

Abstract
The primary cause of mortality for glioblastoma (GBM) is local tumor recurrence following standard-of-care therapies, including surgical resection. With most tumors recurring near the site of surgical resection, local delivery of chemotherapy at the time of surgery is a promising strategy. Herein drug loaded polymer scaffolds with two distinct degradation profiles were fabricated to investigate the effect of local drug delivery rate on GBM recurrence following surgical resection. The novel biopolymer, acetalated dextran (Ace-DEX), was compared to commercially available polyester, poly(L-lactide) (PLA). Steady state doxorubicin (DXR) release from Ace-DEX scaffolds was found to be faster when compared to scaffolds comprised of PLA, in vitro. This increased drug release rate translated to improved therapeutic outcomes in a novel surgical model of orthotopic glioblastoma resection and recurrence. Mice treated with DXR loaded Ace-DEX scaffolds (Ace-DEX/10DXR) resulted in 57% long term survival out to study completion at 120 days compared to 20% survival following treatment with DXR loaded PLA scaffolds (PLA/10DXR). Additionally, all mice treated with PLA/10DXR scaffolds exhibited disease progression by day 38, as defined by a five-fold growth in tumor bioluminescent signal. In contrast, 57% of mice treated with Ace-DEX/10DXR scaffolds displayed a reduction in tumor burden, with 43% exhibiting complete remission. These results underscore the importance of polymer choice and drug release rate when evaluating local drug delivery strategies to improve prognosis for GBM patients undergoing tumor resection.
Keywords: Ace-DEX, poly-lactide (PLA), polyanhidride, carmustine, drug delivery, nanofiber
Graphical Abstract
INTRODUCTION
Glioblastoma (GBM) is an especially aggressive, grade IV central nervous system tumor. GBM is the most common primary brain tumor and accounts for 45–50% of all gliomas,1 and even with surgical resection, radiation, and chemotherapy, the median survival for GBM patients remains only 12 to 15 months.1–3 The primary cause of mortality for GBM is local tumor recurrence, with 90 – 95% of tumors recurring within 2 cm of the original tumor.4–7 As such, patient outcomes could be greatly improved by delaying or preventing local recurrence. Unfortunately, chemotherapeutic options are limited due to the inability of most drugs to cross the blood-brain barrier. One strategy to improve drug delivery to GBM and bypass the blood-brain barrier is interstitial chemotherapy. This can be achieved by lining the resection cavity with a material that releases drug as it degrades.8 This strategy is applied clinically with Gliadel®, a polyester and polyanhydride polymer wafer that is used in some GBM patients to deliver carmustine (BCNU) directly into the resection cavity.9 Unfortunately, Gliadel® only extends median survival by 10–18 weeks over placebo.10, 11 This is likely due to the low tumoricidal activity of BCNU against GBM and its rapid clearance from the brain after release from the polymer wafer.12–14 Additionally, tumor cells easily acquire resistance to alkylating agents like BCNU through the DNA repair mediated by the enzyme O6-methylguanine-DNA methyltransferase (MGMT).15, 16 Low tumoricidal activity, easily acquired drug resistance, and rapid clearance of BCNU from the brain highlight the need for exploration into alternative anticancer drugs for interstitial therapy.
One promising compound to combat GBM is doxorubicin (DXR). This highly potent chemotherapeutic agent induces cell death via multiple mechanisms of action, including DNA intercalation and interaction with topoisomerase II.17, 18 However, DXR is unable to be administered systemically against GBM due to its inability to pass through the blood-brain barrier and reach therapeutic levels in the brain.19 Interstitial delivery of DXR against GBM has been explored previously with some success. Local delivery of DXR was able to extend median survival and delay tumor growth, but ultimately complete remission was not achieved.20, 21 This is likely due to the sub-optimal DXR release kinetics from the polymeric devices utilized for interstitial therapy. Lesniak et al. has shown that interstitial DXR delivered via a polyanhydride polymer wafer was able to extend median survival of Fisher rats with intracranial glioma from 21 to 45 days, despite the fact that DXR release from the polymer wafer plateaued at approximately 14% after the first 50 hours.20 Lin et al. also found that DXR released from polyester polymer device was able to slow tumor growth in a subcutaneous flank model.21 Similar to the polyanhydride wafers, the polyester device displayed an initial release of DXR that plateaued after the first 2 days. This pattern of stagnant DXR release after an initial period of rapid release over the first few days is also seen in multiple polymer drug delivery platforms including polyanhydrides,20 polyesters,21, 22 and polyethylene glycol polyester composites.23, 24 The therapeutic efficacy of interstitial GBM treatment could be greatly improved with sustained DXR release from an optimal polymer matrix.
Aggressive cancers, such as GBM, recur in a matter of weeks. As such, polymer drug delivery platforms capable of rapid and sustained drug release are vital to suppressing local tumor recurrence. Most commonly used polymers for interstitial drug delivery, such as polyesters and polyanhydrides, have relatively slow degradation rates on the order of months to years.25, 26 Additionally, polyesters degrade by bulk degradation, which can lead to spontaneous dumping of drug and potential unforeseen toxicity.27 An example of this can be seen in Manome et al., where poly(D,L-lactide-co-glycolide) (PLGA) sheets exhibited a sudden burst release of DXR around 30 days in vitro.28 Finally, polyesters create an acidic microenvironment as they degrade, which can alter drug activity and cause tissue toxicity.29–31 Taken together, these limitations regarding effective drug release and adverse polymer by-products highlight the need for a tunable drug delivery platform to expand the population of drugs that can be utilized for sustained interstitial drug delivery.
An alternative polymer for drug delivery is acetalated dextran (Ace-DEX). Ace-DEX is a novel polymer with tunable degradation rates ranging from days to months. It is synthesized by chemically modifying dextran with 2-ethyoxypropene to render the polymer hydrophobic.32, 33 This reaction generates acyclic and cyclic acetals on the pendent hydroxyl groups of dextran in a time-dependent manner.34–36 The ratio between cyclic and acyclic acetals can be changed based on the length of reaction time.34–36 Under aqueous conditions, acetal groups are hydrolyzed, revealing the parent hydroxyl groups of dextran and resulting in Ace-DEX degradation. Since cyclic acetals are significantly more stable to hydrolysis than acyclic acetals, the time-dependent nature of acetal coverage allows for tight control of Ace-DEX degradation rate. The degradation by-products of Ace-DEX (dextran, acetone, and ethanol) are pH neutral and occur at exceedingly low concentrations, far below levels that would result in toxicity.34, 35 Our group has previously fabricated drug loaded Ace-DEX nanofibrous scaffolds using electrospinning, and demonstrated tunable drug release and bioactivity.32 The tunability of Ace-DEX degradation rates combined with the safe, pH-neutral degradation products makes it a promising polymer platform for interstitial drug delivery against GBM. To advance this potential therapy towards translation to the clinic, it is imperative that we evaluate GBM treatment in the most relevant animal model.
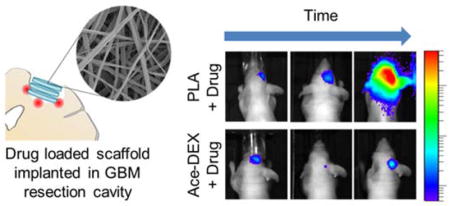
The therapeutic efficacy of interstitial therapy against GBM is often tested in a simple subcutaneous flank or orthotopic tumors that fail to incorporate an essential component of multimodal therapy: surgical resection. As surgery is the standard of care for human patients, a preclinical animal model of GBM resection and recurrence is extremely beneficial. This is particularly important because surgical resection induces changes in the peri-tumoral microenvironment and influences tumor biology, growth, and invasion.37 We previously developed a fluorescent-guided surgical mouse model of GBM resection and recurrence.38, 39 With this model, GBM xenografts are implanted orthotopically and allowed to establish for several days. Under fluorescent guidance, approximately 90% of the tumor is removed by surgical resection and interstitial therapy can then be applied at the time of surgery (Figure 1). Here we describe the fabrication and utilization of electrospun DXR loaded polymer scaffolds comprised of Ace-DEX or poly(L-lactide) (PLA), a polyester commonly used for interstitial drug delivery. We then investigated the effect of polymer on DXR release from scaffolds in vitro. DXR release rates were then evaluated in a clinically relevant murine model of local GBM recurrence following partial surgical resection.
Figure 1.

Schematic of mouse model of glioblastoma resection and recurrence. Tumors xenografts were implanted and allowed to grow for several days as monitored by bioluminescent imaging (BLI). Established tumors were then surgically resected under fluorescent guidance leaving positive tumor margins. Drug-loaded polymer scaffolds were then implanted into the surgical cavity and the wound was sealed. Post-treatment tumor growth was then monitored with BLI.
EXPERIMENTAL METHODS
Chemicals and Reagents
Dextran (molecular weight 450–650 kDa), anhydrous dimethyl sulfoxide (DMSO, >99.9%), hexafluoro-2-propanol (HFIP, >99%), 1-butanol, triethylamine (TEA, >99%), pyridinium p-toluenesulfonate (>98%), poly-L-lactide (PLA, molecular weight 260 kDa), thiazolyl blue tetrazolium bromide (MTT), and BCNU (99%) were purchased from Sigma (St. Louis, MO). Doxorubicin hydrochloride (DXR, >99%) was acquired from LCLabs (Boston, MA). 2-ethoxypropene was purchased from Matrix Scientific (Columbia, SC). High glucose Dulbecco’s Modified Eagle Medium (DMEM) cell culture medium and fetal bovine serum were obtained from Corning (Corning, NY). Penicillin/streptomycin was purchased from Hyclone (Pittsburgh, PA). Rat monoclonal antibodies against CD45 (clone I3/2.3, ab25386), F4/80 (clone BM8, #14-4801-82), and ImmPRESS HRP Anti-Rat Ig (#MP-7444-15) were acquired from Abcam (Cambridge, MA), eBioscience Inc. (San Diego, CA), and Vector Labs (Burlingame, CA), respectively. Rabbit polyclonal antibody against glial fibrillary acidic protein (GFAP, #Z0334) and protein block (#X0909) were both purchased from DAKO/Agilent Technologies (Santa Clara, CA). Bond™ Dewax Solution (AR9222), Bond™ Wash Solution (AR9590), Bond™ Epitope Retrieval Solution 1 (pH 6.0, AR9961), Bond™ Enzyme 1 (RE7160-CE), and Bond™ Polymer Refine Detection (DS9900) were all acquired from Leica Biosystems (Buffalo Grove, IL).
Cell Transfection and Cytotoxicity Assays
Human glioma cell lines U87-MG, LN-18, and LN-229 were purchased from American Type Culture Collection (ATCC; Manassas, VA; #HTB-14, #CRL-2610, and #CRL-2611, respectively). Custom vector synthesis services from Invitrogen (Carlsbad, CA) were utilized to generate mCherry-firefly luciferase (mCh-FL). The construct was packaged as a lentiviral vector in 293T/17 cells using a helper virus-free packaging system as described previously.40
Cells were cultured in DMEM medium supplemented with 1% penicillin/streptomycin and 10% fetal bovine serum and maintained in the exponential phase of growth at 37°C under 5% carbon dioxide. Cells were seeded onto 96-well plates at a density of 5 × 103 cells per well and allowed to grow to 50% confluency. DXR, at concentrations of 0.005, 0.025, 0.05, 0.1, 0.2, 0.5, 1, 2, 5, 10, 20, or 50 μM, was incubated with the cells for 48 hours. MTT powder was dissolved in media at a concentration of 0.6 mg/mL and added to treated cells for 3 hours, allowing for metabolically active cells to reduce the MTT salt to form purple formazan crystals. Crystals were dissolved in isopropanol and the absorption at 560 nm was utilized to quantify cell viability normalizing values to an untreated control, using 670 nm as a reference wavelength. Experiments were repeated in triplicate and a best fit trend-line was used to determine the dose required to reduce cell viability by 50%. This procedure was repeated for BCNU at concentrations of 0.01, 0.05, 0.1, 0.25, 0.5, 0.75, and 1 mM.
Acetalated Dextran Synthesis
Ace-DEX was synthesized and characterized as described previously.33 Briefly, lyophilized dextran was dissolved in DMSO in the presence of an acid catalyst, pyridinium p-toluenesulfonate. Dextran was reacted with 2-ethoxypropene under anhydrous conditions for 2 hours, before the reaction was quenched with TEA. Ace-DEX was precipitated in basic water and lyophilized. Afterwards, the polymer was dissolved in ethanol and centrifuged to remove impurities. Ace-DEX was then re-precipitated in basic water, lyophilized, and stored at -20˚C until it was used. The relative extent of cyclic acetal coverage, which dictates the degradation rate of the polymer, was calculated via nuclear magnetic resonance (NMR, Varian Inova 400) based on a previously developed method.33
Scaffold Fabrication
Ace-DEX or PLA was dissolved in organic solvents at 200 mg/mL with 5% or 10% (wt/wt) DXR and loaded into a glass syringe with a blunt 21 gauge needle. A bias of 20 kV (−10 kV to the collection surface and +10 kV to the needle) was applied over a 13 cm working distance at a flow rate of 1 mL/hr. A tri-solvent system consisting of HFIP, butanol, and TEA was evaluated. Maintaining TEA constant at 1% v/v, HFIP and butanol ratios were varied to create three conditions: 90% HFIP and 10% butanol, 80% HFIP and 20% butanol, and 60% HFIP and 40% butanol.
Scaffold Characterization
Scanning electron microscopy was performed to assess the morphology of the electrospun scaffolds. Scaffolds were mounted on aluminum stubs using carbon tape, and imaged at 2 kV on Hitachi S-4700 Cold Cathode Field Emission Scanning Electron Microscope. DXR encapsulation efficiency was defined as the ratio of empirical DXR loaded to theoretical loading. To quantify this, scaffold samples were weighed, dissolved in DMSO, and evaluated by optical absorption at 480 nm. The amount of DXR loaded was then determined by calibration curve. To evaluate DXR release in vitro, approximately 1 mg of scaffold was placed into dialysis cups with a molecular weight cutoff of 7 kDa and added to a sink of phosphate buffered saline (PBS, pH 7.4) stirring at 37°C. At predetermined time points, scaffolds were removed and stored at -20°C. Samples were then lyophilized, dissolved in DMSO, and evaluated by optical absorption at 480 nm. The amount of DXR retained in each sample was determined by a calibration curve.
Animals
Nude BALB/c mice were housed in groups of five in a vivarium maintained on a 12 hour light/dark schedule at 30°C and 50% relative humidity. Food and water were available ad libitum. For all surgical procedures, mice were anesthetized by vapor isoflurane and immobilized on a three point stereotactic frame (Stoelting, Kiel, WA). For all surgeries, subcutaneous carpofen (5 mg/kg) was administered prior to surgery and twice daily for 3 days following surgery for pain management.
All experimental protocols were approved by the Animal Care and Use Committees at The University of North Carolina at Chapel Hill, and care of the mice was in accordance with the standards set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals, USDA regulations, and the American Veterinary Medical Association.
Maximum Tolerated Dose
An incision was made in the skin to expose the skull of the mouse. A small circular portion of the skull over the right frontal lobe, approximately 3 mm in diameter, was surgically removed using a bone drill (Ideal Microdrill, Harvard Apparatus, Holliston, MA) and forceps. A cranial window was created by leaving this portion of the skull open. The skin was closed with Vetbond Tissue Adhesive (3M, Maplewood, MN). Several days after cranial windows were established, the dura over the cranial window was cut away, and a section of the right frontal lobe was removed by aspiration creating a pseudo resection cavity approximately 1 to 2 mm deep. DXR loaded Ace-DEX scaffolds were placed in the resection cavity and the skin was closed with tissue adhesive. Mice were evaluated daily for two weeks to assess for gross toxicity as evidenced by weight loss or gait abnormalities. After 14 days, mice were euthanized by cardiac perfusion. Brains were extracted and fixed in formalin in preparation for histology.
Therapeutic Efficacy Against Recurrent GBM
The therapeutic efficacy of drug loaded scaffolds against local GBM recurrence was evaluated in an image-guided surgical model of GBM resection and recurrence as described previously (Figure 1).37, 38 After establishment of a cranial window as detailed above, mice were again anesthetized by vapor isoflurane and immobilized on a stereotactic frame. Firefly-luciferase and mCherry transfected U87-MG cells were implanted stereotactically (1 × 105 cells in 3 μL of phosphate buffered saline at a rate of 1 μL per minute) in the right frontal lobe 2 mm lateral to the bregma and 0.5 mm below the dura. Tumor growth was monitored by bioluminescent imaging (BLI, Perkin Elmer IVIS Lumina In Vivo Imaging System). Once tumors were visualized as being well established (1 to 2 weeks after tumor implantation), mice were anesthetized and immobilized. The cranial window was exposed and dura was removed to expose the tumor. Under fluorescent guidance, the tumor was removed by aspiration. Following tumor removal, scaffolds were placed in the surgical cavity and the skin was closed with tissue adhesive. For Ace-DEX/10DXR, this required a total scaffold mass of 1.8 mg (which was equivalent to six, 3 mm circular hole punches). For PLA/10DXR this required a total scaffold mass of 1.65 mg (which was equivalent to four, 3 mm circular hole punches). Equivalent mass for blank polymer scaffolds was implanted. Serial BLI imaging was used to noninvasively quantify tumor growth starting the day after surgical resection and scaffold implantation (Day 1). A total of 42 mice were implanted with U87-MG tumors. Mice were excluded from the study on Day 1 if there was no evidence of tumor by BLI (“complete resection”, n=3), or if the BLI signal was outside two standard deviations of the average (“insufficient resection”, n=3). Mice were also excluded for extra-cranial tumor location, either in the initial tumor placement (n=2), or when the tumors recurred (n=5). Two other mice were also excluded; one began having seizures 41 days after tumor resection despite having no evidence of tumor (statistical significance remained the same regardless of inclusion); another contracted Corynebacterium bovis, which has been shown to affect tumor xenografts and drug sensitivity in nude mice.41 Excluded mice are detailed in Supporting Information Table S1. This resulted in 27 mice being included in the study with the following breakdown for groups: no treatment control (n=5), Ace-DEX/blank (n=5), PLA/blank (n=6), Ace-DEX/10DXR (n=7), PLA/10DXR (n=4). Prior to resection, Day -1, established tumors had an average BLI signal of 4.1×109 ± 2.7×109 ρ/sec/cm2/sr. The average BLI signal after resection, Day 1, was 1.8×108 ± 3.4 ×108 ρ/sec/cm2/sr. Percent of tumor resected was determined by the following equation: . The average percent of tumor resected for all groups was 95.4% ± 9%. A box plot illustrating pre-resection BLI values for each mouse and a table detailing average BLI prior to resection (Day -1), percent tumor resection, and BLI after resection (Day 1) for each treatment group can be found in Supporting Information Figure S1, Table. Mice were evaluated daily and euthanized if they lost more than 15% body weight. The study was conducted for 120 days. After euthanasia by cardiac perfusion, the brain was extracted, formalin fixed and paraffin embedded, and saved for histological evaluation.
Statistical Analysis
Statistical analysis of normalized mouse weights after scaffold implantation was performed by two way analysis of variance with post-hoc Bonferroni test using GraphPad Prism (La Jolla, CA).
Statistical analysis of overall and progression free survival rates was performed by proportional hazards regression with adjustment for the initial tumor size after surgical resection using IBM SPSS Statistics 24 (Chicago, IL).
Histology
Whole mouse brains were fixed in formalin, processed and embedded in paraffin, and step sectioned at 4 μm thickness using 300 μm gaps. Sections collected at each level were stained for histopathology using hematoxylin and eosin (H&E) and immunohistochemistry (IHC) to evaluate for signs of toxicity. H&E staining was performed using an Autostainer XL from Leica Biosystems. For IHC, rat monoclonal antibodies against CD45 and F4/80 and rabbit polyclonal antibody against glial fibrillary acidic protein (GFAP) were utilized. IHC was carried out in the fully automated Bond™ Immunostainer (Leica Biosystems Inc). Slides were dewaxed in Bond™ Dewax Solution and hydrated in Bond™ Wash Solution. Antigen retrieval for CD45 and GFAP was performed for 20 min at 100ºC in Bond™ Epitope Retrieval Solution 1 (pH 6.0) and in Bond™ Enzyme 1 for 5 minutes followed by a 10 minute protein block. After pre-treatment, slides were incubated for 30 minutes with CD45 (1:100) and GFAP (1:2500), and for 1 hour with F4/80 (1:100). Detection of all antibodies was performed using Bond™ Polymer Refine Detection. For CD45 and F4/80, the secondary antibody was replaced with ImmPRESS HRP Anti-Rat Ig. Stained slides were dehydrated and sealed with a glass coverslip. Positive and negative controls (no primary antibody) were included for each antibody. H&E and IHC stained slides were digitally imaged using an Aperio ScanScope XT (Leica Biosystems) with a 20× objective.
RESULTS
GBM Sensitivity to DXR
Sensitivity to DXR was tested in three GBM cell lines: U87-MG, LN-229, and LN-18, and then compared to BCNU (see Supporting Information Figure S2). The concentration required to reduce cell viability by 50% (IC50) after 48 hour incubation is listed in Table 1. All three GBM cell lines were more sensitive to DXR than BCNU by at least 200 fold. These are comparable to literature values.42, 43
Table 1.
Sensitivity of Glioblastoma lines to chemotherapies. Concentration required to reduce cell viability by 50% (IC50) after 48 hour incubation with DXR or BCNU for glioblastoma cell lines U87-MG, LN-18, and LN-229, as measured by MTT assay.
| Cell Line | IC50 (μM) | |
|---|---|---|
| DXR | BCNU | |
| U87-MG | 0.13 | 380 |
| LN-18 | 0.80 | 225 |
| LN-229 | 1.10 | 280 |
Scaffold Fabrication and Characterization
Drug-eluting polymer scaffolds were fabricated by electrospinning. The organic solvent system and drug loading were varied to determine their respective role in DXR release. First, Ace-DEX scaffolds containing a fixed loading of 5% (wt/wt) DXR (Ace-DEX/5DXR) were electrospun varying the solvent system ratio of HFIP to butanol to 90:10, 80:20, and 60:40. The solvent system affected both fiber width and burst release of DXR. Increasing the volume fraction of HFIP generated wider fibers (Figure 2a–c), which correlated with a faster burst release of DXR (Figure 2e), and the scaffolds exhibiting a more vibrant hue of orange, the color of DXR.
Figure 2.

Solvent System Effect on Ace-DEX Scaffolds. Scanning electron micrographs of acetalated dextran 5% (wt/wt) doxorubicin (DXR) (Ace-DEX/5DXR) scaffolds electrospun in a solvent system of hexafluoroisopropanol (HFIP) and butanol with ratios of a) 90:10, b) 80:20, and c) 60:40. d) Picture of 5% wt/wt loaded DXR scaffolds electrospun with HFIP and butanol with ratios of 90:10, 80:20, and 60:40 (from left to right). e) Burst release of DXR from Ace-DEX/5DXR scaffolds electrospun with different HFIP to butanol ratios of 90:10 (black square), 80:20 (gray triangle), and 60:40 (white circle). Data points for all graphs are the mean ± standard error of the mean. Scale bar is the same for all images and represents 5 μm.
The effect of DXR loading on release rate was also investigated. Figure 3a–c illustrates that increasing DXR loading from 5% to 10% (wt/wt) had no effect on fiber morphology. Increasing DXR loading did result in a higher burst release of DXR (Figure 3f); however, afterwards the rate of DXR release is less pronounced (data not shown). To directly compare Ace-DEX to a polyester, one of the most commonly used polymers for drug delivery, we electrospun a scaffold composed of PLA under identical electrospinning conditions with 10% DXR loaded (wt/wt) (PLA/10DXR). Ace-DEX and PLA scaffolds had similar morphology (Figure 3a–e) with high encapsulation efficiencies of 85 ± 5% and 94 ± 12%, respectively. Evaluating DXR release in vitro over time, PLA/10DXR scaffolds had a higher burst release compared to Ace-DEX/10DXR, with 46 ± 7.1% and 28 ± 1.6% of DXR released in the first 24 hours respectively. However, PLA/10DXR scaffolds only released 3% of DXR over the next 34 days. By contrast, Ace-DEX/10DXR released 27% of DXR over the same extended time frame. Although the two scaffolds ultimately released similar amounts of DXR (approximately 50% over 35 days), the steady-state release from Ace-DEX scaffolds is faster than that from PLA, which releases the majority of the DXR within the first 24 hours (Figure 3g, Table).
Figure 3.

Morphology and Drug Release from Scaffolds. Scanning electron micrographs of a) Acetalated dextran blank (Ace-DEX/blank), b) 5% (wt/wt) doxorubicin (DXR) (Ace-DEX/5DXR), c) 10% (wt/wt) DXR (Ace-DEX/10DXR), d) poly lactic acid blank (PLA/blank), and e) 10% (wt/wt) DXR PLA (PLA/10DXR) scaffolds. Scale bar is the same for all images and represents 5 μm. f) Burst release of DXR from Ace-DEX/5DXR (○) and Ace-DEX/10DXR (
 ) electrospun with 60:40 ratio of HFIP to butanol. g) DXR release from Ace-DEX/10DXR (
) and PLA/10DXR scaffolds (◆). Table describes DXR release kinetics from Ace-DEX/10DXR and PLA/10DXR scaffolds. Burst release = DXR released over the first 24 hours. Data points for all graphs are the mean ± standard error of the mean.
) electrospun with 60:40 ratio of HFIP to butanol. g) DXR release from Ace-DEX/10DXR (
) and PLA/10DXR scaffolds (◆). Table describes DXR release kinetics from Ace-DEX/10DXR and PLA/10DXR scaffolds. Burst release = DXR released over the first 24 hours. Data points for all graphs are the mean ± standard error of the mean.
Maximum Tolerated Dose
Prior to testing the efficacy of DXR loaded Ace-DEX scaffolds against GBM, in vivo toxicity was evaluated. Mice were weighed and evaluated daily for gross signs of toxicity. Scaffold mass, DXR dose, and mouse weight are detailed in Supporting Information Table S2. Mice implanted with Ace-DEX/10DXR scaffolds at 200 μg per mouse exhibited a 5% decrease in weight (see Supporting Information Figure S3). This was notable given that other groups gained approximately 5% weight over this same time period. Mice implanted with the highest DXR dose, 200 μg, also displayed delayed surgical wound healing compared to other mice, including the second highest dose, 100 μg (data not shown). When evaluated by histology, dose-dependent toxicity at doses as low as 100 μg were seen with Ace-DEX/10DXR scaffolds after 14 days (see Supporting Information Figure S4). The effect of Ace-DEX/blank scaffold compared to Ace-DEX/10DXR at the 200 μg dose was also evaluated by IHC staining. Concurrent coronal sections were stained for F4/80, GFAP, and CD45 to show presence of macrophages, activated microglia, and immune cell infiltrates, respectively, at the site of scaffold implantation (Figure 4). As demonstrated by IHC staining, higher levels of DXR led to increased glial cell activation and immune cell infiltrates, primarily macrophages, compared to unloaded Ace-DEX scaffolds.
Figure 4.

Histological Toxicity Comparison of unloaded Ace-DEX and Ace-DEX/10DXR Scaffolds. Coronal sections of mice euthanized 14 days after scaffold implantation with Left) Unloaded acetalated dextran (Ace-DEX) scaffold (total scaffold mass of 2.2 mg) and Right) 10% w/w Ace-DEX/10DXR scaffold (total scaffold mass of 2.2 mg) with total amount of doxorubicin (DXR) equal to 200 μg. Images show sections stained with hematoxylin and eosin (H&E), glial fibrillary acidic protein (GFAP, indicating glial reaction), CD45, (immune cell infiltrates), and F4/80 (macrophages). Scale bars are the equivalent for all images at the same magnification. Black boxes indicate location of magnified region of interest presented in subsequent column.
Efficacy Against Recurrent GBM
Treatment with Ace-DEX/10DXR scaffolds statistically improved survival rates compared to Ace-DEX/blank (p < 0.005) and ‘no treatment’ (p < 0.005) with four mice surviving to the end of the study (Day 120) (Figure 5a, 5b). Of these four surviving mice, only one had BLI evidence of tumor at the end of the study. Notably, the quantified BLI for this mouse decreased to just 20% of its original signal the day after resection (Day 1) over the length of the study (120 days) (Figure 5c). In contrast, only one mouse treated with PLA/10DXR survived to the end of the study, and over this time, the tumor BLI signal increased by more than eight fold (Figure 5b, 5c). Treatment with PLA/10DXR scaffolds statistically improved overall survival rates compared to ‘no treatment’ control group (p < 0.05), but was not significant compared to PLA/blank (Figure 5a). Tumor recurrence occurred for all mice in the ‘no treatment’ control group. As expected, unloaded polymer scaffolds, Ace-DEX/blank and PLA/blank, had no effect on GBM recurrence compared to ‘no treatment’ controls. U87-MG tumors regrew at exponential rates for all control mice. BLI measurements throughout the study for each mouse can be found in Supporting Information Figure S5.
Figure 5.

DXR Scaffold Efficacy against Glioblastoma. a) Kaplan-Meier curves of overall survival comparing survival rates of No Treatment (
 , n=5), acetalated dextran blank scaffold (Ace-DEX/blank,
, n=5), acetalated dextran blank scaffold (Ace-DEX/blank,
 , n=5), 10% w/w doxorubicin (DXR) Ace-DEX scaffold (Ace-DEX/10DXR,
, n=5), 10% w/w doxorubicin (DXR) Ace-DEX scaffold (Ace-DEX/10DXR,
 , n=7), polylactic acid blank scaffold (PLA/blank,
, n=7), polylactic acid blank scaffold (PLA/blank,
 , n=6), 10% w/w DXR PLA scaffold (PLA/10DXR,
, n=6), 10% w/w DXR PLA scaffold (PLA/10DXR,
 , n=4). Statistical significance by proportional hazard regression model adjusted for tumor size after resection where *p<0.05 with respect to Ace-DEX/blank, †p<0.05 and ††p<0.005 with respect to No Treatment. b) Quantified BLI the day after tumor resection (Day 1). Mice that survived to the end of the study have markers which are colored green. c) BLI images of mice who survived to the end of the study showing the reduction of BLI signal for Ace-DEX/10DXR compared to the increased signal for PLA/10DXR. BLI scale is in radiance (ρ/sec/cm2/sr). d) Kaplan-Meier curves of progression free survival rates comparing survival of No Treatment (
, n=5), Ace-DEX/blank (
, n=5), Ace-DEX/10DXR (
, n=7), PLA/blank (
, n=6), PLA/10DXR (
, n=4). Tumor progression is defined as a five-fold increase in BLI signal. Statistical significance by proportional hazard regression model adjusted for tumor size after resection where **p<0.01 with respect to Ace-DEX/blank, ††p<0.01 and †††p<0.001 with respect to No Treatment. e) Quantified BLI the day after tumor resection (Day 1) Markers indicating representative mice with comparable starting tumor sizes are highlighted blue. f) Quantified BLI of a representative mouse from each treatment group illustrating tumor growth over time (highlighted in blue in e). No Treatment (
), Ace-DEX/blank (
), Ace-DEX/10DXR (
), PLA/blank (
), PLA/10DXR (
).
, n=4). Statistical significance by proportional hazard regression model adjusted for tumor size after resection where *p<0.05 with respect to Ace-DEX/blank, †p<0.05 and ††p<0.005 with respect to No Treatment. b) Quantified BLI the day after tumor resection (Day 1). Mice that survived to the end of the study have markers which are colored green. c) BLI images of mice who survived to the end of the study showing the reduction of BLI signal for Ace-DEX/10DXR compared to the increased signal for PLA/10DXR. BLI scale is in radiance (ρ/sec/cm2/sr). d) Kaplan-Meier curves of progression free survival rates comparing survival of No Treatment (
, n=5), Ace-DEX/blank (
, n=5), Ace-DEX/10DXR (
, n=7), PLA/blank (
, n=6), PLA/10DXR (
, n=4). Tumor progression is defined as a five-fold increase in BLI signal. Statistical significance by proportional hazard regression model adjusted for tumor size after resection where **p<0.01 with respect to Ace-DEX/blank, ††p<0.01 and †††p<0.001 with respect to No Treatment. e) Quantified BLI the day after tumor resection (Day 1) Markers indicating representative mice with comparable starting tumor sizes are highlighted blue. f) Quantified BLI of a representative mouse from each treatment group illustrating tumor growth over time (highlighted in blue in e). No Treatment (
), Ace-DEX/blank (
), Ace-DEX/10DXR (
), PLA/blank (
), PLA/10DXR (
).
Non-invasive imaging with BLI offered the opportunity to track tumor growth in real time. Figure 5d illustrates progression free survival, defining ‘disease progression’ as a five-fold growth in tumor as measured by BLI. Treatment with Ace-DEX/10DXR scaffolds statistically improved progression-free survival rates compared to Ace-DEX/blank (p < 0.01) and ‘no treatment’ (p < 0.001) with four mice exhibiting no disease progression for the duration of the study. Treatment with PLA/10DXR scaffolds also statistically improved progression-free survival rates compared to ‘no treatment’ (p < 0.01), however all mice exhibited disease progression by day 38 (Table 2).
Table 2.
Statistical Analysis by Proportional Hazard Ratio (Adjusted for Tumor Size), Estimated Hazard Ratios, and 95% Confidence Intervals for Overall and Progression Free Survival Rates Across Treatment Groups.
| Treatment Group | Overall Survival | Progression Free Survival | ||
|---|---|---|---|---|
| Hazard Ratio (95% CI) | P-value | Hazard Ratio (95% CI) | P-value | |
| No Treatment | 1 | - | 1 | - |
| Ace-DEX/blank | 0.81 (0.20–3.27) | 0.766 | 0.74 (0.19–2.86) | 0.661 |
| Ace-DEX/10DXR | 0.09 (0.02–0.46) | 0.004 | 0.04 (0.01–0.25) | 0.001 |
| PLA/blank | 0.46 (0.12–1.75) | 0.254 | 0.41 (0.11–1.59) | 0.200 |
| PLA/10DXR | 0.16 (0.03–0.85) | 0.032 | 0.09 (0.02–0.54) | 0.008 |
As shown in Figure 5e, tumor size after resection was quite variable. To attempt a more uniform comparison between treatment groups, mice with similar tumor sizes are highlighted in blue in Figure 5e and their individual tumor growth is graphed in Figure 5f. In this direct comparison, treatment with Ace-DEX/10DXR led to robust tumor suppression. By comparison, treatment with PLA/10DXR led to an initial decrease in tumor size and control of tumor regrowth for the first 2 weeks, but ultimately the tumor growth rate reached exponential rates similar to untreated control mice.
DISCUSSION
GBM Sensitivity to DXR
Sensitivity to DXR was compared to BCNU, the drug currently used for interstitial GBM therapy in Gliadel® wafers, in three established human GBM cell lines: U87-MG, LN-229, and LN-18 (Table 1). GBM cells lines were found to be approximately 200 fold more sensitive to DXR than BCNU. This illustrates that DXR is highly potent against GBM and could be an enormously beneficial option for GBM patients. However, because systemic treatment with DXR is associated with severe peripheral toxicity, including irreversible cardiotoxicity, combined with the inability of DXR to cross the blood-brain barrier at therapeutic levels, DXR remains an untapped potential for GBM therapy.19, 44
Scaffold Fabrication and Characterization
We next fabricated drug-eluting polymer scaffolds for interstitial DXR therapy. Scaffolds were fabricated by electrospinning, a technique that generates a thin, flexible, fibrous scaffold which is ideal for implantation in the brain.32 Scaffold flexibility allows the implant to contour to the shape of the resection cavity, maximizing contact surface area, while the thin nature of the scaffold reduces the risk of mass effects. This is in contrast to Gliadel®, which is a hard disk that cannot easily cover contoured surfaces. Importantly, electrospinning is an inexpensive and scalable technique that can be used to control drug delivery kinetics by varying parameters such as solvent system, drug loading, and polymer platform. These variables were separately investigated to determine their role in DXR release.
To ensure sustained DXR release over an extended period of time, a slowly degrading Ace-DEX polymer with 60% relative cyclic acetal coverage was utilized for scaffold generation. First, the organic solvent system and drug loading was varied to determine their respective effects on DXR release from Ace-DEX. Varying the organic solvent system affected both fiber width and burst release of DXR. Increased volume fractions of HFIP generated wider fibers (Figure 2a–c), a faster burst release of DXR (Figure 2e), and brighter orange scaffolds (Figure 2d). These effects are likely from two separate phenomena. The increase in fiber diameter is likely due to the higher volatility of HFIP, leading to more rapid solvent evaporation. The increase in burst release is likely dependent on drug and solvent system compatibility and indicates that DXR is concentrated at the surface of the fiber rather than well distributed throughout.22, 45 This is confirmed visually, as scaffolds with higher burst releases were a more vibrant hue of orange, the color of DXR. Increasing DXR loading from 5% to 10% weight loading led to a higher burst release, likely due to the hydrophilicity of DXR. Since high DXR burst release reduces the overall drug reservoir for controlled and sustained release, the solvent system with a lower volume fraction of HFIP, 60% HFIP and 40% butanol, was used for the remainder of the studies.
Polyesters are one of the most commonly used polymers for drug delivery,21, 22, 28, 46–52 however their slow degradation rates can be a limiting factor for effective drug delivery. To investigate the role that polymer platform has on DXR release a scaffold composed of PLA was fabricated under identical electrospinning conditions to Ace-DEX. Controlling for fabrication parameters (polymer concentration, DXR loading, solvent system, and flow rate) allows for a direct comparison between Ace-DEX and PLA. Both scaffolds had nearly identical morphologies as visualized by electron microscopy (Figure 3c,e), however, the release rate of DXR varied quite drastically (Figure 3g). PLA/10DXR scaffolds were found to exhibit a high burst release followed by almost no release over the rest of the 5 week study. This is consistent with the literature, where Zeng et al. showed a rapid release of DXR from an electrospun PLA scaffold that stagnated over time.22 By contrast, Ace-DEX/10DXR released approximately 25% as a burst release and then another 27% of DXR in a controlled manner over the remaining 5 weeks. Although the two scaffolds ultimately released similar amounts of DXR (approximately 50% over 35 days), the steady-state release from Ace-DEX scaffolds is faster than that from PLA, which releases the majority of the DXR within the first 24 hours. This differential release rate will allow investigation into the role of DXR release kinetics as determined by polymer platform in controlling GBM recurrence.
Maximum Tolerated Dose
Prior to testing the efficacy of DXR loaded Ace-DEX scaffolds against GBM, in vivo toxicity was evaluated. To mimic tumor treatment, cranial windows and a resection cavity approximately 3mm in diameter and 1 to 2 mm deep were created in the parenchyma of nude mice. DXR loaded Ace-DEX scaffolds were then implanted into the surgical cavity. To increase the total dose of DXR administered, the scaffold drug loading was maintained and overall mass of scaffold implanted was increased. To achieve the highest DXR dose tested (200 μg), 2.2 mg of total Ace-DEX/10DXR scaffold mass was required. Interestingly, all but the highest dose of DXR, 200 μg, was well tolerated as measured by mouse weight loss. The tolerated dose was higher than expected based on literature where Kooistra et al. found that the highest non-toxic tolerated dose of intrathecal DXR for Sprague Dawley rats was 20 μg.53 This discrepancy may be due to the fact that the dose is not entirely contained intra-cranially (as the cranial window is left open), differences between rats and mice, and the controlled release of drug from the scaffold, compared to bolus delivery. Although by histology, dose-dependent toxicity was seen at doses as low as 100 μg, to maximize therapeutic dose within tolerable toxicity, 150 μg DXR was tested for therapeutic efficacy.
Efficacy Against Recurrent GBM
Surgical resection is part of the standard-of-care for GBM patients. However, the majority of preclinical studies are conducted in mouse or rat models that lack surgical resection of the primary tumor.20, 46, 54–56 To mimic clinical conditions, tumor cells were implanted into the parenchyma of mice. Once tumors were well established, the established primary GBM mass was resected using fluorescent-guided microsurgery. The surgical resection greatly transforms the brain microenvironment with studies showing tumors re-grow more aggressively than the original primary tumor.37 These findings underscore the significance of utilizing this clinically relevant mouse model for pre-clinical GBM therapies.
As expected, tumors for control mice (untreated and blank scaffolds) regrew rapidly. However, treatment with DXR loaded scaffolds composed of both Ace-DEX and PLA led to statistically improved survival rates compared ‘no treatment’ controls (Figure 5a, Table 2). Despite the slow steady state release of DXR from PLA/10DXR scaffolds, treatment with PLA/10DXR extended median survival from 29 to 63 days over PLA/blank. However, the lack of statistical significance differs from previous reports in the literature where Lesniak et al.20 and Lin et al.21 found that DXR loaded polyester or polyanhydride devices were able to significantly extend median survival and delay tumor growth over empty polymer devices. Since overall and progression free survival rates of PLA/10DXR were significant when compared to the ‘no treatment’ control group, the low sample size of our study may be a contributing factor in the lack of statistical significance when compared to PLA/blank. Additionally, the surgical murine model used in this study is unique in its clinical significance, with more aggressive tumors recurrence than primary GBM models. The complexity of this model may have highlighted limitations with PLA delivery of DXR. Moreover, the total therapeutic dose of DXR implanted for interstitial therapy in this work was lower than previous studies.20, 21 This suggests that the ratio of drug to tumor size may be an important factor in interstitial therapy.
As evident in Figure 5e, residual tumor size after resection was quite variable. Unfortunately, because tumors do not grow homogenously and surgical conditions are not equivalent, it is difficult to ensure identical residual tumor size after resection. Although this variation is difficult in a research setting, this model closely mimics the clinical setting where incomplete or insufficient tumor resection outcomes are often a reality. In this way, the range of residual tumors at the start of treatment allows some insight into the robustness of each therapy. Not surprisingly, residual tumor size after surgical resection played a significant role in therapeutic efficacy. For the PLA/10DXR group, mice with larger residual tumors after surgical resection succumbed to tumors faster than those with smaller residual tumors. This is likely due to the slow steady state release of DXR from PLA scaffolds which allowed tumor growth to outpace DXR release. The success of mice treated with Ace-DEX/10DXR was also dependent on residual tumor size. Mice with smaller residual tumors were more responsive to therapy than those with larger tumors. However, in contrast with PLA/10DXR, the size threshold for tumor recurrence was much higher for Ace-DEX/10DXR.
Due to this residual tumor size discrepancy, a more uniform comparison between therapies can be made by comparing representative mice with similar sized tumors after resection (highlighted in blue in Figure 5e). In this case, PLA/10DXR resulted in an initial decrease in tumor burden, possibly due to the burst release of DXR from the scaffold; however, tumor regrowth ultimately outpaced the slow steady state DXR release from the PLA scaffold (Figure 5f). In contrast, the release of DXR from Ace-DEX/10DXR scaffolds was high enough to result in complete remission for a residual tumor of the same size. This direct comparison helps to highlight the benefits of the higher steady state DXR release from Ace-DEX scaffolds.
CONCLUSIONS
Despite current treatments, local GBM recurrence and associated mortality is almost 100%. This is complicated by the fact that the blood-brain barrier limits efficacy of systemic chemotherapy. To combat recurrence and overcome the blood-brain barrier, local drug delivery to the tumor resection site is highly advantageous. Judicious selection of a polymer platform is essential to ensure sustained drug delivery. The most commonly used polymers for drug delivery are limited by slow degradation rates. Previous reports of polymeric delivery of DXR exhibited similar trends: rapid release of DXR over the first few days followed by slow steady state release. Although this led to improved median survival or tumor growth delays, to our knowledge no complete remissions have been achieved.
Here we addressed the need for an improved polymer platform for sustained interstitial drug delivery to prevent GBM recurrence. DXR was incorporated into nanofibrous scaffolds composed of PLA or Ace-DEX to directly compare the effect of polymer on DXR release rate. Although both DXR loaded scaffolds ultimately release approximately the same amount of DXR, PLA/10DXR scaffolds released the full payload within the first 24 hours, whereas Ace-DEX/10DXR scaffolds exhibited controlled and sustained release of DXR over the same period. PLA/10DXR scaffolds extended progression free survival over ‘no treatment’ control; however, all mice in this treatment group exhibited tumor growth by 38 days after treatment. The higher sustained DXR release from Ace-DEX scaffolds led to more robust suppression of tumor recurrence, leading to complete remission in 43% of mice. Future studies will further explore the tunability of the Ace-DEX polymer platform, to determine the optimal degradation rate to maximally suppress GBM recurrence.
Supplementary Material
Figure S1. Tumor Size Prior to Treatment
Figure S2. Sensitivity of Glioblastoma Cell Lines to DXR
Figure S3. Toxicity of Scaffolds In Vivo
Figure S4. Histological Toxicity Increasing Doses of DXR in Ace-DEX scaffolds
Figure S5. Tumor growth after tumor resection
Table S1. Mice Excluded from Therapeutic Efficacy Study
Table S2. Toxicity of Scaffolds In Vivo
Acknowledgments
Funding Sources
This work was supported by the PhRMA Foundation, internal funds from University of North Carolina Chapel Hill, and funding from the NIH (R01NS097507-02). CRM was supported by the UNC University Cancer Research Fund (UCRF), NIH National Center for Advancing Translational Sciences (550KR61332), and NIH (R01CA204136). The UNC Translational and Clinical Sciences Institute (TraCS) is supported by the NCATS and NIH, through grant award number UL1TR001111. The UNC Translational Pathology Laboratory is supported in part, by grants from the NCI (2-P30-CA016086-40), NIEHS (2-P30ES010126-15A1), UCRF, and NCBT (2015-IDG-1007). The UNC Lineberger Comprehensive Cancer Center (LCCC) Animal Studies Core is supported in part by an NCI Center Core Support Grant (CA16086) to the UNC LCCC.
We thank the UNC LCCC Animal Studies Core for assistance with animal studies. We thank Xia Yongjuan in the UNC Translational Pathology Laboratory (TPL) and the Animal Histopathology Core Lab for expert technical assistance. We thank the Chapel Hill Analytical and Nanofabrication Laboratory (CHANL) for SEM usage, the Biomedical Research Imaging Core (BRIC) for IVIS usage, the School of Pharmacy’s NMR Core, and the UNC-Olympus Imaging Research Center for microscope usage.
ABBREVIATIONS
- AceDEX
acetalated dextran
- Ace-DEX/5DXR
5% wt/wt doxorubicin loaded acetalated dextran scaffold
- Ace-DEX/10DXR
10% wt/wt doxorubicin loaded acetalated dextran scaffold
- BCNU
carmustine
- BLI
bioluminescent imaging
- DNA
deoxyribonucleic acid
- DMSO
dimethyl sulfoxide
- DXR
doxorubicin
- DMEM
Dulbecco’s Modified Eagle Medium
- GBM
glioblastoma
- GFAP
glial fibrillary acidic protein
- H&E
hematoxylin and eosin
- HFIP
hexafluoro-2-propanol
- IHC
immunohistochemistry
- mCh-FL
mCherry-firefly luciferase
- MGMT
O6-methylguanine-DNA methyltransferase
- MTT
thiazolyl blue tetrazolium bromide
- NMR
nuclear magnetic resonance
- PLGA
poly(D,L-lactide-co-glycolide)
- PLA
poly(L-lactide)
- PLA/10DXR
10% wt/wt doxorubicin loaded poly(L-lactide) scaffold
- TEA
triethylamine
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Erpolat OP, Akmansu M, Goksel F, Bora H, Yaman E, Buyukberber S. Outcome of newly diagnosed glioblastoma patients treated by radiotherapy plus concomitant and adjuvant temozolomide: a long-term analysis. Tumori. 2009;95(2):191–7. doi: 10.1177/030089160909500210. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The lancet oncology. 2009;10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Kauer TM, Figueiredo J-L, Hingtgen S, Shah K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat Neurosci. 2012;15(2):197–204. doi: 10.1038/nn.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gaspar LE, Fisher BJ, Macdonald DR, Leber DV, Halperin EC, Schold SC, Cairncross JG. Supratentorial malignant glioma: Patterns of recurrence and implications for external beam local treatment. International Journal of Radiation Oncology*Biology*Physics. 1992;24(1):55–57. doi: 10.1016/0360-3016(92)91021-e. [DOI] [PubMed] [Google Scholar]
- 5.Lee SW, Fraass BA, Marsh LH, Herbort K, Gebarski SS, Martel MK, Radany EH, Lichter AS, Sandler HM. Patterns of failure following high-dose 3-D conformal radiotherapy for high-grade astrocytomas: a quantitative dosimetric study. International Journal of Radiation Oncology*Biology*Physics. 1999;43(1):79–88. doi: 10.1016/s0360-3016(98)00266-1. [DOI] [PubMed] [Google Scholar]
- 6.Burger Peter C, Dubois Philip J, Clifford Schold SJ, Smith Kenneth RJ, Odom Guy L, Crafts David C, Giangaspero Felice. Computerized tomographic and pathologic studies of the untreated, quiescent, and recurrent glioblastoma multiforme. Journal of Neurosurgery. 1983;58(2):159–169. doi: 10.3171/jns.1983.58.2.0159. [DOI] [PubMed] [Google Scholar]
- 7.De Bonis P, Anile C, Pompucci A, Fiorentino A, Balducci M, Chiesa S, Lauriola L, Maira G, Mangiola A. The influence of surgery on recurrence pattern of glioblastoma. Clinical Neurology and Neurosurgery. 2013;115(1):37–43. doi: 10.1016/j.clineuro.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Adamson C, Kanu OO, Mehta AI, Di C, Lin N, Mattox AK, Bigner DD. Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin Investig Drugs. 2009;18(8):1061–83. doi: 10.1517/13543780903052764. [DOI] [PubMed] [Google Scholar]
- 9.Bregy A, Shah AH, Diaz MV, Pierce HE, Ames PL, Diaz D, Komotar RJ. The role of Gliadel wafers in the treatment of high-grade gliomas. Expert Rev Anticancer Ther. 2013;13(12):1453–61. doi: 10.1586/14737140.2013.840090. [DOI] [PubMed] [Google Scholar]
- 10.Brem H, Gabikian P. Biodegradable polymer implants to treat brain tumors. J Control Release. 2001;74(1–3):63–7. doi: 10.1016/s0168-3659(01)00311-x. [DOI] [PubMed] [Google Scholar]
- 11.Brem H, Piantadosi S, Burger PC, Walker M, Selker R, Vick NA, Black K, Sisti M, Brem S, Mohr G, et al. Placebo-controlled trial of safety, efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas, The Polymer-brain Tumor Treatment Group. Lancet. 1995;345(8956):1008–12. doi: 10.1016/s0140-6736(95)90755-6. [DOI] [PubMed] [Google Scholar]
- 12.Grossman SA, Reinhard C, Colvin OM, Chasin M, Brundrett R, Tamargo RJ, Brem H. The intracerebral distribution of BCNU delivered by surgically implanted biodegradable polymers. J Neurosurg. 1992;76(4):640–7. doi: 10.3171/jns.1992.76.4.0640. [DOI] [PubMed] [Google Scholar]
- 13.Arifin DY, Lee KY, Wang CH. Chemotherapeutic drug transport to brain tumor. J Control Release. 2009;137(3):203–10. doi: 10.1016/j.jconrel.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 14.Arifin DY, Lee KY, Wang CH, Smith KA. Role of convective flow in carmustine delivery to a brain tumor. Pharm Res. 2009;26(10):2289–302. doi: 10.1007/s11095-009-9945-8. [DOI] [PubMed] [Google Scholar]
- 15.Bobola MS, Silber JR, Ellenbogen RG, Geyer JR, Blank A, Goff RD. O6-Methylguanine-DNA Methyltransferase, O6-Benzylguanine, and Resistance to Clinical Alkylators in Pediatric Primary Brain Tumor Cell Lines. Clin Cancer Res. 2005;11(7):2747–2755. doi: 10.1158/1078-0432.CCR-04-2045. [DOI] [PubMed] [Google Scholar]
- 16.Hermisson M, Klumpp A, Wick W, Wischhusen J, Nagel G, Roos W, Kaina B, Weller M. O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells. Journal of Neurochemistry. 2006;96(3):766–776. doi: 10.1111/j.1471-4159.2005.03583.x. [DOI] [PubMed] [Google Scholar]
- 17.Eom Y-W, Kim MA, Park SS, Goo MJ, Kwon HJ, Sohn S, Kim W-H, Yoon G, Choi KS. Two distinct modes of cell death induced by doxorubicin: apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene. 2005;24(30):4765–4777. doi: 10.1038/sj.onc.1208627. [DOI] [PubMed] [Google Scholar]
- 18.Gewirtz D. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochemical Pharmacology. 1999;57(7):727–741. doi: 10.1016/s0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- 19.Ohnishi T, Tamai I, Sakanaka K, Sakata A, Yamashima T, Yamashita J, Tsuji A. In vivo and in vitro evidence for ATP-dependency of P-glycoprotein-mediated efflux of doxorubicin at the blood-brain barrier. Biochemical Pharmacology. 1995;49(10):1541–1544. doi: 10.1016/0006-2952(95)00082-b. [DOI] [PubMed] [Google Scholar]
- 20.Lesniak MS, Upadhyay U, Goodwin R, Tyler B, Brem H. Local delivery of doxorubicin for the treatment of malignant brain tumors in rats. Anticancer Res. 2005;25(6B):3825–31. [PMC free article] [PubMed] [Google Scholar]
- 21.Lin SY, Cheng LF, Lui WY, Chen CF, Han SH. Tumoricidal Effect of Controlled-release Polymeric Needle Devices Containing Adriamycin Hc1 in Tumor-bearing Mice. Biomaterials, Artificial Cells and Artificial Organs. 1989;17(2):189–203. doi: 10.3109/10731198909118280. [DOI] [PubMed] [Google Scholar]
- 22.Zeng J, Yang L, Liang Q, Zhang X, Guan H, Xu X, Chen X, Jing X. Influence of the drug compatibility with polymer solution on the release kinetics of electrospun fiber formulation. J Control Release. 2005;105(1–2):43–51. doi: 10.1016/j.jconrel.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 23.Lu T, Jing X, Song X, Wang X. Doxorubicin-loaded ultrafine PEG-PLA fiber mats against hepatocarcinoma. J Appl Polym Sci. 2012;123(1):209–217. [Google Scholar]
- 24.Xu X, Yang L, Xu X, Wang X, Chen X, Liang Q, Zeng J, Jing X. Ultrafine medicated fibers electrospun from W/O emulsions. J Control Release. 2005;108(1):33–42. doi: 10.1016/j.jconrel.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 25.Li S, Garreau H, Vert M. Structure-property relationships in the case of the degradation of massive poly(α-hydroxy acids) in aqueous media. Journal of Materials Science: Materials in Medicine. 1990;1(4):198–206. [Google Scholar]
- 26.Li S. Hydrolytic degradation characteristics of aliphatic polyesters derived from lactic and glycolic acids. Journal of Biomedical Materials Research. 1999;48(3):342–353. doi: 10.1002/(sici)1097-4636(1999)48:3<342::aid-jbm20>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 27.Langer R. New Methods of Drug Delivery. Science. 1990;249(4976):1527–1533. doi: 10.1126/science.2218494. [DOI] [PubMed] [Google Scholar]
- 28.Manome Y, Kobayashi T, Mori M, Suzuki R, Funamizu N, Akiyama N, Inoue S, Tabata Y, Watanabe M. Local delivery of doxorubicin for malignant glioma by a biodegradable PLGA polymer sheet. Anticancer Res. 2006;26(5A):3317–26. [PubMed] [Google Scholar]
- 29.Sung HJ, Meredith C, Johnson C, Galis ZS. The effect of scaffold degradation rate on three-dimensional cell growth and angiogenesis. Biomaterials. 2004;25(26):5735–5742. doi: 10.1016/j.biomaterials.2004.01.066. [DOI] [PubMed] [Google Scholar]
- 30.Ding AG, Shenderova A, Schwendeman SP. Prediction of Microclimate pH in Poly(lactic-co-glycolic Acid) Films. Journal of the American Chemical Society. 2006;128(16):5384–5390. doi: 10.1021/ja055287k. [DOI] [PubMed] [Google Scholar]
- 31.Fu K, Pack D, Klibanov A, Langer R. Visual Evidence of Acidic Environment Within Degrading Poly(lactic-co-glycolic acid) (PLGA) Microspheres. Pharmaceutical Research. 2000;17(1):100–106. doi: 10.1023/a:1007582911958. [DOI] [PubMed] [Google Scholar]
- 32.Borteh HM, Gallovic MD, Sharma S, Peine KJ, Miao S, Brackman DJ, Gregg K, Xu Y, Guo X, Guan J, Bachelder EM, Ainslie KM. Electrospun acetalated dextran scaffolds for temporal release of therapeutics. Langmuir. 2013;29(25):7957–65. doi: 10.1021/la400541e. [DOI] [PubMed] [Google Scholar]
- 33.Bachelder EM, Beaudette TT, Broaders KE, Dashe J, Fréchet JMJ. Acetal-Derivatized Dextran: An Acid-Responsive Biodegradable Material for Therapeutic Applications. Journal of the American Chemical Society. 2008;130(32):10494–10495. doi: 10.1021/ja803947s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kauffman KJ, Do C, Sharma S, Gallovic MD, Bachelder EM, Ainslie KM. Synthesis and characterization of acetalated dextran polymer and microparticles with ethanol as a degradation product. ACS Appl Mater Interfaces. 2012;4(8):4149–55. doi: 10.1021/am3008888. [DOI] [PubMed] [Google Scholar]
- 35.Broaders KE, Cohen JA, Beaudette TT, Bachelder EM, Frechet JM. Acetalated dextran is a chemically and biologically tunable material for particulate immunotherapy. Proc Natl Acad Sci U S A. 2009;106(14):5497–502. doi: 10.1073/pnas.0901592106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bachelder EM, Beaudette TT, Broaders KE, Paramonov SE, Dashe J, Frechet JM. Acid-degradable polyurethane particles for protein-based vaccines: biological evaluation and in vitro analysis of particle degradation products. Mol Pharm. 2008;5(5):876–84. doi: 10.1021/mp800068x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okolie O, Bago JR, Schmid RS, Irvin DM, Bash RE, Miller CR, Hingtgen SD. Reactive astrocytes potentiate tumor aggressiveness in a murine glioma resection and recurrence model. Neuro Oncol. 2016;18(12):1622–1633. doi: 10.1093/neuonc/now117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hingtgen S, Figueiredo JL, Farrar C, Duebgen M, Martinez-Quintanilla J, Bhere D, Shah K. Real-time multi-modality imaging of glioblastoma tumor resection and recurrence. J Neurooncol. 2013;111(2):153–61. doi: 10.1007/s11060-012-1008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kauer TM, Figueiredo JL, Hingtgen S, Shah K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat Neurosci. 2011;15(2):197–204. doi: 10.1038/nn.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sena-Esteves M, Tebbets JC, Steffens S, Crombleholme T, Flake AW. Optimized large-scale production of high titer lentivirus vector pseudotypes. J Virol Methods. 2004;122(2):131–9. doi: 10.1016/j.jviromet.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 41.Burr HN, Lipman NS, White JR, Zheng J, Wolf FR. Strategies to Prevent, Treat, and Provoke Corynebacterium-Associated Hyperkeratosis in Athymic Nude Mice. Journal of the American Association for Laboratory Animal Science : JAALAS. 2011;50(3):378–388. [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang S, Han L, Wei J, Shi Z, Pu P, Zhang J, Yuan X, Kang C. Combination treatment with doxorubicin and microRNA-21 inhibitor synergistically augments anticancer activity through upregulation of tumor suppressing genes. Int J Oncol. 2015;46(4):1589–600. doi: 10.3892/ijo.2015.2841. [DOI] [PubMed] [Google Scholar]
- 43.Weller M, Rieger J, Grimmel C, Van Meir EG, De Tribolet N, Krajewski S, Reed JC, von Deimling A, Dichgans J. Predicting chemoresistance in human malignant glioma cells: the role of molecular genetic analyses. Int J Cancer. 1998;79(6):640–4. doi: 10.1002/(sici)1097-0215(19981218)79:6<640::aid-ijc15>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 44.Volkova M, Russell R. Anthracycline Cardiotoxicity: Prevalence, Pathogenesis and Treatment. Current Cardiology Reviews. 2011;7(4):214–220. doi: 10.2174/157340311799960645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seif S, Franzen L, Windbergs M. Overcoming drug crystallization in electrospun fibers--Elucidating key parameters and developing strategies for drug delivery. Int J Pharm. 2015;478(1):390–7. doi: 10.1016/j.ijpharm.2014.11.045. [DOI] [PubMed] [Google Scholar]
- 46.Ranganath SH, Fu Y, Arifin DY, Kee I, Zheng L, Lee HS, Chow PK, Wang CH. The use of submicron/nanoscale PLGA implants to deliver paclitaxel with enhanced pharmacokinetics and therapeutic efficacy in intracranial glioblastoma in mice. Biomaterials. 2010;31(19):5199–207. doi: 10.1016/j.biomaterials.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 47.Ranganath SH, Wang CH. Biodegradable microfiber implants delivering paclitaxel for post-surgical chemotherapy against malignant glioma. Biomaterials. 2008;29(20):2996–3003. doi: 10.1016/j.biomaterials.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 48.Lin SY, Cheng LF, Lui WY, Wu LH, Kao SJ, Han SH. Controlled release of adriamycin HCl from polymeric needle devices. Biomater Artif Cells Artif Organs. 1988;16(4):801–14. doi: 10.3109/10731198809117570. [DOI] [PubMed] [Google Scholar]
- 49.Lee LY, Ranganath SH, Fu Y, Zheng JL, Lee HS, Wang C-H, Smith KA. Paclitaxel release from micro-porous PLGA disks. Chemical Engineering Science. 2009;64(21):4341–4349. [Google Scholar]
- 50.Ong BY, Ranganath SH, Lee LY, Lu F, Lee HS, Sahinidis NV, Wang CH. Paclitaxel delivery from PLGA foams for controlled release in post-surgical chemotherapy against glioblastoma multiforme. Biomaterials. 2009;30(18):3189–96. doi: 10.1016/j.biomaterials.2009.02.030. [DOI] [PubMed] [Google Scholar]
- 51.Xie J, Wang CH. Electrospun micro- and nanofibers for sustained delivery of paclitaxel to treat C6 glioma in vitro. Pharm Res. 2006;23(8):1817–26. doi: 10.1007/s11095-006-9036-z. [DOI] [PubMed] [Google Scholar]
- 52.Huang HH, He CL, Wang HS, Mo XM. Preparation of core-shell biodegradable microfibers for long-term drug delivery. J Biomed Mater Res A. 2009;90(4):1243–51. doi: 10.1002/jbm.a.32543. [DOI] [PubMed] [Google Scholar]
- 53.Kooistra KL, Rodriguez M, Powis G. Toxicity of intrathecally administered cytotoxic drugs and their antitumor activity against an intrathecal Walker 256 carcinosarcoma model for meningeal carcinomatosis in the rat. Cancer Res. 1989;49(4):977–82. [PubMed] [Google Scholar]
- 54.Walter KA, Cahan MA, Gur A, Tyler B, Hilton J, Colvin OM, Burger PC, Domb A, Brem H. Interstitial Taxol Delivered from a Biodegradable Polymer Implant against Experimental Malignant Glioma. Cancer Res. 1994;54(8):2207–2212. [PubMed] [Google Scholar]
- 55.Ruan S, Yuan M, Zhang L, Hu G, Chen J, Cun X, Zhang Q, Yang Y, He Q, Gao H. Tumor microenvironment sensitive doxorubicin delivery and release to glioma using angiopep-2 decorated gold nanoparticles. Biomaterials. 2015;37:425–35. doi: 10.1016/j.biomaterials.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 56.Cui Y, Xu Q, Chow PK, Wang D, Wang CH. Transferrin-conjugated magnetic silica PLGA nanoparticles loaded with doxorubicin and paclitaxel for brain glioma treatment. Biomaterials. 2013;34(33):8511–20. doi: 10.1016/j.biomaterials.2013.07.075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Tumor Size Prior to Treatment
Figure S2. Sensitivity of Glioblastoma Cell Lines to DXR
Figure S3. Toxicity of Scaffolds In Vivo
Figure S4. Histological Toxicity Increasing Doses of DXR in Ace-DEX scaffolds
Figure S5. Tumor growth after tumor resection
Table S1. Mice Excluded from Therapeutic Efficacy Study
Table S2. Toxicity of Scaffolds In Vivo