

Abstract
The multicomponent synthesis of a mini library of histone deacetylase inhibitors with imidazo[1,2-a]pyridine-based cap groups is presented. The biological evaluation led to the discovery of the hit compound MAIP-032 as a selective HDAC6 inhibitor with promising anticancer activity. The X-ray structure of catalytic domain 2 from Danio rerio HDAC6 complexed with MAIP-032 revealed a monodentate zinc-binding mode.
Graphical Abstract
The acetylation/deacetylation interplay is a key modification in human cells to control and regulate many important biological processes, such as gene transcription and protein functions.1 Histone deacetylases (HDACs) play an important role in these posttranslational modifications by cleaving acetyl groups from histone and non-histone proteins possessing N-acetyl-lysine residues.2 HDACs are also involved in many diseases such as cancer, neurodegenerative diseases, and inflammation, making these enzymes promising targets for therapeutic approaches.3 Classical HDAC inhibitors (HDACi) can be described by a widely accepted cap-linker-chelator pharmacophore model (Figure 1).3b To date, there are four FDA-approved anticancer HDACi (vorinostat, belinostat, romidepsin and panobinostat).3b These inhibitors target multiple HDAC isoforms, leading to alteration of the acetylation status of many different substrates, which can result in serious unwanted side effects.3a,b,e Therefore, it has been hypothesized that isoform-selective HDACi may enable a more precise regulation, and thus, offer decreased side effects.3a,e HDAC6 modulates the function of many non-histone proteins, including α-tubulin, Hsp90, cortactin and many more, thus participating in numerous diseases.4 For instance, HDAC6 is highly expressed in several cancer types such as oral squamous cell cancer, advanced stage breast cancer and acute myeloid leukemia, ensuring migration of cancer cells and angiogenesis.3a,4 The knock out of HDAC6 in mice produces a viable phenotype with no significant defects, which may indicate an improved safety profile for HDAC6 selective inhibitors.3a Furthermore, it has recently been reported that HDAC6 differs distinctly in its structural features compared to other isoforms, giving space for bulky rigid cap groups.5 In addition, the structural analysis suggests that HDAC6 uses HDAC6-specific loops for the recognition of HDAC6-selective substrates and inhibitors.5 Consequently, designing HDAC6i seems to be more approachable compared to other isoforms and several HDAC6 selective inhibitors have been developed (Figure 1).4,5 In this context, we report here on the rational design and synthesis of a series of preferential HDAC6 inhibitors containing imidazo[1,2-a]pyridine-based cap groups.
Figure 1.

Selected HDAC6 preferential inhibitors and the imidazo[1,2-a]pyridine-based target compounds.
Selective HDAC6 inhibitors typically possess a rigid (hetero)cyclic or branched cap group combined with a short benzyl or 4-aminophenyl linker (Figure 1).4,5 In this project, we rationally designed bicyclic imidazo[1,2-a]pyridine-based hydroxamic acids (Figure 1) and first investigated the potential of these new compounds to inhibit HDAC6 by molecular docking utilizing AutoDock4.2.6 Docking studies were performed in the active site of the catalytic domain 2 of human HDAC6 (PDB ID: 5EDU) using an aryl-substituted (4a), alkyl-substituted (4l) and unsubstituted (4i) ligand as representative examples (see Figure S1, Supporting Information (SI)). All three ligands were found to fit smoothly in the active site and we thus decided to synthesize a mini library of imidazo[1,2-a]pyridine-capped HDACi. The target compounds 4a–m are accessible by the Groebke-Blackburn-Bienaymé three-component reaction as the key step, followed by hydroxylaminolysis (Scheme 1).7 In addition to the widely commercially available aldehydes and 2-aminopyridines, the isocyanide 2 is required for the synthesis of our target compounds. Thus, we first synthesized isocyanide 2 in a two-step synthesis, starting with the formamidation of methyl 4-aminobenzoate, followed by dehydration (2, SI). Subsequently, we optimized the preparative conditions for the Groebke-Blackburn-Bienaymé three-component reaction using intermediate 3a (R1 = 4-Me2N-Ph; R2 = H) as model compound. We investigated different catalysts, solvents, temperatures and reaction times that are described in the literature to identify an efficient procedure for the synthesis of our target compounds.7 The best result was achieved by microwave irradiation (150 W) at 85 °C for 3 hours with acetic acid as catalyst and methanol as solvent with 52 % yield for the model compound 3a (see SI). Furthermore, we were able to synthesize the intermediate 3a using an isocyanide-less protocol (see SI, 39 % yield) adapting the recently published method by Dömling and co-workers.8 Due to the slightly higher yield and the efficient access to 2, we decided to use the classical isocyanide-based protocol for the synthesis of esters 3a–m (Scheme 1). For the synthesis of the 2-unsubstituted esters 3i–k, glyoxylic acid was used as a formaldehyde source.7c Pure compounds were obtained either by simple filtration of the crude reaction mixture, if crystallization of the products occurred, or by flash column chromatography in 33–73% yields. Although the possible formation of regioisomers during the Groebke-Blackburn-Bienaymé three-component reaction has been reported, the desired regioselective formation of imidazo[1,2-a]pyridines 3a–m was confirmed for selected compounds through 2D-NOESY-NMR (Figure S2, SI).7b The final step of the synthesis included the hydroxylaminolysis of the esters, which was accomplished in 43–90% yield (4a–m) using an excess of aqueous hydroxylamine solution in the presence of sodium hydroxide in methanol/dichloromethane (3:1).
Scheme 1.

Synthesis of imidazo[1,2-a]pyridine-based HDACi 4a–m.
All synthesized hydroxamic acids were evaluated in regards to their inhibitory activity in a biochemical assay against HDAC1 and HDAC6 using ZMAL (Z-(Ac)Lys-AMC)9 as a substrate, as previously published (Table 1).10 First, the HDAC isoform profiling revealed that the majority of compounds showed potent double-digit nanomolar activity against HDAC6. In particular, the nature of substituent R1 seems to be important for the selectivity profile. Aryl-substituted compounds revealed either moderate (4b–c; selectivity factor (SF) ≥ 10) or no noteworthy (4a,e–h; SF < 10) preference over HDAC1, depending on further substitutions on the phenyl residue. In particular, compounds with a 4-dimethylamino-substituted aryl ring tend to be non-selective inhibitors (see compounds 4a and 4e). Compounds 4i–k with no R1 substitution displayed moderate preference with selectivity factors of 11–15. Interestingly, compounds 4l and 4m, which bear an alkyl-group as R1 substituent, showed the highest preference over HDAC1. Compound 4l exhibited the best selectivity factor (SF = 38), comparable to other well-known preferential HDAC6 inhibitors such as HPOB (SF = 25). In order to evaluate its selectivity profile further, 4l was selected for a screening against the remaining class I isoforms HDAC2, HDAC3 and HDAC8. Strikingly, 4l was inactive against HDAC2 and HDAC3 (IC50 > 10 μM) and displayed only moderate activity against HDAC8 (IC50: 1.58±0.21 μM; SFHDAC8/HDAC6: 27).
Table 1.
Activities of 4a–m, vorinostat, HPOB and Tubastatin A against HDAC1 and HDAC6.
| entry | R1 | R2 | HDAC1 IC50 [μM] | HDAC6 IC50 [μM] | SFa |
|---|---|---|---|---|---|
| 4a |
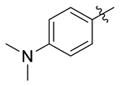
|
H | 0.13(±0.014) | 0.050(±0.00035) | 3 |
| 4b |
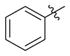
|
H | 0.89(±0.012) | 0.074(±0.0063) | 12 |
| 4c |
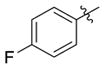
|
H | 0.51(±0.057) | 0.051(±0.0033) | 10 |
| 4d |
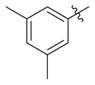
|
H | 0.66(±0.033) | 0.12(±0.036) | 6 |
| 4e |

|
H | 0.18(±0.018) | 0.14(±0.024) | <2 |
| 4f |
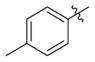
|
H | 0.17(±0.029) | 0.064(±0.0047) | 3 |
| 4g |
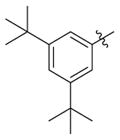
|
H | >3.333 | 1.33(±0.22) | >3 |
| 4h |
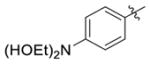
|
H | 0.27(±0.019) | 0.10(±0.0041) | 3 |
| 4i | H | H | 0.84(±0.062) | 0.076(±0.0036) | 11 |
| 4j | H | 7-Me | 1.01(±0.0035) | 0.069(±0.0039) | 15 |
| 4k | H | 6-Me | 0.75(±0.043) | 0.071(±0.0054) | 11 |
| 4l |
|
H | 2.20(±0.33) | 0.058(±0.012) | 38 |
| 4m |
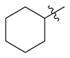
|
H | 1.31(±0.086) | 0.057(±0.0034) | 23 |
|
| |||||
| vorinostat | 0.10(±0.0058) | 0.030(±0.0040) | 3 | ||
| HPOB | 2.10(±0.23) | 0.085(±0.0090) | 25 | ||
| Tubastatin A | 2.49(±0.14) | 0.014(±0.00061) | 178 | ||
Selectivity factor (SF = IC50 (HDAC1)/IC50 (HDAC6)).
In addition, the preferential inhibition of HDAC6 was studied by western blotting experiments using the human tongue squamous cell carcinoma cell line Cal27 (Figure S3, SI). Compounds 4a and 4l were selected as representative compounds for aryl and alkyl (R1) substituted derivatives, whereas the pan-inhibitor vorinostat, as well as the HDAC6 selective HDACi nexturastat and tubastatin A were used as reference compounds. As expected, vorinostat induced an increase in acetylation of both α-tubulin and histone H3 compared to control, indicating the inhibition of HDAC6 and class I HDACs. In contrast, compounds 4a, 4l, nexturastat and tubastatin A showed preferential hyperacetylation of α-tubulin. Notably, compound 4l only induced the acetylation of α-tubulin but not of histone H3, demonstrating that its selective HDAC6 inhibition is retained in a cellular environment.
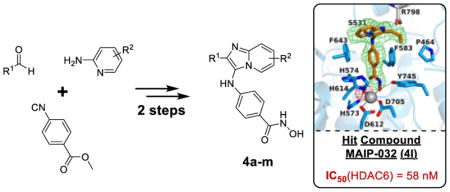
To investigate the binding mode of 4l, co-crystallization experiments were performed with the CD2 domain of Danio rerio HDAC6 (henceforth, simply “HDAC6”). The 2.50 Å-resolution crystal structure of the HDAC6-4l complex (Figure 2) reveals no major conformational changes between the inhibitor-bound and unliganded states of the enzyme, and the root-mean square deviation is 0.20 Å for 311 Cα atoms between the two structures (unliganded HDAC6, PDB accession code 5EEM) (Figure S4, SI). The catalytic Zn2+ ion exhibits square pyramidal coordination geometry, with D612, D705, H2O, and the ionized hydroxamate N-O− group serving as equatorial ligands and H614 serving as an apical ligand (coordination distances range 2.0–2.4 Å). Several hydrogen bond interactions stabilize the bound inhibitor: the Zn2+-bound hydroxamate N-O− group accepts a hydrogen bond from Y745, the Zn2+-bound water molecule forms hydrogen bonds with H573 and H574, and the hydroxamate carbonyl group accepts a hydrogen bond from the Zn2+-bound water molecule. Interestingly, the monodentate hydroxamate-Zn2+ coordination mode observed for 4l is also observed for other sterically bulky inhibitors such as HPOB, HPB, and ACY-1083.5a,5c The monodentate hydroxamate-Zn2+ coordination mode is only 0.5 kcal/mol less stable than the canonical bidentate hydroxamate-Zn2+ coordination mode observed for inhibitors with less steric bulk adjacent to the hydroxamate moiety, such as rico-linostat.5c The aromatic ring of the phenylhydroxamate is nestled in an aromatic crevice formed by F583 and F643. The para-substituted secondary amino group of 4l forms a hydrogen bond with S531 on the L2 loop (N—O separation = 3.1 Å).
Figure 2.

Polder omit maps (stereoview, contoured at 3.0 σ each) for 4l (green) and the Zn2+-bound water molecule (magenta) in the active site of HDAC6. Atom color codes are as follows: C = orange (4l) or light blue (protein), N = blue, O = red; Zn2+ appears as a grey sphere, and the Zn2+-bound water molecule is shown as a small red sphere. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively.
S531 plays an important role in substrate binding to HDAC6 by accepting a hydrogen bond from the backbone NH group of substrate acetyl-L-lysine.5a Since S531 is unique to HDAC6, a hydrogen bond with this residue will contribute to HDAC6 inhibitor selectivity. The pendant propyl substituent on the capping group of 4l is oriented toward P464, and the bicyclic aromatic ring of the capping group is accordingly oriented in the opposite direction toward F643 (Figure 2). Interestingly, aromatic capping groups of many hydroxamate inhibitors are observed to bind adjacent to P464.5a,5c However, the region occupied by the bicyclic aromatic ring of 4l can be exploited further in the design of HDAC6-selective inhibitors, due to the region being occupied by HDAC6 bound substrates. We note that the imidazo nitrogen atom in the heteroaromatic ring of the capping group is presumably protonated, since it is within hydrogen bonding distance to the C-terminal carboxylate of R798 from another HDAC6 molecule in the crystal lattice (Figure 2). Although this interaction is poorly oriented for a hydrogen bond, it could still comprise an electrostatic interaction. Since this interaction could conceivably occur regardless of the orientation of the capping group of 4l, this interaction is unlikely to significantly influence the binding conformation of 4l.
To explore the biological activity on a cellular level, all compounds were tested for antiplasmodial as well as anti-cancer activity (Table S1, SI and Table 2). Besides their promising potency, antiplasmodial HDACi are often disadvantageous as a result of their toxicity against human cells. Therefore, we, and others, hypothesized that selective human HDAC6 inhibitors might be a better starting point for the development of parasite-selective antiplasmodial HDACi due to a usually lower toxicity.10,11 Compared to the reference HDACi vorinostat (Pf3D7 IC50: 0.209 μM; PfDd2 IC50: 0.297 μM), the majority of compounds revealed only moderate activity against the drug sensitive 3D7 (Pf3D7 IC50: 0.9–1.9 μM) and the multi-drug resistant Dd2 (PfDd2 IC50: 1.4–6.8 μM) strain. Nonetheless, compounds 4e (0.524 μM) and 4m (0.517 μM) showed promising submicromolar antiplasmodial activity against the 3D7 strain.
Table 2.
Whole-cell HDAC inhibition (HDAC) and cytotoxic activity (MTT) against the human tongue squamous cell carcinoma cell line Cal27 of 4a–m, vorinostat, Nexturastat A and Tubastatin A.
| entry | R1 | R2 | HDAC (Cal27) IC50 [μM] | MTT (Cal27) IC50 [μM] |
|---|---|---|---|---|
| 4a |
|
H | 14.0(±1.40) | 3.64(±1.24) |
| 4b |
|
H | 14.8(±3.14) | 4.78(±1.70) |
| 4c |
|
H | 13.9(±2.93) | 4.53(±1.93) |
| 4d |
|
H | 14.0(±1.41) | 11.9(±2.72) |
| 4e |
|
H | 1.75(±0.27) | 3.22(±0.87) |
| 4f |
|
H | 14.8(±3.13) | 4.60(±1.60) |
| 4g |
|
H | >100 | 27.7(±8.23) |
| 4h |
|
H | >100 | >100 |
| 4i | H | H | 13.9(±2.93) | 4.61(±1.45) |
| 4j | H | 7-Me | 8.13(±1.08) | 3.92(±1.20) |
| 4k | H | 6-Me | 17.5(±1.85) | 4.21(±1.95) |
| 4l |
|
H | 27.6(±4.68) | 3.87(±1.15) |
| 4m |
|
H | 9.38(±3.03) | 3.83(±1.40) |
|
| ||||
| vorinostat | 0.87(±0.10) | 2.64 (±0.10)a | ||
| Nexturastat A | 5.87(±1.70) | 7.75(±1.56) | ||
| Tubastatin A | 11.4(±2.16) | 5.34(±0.74) | ||
Next, all synthesized HDACi 4a–m were assessed in a whole cell HDAC assay using the cell line Cal27 and the class I/IIb selective substrate Boc-Lys(Ac)-AMC (Table 2). All compounds, except 4g and 4h, exhibited HDAC inhibition in the whole cell assay, thus confirming the inhibition of cellular histone deacetylase activity. In agreement with the cellular HDAC data, all compounds, except 4g and 4h, showed encouraging cytotoxicity in a MTT cytotoxicity assay against Cal27 cells with IC50 values ranging from 3.22 to 11.9 μM (Table 2). The highest anticancer activity was observed for compounds 4a, 4e, 4j, 4l and 4m (IC50: 3–4 μM). Flow cytometric analysis using propidium iodide (PI) staining showed that the cytotoxic effect was mediated by induction of apoptosis (Fig. S5, SI). Compounds 4a and 4l were chosen as representative HDACi. Both compounds increased the amount of apoptotic nuclei in a concentration-dependent manner after 48h incubation. Notably, the apoptotic effect was more pronounced for the alkyl substituted HDACi 4l, even at 1 μM, which is approx. 4-fold below IC50 of the MTT assay.
In conclusion, we have developed a multicomponent approach for the synthesis of a minilibrary of novel imidazo[1,2-a]pyridine-based HDAC6i. Most notably, we show that 4l, hereafter named MAIP-032, is a selective HDAC6 inhibitor with promising anticancer activity. In addition, the crystal structure of MAIP-032 bound to the second catalytic domain of zebrafish HDAC6 demonstrates a monodentate binding mode. Taken together, the results suggest that MAIP-032 is a promising candidate for further development of selective HDAC6 inhibitors with anticancer properties.
Supplementary Material
Acknowledgments
We thank Nicholas Porter of the University of Pennsylvania for helpful scientific discussions. Additionally, we thank Tzanko Doukov, Ana Gonzalez, and the beamline 12-2 staff at the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, for assistance with data collection. SLAC is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (grant P41GM103393). This research was supported by NIH Grant GM49758 to D.W.C. and by the Deutsche Forschungsgemeinschaft (DFG) (HA 7783/1-1 to FKH and HE 7607/1-1 to JH).
Footnotes
Accession Code
The atomic coordinates and crystallographic structure factors of the HDAC6-4l complex have been deposited in the Protein Data Bank (www.rcsb.org) with accession code 6CGP.
Notes
There are no conflicts to declare.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1039/x0xx00000x.
Experimental details (synthetic protocols, bioassays, X-Ray crystallography) characterization data of new compounds, and copies of NMR spectra (PDF).
References
- 1.Drazic A, Myklebust LM, Ree R, Arnesen T. Biochim Biophys Acta. 2016;1864:1372. doi: 10.1016/j.bbapap.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Biel M, Wascholowski V, Giannis A. Angew Chem, Int Ed. 2005;44:3186. doi: 10.1002/anie.200461346. [DOI] [PubMed] [Google Scholar]
- 3.(a) Witt O, Deubzer HE, Milde T, Oehme I. Cancer Lett. 2009;277:8. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]; (b) Mottomal M, Zheng S, Huang TL, Wang G. Molecules. 2015;20:3898. doi: 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Didonna A, Opal P. Ann Clin Transl Neurol. 2015;2:79. doi: 10.1002/acn3.147. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Villagra A, Sotomayor EM, Seto E. Oncogene. 2010;29:157. doi: 10.1038/onc.2009.334. [DOI] [PubMed] [Google Scholar]; (e) Su H, Altucci L, You Q. Mol Cancer Ther. 2008;5:1007. doi: 10.1158/1535-7163.MCT-07-2289. [DOI] [PubMed] [Google Scholar]
- 4.(a) Lin X, Chen W, Qiu Z, Guo L, Zhu W, Li W, Wang Z, Zhang W, Zhang Z, Rong Y, Zhang M, Yu L, Zhong S, Zhao R, Wu X, Wong JC, Tang G. J Med Chem. 2015;58:2809. doi: 10.1021/jm502011f. [DOI] [PubMed] [Google Scholar]; (b) Diedrich D, Hamacher A, Gertzen CGW, Alves Avelar LA, Reiss GJ, Kurz T, Gohlke H, Kassack MU, Hansen FK. Chem Commun. 2016;52:3219. doi: 10.1039/c5cc10301k. [DOI] [PubMed] [Google Scholar]; (c) Shen S, Benoy V, Bergman JA, Kalin JH, Frojuello M, Vistoli G, Haeck W, Van den Bosch L, Kozikowski AP. ACS Chem Neurosci. 2016;7:240. doi: 10.1021/acschemneuro.5b00286. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Simões-Pires CA, Bertrand P, Cuendet M. Expert Opin Ther Patents. 2017;27:229. doi: 10.1080/13543776.2017.1282945. [DOI] [PubMed] [Google Scholar]
- 5.(a) Hai Y, Christianson DW. Nat Chem Biol. 2016;12:741. doi: 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Miyake Y, Keusch JJ, Wang L, Saito M, Hess D, Wang X, Melancon BJ, Helquist P, Gut H, Matthias P. Nat Chem Bio. 2016;12:748. doi: 10.1038/nchembio.2140. [DOI] [PubMed] [Google Scholar]; (c) Porter NJ, Mahendran A, Breslow R, Christianson DW. Proc Natl Acad Sci USA. 2017;114:13459. doi: 10.1073/pnas.1718823114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. J Computational Chemistry. 2009;16:2785. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Shaaban S, Abdel-Wahab BF. Mol Divers. 2015;20:233. doi: 10.1007/s11030-015-9602-6. [DOI] [PubMed] [Google Scholar]; (b) Buscató E, Wisniewska JM, Rödl CB, Brüggerhoff A, Kaiser A, Rörsch F, Kostewicz E, Wurglics M, Schubert-Zsilavecz M, Grösch S, Steinhilber D, Hofmann B, Proschak E. Future Med Chem. 2013;5:865. doi: 10.4155/fmc.13.72. [DOI] [PubMed] [Google Scholar]; (c) Sharma A, Li H. Synlett. 2011;10:1407. [Google Scholar]
- 8.Neochoritis CG, Stotani S, Mishra B, Dömling A. Org Lett. 2015;17:2002. doi: 10.1021/acs.orglett.5b00759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heltweg B, Dequiedt F, Verdin E, Jung M. Anal Biochem. 2003;319:2003. doi: 10.1016/s0003-2697(03)00276-8. [DOI] [PubMed] [Google Scholar]
- 10.Stenzel K, Chua MJ, Duffy S, Antonova-Koch Y, Meister S, Hamacher A, Kassack MU, Winzeler E, Avery VM, Kurz T, Andrews KT, Hansen FK. ChemMedChem. 2017;12:1627. doi: 10.1002/cmdc.201700360. [DOI] [PubMed] [Google Scholar]
- 11.De Vreese R, de Kock D, Smith PJ, Chibale K, D’hooghe M. Fut Med Chem. 2017;9:357. doi: 10.4155/fmc-2016-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.