

Abstract
Background
Phenotypic modulation or switching of Vascular Smooth Muscle Cells (VSMC) from contractile/quiescent to a proliferative/synthetic phenotype plays a key role in vascular proliferative disorders, such as atherosclerosis and restenosis. Although several calcium handling proteins that control differentiation of SMCs have been identified, the role of protein phosphatase inhibitor 1 (I-1) in the acquisition and/or maintenance of the contractile phenotype modulation remain unknown.
Methods and Results
In human coronary arteries, I-1 and Sarco/endoplasmic Reticulum Ca2+-ATPase (SERCA2a) expression is specific to contractile VSMCs. In synthetic cultured human coronary artery SMC (hCASMCs), protein phosphatase inhibitor 1 (PP1; I-1 target) is highly expressed, leading to a decrease in Phospholamban (PLB) phosphorylation, SERCA2 and cAMP Responsive Element Binding (CREB) activity. I-1 knock-out mice lack PLB phosphorylation and exhibit VSMC arrest in the synthetic state with excessive neointimal proliferation following carotid injury, and significant modifications of contractile properties and relaxant response to acetylcholine (ACh) of femoral artery in vivo. Constitutively active I-1 (I-1c) gene transfer decreased neointimal formation in an angioplasty rat model by preventing VSMC contractile to synthetic phenotype change
Conclusions
I-1 and SERCA2a synergistically induce the VSMC contractile phenotype. Gene transfer of I-1c is a promising therapeutic strategy for preventing vascular proliferative disorders.
Keywords: Vascular disease, remodeling, muscle, Smooth, calcium, gene therapy
Introduction
Vascular proliferative disorders, such as primary atherosclerosis, restenosis after balloon angioplasty, vein-graft disease, and coronary arteriosclerosis are the most common causes of severe cardiovascular diseases, the current leading cause of death in the United States and the predicted number one killer worldwide by 20201. Although vascular smooth muscle cells (VSMCs) are normally located in the arterial media and maintained in a contractile non-proliferative state in vivo, injury or mechanical stress of arteries causes migration of VSMCs into the intima layer of the arterial wall, where the VSMCs switch their phenotype and start to proliferate and synthesize extracellular matrix proteins, resulting in expansion of the arterial intima2–5.
VSMC phenotype e switch from a contractile/quiescent to a proliferative/synthetic one is associated with alterations of many components of the VSMC Ca2+ signaling network6. These changes in VSMC phenotype have been well characterized at the level of contractile proteins7, and more recently at the level of Ca2+ handling proteins8. Indeed, expression levels of the L-type Ca2+ channel, sarco/endo plasmic reticulum (SR) Ca2+-ATPase (SERCA2a) and ryanodine receptor are decreased in synthetic VSMCs8–12. Importantly, a sustained increase in cytosolic Ca2+ is necessary to activate calcineurin, a Ca2+/calmodulin-dependent serine/threonine-specific protein phosphatase 2B (PP2B), which dephosphorylates many proteins including NFAT that may translocate to the nucleus13, and induce VSMC proliferation14.
Recently, we demonstrated that Ca2+ cycling in contractile VSMCs requires the expression of the SERCA2a isoform, whereas the Ca2+ cycling in synthetic VSMCs is only associated with the expression of the ubiquitous isoform SERCA2b15. Phospholamban (PLB), a negative regulator of SERCA2 activity, is inhibited by protein kinase A (PKA) phosphorylation and activated by protein phosphatase 1 (PP1)-dependent dephosphorylation16. However, inhibitor-1 (I-1), a ubiquitously expressed 28 kDa protein, is a highly specific inhibitor of PP1, that enhances both PKA-dependent PLB phosphorylation and therefore SERCA2 activity17–21. I-1 is a major player in multiple neurohormonal pathways associated with Ca2+ homeostasis and contractile function. Upon stimulation, PKA phosphorylates Thr35 in I-1, resulting in PP1 inhibition and amplification of the contractile response17, 21, 22. Inactivation of I-1 occurs by dephosphorylation of Thr35 by PP2A and PP2B, leading to the reversal of PP1 inhibition and restoration of basal function23, 24. I-1 can also be phosphorylated at Ser67 and Thr75 by PKC, but these phosphorylations enhance PP1 activity and diminish contractility25–27. In VSMCs, regulation of phosphoprotein phosphatases has been shown to be a major component in the Ca2+ sensitization of contraction28. Of relevance, I-1 is present in VSMCs29, however, studies of its role in modulating VSMC function have provided conflicting results20, 30. In light of the high expression of I-1 in these cells, other functions for I-1 in VSMCs are a strong possibility.
The goal of this study was to elucidate the role of PKA signaling enhancer I-1 in the control of VSMC Ca2+ cycling and phenotypic modulation. Herein, we demonstrate that I-1 and SERCA2a act synergistically on Ca2+ cycling and contribute directly to the VSMC contractile phenotype. Gene transfer of constitutively active I-1 (I-1c) may be a promising therapeutic strategy for preventing vascular proliferative disorders.
Methods
Please refer to the expanded Methods section in the online-only Data Supplement for a detailed description.
Human samples
Fragments of the left anterior descending coronary artery were dissected from human explanted hearts and unused segments of left internal mammary artery from 5 patients were obtained during coronary artery bypass from the Surgical Department of the Cardiology Institute at Pitié-Salpêtrière Hospital, Paris, France, in accordance with French bioethical laws (L.1211-3-9). The artery segments were immediately immersed in physiological saline solution, placed at 4°C and processed within 2 – 3 hours.
Animals
Genetically modified I-1−/− mice31 were used. C57 Bl-6 mice (Charles River) of the same age were used as a non-littermate control. Adult male Sprague-Dawley rats (Charles River) were used for the in vivo carotid injury model and for isolation of aortic VSMCs. Animals were treated according to institutional guidelines.
Measurement of I-I KO femoral artery contraction and relaxation
Mice were anesthetized with mixture of Ketamine (65mg/Kg) + Xylazine (13 mg/Kg, IP). The left femoral artery was dissected gently and a 7–0 nylon suture (Ethicon, J&J) was placed under the vessel to serve as reference to calculate vessel diameter. The vessel was equilibrated by adding 100 μl of phosphate buffered saline (PBS) to the perivascular site for 5 min. Prostaglandin F (2alpha) (PGF2α, 1 mM, 100 μl, Sigma P0314) was locally applied on the vessel to contract the artery. After 10 min, the PGF2α solution was removed and 100 μl of acetylcholine (Ach, 10−9 to 10−3 M, Sigma: A6625-25G) was added cumulatively to the vessel with 5 min interval. The change of vessel diameter was recorded using a pixel link camera and measurements were performed using Image-J.
Measurement of intracellular free calcium concentration ([Ca2+]i)
Cells were loaded with 2μM Fura-2-AM for 45 min at 37°C and kept in serum free medium for 30 min prior to experimentation. HEPES buffer (in mmol/L: 116 NaCl, 5.6 KCl, 1.2 MgCl2, 5 NaHCO3, 1 NaH2PO4, 20 HEPES pH 7.4) was used for the experiments. Single images of fluorescent emission at 510 nm under excitation at 340 and 380 nm were taken every 5 sec. Changes in [Ca2+]i in response to the indicated agonist were calculated using the Fura-2 340/380 fluorescence ratio according to the equation of Grynkeiwicz or using the ratio 380em/380em (basal). For each recorded images, 3 distinct ROI (Region of Interest) with no cells were selected and mean of fluorescence measured for these 3 areas were used as a background and subtraction was applied for each cell measurement before any further calculation.
Statistical analysis
All quantitative data are presented as mean of at least 3 independent experiments ± standard error of the mean (SEM). Data were analyzed using Kruskal–Wallis test with Dunn’s adjustment for multiple comparisons. Statistical comparisons of 2 groups were analyzed by Wilcoxon matched-pairs signed rank test or Mann Whitney nonparametric test. Linear mixed-effect models with random intercepts were used to analyze the femoral artery contraction and relaxation percent change. Differences were considered significant when P<0.05. Data were analyzed by using GraphPad Prism 5 software and IBM SPSS Statistics software.
Results
Regulation of the I-1-PP1-PLB-SERCA signaling axis in contractile vs. synthetic human VSMCs
Since VSMCs have the capacity for phenotypic dedifferentiation from a contractile to synthetic phenotype, we assessed whether the I-1/PP1 signaling pathway was modified during this process. First, we analyzed I-1 expression in contractile and synthetic VSMCs from healthy segments of human coronary arteries (hCAs) obtained from 5 patients with dilated (non-ischemic) cardiomyopathy. hCAs contain a subendothelial layer of synthetic VSMCs and connective tissue to support the endothelium, while the medial layer is mainly composed of contractile VSMCs15. I-1 protein was expressed only in the medial layers of hCAs (Figure 1A and Supplementary Figure 1A) and mammary arteries (Supplementary Figure 1B) and was down-regulated in the subendothelial layer (Figure 1A) and in the media of atherosclerotic vessels (Supplementray Figure 1C), suggesting I-1 is specifically highly expressed in contractile VSMCs15. Immunoblot analysis performed on VSMCs of the medial layer of hCAs (contractile cells) and cultured hCASMCs (synthetic) confirmed a higher expression of I-1 in the contractile cells compared to hCASMCs (Figure 1B and 1C), whereas its target, total PP1 was up-regulated and highly detectable by immunofluorescence in synthetic hCASMC (Figure 1B and 1D and Supplementary Figure 2A). Next, we looked to PP1 isoforms expression in synthetic vs contractile VSMCs by co-immunostaining with SMA. PP1α was detected only in the nucleus, however PP1β was abundantly expressed in synthetic VSMCs with higher level in the nucleus and in the cytosol compared to the contractile ones (Supplementary Figure 2B and 2C, respectively). Consequently, PLB phosphorylation (pPLB) was higher in contractile VSMCs as compared to synthetic VSMCs (Figure 1B and 1E), without change in total PLB expression (Figure 1B and 1F). Confirming previous observations, SERCA2a was only expressed in contractile VSMCs15, whereas SERCA2b expression was similar between contractile and synthetic VSMCs (Figure 1B and 1G). In agreement with this pattern of activity in the I-1/PP1/PLB/SERCA signaling pathway, global SR Ca2+-ATPase activity was significantly decreased in synthetic VSMCs compared to contractile ones (Figure 1H). The decrease of SR Ca2+-ATPase activity in synthetic VSMCs can be linked to the lack of SERCA2a expression, and to the inhibitory effect of unphosphorylated PLB on SR Ca2+-ATPase. In addition, the expression of other markers of quiescent status, such as CREB (Figure 1B and 1I), p53 (tumor suppressor gene) and p21 were also markedly reduced in synthetic SMCs (Figure 1B).
Figure 1.
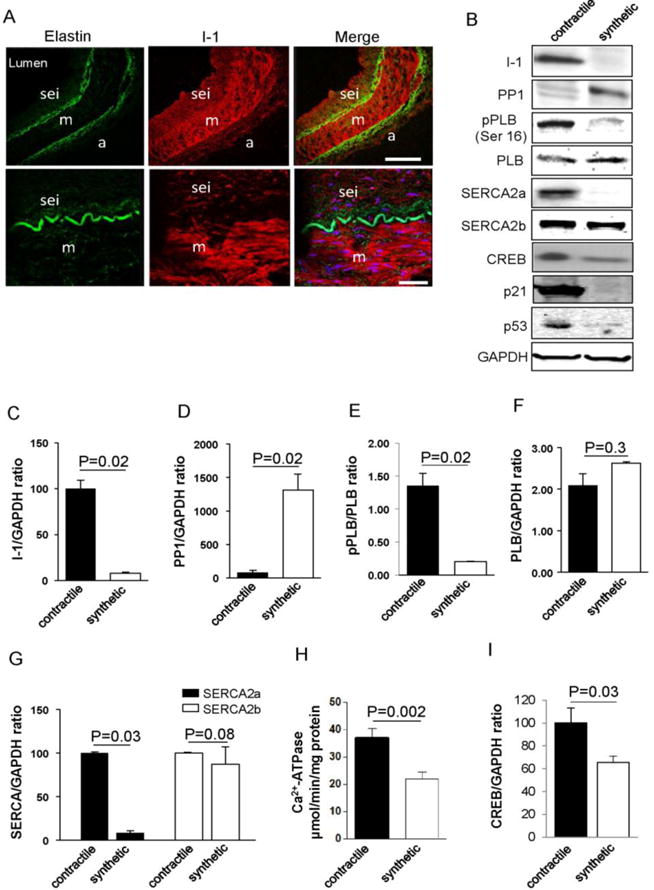
Modulation of I-1/PP1/PLB/SERCA signaling pathway within contractile and synthetic hCASMC. (A) Confocal immunofluorescence microscopy of healthy human coronary artery (hCA) cross sections showing I-1 (red) and elastin autofluorescence (green); a (adventitia), m (media), sei (subendothelial neointima). Bar scale = 20μm and 50μm. (B). Immunoblot analysis of indicated proteins in contractile hCAs and synthetic (cultured) hCASMCs. I-1 (Inhibitor 1); PP1 (Protein Phosphatase 1); PLB (phospholamban); pPLB (PLB phosphorylated on Ser16); SERCA2a or SERCA2b (Sarco/Endoplasmic Reticulum Calcium ATPase 2a or 2b); CREB (cAMP responsive element binding), and (C–I) Histograms showing the relative ratio of I-1, PP1,SERCA2a and SERCA2b, and CREB expression normalized to GAPDH in three independent experiments. (E) Relative pPLB level was determined through normalization to total PLB level. (H) Sarco/endoplasmic reticulum Ca2+-ATPase activity measured in contractile and synthetic (cultured) VSMCs.
Loss of I-1 leads to a shift from contractile to synthetic VSMC phenotype
To study the impact of the loss of I-1 on the VSMC phenotype, thoracic aortas of adult (6 month old) I-1−/− mice (I-1 KO) and control mice (WT) were analyzed by immunoblot with specific markers. Immunoblot analysis and immunostaining in aortic cross sections confirmed the loss of I-1 expression in the thoracic aortas from I-1 KO compared to WT mice (Figure 2A–C). Interestingly, PP1 expression was markedly increased in I-1 KO compared to WT mice (Figure 2A and 2D), which was consistent with reduced PLB phosphorylation (Figure 2A and 2E). SERCA2a protein expression was also significantly decreased in I-I KO aortic VSMCs (Figure 2A and 2F), whereas SERCA2b level remained unchanged (Figure 2A and 2G). A marked decrease of SR Ca2+-ATPase activity in I-1 KO mice thoracic aorta compared to WT mice was observed (Figure 2H), recapitulating the observations in human samples (Figure 1).
Figure 2.
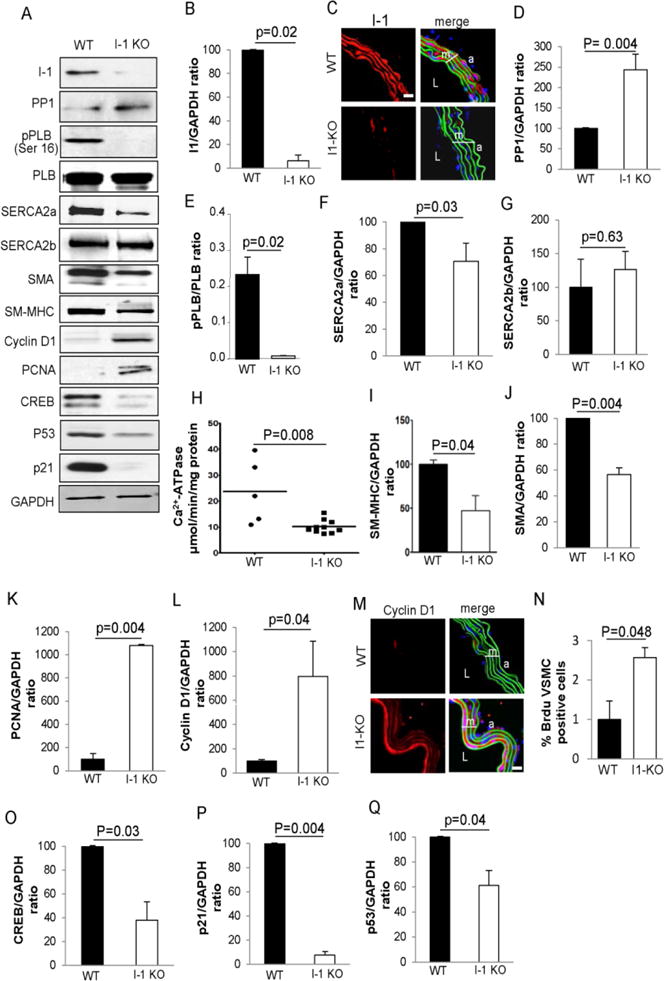
Phenotype analysis and SR Ca2+-ATPase activity of VSMC from thoracic aortas of WT and I-1 KO mice. (A) Immunoblotblot analysis of the indicated proteins in the media of thoracic aortas from adult (6 month old) WT and I-1 KO mice. Three aortas were used for preparation of each sample. (B, D, F–L, O–Q) Histograms showing the relative ratio of I-1, PP1, SERCA2a, SERCA2b, SM α-actin (SMA), SM-MHC, PCNA, Cyclin D1, CREB, p53, and p21 expression normalized to GAPDH in three independent experiments. (E) Relative pPLB levels were determined by normalization to total PLB. Data represent mean ± SEM of three independent experiments. (H) Sarco/endoplasmic reticulum Ca2+-ATPase activity measured on thoracic aortas from WT (n = 5) and I-1 KO (n = 10) mice. (C, M) Immunostaning of I-1 and Cyclin D1 in the media of thoracic aortas from WT and I-1 KO mice, respectively. Scale bar = 20 μm. m (media), L (Lumen), a (adventitia). (N) Quantification of proliferating aorta VSMC BrdU positive cells from adult 6 months old WT (n = 3) and I-1 KO mice (n = 4). Data represent mean ± SEM.
Next, we analyzed the expression of markers of differentiated SMCs. We found that by immunoblot and immunostaining analysis, smooth muscle myosin heavy chain (SM-MHC) and smooth muscle α-actin (SMA), markers of contractile VSMCs, were highly expressed in WT aortic VSMCs, but significantly decreased in I-1 KO aortic VSMCs (Figure 2A, 2I and 2J, respectively) and (Supplementary Figure 3A and 3C, respectively). In I-1 KO aortic VSMCs, the few I-1 positive cells (green) were SM-MHC-positive cells (red) (Supplementary Figure 3B). Calponin was also decreased in the medial I-1 KO aortic VSMCs (Supplementary Figure 3D). Markers of proliferating cells, such as PCNA (Proliferating Cell Nuclear Antigen), were absent in the aortic VSMC of WT mice, in agreement with their quiescent status compared to I-1 KO mice (Figure 2A and 2K). Also, Cyclin D1 was significantly increased in I-1 KO mice aortic VSMC (Figure 2A and 2L). Immunolabelling performed on thoracic aortic cross sections confirmed the presence of abundant Cyclin D1 expressing proliferating cells on the luminal vessel border (Figure 2M), with an increase of NFATC3 expression compared to WT (Supplementary Figure 3E) and significantly higher proportion of BrdU positive VSMCs in I-1 KO compared to WT mice (Figure 2N). Furthermore, expression levels of CREB, p53 and p21 were significantly decreased in the aortic VSMCs of I-1 KO mice (Figure 2A and 2O–Q, respectively). These data likely indicate that the downregulation of these key modulators of SMC contraction in the aorta of I-1 KO mice reflects the shift between contractile and synthetic phenotypes and suggests that the loss of I-1 affects the differentiation of VSMCs and their ability to adjust the phenotype to the physiologically required contractile state. Apoptosis was also evaluated in aortic cross sections from I-1 KO and WT aortas using TUNEL. Numerous apoptotic cells were detected in the media of I-1 KO aortas compared to WT (Supplementary Figure 3F).
Moreover, morphometric analysis revealed a marked increase of thoracic aortic thickness in I-1 KO mice (Figure 3A and 3B). The phenotype observed in the vascular cells of I-1 KO mice, characterized by anarchic proliferation and apoptosis, can be described as a vascular proliferative disorder. We further investigated the proliferative capacities of VSMCs lacking I-1. To achieve this, left carotid artery injury with and without I-1c gene transfer was performed in adult (12 weeks old) WT and I-1 KO mice. The right carotid from WT was used as sham-operated (no wire injury). One month after surgery, histological analysis revealed the presence of neointimal lesions in injured arteries of both mouse strains. Of note, the medial thickness of the carotid artery was already increased in non-injured carotids from I-1 KO mice (Figure 3C and 3D), confirming the observations made from thoracic aortas (Figure 3B). As expected, carotid injury induced medial and neointimal proliferation in both strains (Figure 3D and 3E), but the thickness was significantly greater in I-1 KO animals (Figure 3E). Overexpression of a truncated, constitutively active form of I-1 by an adenovirus (Ad. I-1c) by gene transfer markedly reduced the neointimal thickness in the injured WT carotid artery compared to WT non-transduced animals (Figure 3D and 3E). However, I-1c infected I-1 KO did not significantly reduce post-injury neointimal proliferation, nor did it reduce medial cell proliferation when compared to I-1 KO injured non-infected mice (Figure 3D and 3E), probably as a consequence of VSMC dedifferentiation. Analysis by immunofluorescence confirmed that in injured left carotids transduced with I-1c showed downregulation of markers of the contractile VSMCs phenotype such as SM-MHC and SERCA2a in I-1 KO mice compared to sham non-injured and WT infected carotid after injury (Supplementary Figure 4). These data demonstrate that VSMCs lacking I-1 proliferate readily and appear unable to adopt a quiescent state. Consistent with this, adult I-1 deficient mice develop a vascular proliferative disorder with excessive neointimal proliferation after vascular injury.
Figure 3.
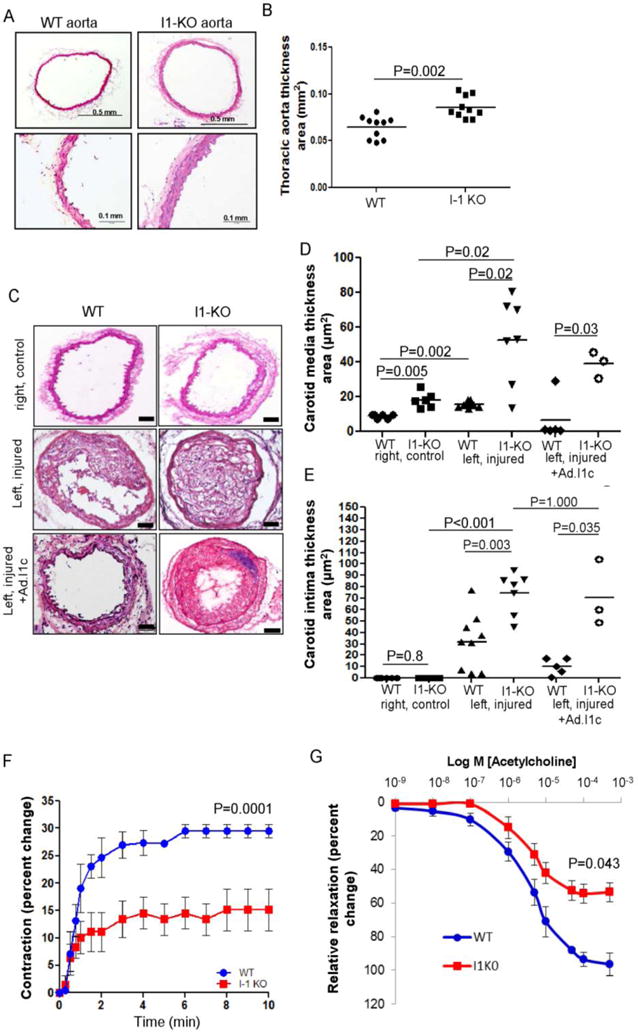
Enhanced proliferative capacity of aortic VSMCs from I-1 KO mice. (A) Bright-field photomicrograph of vascular hyperplasia in the aorta of I-1 KO compared to WT mice. (B) Quantification of thoracic aortic thickness in WT (n = 10) and I-1 KO mice (n = 10). (C) Bright-field photomicrograph of cross sections harvested one month post-injury and stained with hematoxylin and eosin from non-injured control arteries (sham group) of WT (n=6), I-1 KO (n=6) and non-infected left injured carotid of WT (n = 9) and I-1 KO (n = 7) and infected with Ad.I-1c; WT (n=5) and I-1 KO (n=3). Scale bar = 50 μm. (D, E) Dot plots showing morphometric measurements of media and intima thickness area respectively in control non-injured, injured non transduced and injured-infected I-1c carotid arteries. (F) Time-response curve for prostaglandin F2α (1μM) and (G) dose-response curve for Acetylcholine-mediated relaxation of femoral arteries from WT (blue, n=3) and I-1 KO (red, n=5). Percent changes in vessel diameter were calculated by the difference from baseline. For relaxation, baseline was the pre-contraction state after administration of prostaglandin F2α (1μM). Values represent mean ± SEM. The maximum strength of contraction and relaxation was significantly lower in I-1 KO.
To assess the effects of I-1 gene ablation on endothelium-dependent contraction, mouse femoral arteries were stimulated with 1 μmol/L of prostaglandin (PGF2α) and measurement of outer vessel diameter were performed at different time points and compared to baseline (Figure 3F). PGF2α, which is known to induce endothelium-dependent contractions32, resulted in significant modifications of contractile properties of femoral arteries of I-KO mice compared to WT (Figure 3F). Furthermore, analysis of the dose-response relationships to acetylcholine indicated a decrease in the strength of the response of the I-1.KO femoral arteries to endothelium-dependent relaxation at higher concentrations compared to WT mice (Figure 3G).
The effect of I-1 on calcium signaling in VSMCs is SERCA2 isoform-dependent
To determine whether I-1 dependent PLB phosphorylation plays a role in the control of SR Ca2+ uptake and intracellular Ca2+ transient, we overexpressed Ad. I-1c in synthetic cultured hCASMCs21. Of note, the ubiquitous SERCA2b is the only isoform expressed in these cells (Figure 1B). The expression of I-1c protein was observed (7 kDa) (Figure 4A, left panel), whereas endogenous I-1 (28 kDa) was not detectable in synthetic hCASMCs overexpressing I-1c (not shown). Adenovirus-mediated gene transfer of I-1c (Ad. I-1c) significantly increased PLB phosphorylation at Ser16 (Figure 4A). However, I-1c overexpression had no effect on hCASMC proliferation (Figure 4B), Cyclin D1 expression (Supplementary Figure 5B) and cell survival (Supplementary Figure 5C and 5D). Remarkably, when the cells were co-infected with Ad.I-1c and an adenovirus encoding for SERCA2a, PLB phosphorylation was significantly increased (Supplemental Figure 5A) with a marked decrease of hCASMC proliferation (Figure 4B). These data indicate that to inhibit proliferation, I-1 needs the presence of SERCA2a.
Figure 4.
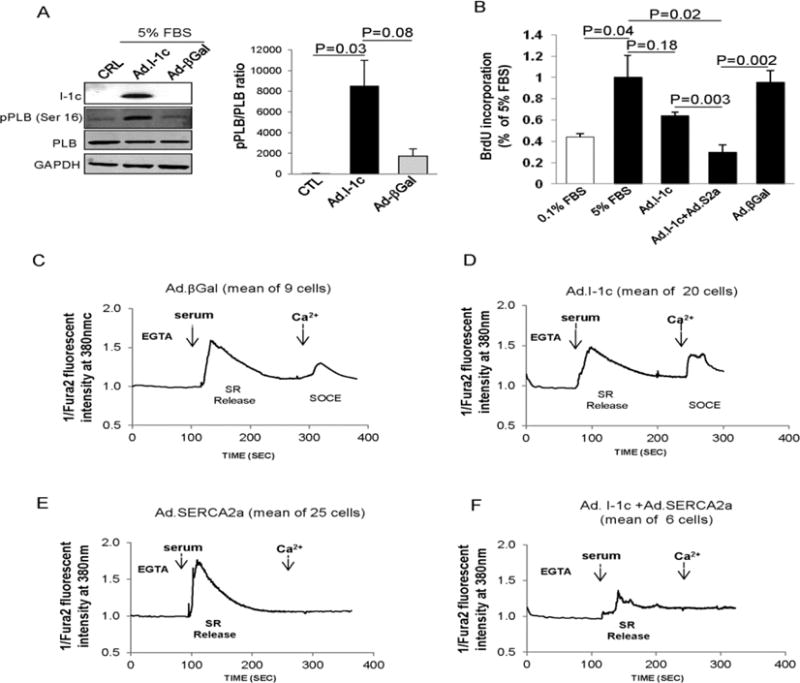
Effect of I-1c overexpression on hCASMC. (A) Immunoblot analysis (left panel) for indicated proteins and relative quantification of pPLB expression (right panel) in hCASMCs infected with the indicated adenovirus (B) Effect of Ad.I-1c, Ad.βGal, and Ad.I-1c together with Ad.SERCA2a on hCASMC proliferation. Cells were infected with the aforementioned virus for 48h and then cultured for 48h in virus-free BrdU in 5% FBS medium. Bars represent the mean ± SEM of BrdU incorporation of 4 independent experiments. Data are expressed as a percentage of control cells cultured in 5% FBS. Values represent mean ± SEM. (C–F) Analysis of I-1c overexpression on Ca2+ transients in synthetic hCASMCs. Intracellular Ca2+ imaging recorded in FURA-2 loaded cells representative of 3 experiments obtained from 3 independent infections. Cells were infected with Ad.βGal (C) Ad.I-1c (D), Ad.SERCA2a (E) or both Ad.I-1c and Ad.SERCA2a (F) for 2 days and then cultured for 24h in virus-free and serum-free medium before Ca2+ transient recording. Fluorescence intensity was only recorded in response to one excitation wavelength (380 nm). Traces represent the mean of several cells recordings. To record SR Ca2+ release and SOC activation, cells were treated with 5% serum buffered with EGTA in absence of extracellular Ca2+ (EGTA, 100μM); extracellular Ca2+ (CaCl2, 300μM) was then added at the indicated time.
Regulation of gene expression by Ca2+ can be mediated by Ca2+ dependent phosphorylation of the transcription factor CREB33. Therefore, we hypothesized that I-1/CREB signaling might regulate the VSMC phenotype. In testing this hypothesis, we found that CREB phosphorylated on Ser133 (pCREB) and p21 expression levels were highly elevated in synthetic VSMCs co-overexpressing I-1c and SERCA2a (Supplemental Figure 5A).
Next, we analyzed the effect of I-1c expression on Ca2+ transients. Since in VSMCs the Ca2+ signal required for proliferation depends both on SR Ca2+ release and Store Operated Ca2+ Entry (SOC)15, we performed Ca2+ transient recording in the absence of extracellular Ca2+ (EGTA). We have previously shown that during proliferation of VSMC, the caffeine-induced Ca2+ transient, normally observed in contractile VSMC, is progressively lost in favor of an IP3-induced Ca2+ transient10. Here, we examined the IP3-induced Ca2+ transients generated by agonist. In hCASMCs infected with Ad.βGal, serum induced a long lasting increase in cytosolic Ca2+ due to SR Ca2+ release (Figure 4C). Addition of Ca2+ to the extracellular medium triggered SOC in these cells (Figure 4C). Similar transients were observed in control non-infected cells (not shown). Surprisingly, I-1c expression had no effect on Ca2+ transients in synthetic hCASMCs (Figure 4D and Supplementary Figure 6A–D). In line with this, the increase of PLB phosphorylation by I-1c expression (Figure 4A) had no effect on Ca2+-ATPase activity, since the overall store load was unchanged in I-1c-expressing cells compared to cells infected with control virus (Supplementary Figure 6A). Meanwhile, when cells were infected with an adenovirus encoding SERCA2a, SOC was not observed after Ca2+ addition to the extracellular medium (Figure 4E), confirming previous observations15. Remarkably, in cells co-expressing SERCA2a and I-1c, the serum-induced SR Ca2+ transient was significantly reduced, and SOC following the addition of Ca2+ to the extracellular medium was not observed (Figure 4F and Supplementary Figure 6A–D). These data suggest that I-1c increased SERCA2a activity and that the type of Ca2+ transient is a SERCA2a isoform-dependent characteristic, and that the effects of I-1c and pPLB on Ca2+ transients depend on SERCA2 isoform expression in VSMCs. When thapsigargin (Tg), a Ca2+-mobilizing agent from SR was added, the quantity of Ca2+ that was re-mobilized was higher in cells co-expressing SERCA2a and I-1c compared to βGal and I-1c expressing cells (Supplementary Figure 6E and 6F), demonstrating that I-1c and SERCA2a overexpression increased the SR Ca2+ load and Ca2+ uptake.
To validate the role of I-1 on pPLB, pCREB and CRE (CREB Responsive Element) and also on NFAT activity through PP1, we performed siRNA knockdown to silence PP1 expression in hCASMCs. Immunoblot analysis of PP1 expression in hCASMCs cultured in 0.1% FBS for 4 days and transfected with pan PP1 or control siRNA showed that PP1 protein levels were decreased by 60–80% in PP1 siRNA transfected hCASMCs (Supplementary Figure 7A). CREB and PLB phosphorylation on Ser 16 was increased in PP1 siRNA-transfected cells compared to control (Supplementary Figure 7B). CRE transcriptional activity was also significantly increased in PP1 siRNA- transfected cells compared to control. (Supplementary Figure 7C). However, NFAT-driven luciferase activity was significantly decreased in PP1 siRNA-transfected cells compared to control (Supplementary Figure 7D). These results demonstrate that I-1 through PP1 controls CREB and NFAT transcriptional activity. NFAT activity was also measured in contractile and synthetic VSMCs overexpressing I-1c. Furthermore, I-1c overexpression in contractile VSMCS significantly decreased NFAT activity as compared to synthetic VSMCs. However, in cells co-expressing I-1c and SERCA2a, NFAT activity was significantly decreased in synthetic VSMCs. Infection with Ad.βGal had no effect on NFAT activity in either contractile or synthetic VSMCs (Supplementary Figure 7E).
Effect of I-1 on Ca2+ cycling and signaling in contractile and synthetic rat aortic VSMCs
In the aforementioned adenovirus transduction experiments we used a GFP tag to identify infected cells. Since both viruses (Ad.SERCA2a and Ad.I-1c) were tagged with GFP, we were unable to ascertain if any given cell was infected with one or both viruses, even though the rate of infection for each virus was approximately 80%. To avoid this uncertainty, we performed experiments using freshly dissociated contractile rat aortic VSMCs, expressing SERCA2a, and synthetic cultured rat aortic VSMCs, expressing mainly SERCA2b11, 14. The phenotypic status of the rat aortic VSMCs was verified by analyzing the expression of contractile proteins; SMA, SM-MHC and calponin (Supplementary Figure 8A). In parallel, the Cyclin D1 protein level was increased in synthetic VSMCs (Supplementary Figure 8A). By immunostaining, Cyclin D1 was not detected in freshly dissociated VSMCs, confirming their non-proliferative contractile phenotype. Three days after induction of proliferation by serum (10% FBS), Cyclin D1 was expressed demonstrating the proliferative synthetic phenotype (Supplementary Figure 8B). As expected, SERCA2a andI-1 were expressed in contractile VSMCs and down-regulated in synthetic VSMCs (Figure 5A and Supplementary Figure 8B). p21 and p53 expression as well as pCREB and pPLB were also decreased in rat synthetic VSMCs (Figure 5A).
Figure 5.
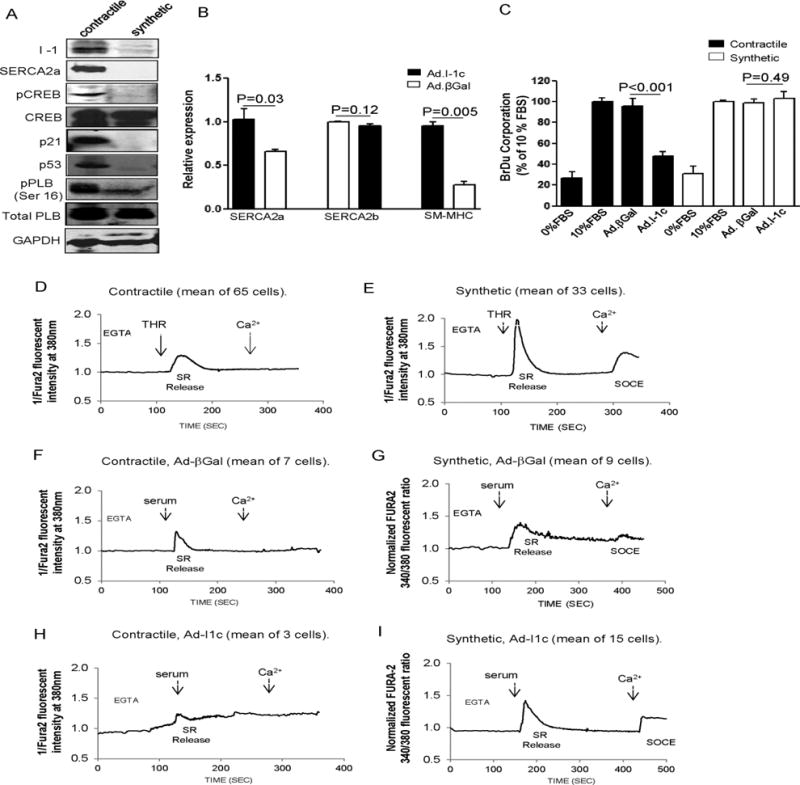
Effect of I-1c expression on contractile and synthetic rat aortic VSMCs. (A) Immunoblot analysis with indicated proteins in contractile and synthetic aortic VSMCs from adult male rats. (B) Real-time RT-PCR analysis showing relative expression of SERCA2a, SERCA2b and SM-MHC in freshly dissociated contractile rat VSMCs infected either with Ad.I-1c or Ad.βGal. The cells were cultured in the presence of serum (10%) and were collected 4 days after infection. (C) Effect of Ad.I-1c and Ad.βGal infection on proliferation of contractile and synthetic rat aortic VSMCs. Cells were infected with the aforementioned virus for 48h and then cultured for 48h in virus-free BrdU containing medium supplemented with serum (10%). Bars represent the mean ± SEM of BrdU incorporation of 4 independent experiments. Data are expressed as a percentage of control cells cultured in 10% serum. (D–E) Analysis of Ca2+ transients in contractile and synthetic rat VSMCs. Representative intracellular Ca2+ imaging recorded in FURA-2 loaded cells, from a total of 3 experiments obtained from 3 independent infections. Contractile cells were cultured without serum and used within 3 days after dissociation from the aorta. The cells were infected with Ad.βGal (F, G) or Ad. I-1c (H, I) for 2 days and then cultured for 24h in virus-free and serum-free medium before recording. Fluorescence intensity was only recorded in response to one excitation wavelength (380 nm). Traces represent the mean of several cells recordings. To record SR Ca2+ release and SOC activation, cells were treated with 5% serum buffered with EGTA (100μM) in absence of extracellular Ca2+ (CaCl2, 300μM) was then added at the indicated time.
The initial Ca2+ event of VSMC dedifferentiation is the down-regulation of Ca2+-ATPase activity, which results in a sustained increase of cytosolic Ca2+ and increased calcineurin-NFAT activity11. Since I-1 down-regulation is a part of the dedifferentiation process, we assessed the effect of I-1c overexpression on the dedifferentiation, proliferation, and Ca2+ transient of contractile VSMCs. Immediately after dissociation of rat aortic VSMCs, contractile aortic VSMCs were infected with Ad. I-1c or βGal virus and analyzed at day 4 post-infection in a high concentration of FBS (10%). Remarkably, expression of I-1c prevented dedifferentiation of contractile VSMCs induced by serum, as attested by a sustained increase in SERCA2a and SM-MHC mRNA expression (Figure 5B). As expected, exposure to serum induced proliferation of contractile VSMCs, with the percentage of proliferation being similar to that in synthetic VSMCs (Figure 5C). Infection of VSMCs with Ad.I-1c significantly reduced proliferation of contractile VSMCs without any effect on synthetic VSMC proliferation (Figure 5C). Of note, infection of contractile VSMCs with Ad.βGal had no effect on VSMC proliferation, nor on serum-induced dedifferentiation (Figure 5B and 5C). Next, we examined Ca2+ transients in contractile and synthetic rat aortic VSMCs (Figure 5D–I). As expected, thrombin (THR) induced SR Ca2+ release in contractile VSMCs with the inhibition of Ca2+ entry by SOC after addition of Ca2+ in the extracellular medium (Figure 5D). In contrast, in synthetic VSMCs, THR induced a large SR released Ca2+ transient followed by SOC after addition of Ca2+ (Figure 5E). Infection with control virus (Ad.βGal) had no effect on Ca2+ transients in either contractile or synthetic VSMCs (Figure 5F and 5G). Ad.I-1c overexpression had no effect on Ca2+ transients in synthetic rat VSMCs stimulated with serum compared to controls (Figure 5I and Supplementary Figure 9B) and in Ca2+ re-mobilized when Tg was added (Supplementary Figure 9C), confirming the observations made for hCASMCs (Figure 4D). However, I-1c overexpression significantly decreased overall Ca2+ transients in contractile rat VSMCs stimulated with serum (Figure 5H and Supplementary Figure 9A), confirming that the effect of I-1 on Ca2+ transients depends on SERCA2a isoform expression in contractile vs. synthetic VSMCs.
Constitutively active I-1 prevents post-injury remodeling and neointimal proliferation in a rat carotid injury model
Since I-1c overexpression prevents dedifferentiation and proliferation of contractile VSMCs in vitro, we next tested the possibility that I-1c gene transfer may prevent post-angioplasty neointimal proliferation. For this purpose we performed balloon injury in the left carotid arteries of adult male rats, followed immediately by gene transfer of Ad.I-1c or Ad.βGal. The right carotid was used as sham-operated, non-injured. Animals were sacrificed 2 weeks after injury, and neointimal thickening was analyzed on hematoxylin/eosin stained cross sections (Figure 6A). Infection was confirmed by PCR detection of GFP DNA in the carotid arteries infected with Ad. I-1c compared to non-infected arteries (Supplementary Figure 10). Injury induced abundant neointimal proliferation and an increase in the medial thickness in non-infected (saline) or Ad.βGal-infected arteries, whereas infection with Ad. I-1c significantly reduced medial thickening and prevented neointimal proliferation (Figure 6B). Analysis by immunofluorescence microscopy confirmed that in saline and Ad.βGal infected arteries, there was downregulation of I-1 in the injured carotid segments, as well as downregulation of markers of the contractile phenotype such as SM-MHC and SERCA2a (Figure 7A). In contrast, in Ad.I-1c infected arteries, I-1 expression was detected; however our antibody does not allow us to discriminate between the endogenous and the transgene I-1 protein. Nevertheless, expression of SERCA2a and SM-MHC was preserved in I-1c infected arteries (Figure 7A). In addition, the expression of CREB and p21 was lower in the media and adventitia of βGal-infected carotid arteries compared to arteries treated with Ad.I-1c. However, NFAT expression was highly increased in the βGal compared to I-1c infected infected-injured carotid (Figure 7B). Thus, I-1c prevented dedifferentiation and proliferation of contractile VSMCs expressing SERCA2a, but had no effect on synthetic VSMCs, which predominantly express the SERCA2b isoform and NFATC3.
Figure 6.
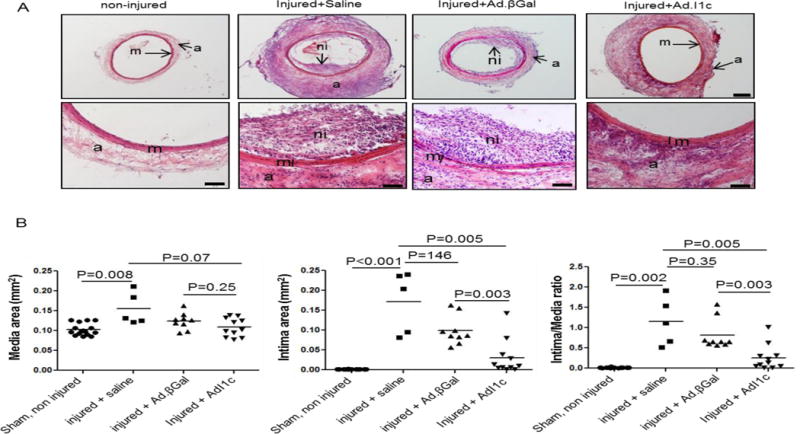
I-1c gene transfer prevents balloon angioplasty-associated restenosis in a rat carotid injury model. (A) Bright-field photomicrograph of hematoxylin and eosin stained cross sections from sham control (non-injured carotid, n=16) and injured-treated-saline (n=5), Ad.βGal (n=9) and Ad.I1c (n=11) arteries 14 days after surgery; m (media), ni (neointima), ad (adventitia). Scale bar = 200 μm in upper panels and 100 μm in lower panels. (B) Measurements of media and intima area size, and intima/media area ratio of the 4 groups from (A). Values represent mean ± SEM.
Figure 7.
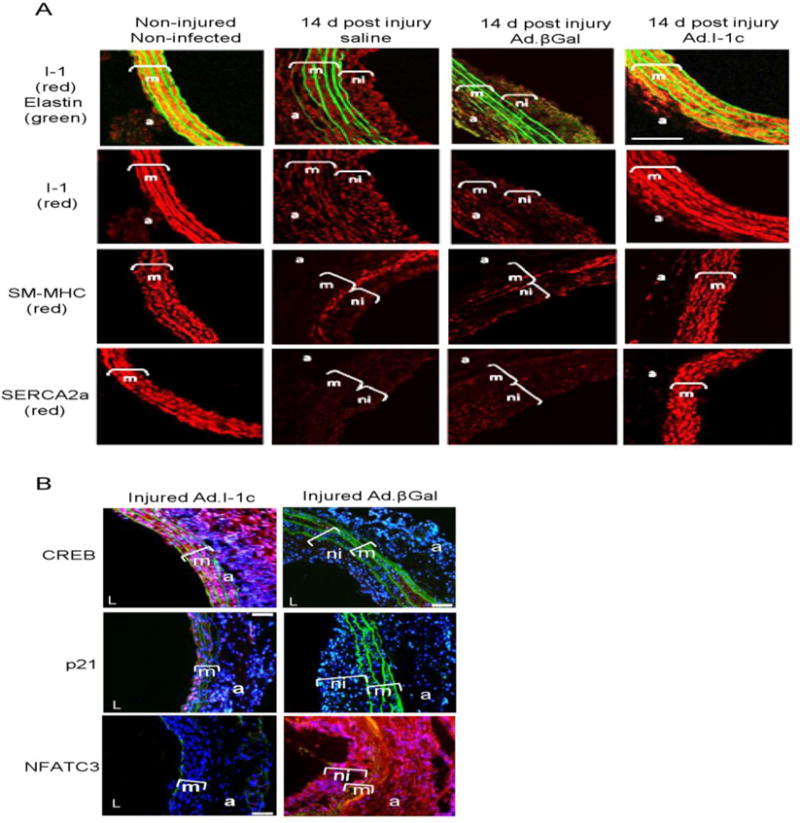
I-1c overexpression preserves VSMCs contractile phenotype in balloon-injured rat carotid arteries. (A) Immunolabeling of control or balloon-injured carotid arteries with anti-I-1, anti-SERCA2a or anti-SM-MHC (red). Elastin autofluorescence is shown in green. Bar scale =20 μm. (B) Representative CREB, p21 and NFATC3 immunostaining of cross-sections from βGal and I-1c infected arteries at 2 weeks after surgery, m (media), ni (neointima), a (adventitia). Red color shows CREB, p21 and NFAT positive staining.
Discussion
The important finding of the present study is that PKA/I-1 downstream signaling pathways are organized differently in contractile and synthetic VSMCs (Figure 8). This signaling specificity is supported by altered expression of molecules involved in the regulation of the downstream of cAMP/PKA signaling cascade; with I-1 and PP1 possibly providing of dual control of the signaling pathway at both the protein expression and phosphorylation levels. We demonstrated mirrored expression of I-1 and PP1 within contractile and synthetic VSMCs leading to the enhancement of the PKA pathway in contractile VSMCs. in the media of human coronary and freshly dispersed rat aortic VSMCs, I-1 is more highly expressed than in their cultured counterparts. The expression of I-1 correlated closely with the expression of genes for contractile proteins: α-SMC, calponin, SERCA2a, and the SM-MHC proteins, which are also downregulated in synthetic dedifferentiated VSMCs15.Therefore, these results establish a clear link between the differentiated, contractile phenotype of VSMCs and the dowregulation of I-1 in rat and human VSMCs, suggesting a possible role for I-1 in the maintenance or function of the differentiated VSMC phenotype.
Figure 8.
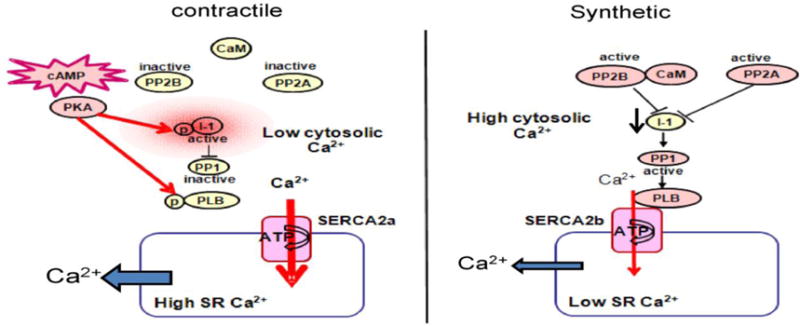
Schematic representation of PKA/I-1/PP1 signaling pathways in contractile (left panel) and synthetic (right panel) VSMCs. Abbreviations: SR (sarco/endoplasmic reticulum), SERCA (Sarco/Endo Plasmic Reticulum Ca2+-ATPase), PLB (phospholamban); p (phosphate); PP1 (protein phosphatase 1); I-1 (Inhibitor 1); PP2A (protein phosphatase 2A), PP2B (protein phosphatase 2B), CaM (calmodulin), PKA (protein kinase A).
Moreover, we showed that VSMCs obtained from I-1KO mice are in a proliferative state and display the characteristics of immature or dedifferentiated phenotypes compared to WT SMCs. The expressed minimal differentiation marker proteins such as SMA and SM-MHC and showed increased of proliferation marker as Cyclin D1 and PCNA. More interestingly, in contrast to PP1, SERCA2a protein levels together with PLB phosphorylation and therefore Ca2+-ATPase were reduced. However, when constitutively active I-1 is overexpressed, SERCA2a activity can be further increased. We also demonstrated that in vivo gene transfer of constitutively active I-1c prevents VSMC dedifferentiation and neointimal formation in a rat and mouse carotid injury model. However, in I-1 KO mice, since the VSMCs are dedifferentiated, neointimal formation was not decreased following I-1c overexpression. Thus, I-1 and SERCA2a are necessary and act synergistically to regulate the VSMC phenotype. A further interesting observation that can be drawn from the I-1 KO mice is the increase in aortic thickness due to hyperplasia and the excessive neointimal formation with vascular injury in vivo. The results presented here demonstrate that loss of I-1 in a mouse model increases SMC proliferation. These data support the hypothesis that a hyperplastic response modulated by I-1 in VSMCs contributes to vascular disease. In the heart, I-1 was found to be down-regulated and hypo-phosphorylated in human and experimental heart failure but hyperactive in human atrial fibrillation, implicating I-1 in the pathogenesis of heart failure and arrhythmias34. In addition, constitutive I-1c expression by gene transfer restored contractile properties in failing rat hearts and failing human cardiac myocytes21, 22. In our study we showed that I-1c overexpression in carotid artery injury models prevented the downregulation of the expression of VSMCs contractile markers such as SERCA2a and SM-MHC, as well as CREB and p21 expression. Several studies have also shown that dowregulation of CREB expression and/or phosphorylations are associated with elevated cardiovascular risk markers in rodent models35. Indolfi et al. have reported that PKA signaling inhibits neointimal formation after vascular injury in vivo as a model of restenosis after angioplasty36. In addition, cyclic AMP signaling in SMCs has been shown to decrease the expression of Cyclin D137 and increase the expression of antiproliferative regulators like p53 and p2138. Furthermore, I-1c overexpression in contractile VSMCs also decreased transcription factor NFAT activity required for proliferation and migration of VSMCs that express SERCA2a13, 15. We have shown that the restoration of SERCA2a expression by gene transfer in synthetic VSMCs was sufficient to block their proliferation and migration via inhibition of NFAT15, however, it will be interesting to assess whether SERCA2a gene transfer restore I-1 expression level. The molecular mechanisms of this effect are related to the prevention of functional association between STIM1 and OraI-1 (CRAC protein entity) which lead to the suppression of SOC channel influx15. It is worth mentioning that SOC influx following agonist stimulation is not observed in contractile VSMCs, which express SERCA2a and I-1 compared to synthetic VSMCs. This suggests that I-1 exerts its effects through SERCA2a.
In addition, I-1 KO femoral arteries in vivo displayed a significantly lower vasoconstriction and vasorelaxation response compared to WT. These results suggest that changes in I-1 expression can modify the VSMC phenotype and therefore VSMC contractility. Indeed, the loss of I-1 in synthetic VSMCs and in the mouse model is associated with a reduced SR Ca2+-ATPase activity. Therefore, I-1 is a critical determinant of SR Ca2+ uptake function. These results are particularly interesting when considered along with recent results demonstrating reduced endothelium-dependent relaxation in mice deficient in SERCA3 isoform39. Another study showed that deletion of PLB can also reduce endothelium-dependent relaxation40. However yet another study has suggested that ablation of I-1 does not play a major role in either tonic or phasic SM contractility in vivo. In that report, while I-1 did not have any major effects on the aorta, there was a significant, rightward-shifted (desensitized) response to isoproterenol in I-1 KO portal veins21. The difference between that study and our findings may potentially be due to the fact that our experiments were carried out in femoral arteries in vivo and the published study was done on mounted isolated denuded aortic rings21. In theory, the organization of the PKA/I-1 signaling pathway should increase SERCA2a activity in contractile VSMCs while inhibiting SERCA2a activity in synthetic cells. However, in reality, SERCA2a activity is further distinguished between contractile and synthetic VSMCs by SERCA2 isoform expression levels. In accordance with our previous observations10, 14, 15, here we confirm that the SERCA2a isoform is expressed exclusively in contractile VSMCs, whereas SERCA2b is the only SR Ca2+-ATPase present in synthetic VSMCs. Furthermore, manipulation of these signaling pathways resulted in high SR Ca2+-ATPase activity in contractile cells and reduced SR Ca2+-ATPase activity in synthetic cells. In this case, we hypothesized that this reduction was attributable to enhanced endoplasmic reticulum Ca2+ uptake leading to lower [Ca2+]i. Therefore, the interplay between Ca2+ release and uptake systems is an important determinant of endothelial steady-state Ca2+ levels that elicits endothelium-dependent relaxation.
Our study also shows that knockdown of PP1 enhances hyperphosphorylation of PLB and CREB and decreases NFAT activity. Our results provide further evidence for a role of PP1 pathway in regulating the dephosphorylation of Ser-133 and thereby limiting CREB transcriptional activity in synthetic VSMCs. Many studies have shown that increased PP1 activity is closely associated with the progression of heart failure16, 22, 41. PP1, a haloenzyme, consists of three distinct genes, namely: PP1c-alpha (PP1α), PP1c-gamma (PP1λ, which gives rise to two splice variants (PP1c-λ1 and PP1c-λa2) and PP1c-beta/delta (PP1β)42–44, whose functions are not well characterized in cardiovascular cells. In our study we found that PP1α and PP1β are increased in synthetic VSMCs. PP1β expression has been shown to gradually increase during the progressive time course of cardiac dysfunction in cardiomyopathic hamsters41. The expression levels of PP1β may have a crucial impact on SR Ca2+ cycling. PP1β isoform was shown to be highly expressed in the longitudinal SR, and formed a molecular complex with PLB and SERCA2a. Moreover, PP1β is the most significantly involved PP1 isoform in regulating PLB phosphorylation at Ser16 in the longitudinal SR, thereby controlling SR-mediated Ca2+ cycling in cardiomyocytes45
In summary, we have demonstrated for the first time that Ca2+ signaling pathways via the I-1/PP1 signaling pathway are altered in synthetic VSMCs. I-1 expression appears to be a robust marker for contractile VSMCs, whereas PP1 expression is specific for synthetic VSMCs, thus reinforcing our finding of altered Ca2+ signaling. Furthermore, the synergistic action of I-1 and SERCA2a is necessary for the acquisition of high SERCA2a activity required for the contractile phenotype. Gene transfer of constitutive active I-1c to contractile VSMCs prevents injury-induced dedifferentiation and neointimal proliferations of VSMCs. Collectively, these data suggest that I-1c gene transfer may be considered as a therapeutic strategy for preventing vascular proliferative disorders.
Supplementary Material
Acknowledgments
Funding Sources: This work was supported by the American Heart Association SDG 0930116N (LL); by the National Heart Lung and Blood Institute K01 HL1031176-01 (LH), K08HL111330 (JCK), R01 HL080498 and R01 HL083156 (RJH); by the Association Française Contre les Myopathies (RB); AFM16442 (RB, LL, JJL and RJH), by MEC-FEDER (BFU2010-C02-01) (RB and JJL), and JJL was supported by a postdoctoral fellowship from the Junta de Extremadura (POS0922).
Footnotes
Journal Subject Codes: [162] Smooth muscle proliferation and differentiation, [130] Animal models of human disease, [123] Restenosis, [88] Gene therapy
Conflict of Interest Disclosures: RJH is a scientific founder of Celladon Corp., which plans to commercialize AAV1.SERCA2a for the treatment of heart failure. The other authors declare no competing financial interests.
References
- 1.Novak K. Cardiovascular disease increasing in developing countries. Nat Med. 1998;4:989–990. doi: 10.1038/1965. [DOI] [PubMed] [Google Scholar]
- 2.Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: New perspectives and therapeutic strategies. Nat Med. 2002;8:1249–1256. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 3.Forrester JS, Fishbein M, Helfant R, Fagin J. A paradigm for restenosis based on cell biology: Clues for the development of new preventive therapies. J Am Coll Cardiol. 1991;17:758–769. doi: 10.1016/s0735-1097(10)80196-2. [DOI] [PubMed] [Google Scholar]
- 4.Braun-Dullaeus RC, Mann MJ, Dzau VJ. Cell cycle progression: New therapeutic target for vascular proliferative disease. Circulation. 1998;98:82–89. doi: 10.1161/01.cir.98.1.82. [DOI] [PubMed] [Google Scholar]
- 5.Chen KH, Guo X, Ma D, Guo Y, Li Q, Yang D, Li P, Qiu X, Wen S, Xiao RP, Tang J. Dysregulation of hsg triggers vascular proliferative disorders. Nat Cell Biol. 2004;6:872–883. doi: 10.1038/ncb1161. [DOI] [PubMed] [Google Scholar]
- 6.Marchand A, Abi-Gerges A, Saliba Y, Merlet E, Lompre AM. Calcium signaling in vascular smooth muscle cells: From physiology to pathology. Adv Exp Med Biol. 2012;740:795–810. doi: 10.1007/978-94-007-2888-2_35. [DOI] [PubMed] [Google Scholar]
- 7.Regan CP, Adam PJ, Madsen CS, Owens GK. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. J Clin Invest. 2000;106:1139–1147. doi: 10.1172/JCI10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.House SJ, Potier M, Bisaillon J, Singer HA, Trebak M. The non-excitable smooth muscle: Calcium signaling and phenotypic switching during vascular disease. Pflugers Arch. 2008;456:769–785. doi: 10.1007/s00424-008-0491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Golovina VA, Platoshyn O, Bailey CL, Wang J, Limsuwan A, Sweeney M, Rubin LJ, Yuan JX. Upregulated trp and enhanced capacitative ca(2+) entry in human pulmonary artery myocytes during proliferation. Am J Physiol Heart Circ Physiol. 2001;280:H746–755. doi: 10.1152/ajpheart.2001.280.2.H746. [DOI] [PubMed] [Google Scholar]
- 10.Vallot O, Combettes L, Jourdon P, Inamo J, Marty I, Claret M, Lompre AM. Intracellular ca(2+) handling in vascular smooth muscle cells is affected by proliferation. Arterioscler Thromb Vasc Biol. 2000;20:1225–1235. doi: 10.1161/01.atv.20.5.1225. [DOI] [PubMed] [Google Scholar]
- 11.Lipskaia L, Pourci ML, Delomenie C, Combettes L, Goudouneche D, Paul JL, Capiod T, Lompre AM. Phosphatidylinositol 3-kinase and calcium-activated transcription pathways are required for vldl-induced smooth muscle cell proliferation. Circ Res. 2003;92:1115–1122. doi: 10.1161/01.RES.0000074880.25540.D0. [DOI] [PubMed] [Google Scholar]
- 12.Gomez MF, Stevenson AS, Bonev AD, Hill-Eubanks DC, Nelson MT. Opposing actions of inositol 1,4,5-trisphosphate and ryanodine receptors on nuclear factor of activated t-cells regulation in smooth muscle. J Biol Chem. 2002;277:37756–37764. doi: 10.1074/jbc.M203596200. [DOI] [PubMed] [Google Scholar]
- 13.Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- 14.Lipskaia L, del Monte F, Capiod T, Yacoubi S, Hadri L, Hours M, Hajjar RJ, Lompre AM. Sarco/endoplasmic reticulum ca2+-atpase gene transfer reduces vascular smooth muscle cell proliferation and neointima formation in the rat. Circ Res. 2005;97:488–495. doi: 10.1161/01.RES.0000180663.42594.aa. [DOI] [PubMed] [Google Scholar]
- 15.Bobe R, Hadri L, Lopez JJ, Sassi Y, Atassi F, Karakikes I, Liang L, Limon I, Lompre AM, Hatem SN, Hajjar RJ, Lipskaia L. Serca2a controls the mode of agonist-induced intracellular ca2+ signal, transcription factor nfat and proliferation in human vascular smooth muscle cells. J Mol Cell Cardiol. 2011;50:621–633. doi: 10.1016/j.yjmcc.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicolaou P, Kranias EG. Role of pp1 in the regulation of ca cycling in cardiac physiology and pathophysiology. Front Biosci. 2009;14:3571–3585. doi: 10.2741/3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Armouche A, Rau T, Zolk O, Ditz D, Pamminger T, Zimmermann WH, Jackel E, Harding SE, Boknik P, Neumann J, Eschenhagen T. Evidence for protein phosphatase inhibitor-1 playing an amplifier role in beta-adrenergic signaling in cardiac myocytes. FASEB J. 2003;17:437–439. doi: 10.1096/fj.02-0057fje. [DOI] [PubMed] [Google Scholar]
- 18.Aitken A, Cohen P. Isolation and characterisation of active fragments of protein phosphatase inhibitor-1 from rabbit skeletal muscle. FEBS letters. 1982;147:54–58. doi: 10.1016/0014-5793(82)81010-7. [DOI] [PubMed] [Google Scholar]
- 19.Oliver CJ, Shenolikar S. Physiologic importance of protein phosphatase inhibitors. Front Biosci. 1998;3:D961–972. doi: 10.2741/a336. [DOI] [PubMed] [Google Scholar]
- 20.Tokui T, Brozovich F, Ando S, Ikebe M. Enhancement of smooth muscle contraction with protein phosphatase inhibitor 1: Activation of inhibitor 1 by cgmp-dependent protein kinase. Biochem Biophys Res Commun. 1996;220:777–783. doi: 10.1006/bbrc.1996.0480. [DOI] [PubMed] [Google Scholar]
- 21.Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C, Breeden K, Jing SL, Allen PB, Greengard P, Yatani A, Hoit BD, Grupp IL, Hajjar RJ, DePaoli-Roach AA, Kranias EG. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol. 2002;22:4124–4135. doi: 10.1128/MCB.22.12.4124-4135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pathak A, del Monte F, Zhao W, Schultz JE, Lorenz JN, Bodi I, Weiser D, Hahn H, Carr AN, Syed F, Mavila N, Jha L, Qian J, Marreez Y, Chen G, McGraw DW, Heist EK, Guerrero JL, DePaoli-Roach AA, Hajjar RJ, Kranias EG. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res. 2005;96:756–766. doi: 10.1161/01.RES.0000161256.85833.fa. [DOI] [PubMed] [Google Scholar]
- 23.Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 24.El-Armouche A, Bednorz A, Pamminger T, Ditz D, Didie M, Dobrev D, Eschenhagen T. Role of calcineurin and protein phosphatase-2a in the regulation of phosphatase inhibitor-1 in cardiac myocytes. Biochem Biophys Res Commun. 2006;346:700–706. doi: 10.1016/j.bbrc.2006.05.182. [DOI] [PubMed] [Google Scholar]
- 25.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, DePaoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. Pkc-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10:248–254. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez P, Mitton B, Waggoner JR, Kranias EG. Identification of a novel phosphorylation site in protein phosphatase inhibitor-1 as a negative regulator of cardiac function. J Biol Chem. 2006;281:38599–38608. doi: 10.1074/jbc.M604139200. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez P, Mitton B, Nicolaou P, Chen G, Kranias EG. Phosphorylation of human inhibitor-1 at ser67 and/or thr75 attenuates stimulatory effects of protein kinase a signaling in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2007;293:H762–769. doi: 10.1152/ajpheart.00104.2007. [DOI] [PubMed] [Google Scholar]
- 28.Somlyo AP, Somlyo AV. Signal transduction by g-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin ii. J Physiol. 2000;522(Pt 2):177–185. doi: 10.1111/j.1469-7793.2000.t01-2-00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elbrecht A, DiRenzo J, Smith RG, Shenolikar S. Molecular cloning of protein phosphatase inhibitor-1 and its expression in rat and rabbit tissues. J Biol Chem. 1990;265:13415–13418. [PubMed] [Google Scholar]
- 30.Alessi D, MacDougall LK, Sola MM, Ikebe M, Cohen P. The control of protein phosphatase-1 by targetting subunits. The major myosin phosphatase in avian smooth muscle is a novel form of protein phosphatase-1. Eur J Biochem. 1992;210:1023–1035. doi: 10.1111/j.1432-1033.1992.tb17508.x. [DOI] [PubMed] [Google Scholar]
- 31.Allen PB, Hvalby O, Jensen V, Errington ML, Ramsay M, Chaudhry FA, Bliss TV, Storm-Mathisen J, Morris RG, Andersen P, Greengard P. Protein phosphatase-1 regulation in the induction of long-term potentiation: Heterogeneous molecular mechanisms. J Neurosci. 2000;20:3537–3543. doi: 10.1523/JNEUROSCI.20-10-03537.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang EH, Jensen BL, Skott O, Leung GP, Feletou M, Man RY, Vanhoutte PM. The role of prostaglandin e and thromboxane-prostanoid receptors in the response to prostaglandin e2 in the aorta of wistar kyoto rats and spontaneously hypertensive rats. Card Res. 2008;78:130–138. doi: 10.1093/cvr/cvm112. [DOI] [PubMed] [Google Scholar]
- 33.Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an l-type calcium channel-calmodulin complex through the map kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- 34.Wittkopper K, Dobrev D, Eschenhagen T, El-Armouche A. Phosphatase-1 inhibitor-1 in physiological and pathological beta-adrenoceptor signalling. Card Res. 2011;91:392–401. doi: 10.1093/cvr/cvr058. [DOI] [PubMed] [Google Scholar]
- 35.Schauer IE, Knaub LA, Lloyd M, Watson PA, Gliwa C, Lewis KE, Chait A, Klemm DJ, Gunter JM, Bouchard R, McDonald TO, O’Brien KD, Reusch JE. Creb downregulation in vascular disease: A common response to cardiovascular risk. Arterioscler Thromb Vasc Biol. 2010;30:733–741. doi: 10.1161/ATVBAHA.109.199133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Indolfi C, Avvedimento EV, Di Lorenzo E, Esposito G, Rapacciuolo A, Giuliano P, Grieco D, Cavuto L, Stingone AM, Ciullo I, Condorelli G, Chiariello M. Activation of camp-pka signaling in vivo inhibits smooth muscle cell proliferation induced by vascular injury. Nat Med. 1997;3:775–779. doi: 10.1038/nm0797-775. [DOI] [PubMed] [Google Scholar]
- 37.Vadiveloo PK, Filonzi EL, Stanton HR, Hamilton JA. G1 phase arrest of human smooth muscle cells by heparin, il-4 and camp is linked to repression of cyclin d1 and cdk2. Atherosclerosis. 1997;133:61–69. doi: 10.1016/s0021-9150(97)00116-0. [DOI] [PubMed] [Google Scholar]
- 38.Hayashi S, Morishita R, Matsushita H, Nakagami H, Taniyama Y, Nakamura T, Aoki M, Yamamoto K, Higaki J, Ogihara T. Cyclic amp inhibited proliferation of human aortic vascular smooth muscle cells, accompanied by induction of p53 and p21. Hypertension. 2000;35:237–243. doi: 10.1161/01.hyp.35.1.237. [DOI] [PubMed] [Google Scholar]
- 39.Liu LH, Paul RJ, Sutliff RL, Miller ML, Lorenz JN, Pun RY, Duffy JJ, Doetschman T, Kimura Y, MacLennan DH, Hoying JB, Shull GE. Defective endothelium-dependent relaxation of vascular smooth muscle and endothelial cell ca2+ signaling in mice lacking sarco(endo)plasmic reticulum ca2+-atpase isoform 3. J Biol Chem. 1997;272:30538–30545. doi: 10.1074/jbc.272.48.30538. [DOI] [PubMed] [Google Scholar]
- 40.Sutliff RL, Hoying JB, Kadambi VJ, Kranias EG, Paul RJ. Phospholamban is present in endothelial cells and modulates endothelium-dependent relaxation. Evidence from phospholamban gene-ablated mice. Circ Res. 1999;84:360–364. doi: 10.1161/01.res.84.3.360. [DOI] [PubMed] [Google Scholar]
- 41.Yamada M, Ikeda Y, Yano M, Yoshimura K, Nishino S, Aoyama H, Wang L, Aoki H, Matsuzaki M. Inhibition of protein phosphatase 1 by inhibitor-2 gene delivery ameliorates heart failure progression in genetic cardiomyopathy. FASEB J. 2006;20:1197–1199. doi: 10.1096/fj.05-5299fje. [DOI] [PubMed] [Google Scholar]
- 42.Cohen P, Alemany S, Hemmings BA, Resink TJ, Stralfors P, Tung HY. Protein phosphatase-1 and protein phosphatase-2a from rabbit skeletal muscle. Methods Enzymol. 1988;159:390–408. doi: 10.1016/0076-6879(88)59039-0. [DOI] [PubMed] [Google Scholar]
- 43.Sasaki K, Shima H, Kitagawa Y, Irino S, Sugimura T, Nagao M. Identification of members of the protein phosphatase 1 gene family in the rat and enhanced expression of protein phosphatase 1 alpha gene in rat hepatocellular carcinomas. Jpn J Cancer Res. 1990;81:1272–1280. doi: 10.1111/j.1349-7006.1990.tb02690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dombradi V, Axton JM, Brewis ND, da Cruz e Silva EF, Alphey L, Cohen PT. Drosophila contains three genes that encode distinct isoforms of protein phosphatase 1. Eur J Biochem. 1990;194:739–745. doi: 10.1111/j.1432-1033.1990.tb19464.x. [DOI] [PubMed] [Google Scholar]
- 45.Aoyama H, Ikeda Y, Miyazaki Y, Yoshimura K, Nishino S, Yamamoto T, Yano M, Inui M, Aoki H, Matsuzaki M. Isoform-specific roles of protein phosphatase 1 catalytic subunits in sarcoplasmic reticulum-mediated ca(2+) cycling. Card Res. 2011;89:79–88. doi: 10.1093/cvr/cvq252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.