a larger number of E2 ligases (35 known), by analogy to the transformation of NO into SNO by Hcp and other “SNO synthases” (Angelo et al., 2006; Boffi et al., 2002; Gow and Stamler, 1998; Koppenol, 2012; Luchsinger et al., 2003; Pawloski et al., 2001; Weichsel et al., 2005). E2 ligases transfer to a much larger set of E3 ligases (hundreds identified) (Chaugule and Walden, 2016; Morreale and Walden, 2016), by analogy to the cascade of trans-nitrosylation from SNO-synthase (Hcp) to trans-nitrosylase e.g. GAPDH (multiple trans-nitrosylases have been identified and hundreds are predicted) (Anand and Stamler, 2012; Jia et al., 2014; Kornberg et al., 2010; Mitchell and Marletta, 2005; Nakamura et al., 2010; Pawloski et al., 2001; Seth and Stamler, 2011; Wu et al., 2011) that greatly increases the set of S-nitrosylated substrates and thereby the signaling repertoire of NO. Ubiquitinylation takes place within multi-protein complexes that include ligases and substrates by analogy to the mechanism we have described for S-nitrosylation, which entails assembly of a signaling complex that includes S-nitrosylases and substrates. However, other features we describe are unique to S-nitrosylation, in particular the unprecedented dependence on NO for assembly of a multi-protein complex (>100 proteins; Figure 3), requiring modification by NO of Hcp. Thus, our finding that the mechanism of protein S-nitrosylation in E. coli is essentially enzymatic and that the operation of that mechanism involves formation of an NO-dependent multi-protein complex that includes an enzymatic source of NO, an essential SNO-protein synthase, and substrates and propagators of trans-nitrosylation provides a new paradigm in NO biology.
Components of the machinery described here have been observed individually in higher organisms in limited and separate contexts, supporting the possible generality of our findings. In particular, metazoan NO synthases generate NO by analogy to NarGHI, and iron-dependent formation of SNO from NO that mediates auto-S-nitrosylation has been shown for both mammalian (Gow and Stamler, 1998; Luchsinger et al., 2003) and molluscan (Boffi et al., 2002) hemoglobin and for insect nitrophorin (Weichsel et al., 2005). Moreover, mammalian hemoglobin, combines SNO synthase and S-nitrosylase activities (Angelo et al., 2006; Luchsinger et al., 2003; Pawloski et al., 2001) and may interact with components of a supercomplex that includes GAPDH (Pawloski et al., 2001; Puchulu-Campanella et al., 2013), analogous to Hcp. Further, multiple proteins have been shown to function as trans-nitrosylases capable of NO group transfer to interacting substrates (Anand and Stamler, 2012; Jia et al., 2014; Kornberg et al., 2010; Mitchell and Marletta, 2005; Seth and Stamler, 2011), including examples that depend on S-nitrosylation for inducing the interactions that subserve trans-nitrosylation (Kornberg et al., 2010).
Our findings emphasize the potentially broad role of metalloproteins in catalyzing S-nitrosylation, and in particular the role of iron-sulfur species. It is well established that iron-sulfur clusters in proteins are major targets of modification by endogenous NO, including those forming protein-associated dinitrosyl iron complexes (DNIC), which can support the oxidative requirements for S-nitrosylation by NO (Bosworth et al., 2009; Lancaster, 2017; Stojanovic et al., 2004; Vanin et al., 1997). We have previously shown that treatment of E. coli with a divalent metal chelator decreased formation of SNOs during ARN (Seth et al., 2012). Similarly, analysis of the decrement of cellular SNOs induced by iron chelation in mammalian cells indicated that levels of DNICs and SNO co-vary (Bosworth et al., 2009). Thus, our finding that iron-sulfur centers within Hcp mediate protein S-nitrosylation is consistent in general with a role for transition metal-thiol coupling in the mechanism of redox-based enzymatic protein S-nitrosylation that has been observed previously in mammalian hemoglobin (Luchsinger et al., 2003; Pawloski et al., 2001) and may therefore operate across cell types.
Our results are also consistent with the current understanding that trans-nitrosylation is a principal mechanism for the propagation of SNO-based cellular signals (Anand and Stamler, 2012; Kornberg et al., 2010; Seth and Stamler, 2011). The trans-nitrosylation cascade we have described (summarized in Figure 5G), which entails assembly of a complex comprising (1) an enzymatic source of NO, (2) an S-nitrosylase that generates SNO-proteins, and (3) trans-nitrosylases that propagate SNO-based signaling, is broadly congruent with the finding that mammalian inducible NO synthase (iNOS) can be found in a complex with S100A9, which transfers iNOS-derived NO to multiple substrates including the trans-nitrosylase GAPDH (Jia et al., 2014). However, coordinated operation of the enzymatic activities we describe, including a central role for de novo synthesis of SNO, has not heretofore been incorporated into a more general paradigm.
Functional Ramifications of S-nitrosylation in E. coli
The ability to respire on nitrate, where nitrate reductase is induced, provides facultative anaerobes with an advantage in microaerobic/anaerobic environments. Our previous (Seth et al., 2012) and present findings show that production of NO during ARN by nitrate reductase (NarGHI) (Ji and Hollocher, 1988; Ralt et al., 1988) mediates the Hcp-dependent S-nitrosylation of a broad spectrum of proteins, including multiple metabolic enzymes and the transcription factor OxyR whose S-nitrosylation activates a nitrosative stress regulon of ~ 100 genes (Figure 1), with demonstrable benefits on organismal fitness (Figures 5D, 5E and S5B). Increasing evidence indicates that S-nitrosylation plays manifold roles in plants where nitrate reductase also serves as a principal source of NO (Hichri et al., 2015; Zaffagnini et al., 2016). Our results thus suggest that endogenous S-nitrosylation may contribute to the advantages conferred by ARN. In this regard, deletion of Hcp profoundly inhibited the motility of E. coli respiring anaerobically on nitrate but not on fumarate. Because motility is the default state, S-nitrosylation-dependent motility during ARN would presumably preserve a functional benefit, perhaps in the acquisition of nutrients.
In addition, infection and inflammation can create microaerobic/anaerobic tissue environments, which are also characterized by high levels of both NO and nitrate as a result of NO production by innate immune cells (Roos and Klemm, 2006). Our findings here and earlier (Seth et al., 2012) that Hcp protects E. coli against exogenous nitrosative stress, which is in fact conveyed in substantial part by S-nitrosylation (Hausladen et al., 1996), suggest that specific and targeted enzymatic S-nitrosylation by Hcp protects against diffuse and aberrant NO-based modification of proteins. Moreover, the protection conferred by Hcp-mediated S-nitrosylation in E. coli may provide insight into the consequences of S-nitrosylation in mammalian cells under heightened NO levels that characterize infection/inflammation, where cellular protein S-nitrosylation is greatly enhanced without deleterious effects (Eu et al., 2000). Thus, our findings provide a new perspective on the molecular mechanisms of protection against nitrosative stress, under which specific and targeted S-nitrosylation may confer functional benefit.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact, Dr. Jonathan Stamler at jonathan.stamler@case.edu.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Bacterial Strains and Plasmids
E. coli strain BW25113 and the isogenic Δhcp, Δhmp, and ΔnorV strains were from the Keio collection (Baba et al., 2006) and MG1655 and the isogenic strain ΔgapA (Seta et al., 1997) were obtained from the Coli Genetic Stock Center (Yale University). To prepare plasmids overexpressing OxyR, Hcp or LpdA, the gene was PCR-amplified from MG1655 genomic DNA using primers (Table S5) that add a ribosome binding site to the 5' end of the gene and a FLAG tag to the 3' terminus. The resulting DNA fragment was cloned into the pUC19 vector.
In order to express 6xHis tagged recombinant proteins, the genes for the respective proteins were cloned into pET21b vector (Novagen, Merck Biosciences). The primers used for amplification of the genes of interest from MG1655 genomic DNA are listed in Table S5.
Generation of Δhcp/hmp, Δhcp/norV, Δhmp/norV and Δhcp/hmp/norV Strains
The kanamycin resistant gene was eliminated from Δhcp and ΔnorV using pCP20 plasmid that allows thermal induction of FLP synthesis. Electro-competent cells of Δhcp and ΔnorV strains were prepared by standard methods, transformed with pCP20 and ampicillin-resistant transformants were selected at 30°C. pCP20 shows temperature sensitive growth and hence non-selective colony purification was done at 43°C to eliminate pCP20. The colonies were then tested for loss of all antibiotic resistances.
P1 transduction was used to transfer the Δhmp mutation into Δhcp and ΔnorV strains to generate Δhcp/hmp and Δhmp/norV strains (Thomason et al., 2007). Similarly ΔnorV mutation was transferred into the Δhcp strain to generate Δhcp/norV strain. The kanamycin-resistant transformants were purified twice on kanamycin plates (25 mg/L kanamycin). Genomic DNA was extracted from the selected colonies and PCR-amplified using primers flanking the hcp, hmp and norV genes, followed by gel purification and sequencing to verify the mutations. Δhmp mutation was similarly transferred into the Δhcp/norV mutant to obtain the Δhcp/hmp/norV strain, which was also verified by sequencing.
Bacterial Culture and Manipulations
Bacteria were grown in LB medium or on LB agar plates for cloning and storage. For the various assays, bacteria were grown in MOPS-buffered minimal media containing 80 mM MOPS, 4 mM tricine, 9.95 mM NH4Cl, 0.276 mM K2SO4, 0.1 mM CaCl2, 0.528 mM MgCl2, 0.01 mM FeSO4 and 50 mM NaCl in MQ water (pH 7.0), 0.013 mM K2HPO4, 0.2% glucose and trace metals. The ΔgapA strain was grown in MOPS-buffered minimal media containing 80 mM MOPS, 4 mM tricine, 9.95 mM NH4Cl, 0.276 mM K2SO4, 0.1 mM CaCl2, 0.528 mM MgCl2, 0.01 mM FeSO4 and 50 mM NaCl in MQ water (pH 7.0), 0.013 mM K2HPO4, 0.1% glycerol, 1% sodium succinate and trace metals. Ampicillin (100 mg/L) was added when required. Anaerobic bacterial growth was carried out without shaking in a 37°C incubator placed inside a glove box with an atmosphere of 85% N2, 5% CO2 and 10% H2 (Coy Laboratory Products, Grass Lake, MI). All culture media, buffers and cultures were equilibrated in the glove box for 18 hr prior to use. Any possible O2 traces were continuously removed with a palladium catalyst.
Cultures were started by inoculation of the MOPS-buffered minimal media containing 10 mM sodium fumarate and incubated overnight in the glove box at 37°C. Cultures were centrifuged, spent medium was removed and the cell pellets were re-suspended in media at A600 of 0.1–0.2 and grown for 6 hr anaerobically. Where indicated, sodium nitrate or sodium fumarate was added to the growth media to a final concentration of 10 mM, or cells were treated with specified concentrations of NO donors before harvesting. All cells were chilled on ice, harvested by centrifugation and washed once in phosphate buffered saline (PBS, pH 7.4) containing 100 µM DTPA. Pellets were stored at −80°C. For RNA extractions, pellets were stored overnight at 4°C in RNAlater solution (Ambion).
METHOD DETAILS
Photolysis-chemiluminescence
For screening E. coli mutants for differential regulation of protein S-nitrosylation, cells were grown on nitrate as described above. Cell pellets were lysed in 2× Cellytic buffer (Sigma Aldrich) with 1 mM PMSF according to the manufacturer’s instructions. Measurements of XNO/SNO (where XNO is predominantly metal-NO (MNO)) in lysates were done using photolysis/chemiluminescence essentially as described (Stamler et al., 1992). Briefly, nitric oxide (NO) released from MNO/SNO by UV-photolysis is detected by chemiluminescence generated by the reaction of NO with ozone. Pre-treatment of samples with HgCl2 (1 mM) (Hg2+-coupled photolysis/chemiluminescence) removes SNO specifically and allows differentiation between SNO and other UV-photolyzable bound NO species (predominantly MNO).
To measure SNO/MNO in NO treated Hcp, recombinant Hcp (1 µM) was treated with PROLINONOate (20 µM) anaerobically for 15 min at 37°C in 100 mM sodium phosphate buffer, pH 7.4, containing 100 µM EDTA and 100 µM DTPA (500 µL reaction volume). A reaction without Hcp added was included to correct for residual NO in the reaction mix. The samples were centrifuged through a 30 kDa cutoff filter (Amicon, EMD Millipore, Billireca, MA) for 10 min. at 14,000 × g to remove unreacted NO. The >30 kDa protein fraction was collected by inverting the filter into a fresh tube and centrifugation for 2 min at 1000 × g. Buffer was added to restore the volume to 500 µL. 100 µL was analyzed by photolysis-chemiluminescence with or without the addition of HgCl2 (1 mM).
Griess Assay for Nitrite
To measure nitrite by Griess assay, 100 µl of sample was added to 100 µl of 1% sulfanilamide (w/v) in 0.5 N HCl. Following addition of 100 µl of 0.1% N-(1-napthyl)ethylenediamine, reaction was carried out at room temperature for 10 min. The chromophoric azo product formed is read spectrophotometrically at 540 nm. Sodium nitrite (3.125 µM to 100 µM) was used to generate a standard curve.
Biotin-switch and SNO-RAC Assays
Total S-nitrosylated proteins were detected with the biotin-switch assay (Jaffrey et al., 2001) or by SNO-RAC (resin-assisted capture of S-nitrosylated proteins) (Forrester et al., 2009), described briefly here. For biotin-switch, 300 µL of lysate (1–2 mg total protein) were incubated in HEN buffer containing 100 mM HEPES, 1 mM EDTA, 0.1 mM neocuproine (pH 7.7) with SDS (1% final concentration) and 0.1% methyl methanethiosulfonate (MMTS) to block free thiols in a final volume of 2 ml at 50°C for 20 min. Proteins were acetone precipitated, washed three times in 70% acetone, and mixed with 0.2 mM biotin-HPDP (Pierce; Rockford, IL), with or without 50 mM sodium ascorbate to selectively remove the NO group from SNOs, at room temperature for 1 hr. Following a second acetone precipitation and re-solubilization, biotinylated proteins were pulled down using 50 µL streptavidin-agarose beads (Sigma-Aldrich). S-nitrosylated proteins were separated by non-reducing SDS-PAGE and transferred to nitrocellulose membranes, and blotted proteins were detected using neutravidin-HRP.
For SNO-RAC, samples containing affinity tagged target proteins were initially treated as in the biotin-switch method. Following blocking with MMTS and acetone precipitation, pellets were re-suspended in HEN buffer containing 1% SDS. 50 µl Thiopropyl sepharose (GE Lifesciences, Pittsburgh, PA) and 50 mM sodium ascorbate were added followed by rotation in the dark for 4 hr. Bound proteins were eluted in 4×SDS-PAGE loading buffer containing 10% β- mercaptoethanol and SNO-proteins were detected by western blotting using anti-tag antibodies (anti-FLAG or anti-His) following separation on reducing SDS-PAGE.
Reverse Transcription and Real-time PCR
RNA was extracted using the Masterpure RNA purification kit (Epicenter Biotechnologies, Madison, WI) and 3 µg of RNA was used for preparing cDNA with random hexamer oligonucleotide primers (Integrated DNA Technologies, Coralville, IA) using MuMLV reverse transcriptase enzyme (Invitrogen). Gene specific primers were used for real-time PCR in an Applied Biosystems StepOnePlus instrument using 2× iQ SYBR green supermix (BioRad, Hercules, CA). The expression of 16s rRNA in each sample was used to normalize the expression of the gene(s) of interest. Real-time PCR primers are listed in Table S5. Fold-change in expression was calculated using the comparative Ct method.
Purification of 6xHis Tagged Recombinant Proteins
The genes for OxyR, Hcp, Hcr, LpdA, GAPDH, CodA and NapA were cloned into pET21b (Novagen) to introduce a C-terminal 6xHis tag on the expressed protein. Protein purifications were carried out in a glove box under anaerobic conditions. All buffers and media were equilibrated in the glove box for at least 18 hr before use. Overnight cultures were sub-cultured into 3 L of LB medium at 4%. At A600 of 0.4, cultures were induced with 100 µM IPTG and grown for a further 4 hr at 37°C. Since Hcr did not express well under these conditions, cultures for Hcr purification were grown at 20°C following IPTG induction aerobically (all subsequent steps were under anaerobic conditions). Cultures were centrifuged at 4500 × g for 15 min to harvest the cells. Cell pellets from 1 L cultures were lysed in 2 mL of 2× Cellytic buffer and 2 mL of 50 mM NaH2PO4, 300 mM NaCl buffer containing 0.2 mg/ml lysozyme, 5 µg/ml DNase and 1 mM PMSF. Following rotation at room temperature for 30 min and centrifugation at 16,000 × g for 12 min, the supernatant was collected. The lysate was diluted 4-fold in 50 mM NaH2PO4, 300 mM NaCl buffer containing 10 mM imidazole and incubated with 1.5 ml of Ni-NTA agarose (equilibrated with 50 mM NaH2PO4, 300 mM NaCl buffer) at room temperature for 2 hr with rotation. The slurry was then poured into empty PD-10 columns (GE Healthcare, Chicago, IL). The beads were washed with 100 ml of 50 mM NaH2PO4, 300 mM NaCl buffer containing 20 mM imidazole. Elution was done in 20 ml of 50 mM NaH2PO4, 300 mM NaCl buffer with 250 mM imidazole. 1 ml fractions were collected and 15 µl from each was analyzed by SDS-PAGE. Fractions containing the pure protein of interest were pooled and stored at −80°C in 30% glycerol. Anaerobic conditions were maintained throughout the purification process and storage.
Hcp Dimerization and Interactome Formation
Overnight cultures of WT/Hcp-FLAG cells were sub-cultured at A600 0.1 – 0.2 and grown for 6 hr in the presence of nitrate (10 mM). For DEANO treatment, cells were grown on minimal medium for 6 hr prior to treatment with DEANO (100 µM). Cells were harvested and lysates were prepared by sonication (30s × 3) in HEN buffer containing 0.1% MMTS. After addition of non-reducing or reducing (2.5% β-mercaptoethanol) loading buffer, lysates were heated for 2 min at 42°C. Equal amounts of protein were analyzed by SDS-PAGE.
For analyzing disulfide formation in purified Hcp, Hcp (1.8 µM) was treated with DEANO or CysNO (200 µM) anaerobically. CysNO was freshly synthesized by mixing equimolar volumes of L-Cysteine prepared in 0.5 M HCl (containing 100 µM EDTA) and sodium nitrite.
Where indicated, ascorbate or GSH (50 mM) was added prior to DEANO treatment. Reaction was stopped by the addition of SDS-PAGE loading buffer followed by SDS-PAGE and Coomassie staining.
Immunoprecipitation of Hcp
Overnight cultures of WT/Hcp-FLAG were sub-cultured into minimal media and grown for 6 hr on either fumarate or nitrate (10 mM). Where indicated, cells grown on fumarate were treated with 100 µM DEANO for 5 min before harvesting. Cell pellets from 50 ml cultures were re-suspended in 0.5 mL of buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA (TNE buffer) and 1 mM PMSF. Lysates were prepared by sonication (3 cycles, 30 sec), followed by centrifugation at 18,000 g for 15 min. Protein concentration was measured in the supernatant using the Bradford protein assay, and 5 – 7.5 mg protein in TNE buffer containing 1% Triton X-100 (TNET buffer) was incubated with 50 µL of anti-FLAG agarose that had been washed with TNET buffer. Binding was allowed to proceed for 2 hr at 4°C with rotation. Beads were washed three times with TNET buffer and twice with TNE buffer. Bound protein was eluted by incubating with 45 µL 0.1 M glycine HCl, pH 3.5 for 10 min at room temperature with shaking. Following centrifugation at 1000 × g for 2 min, the eluate was neutralized by the addition of 5 µL 1.0 M TrisHCl, pH 8.0. Samples were heated at 42°C for 2 min and analyzed by non-reducing SDS-PAGE.
Mass Spectrometric Analysis
To identify the Hcp interactome, Flag-tagged Hcp was immunoprecipitated from whole cell lysates as described above. Following SDS-PAGE and Coomassie staining, bands were excised and washed in 50% ACN/50% ammonium bicarbonate (200 µL) for 5 hr with vortexing. The supernatant was removed and 100% ACN (200 µL) was added to gel bands and vortexed for 10 min. The supernatant was removed and the gel band was dried under vacuum for 10 min. Free cysteines were labeled in-gel with 55 mM IAA (200 µL) and vortexing for 45 min. Bands were then washed with 200 µL each of 100 mM ammonium bicarbonate followed by 100% ACN for 10 min, and the washes were repeated. The supernatant was removed and bands dried under vacuum for 10 min. Proteins were digested in-gel with trypsin as described above. From the same gel, the Hcp monomer (60 kDa) and dimer (120 kDa) bands were processed as described above and analyzed for the presence of intra-molecular disulfide bonds.
iTRAQ Labeling for Identification of the Hcp SNOome
SNO-RAC samples were run on SDS-PAGE and Coomassie stained. Gel bands were excised and washed with 50% acetonitrile (ACN)/50% Ammonium bicarbonate (200 µl) for >5 hr with vortexing followed by 100% ACN (200 µl) for 10 min. Bands were then dried under vacuum for 10 min, and samples were reduced with 10 mM DTT (200 µl) for 45 min with vortexing. Free cysteines were labeled by adding 55 mM IAA (200 µl) to the gel pieces and vortexing for 45 min followed by washing (10 min/wash with vortexing) sequentially with 200 µl each of 1× iTRAQ dissolution solution, 100% ACN, 1× iTRAQ dissolution solution and 100% ACN. Following drying under vacuum for 10 min, proteins were digested in-gel with trypsin (1 µl of 0.30 µg/µl trypsin and 2 µl of 100 mM CaCl2 added to 47 µl of 1× iTRAQ dissolution solution; 50 µL/band) on ice for 30 min. Bands were then incubated in 50 mM iTRAQ buffer overnight at 37°C. Peptide extraction solution (60%ACN/5% formic acid; 200 µl) was then added to gel bands, followed by vortexing for 30 min and sonication for 15 min. Gel extraction was repeated twice The digest solution was dried under vacuum.
Extracted peptides were iTRAQ labeled with the iTRAQ reagents multiplex kit (Sciex, Concord, ON). 10× iTRAQ dissolution buffer (30 µl) was added to each sample (samples 1–4) and vortexed and centrifuged at room temperature. The pH was measured to ensure pH ≥7. 70 µl of ethanol was added to each iTRAQ reagent vial (114, 115, 116, 117) and vortexed. The contents of one iTRAQ reagent vial was transferred to one sample tube (114, 115, 116, 117 to sample 1, 2, 3 and 4 respectively), and the mixtures were vortexed for >5 hr at room temperature. 60 µl from each sample was combined to prepare the final sample, which was dried under vacuum. Samples were cleaned by Ziptip (C18 resin, Millipore) before analysis by LC/MS-MS.
LC/MS-MS
Digested peptides were separated by a UPLC (Waters, Milford, MA) with a 5-cm × 75-µm Pico Frit C18 column (New Objective, Woburn, MA). A linear gradient was used in chromatography by using mobile phase A (0.1% formic acid in water) and B (100% ACN) at a flow rate of 0.3 µl/min. Gradients started with 1% mobile phase B that was gradually increased to 40% at 65 min for protein interaction and disulfide bond identification, or increased to 40% at 130 min for iTRAQ experiments, then increased to 90% within 2 min and maintained at 90% for 10 min to clean up the column. Separated peptides were continuously injected into an Orbitrap Elite hybrid mass spectrometer (Thermo Finnigan, San Jose, CA) by a nanospray emitter (10 µm, New Objective). All mass spectrometry data were acquired in positive ion mode. For identification of Hcp-interacting proteins, a full MS scan (m/z 350–1800) at resolution of 120,000 was conducted; twenty MS2 scans (m/z 350–1800) were selected from the twenty most-intense peptide peaks of full MS scans. CID cleavage mode was performed at normalized collision energy of 35%. For iTRAQ experiments, a full MS scan (m/z 300–1800) at resolution of 120,000 was conducted, ten MS2 scans (m/z 100–1600) were activated from five most intense peptide peaks of full MS scans. CID and HCD cleavage modes were performed alternatively of same peptides selected from full MS scans. MS2 resolution of HCD is 15,000.
LC-MS/MS: Data Analysis
MassMatrix bioinformatics software (Xu and Freitas, 2007) was used to search the acquired MS data against a database composed of sequences of bacteria E. coli K12 proteins from Uniprot and their reversed sequences were used as a decoy database. Modifications including oxidation of methionine and labeling of cysteine (N-methyl maleimide, N-ethyl maleimide or IAA modifications) were selected as variable modifications in searching. For iTRAQ labeling, tagging of N terminus Lys and/or Tyr were selected as variable modification to test labeling efficiency and fixed modification for iTRAQ quantification. Trypsin was selected as an in-silico enzyme to cleave proteins after Lys and Arg. Precursor ion searching was within 10 ppm mass accuracy and product ions within 0.8 Da for CID cleavage mode and 0.02 Da for HCD cleavage mode. 95% confidence interval was required for protein identification.
For identification of proteins that interacted with Hcp following NO treatment, or during anaerobic respiration on nitrate (ARN) (versus fumarate), if a hit was exclusive in the NO-treated or ARN-grown sample, then proteins with spectral count ≥ 5 were included in the positive hit set. If a hit appeared in both fumarate-grown and treated (NO-treated or ARN-grown) samples, then the treated sample was required to have a spectral count ≥20 and fold-change of treated versus fumarate-grown ≥ 2.0 to be included.
For disulfide bond identification, MS data were searched against a database composed of sequences of Hcp and its reversed sequences as a decoy database. Crosslinking of disulfide bonds between cysteine residues was selected as search criteria. Oxidation of methionine, labeling of cysteine by IAA and NEM and deamidation of asparagine and glutamine were selected as variable modifications.
Circular Dichroism spectroscopy
Recombinant WT or C102/155S Hcp (2.9 µM) was treated with DEANO (200 µM) anaerobically for 15 min at 37°C in 100 mM sodium phosphate buffer, pH 7.4, containing 100 µM EDTA and 100 µM DTPA. Where indicated, sodium ascorbate (2 mM) was added to the reaction. The samples were centrifuged through a 30 kDa cutoff filter (Amicon, EMD Millipore, Billireca, MA) for 10 min at 14,000 × g to remove unreacted NO. An additional 500 µL of buffer was added to the filter and samples were centrifuged to ensure removal of residual NO or ascorbate. The >30 kDa protein fraction was collected by inverting the filter into a fresh tube and centrifugation for 2 min at 1000 × g. Buffer was added to restore the volume to 400 µL.
CD analysis was done on an Aviv-215 CD spectrometer in a 1 mm path length quartz cuvette at 25°C. Three scans were acquired over the wavelength range of 340 – 190 nm at 1 nm intervals. The spectra were smoothed using Savitsky-Golay smoothing, using a polynomial order of 3 and a smoothing window of 11 points. The smoothed spectra were averaged and corrected for buffer contributions.
In vitro S-nitrosylation Assay
Under anaerobic conditions, substrate protein (LpdA: 1.6 µM, GAPDH: 12.8 µM, OxyR: 1.6 µM) were incubated with Hcp (1.2 µM) and DEANO (50 µM), and Hcr (1.2 µM) where indicated, in 600 µL of 100 mM sodium phosphate buffer, pH 7.4 containing 100 µM EDTA and 100 µM DTPA for 15 min at room temperature. The reaction was stopped by addition of 1.8 mL cold acetone and 100 µg BSA. All subsequent steps were carried out in room air. After incubation at −20° C for 30 min, the samples were centrifuged at 4500 × g for 15 min. The pellet was re-suspended in 600 µL HEN buffer containing 2.5% SDS and 0.1% MMTS. Samples were then subjected to SNO-RAC as described above and analyzed by western blotting with anti-His- tag antibodies.
Where indicated, Hcp was treated with 2’, 2’- bipyridyl (50 µM or 200 µM) in 100 mM sodium phosphate buffer pH 7.4 for 30 min at room temperature under anaerobic conditions, prior to use in the S-nitrosylation assay. Treatment of Hcp with iodoacetamide (IAA) (1 mM) was done anaerobically for 30 min at 37 °C.
Ferene S Assay for Iron Content
Hcp was left untreated or treated with 50 µM or 200 µM 2’, 2’- bipyridyl in 100 mM sodium phosphate buffer pH 7.4 for 30 min at room temperature under anaerobic conditions. The reaction was stopped by addition of 3 volumes of ice-cold acetone. Subsequent steps were carried out in room air. Samples were incubated at −20°C for 20 min and centrifuged at 4500 × g for 15 min, and the pellet was re-suspended in 0.4M HCl and heated at 95°C for 15 min. Following incubation for 5 min on ice, proteins were precipitated by the addition of 3.5% TCA (w/v). The samples were centrifuged (12,000 × g, 15 min, 4°C) to remove any precipitate. 100 µL of supernatant were incubated for 30 min at room temperature with 100 µL of 22.5% (w/v) ammonium acetate, 50 µL of 12.5% (w/v) ascorbate and 50 µL of 10 mM Ferene S. Absorbance of samples at 593 nm was measured spectrophotometrically. A standard curve was generated with iron standards.
Acetyl CoA Assay
WT and Δhcp cells were grown under anaerobic conditions in 10 mM fumarate or nitrate for 6 hr. Cells were harvested and 1.4 × 1010 cells were assayed for acetyl CoA by the CWRU Mouse Metabolic Phenotyping Center. Acetyl-CoA was measured by LC-MS/MS as described previously (Harris et al., 2013; Zhang et al., 2009). Briefly, the samples were spiked with 0.4 nmol of [2H5] pentanoyl-CoA as an internal standard prior to homogenization with 4 ml of 1:1 methanol-H2O with 5% acetic acid. The supernatant was loaded onto a Supelco solid-phase extraction cartridge [2-(pyridyl)-ethyl functionalized silica gel] preconditioned with 3 ml of methanol followed by 3 ml of 1:1 methanol-H2O/5% acetic acid. The cartridge was then washed with 3 ml each of 1:1 methanol-H2O/5% acetic acid to elute impurities, followed sequentially by 1:1 methanol-H2O/50 mM ammonium formate, 3:1 methanol-H2O/50 mM ammonium formate, and methanol to elute acyl-CoAs. The eluent was evaporated under nitrogen. The LC was coupled to an API4000 Qtrap Mass Spectrometer (Applied Biosystems, Foster City, CA) operated under positive ionization mode (Harris et al., 2013). Acetyl-CoA was measured at the14.2 minute elution time and the ions 810/303 – 812/305 were monitored.
Nitrate Reductase Activity Assay
WT and Δhcp cells grown overnight anaerobically in minimal media containing 10 mM fumarate were sub-cultured at A600 0.15 into minimal media containing 10 mM nitrate. Following growth for 4 hr to induce nitrate reductase (NarGHI) activity, cells (100 µl) were harvested and re-suspended in 100 mM sodium phosphate buffer, pH 7.4, containing 5 mM nitrate, 5 mM formate, 100 µM EDTA and 100 µM DTPA, and incubated at 37°C. At indicated time points, the reaction was stopped by the addition of 25 µl of 1% formaldehyde to 25 µl of reaction mix. Nitrite formed was measured by Griess assay.
In vitro LpdA Activity Assay
In vitro S-nitrosylation of LpdA was carried out under anaerobic conditions in a glove box. LpdA (320 nM) was incubated for 15 min at room temperature with Hcp (30 nM) and DEANO (12.5 µM) in 600 µL of 100 mM sodium phosphate buffer, pH 7.4, containing 100 µM EDTA and 100 µM DTPA. The reaction was stopped by placing the samples on ice, and the reaction mixture was added to 200 mM potassium phosphate buffer, pH 8.0 containing 120 mM dihydrolipoamide, 120 mM NAD+ and 100 mM EDTA. Production of NADH was followed spectrophotometrically at 340 nm.
Trans-nitrosylation Assay
His-tagged GAPDH (50 µM) was treated with CysNO (1 mM) in 100 mM sodium phosphate buffer, pH 7.4 containing 100 µM EDTA and 100 µM DTPA for 30 min at 37°C to generate SNO-GAPDH. The reaction mixture was centrifuged at 14,000 × g for 10 min through a 10 kDa- cutoff centrifugal filter (Amicon Ultra) to remove residual CysNO. A separate sample containing CysNO alone (without GAPDH) served as a control to assure that S-nitrosylation by SNO-GAPDH did not reflect a contribution from residual CysNO (see Figures 4B and 4C). The flow-through was discarded, 400 µL buffer was added and the filters were re-centrifuged, and washes were repeated 4 times. GAPDH concentration was measured by protein assay. Trans-nitrosylation reactions employed 10 µM SNO-GAPDH and 2.4 µM substrate (His-tagged CodA or His-tagged NapA) in 100 mM sodium phosphate buffer, pH 7.4, containing 100 µM EDTA and 100 µM DTPA, and were incubated for 30 min at 37°C. The reaction was stopped by the addition of 3 volumes of ice-cold acetone. Following precipitation at −20°C for 30 min, samples were centrifuged at 4500 × g for 15 min. The pellet was washed once in 70% acetone and re- suspended in HEN buffer containing 0.15% MMTS, 2.5% SDS and 50 µM 2’, 2’-bipyridyl. Blocking of free cysteines and SNO-RAC were performed as described above, followed by western blotting using anti-His tag antibody.
Co-immunoprecipitation of GAPDH
GAPDH (10 µM) and substrate (CodA or NapA) (2.4 µM) in 100 mM sodium phosphate buffer, pH 7.4 containing 100 µM EDTA and 100 µM DTPA was incubated for 30 min at 37°C in the presence or absence of CysNO (1 mM). GAPDH was immunoprecipitated with anti-GAPDH antibody (3 µg) for 2.5 hrs at 4°C with rotation. 30 µL of Protein A/G sepharose in sodium phosphate buffer was added and samples were incubated for 2 hr. Following five washes with sodium phosphate buffer, bound proteins were eluted in 20 µL glycine-HCl, pH 3.5. The eluate was neutralized with 3 µL of 1M Tris-HCl, pH 8.0 and analyzed by western blotting with anti- His-tag antibody.
Motility Assays
Single colonies of E. coli grown overnight on LB agar plates were introduced onto soft agar plates that were supplemented with either fumarate or nitrate (20 mM). Following growth at 37°C for 18 hrs under anaerobic conditions, the plates were photographed under white light. The diameter of spread was measured.
SDS-PAGE and Western Blot Quantification
Coomassie-stained SDS-PAGE gels were imaged using Licor Odyssey Imaging system in the 700 nm channel. IRDye-labeled secondary antibodies (Licor) were used for western blotting and blots were visualized on the Licor Odyssey Imaging system.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of all western blot and SDS-PAGE data was done using the software ImageJ. Data in figures are represented as mean ± SEM. p-values were calculated using either Student’s t-test, a two-way ANOVA or repeated measures ANOVA with Dunnett’s post-test statistic. A value of p<0.05 was considered significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-FLAG | Sigma-Aldrich | Cat#F1804 |
| Rabbit polyclonal anti-His | Cell Signaling | Cat#2365 |
| Mouse monoclonal anti-GAPDH | Epitope Biotech Inc. | Cat#L001 |
| Mouse monoclonal anti-E. coli RNA polymerase β′ | Biolegend | Cat#662903 |
| Bacterial and Virus Strains | ||
| E. coli BW25113- F-, Δ(araD-araB)567, ΔlacZ4787(::rrnB-3), λ-,rph-1, Δ(rhaD-rhaB)568, hsdR514 | Coli Genetic Stock Center, Yale University | CGSC#7636 |
| E. coli Δhcp hcp deletion mutant, BW25113 hcp-774(del)::kan | Coli Genetic Stock Center, Yale University | CGSC#8890 |
| E. coli Δhmp hmp deletion mutant, BW25113 hmp-726(del)::kan | Coli Genetic Stock Center, Yale University | CGSC#10019 |
| E. coli ΔnorV norV deletion mutant, BW25113 norV785(del)::kan | Coli Genetic Stock Center, Yale University | CGSC#10112 |
| E. coli MG1655 | Laboratory strain | ATCC700926 |
| E. coli ΔgapA gapA deletion mutant MG1655 gapA12(del)::Cm | Coli Genetic Stock Center, Yale University | CGSC#7563 |
| Phage P1vir | Coli Genetic Stock Center, Yale University | CGSC#12133 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sodium ascorbate | Sigma-Aldrich | Cat#11140 |
| S-Methyl thiomethanesulfonate, MMTS | Sigma-Aldrich | Cat#64306 |
| Thiopropyl sepharose 6B | GE Lifesciences | Cat#17-0420-01 |
| 2,2′-Bipyridyl | Sigma-Aldrich | Cat#D216305 |
| Cysteine | Sigma-Aldrich | Cat#C6852 |
| Ferene S | Sigma-Aldrich | Cat#82940 |
| Iodoacetamide | Sigma-Aldrich | Cat#I1149 |
| Critical Commercial Assays | ||
| iTRAQ | Sciex | Cat#4352135 |
| Experimental Models: Organisms/Strains | ||
| BW25113 E. coli Δ hcp/hmp hcp/hmp deletion mutant | This study | N/A |
| BW25113 E. coli Δ hcp/norV hcp/norV deletion mutant | This study | N/A |
| BW25113 E. coli Δ hmp/norV hmp/norV deletion mutant | This study | N/A |
| BW25113 E. coli Δ hcp/hmp/norV hcp/hmp/norV deletion mutant | This study | N/A |
| Oligonucleotides | ||
| See Table S5 | ||
| Recombinant DNA | ||
| pUC19 | NEB | Cat#N3041 |
| pET21b | EMD Millipore | Cat#69741 |
| pCP20 | Coli Genetic Stock Center, Yale University | Cat#7629 |
HIGHLIGHTS.
De novo endogenous protein S-nitrosylation in E. coli is essentially enzymatic.
The hybrid cluster protein Hcp is an essential enzyme required for S-nitrosylation.
S-nitrosylation entails three classes of enzymes within a multi-protein complex.
S-nitrosylation regulates metabolism and motility and counters nitrosative stress.
Acknowledgments
This work was supported by NIH grant R01-GM099921. We thank P. McLaughlin for invaluable technical assistance.
Footnotes
Supplemental Information includes five figures and six tables and can be found with this article online at https://doi.org/10.1016/j.molcel.2017.12.025.
AUTHOR CONTRIBUTIONS
D.S., D.T.H. and J.S.S. designed the study; D.S. and A.H. performed experiments and analyzed results; L.W. and Y.W. carried out mass spectrometric analysis; D.S., D.T.H. and J.S.S. wrote the manuscript.
References
- Anand P, Stamler JS. Enzymatic mechanisms regulating protein S-nitrosylation: implications in health and disease. J. Mol. Med. 2012;90:233–244. doi: 10.1007/s00109-012-0878-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelo M, Singel DJ, Stamler JS. An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizes nitrite as a substrate. Proc. Natl. Acad. Sci. U.S.A. 2006;103:8366–8371. doi: 10.1073/pnas.0600942103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnelle DR, Stamler JS. NO+, NO, and NO− donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Arch. Biochem. Biophys. 1995;318:279–285. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single- gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100050. 2006 0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boffi A, Sarti P, Amiconi G, Chiancone E. The interplay between heme iron and protein sulfhydryls in the reaction of dimeric Scapharca inaequivalvis hemoglobin with nitric oxide. Biophys. Chem. 2002;98:209–216. doi: 10.1016/s0301-4622(02)00094-7. [DOI] [PubMed] [Google Scholar]
- Bosworth CA, Toledo JC, Zmijewski JW, Li Q, Lancaster JR. Dinitrosyliron complexes and the mechanism(s) of cellular protein nitrosothiol formation from nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 2009;106:4671–4676. doi: 10.1073/pnas.0710416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaugule VK, Walden H. Specificity and disease in the ubiquitin system. Biochem. Soc. T. 2016;44:212–227. doi: 10.1042/BST20150209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eu JP, Liu LM, Zeng M, Stamler JS. An apoptotic model for nitrosative stress. Biochemistry. 2000;39:1040–1047. doi: 10.1021/bi992046e. [DOI] [PubMed] [Google Scholar]
- Fang FC, Frawley ER, Tapscott T, Vazquez-Torres A. Discrimination and integration of stress signals by pathogenic bacteria. Cell Host & Microbe. 2016;20:144–153. doi: 10.1016/j.chom.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeney MA, Veeravalli K, Boyd D, Gon S, Faulkner MJ, Georgiou G, Beckwith J. Repurposing lipoic acid changes electron flow in two important metabolic pathways of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2011;108:7991–7996. doi: 10.1073/pnas.1105429108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filenko NA, Browning DF, Cole JA. Transcriptional regulation (prismane) protein of a hybrid cluster. Biochem. Soc. T. 2005;33:195–197. doi: 10.1042/BST0330195. [DOI] [PubMed] [Google Scholar]
- Forrester MT, Foster MW, Stamler JS. Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J. Biol. Chem. 2007;282:13977–13983. doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, Stamler JS. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol. 2009;27:557–559. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes CM, Giuffrè A, Forte E, Vicente JB, Saraiva LgM, Brunori M, Teixeira M. A novel type of nitric-oxide reductase Escherichia coli flavorubredoxin. J. Biol. Chem. 2002;277:25273–25276. doi: 10.1074/jbc.M203886200. [DOI] [PubMed] [Google Scholar]
- Gould N, Doulias PT, Tenopoulou M, Raju K, Ischiropoulos H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J. Biol. Chem. 2013;288:26473–26479. doi: 10.1074/jbc.R113.460261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow AJ, Stamler JS. Reactions between nitric oxide and haemoglobin under physiological conditions. Nature. 1998;391:169–173. doi: 10.1038/34402. [DOI] [PubMed] [Google Scholar]
- Grossi L, D'Angelo S. Sodium nitroprusside: Mechanism of NO release mediated by sulfhydryl-containing molecules. J. Med. Chem. 2005;48:2622–2626. doi: 10.1021/jm049857n. [DOI] [PubMed] [Google Scholar]
- Harris SR, Zhang GF, Sadhukhan S, Wang H, Shi C, Puchowicz MA, Anderson VE, Salomon RG, Tochtrop GP, Brunengraber H. Metabolomics and mass isotopomer analysis as a strategy for pathway discovery: pyrrolyl and cyclopentenyl derivatives of the pro-drug of abuse, levulinate. Chem. Res. Toxicol. 2013;26:213–220. doi: 10.1021/tx3003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausladen A, Gow AJ, Stamler JS. Nitrosative stress: Metabolic pathway involving the flavohemoglobin. Proc. Natl. Acad. Sci. U.S.A. 1998;95:14100–14105. doi: 10.1073/pnas.95.24.14100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausladen A, Privalle CT, Keng T, DeAngelo J, Stamler JS. Nitrosative stress: activation of the transcription factor OxyR. Cell. 1996;86:719–729. doi: 10.1016/s0092-8674(00)80147-6. [DOI] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S- nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- Hichri I, Boscari A, Castella C, Rovere M, Puppo A, Brouquisse R. Nitric oxide: a multifaceted regulator of the nitrogen-fixing symbiosis. J. Exp. Bot. 2015;66:2877–2887. doi: 10.1093/jxb/erv051. [DOI] [PubMed] [Google Scholar]
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat. Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- Ji XB, Hollocher TC. Mechanism for nitrosation of 2,3-diaminonaphthalene by Escherichia coli: enzymatic production of NO followed by O2-dependent chemical nitrosation. Appl. Environ. Microbiol. 1988;54:1791–1794. doi: 10.1128/aem.54.7.1791-1794.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J, Arif A, Terenzi F, Willard B, Plow EF, Hazen SL, Fox PL. Target- selective protein S-nitrosylation by sequence motif recognition. Cell. 2014;159:623–634. doi: 10.1016/j.cell.2014.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol WH. Nitrosation, thiols, and hemoglobin: energetics and kinetics. Inorg. Chem. 2012;51:5637–5641. doi: 10.1021/ic202561f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, Law L, Hester LD, Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 2010;12:1094–1100. doi: 10.1038/ncb2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster JR., Jr How are nitrosothiols formed de novo in vivo? Arch. Biochem. Biophys. 2017;617:137–144. doi: 10.1016/j.abb.2016.10.015. [DOI] [PubMed] [Google Scholar]
- Luchsinger BP, Rich EN, Gow AJ, Williams EM, Stamler JS, Singel DJ. Routes to S-nitroso-hemoglobin formation with heme redox and preferential reactivity in the beta subunits. Proc. Natl. Acad. Sci. U.S.A. 2003;100:461–466. doi: 10.1073/pnas.0233287100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macedo S, Mitchell EP, Romao CV, Cooper SJ, Coelho R, Liu MY, Xavier AV, LeGall J, Bailey S, Garner DC, et al. Hybrid cluster proteins (HCPs) from Desulfovibrio desulfuricans ATCC 27774 and Desulfovibrio vulgaris (Hildenborough): X-ray structures at 1.25 Å resolution using synchrotron radiation. J. Biol. Inorg. Chem. 2002;7:514–525. doi: 10.1007/s00775-001-0326-y. [DOI] [PubMed] [Google Scholar]
- Membrillo-Hernandez J, Coopamah MD, Anjum MF, Stevanin TM, Kelly A, Hughes MN, Poole RK. The flavohemoglobin of Escherichia coli confers resistance to a nitrosating agent, a "nitric oxide releaser," and paraquat and is essential for transcriptional responses to oxidative stress. J. Biol. Chem. 1999;274:748–754. doi: 10.1074/jbc.274.2.748. [DOI] [PubMed] [Google Scholar]
- Mitchell DA, Marletta MA. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat. Chem. Biol. 2005;1:154–158. doi: 10.1038/nchembio720. [DOI] [PubMed] [Google Scholar]
- Morreale FE, Walden H. SnapShot: Types of ubiquitin ligases. Cell. 2016;165:248–248.e1. doi: 10.1016/j.cell.2016.03.003. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Wang L, Wong CC, Scott FL, Eckelman BP, Han X, Tzitzilonis C, Meng F, Gu Z, Holland EA, et al. Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol. Cell. 2010;39:184–195. doi: 10.1016/j.molcel.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409:622–626. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- Puchulu-Campanella E, Chu HY, Anstee DJ, Galan JA, Tao WA, Low PS. Identification of the components of a glycolytic enzyme metabolon on the human red blood cell membrane. J. Biol. Chem. 2013;288:848–858. doi: 10.1074/jbc.M112.428573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralt D, Wishnok JS, Fitts R, Tannenbaum SR. Bacterial catalysis of nitrosation: involvement of the nar operon of Escherichia coli. J. Bacteriol. 1988;170:359–364. doi: 10.1128/jb.170.1.359-364.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson AR, Payne EC, Younger N, Karlinsey JE, Thomas VC, Becker LA, Navarre WW, Castor ME, Libby SJ, Fang FC. Multiple targets of nitric oxide in the tricarboxylic acid cycle of Salmonella enterica serovar typhimurium. Cell Host & Microbe. 2011;10:33–43. doi: 10.1016/j.chom.2011.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos V, Klemm P. Global gene expression profiling of the asymptomatic bacteriuria Escherichia coli strain 83972 in the human urinary tract. Infect. Immun. 2006;74:3565–3575. doi: 10.1128/IAI.01959-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seta FD, Boschi-Muller S, Vignais ML, Branlant G. Characterization of Escherichia coli strains with gapA and gapB genes deleted. J. Bacteriol. 1997;179:5218–5221. doi: 10.1128/jb.179.16.5218-5221.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth D, Hausladen A, Wang YJ, Stamler JS. Endogenous protein S-nitrosylation in E. coli: regulation by OxyR. Science. 2012;336:470–473. doi: 10.1126/science.1215643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth D, Stamler JS. The SNO-proteome: causation and classifications. Curr. Opin. Chem. Biol. 2011;15:129–136. doi: 10.1016/j.cbpa.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showe MK, DeMoss JA. Localization and regulation of synthesis of nitrate reductase in Escherichia coli. J. Bacteriol. 1968;95:1305–1313. doi: 10.1128/jb.95.4.1305-1313.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva CM, Cidlowski JA. Direct evidence for intramolecular and intermolecular disulfide bond formation in the human glucocorticoid receptor - Inhibition of DNA-binding and identification of a new receptor-associated protein. J. Biol. Chem. 1989;264:6638–6647. [PubMed] [Google Scholar]
- Stamler JS, Jaraki O, Osborne J, Simon DI, Keaney J, Vita J, Singel D, Valeri CR, Loscalzo J. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7674–7677. doi: 10.1073/pnas.89.16.7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovic S, Stanic D, Nikolic M, Spasic M, Niketic V. Iron catalyzed conversion of NO into nitrosonium (NO+) and nitroxyl (HNO/NO−) species. Nitric Oxide. 2004;11:256–262. doi: 10.1016/j.niox.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Stoyanovsky DA, Tyurina YY, Tyurin VA, Anand D, Mandavia DN, Gius D, Ivanova J, Pitt B, Billiar TR, Kagan VE. Thioredoxin and lipoic acid catalyze the denitrosation of low molecular weight and protein S-nitrosothiols. J. Am. Chem. Soc. 2005;127:15815–15823. doi: 10.1021/ja0529135. [DOI] [PubMed] [Google Scholar]
- Thomason LC, Costantino N, Court DL. E. coli genome manipulation by P1 transduction. In: Ausubel FM, editor. Curr. Protoc. Mol. Biol. Unit 1. Chapter 1. 2007. p. 17. [DOI] [PubMed] [Google Scholar]
- Unden G, Bongaerts J. Alternative respiratory pathways of Escherichia coli: energetics and transcriptional regulation in response to electron acceptors. Biochim. Biophys. Acta. 1997;1320:217–234. doi: 10.1016/s0005-2728(97)00034-0. [DOI] [PubMed] [Google Scholar]
- van den Berg WA, Hagen WR, van Dongen WM. The hybrid-cluster protein ('prismane protein') from Escherichia coli Characterization of the hybrid-cluster protein, redox properties of the [2Fe-2S] and [4Fe-2S-2O] clusters and identification of an associated NADH oxidoreductase containing FAD and [2Fe-2S] Eur. J. Biochem. 2000;267:666–676. doi: 10.1046/j.1432-1327.2000.01032.x. [DOI] [PubMed] [Google Scholar]
- Vanin AF, Malenkova IV, Serezhenkov VA. Iron catalyzes both decomposition and synthesis of S-nitrosothiols: Optical and electron paramagnetic resonance studies. Nitric Oxide: Biol. Ch. 1997;1:191–203. doi: 10.1006/niox.1997.0122. [DOI] [PubMed] [Google Scholar]
- Wang J, Vine CE, Balasiny BK, Rizk J, Bradley CL, Tinajero-Trejo M, Poole RK, Bergaust LL, Bakken LR, Cole JA. The roles of the hybrid cluster protein, Hcp and its reductase, Hcr, in high affinity nitric oxide reduction that protects anaerobic cultures of Escherichia coli against nitrosative stress. Mol. Microbiol. 2016;100:877–892. doi: 10.1111/mmi.13356. [DOI] [PubMed] [Google Scholar]
- Weichsel A, Maes EM, Andersen JF, Valenzuela JG, Shokhireva TK, Walker FA, Montfort WR. Heme-assisted S-nitrosation of a proximal thiolate in a nitric oxide transport protein. Proc. Natl. Acad. Sci. U.S.A. 2005;102:594–599. doi: 10.1073/pnas.0406549102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojdyla K, Williamson J, Roepstorff P, Rogowska-Wrzesinska A. The SNO/SOH TMT strategy for combinatorial analysis of reversible cysteine oxidations. J. Proteomics. 2015;113:415–434. doi: 10.1016/j.jprot.2014.10.015. [DOI] [PubMed] [Google Scholar]
- Wu CG, Parrott AM, Liu T, Jain MR, Yang YF, Sadoshima J, Li H. Distinction of thioredoxin transnitrosylation and denitrosylation target proteins by the ICAT quantitative approach. J. Proteomics. 2011;74:2498–2509. doi: 10.1016/j.jprot.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Freitas MA. A mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinformatics. 2007;8:133. doi: 10.1186/1471-2105-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LJ, Liu L, Forster MJ. Reversible inactivation of dihydrolipoamide dehydrogenase by Angeli's salt. Sheng Wu Wu Li Hsueh Bao. 2012;28:341–350. [PMC free article] [PubMed] [Google Scholar]
- Zaffagnini M, De Mia M, Morisse S, Di Giacinto N, Marchand CH, Maes A, Lemaire SD, Trost P. Protein S-nitrosylation in photosynthetic organisms: A comprehensive overview with future perspectives. Biochim. Biophys. Acta - Proteins Proteomics. 2016;1864:952–966. doi: 10.1016/j.bbapap.2016.02.006. [DOI] [PubMed] [Google Scholar]
- Zhang GF, Kombu RS, Kasumov T, Han Y, Sadhukhan S, Zhang J, Sayre LM, Ray D, Gibson KM, Anderson VA, et al. Catabolism of 4-hydroxyacids and 4-hydroxynonenal via 4-hydroxy-4-phosphoacyl-CoAs. J. Biol. Chem. 2009;284:33521–33534. doi: 10.1074/jbc.M109.055665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.