

Abstract
Rationale
GLT-1 is the major glutamate transporter in the brain and is expressed predominantly in astrocytes but is also present in excitatory axon terminals. To understand the functional significance of GLT-1 expressed in neurons, we generated a conditional GLT-1 knockout mouse and inactivated GLT-1 in neurons using Cre-recombinase expressed under the synapsin 1 promoter, (synGLT-1 KO).
Objectives
Abnormalities of glutamate homeostasis have been shown to affect hippocampal-related behaviors including learning and memory as well as responses to drugs of abuse. Here, we asked whether deletion of GLT-1 specifically from neurons would affect behaviors that assessed locomotor activity, cognitive function, sensorimotor gating, social interaction, as well as amphetamine-stimulated locomotor activity.
Methods/Results
We found that the neuronal GLT-1 KO mice performed similarly to littermate controls in the behavioral tests we studied. Although performance in open field testing was normal, the acute locomotor response to amphetamine was significantly blunted in the synGLT-1 KO (40% of control). We found no change in amphetamine-stimulated extracellular dopamine in the medial shell of the nucleus accumbens, no change in electrically stimulated or amphetamine-induced dopamine release, and no change in dopamine tissue content.
Conclusions
These results support the view that GLT-1 expression in neurons is required for amphetamine-induced behavioral activation, and suggest that this phenotype is not produced through a change in dopamine uptake or release. Although GLT-1 is highly expressed in neurons in the CA3 region of the hippocampus, the tests used in this study were not able to detect a behavioral phenotype referable to hippocampal dysfunction.
Keywords: glutamate transporter, glutamate homeostasis, glutamate uptake, behavior, amphetamine, locomotor response, dopamine
Introduction
Glutamate homeostasis is regulated by cellular processes releasing and clearing glutamate from the extracellular space. Glutamate clearance is attained via a family of Na+-dependent glutamate transporters (GLAST, GLT-1, EAAC1, EAAT4, and EAAT5). Of these, GLT-1 is responsible for >90% of synaptosomal glutamate uptake (Tanaka et al., 1997; Danbolt, 2001). While previously thought to be expressed solely in astrocytes (Rothstein et al., 1994; Danbolt, 2001), GLT-1 has been identified as the major, if not the only, glutamate transporter associated with excitatory terminals (Chen et al., 2004; Berger et al., 2005; Furness et al., 2008). In order to understand the specific functions of GLT-1 expressed in excitatory terminals, we generated a conditional knockout mouse line whereby GLT-1 could be deleted specifically from astrocytes using GFAP-CreERT2 or from neurons using synapsin-Cre (synGLT-1 KO) (Petr et al., 2015). We recently reported that knockout of neuronal, but not astrocytic, GLT-1 produces a significant reduction of 3H-L-glutamate uptake into forebrain synaptosomes (Petr et al., 2015), which is remarkable given that neuronal GLT-1 accounts for only 5–10% of total GLT-1 protein expression (Furness et al., 2008). Mice lacking GLT-1 in neurons were found to be grossly normal, with normal weight gain, survival up to one year, and no seizures. Thus, the function of GLT-1 protein expressed in neurons remains an unanswered question and one of considerable interest (Danbolt et al., 2016; Rimmele and Rosenberg, 2016).
Here we have utilized the conditional GLT-1 knockout to determine whether loss of GLT-1 expressed in neurons is associated with behavioral abnormalities. We carried out a wide array of behavioral tests to assess locomotor activity, cognitive function, sensorimotor gating, and social interaction. Additionally, since responses to the monoaminergic psychostimulant amphetamine (AMP) are highly modulated by glutamate receptors (Wolf, 1998; Luscher and Malenka, 2011; Wolf and Tseng, 2012) and by changes in glutamate homeostasis (Underhill et al., 2014; Scofield et al., 2016; Mingote et al., 2017) it seemed likely that such responses would be affected by deletion of GLT-1 if neuronal GLT-1 were to contribute significantly to glutamate homeostasis. Therefore, we also tested the synGLT-1 KO mice for their locomotor response to AMP. We found that synGLT-1 KO mice were relatively resistant to the locomotor effects of AMP, and so assayed dopamine (DA) release by microdialysis and cyclic voltammetry, as well as total DA in various regions in synGLT-1 KO and littermate controls to characterize possible differences in DA dynamics that might help explain the observed phenotype.
Materials and Methods
Animals
We crossed GLT-1flox/flox mice (on a mixed 129SvJ-C57BL/6 background) with a synapsin 1-cre (SynCre) transgenic mouse line (on a C57BL/6 background; JAX Stock No. 003966) that expresses Cre-recombinase under the neuron-specific promoter, synapsin 1, as described previously (Petr et al., 2015). From this pairing, we obtained SynCre(+); GLT-1 floxed mice (neuron specific GLT-1 knockout mice), and SynCre(−); GLT-1 floxed mice (WT littermate control mice). SynCre was introduced only through females, due to known synapsin expression in the testes (Rempe et al., 2006). The GLT-1 floxed mice were bred to C57BL/6J mice for 10 generations. Congenicity with C57BL/6J was confirmed by Jackson Laboratory’s C57BL/6 substrain characterization SNP panel. Experiments with synapsin-Cre were performed with mice on a 129XC57BL/6J background that was 87.5% C57BL/6J. In order to determine that our observations were not due to cre-recombinase, per se, we generated and used synapsin-Cre control mice in the experiments by which significant differences between genotypes were obtained. All animal experiments were carried out in accordance with NIH guidelines, and were approved by the Children’s Hospital Boston Institutional Animal Care and Use Committee. Animals were maintained on a 12-h light-dark cycle; all experiments were conducted during the light phase. All experiments were conduced on adult male mice 12–16 weeks of age, using age matched littermates as controls. Food and water were available ad libitum. For behavioral experiments, separate groups of mice were used for each type of behavioral experiment with the exception of one group of mice that was first tested in an open field followed by fear conditioning.
Behavioral Testing
Open Field
Locomotor activity and habituation to a novel open field environment were assessed using circular, open field arenas [15 cm (radius) × 10 cm (height)]. The lights remained on in the testing room. In the open field, the following parameters were measured: total distance traveled, thigmotaxis, and number of movement initiations. Mice were exposed to the open field arena for 60 min and behavior was recorded with Ethovision XT v10 video tracking software (Noldus, The Netherlands) using center-point detection with dynamic subtraction. For total distance traveled, minimum distance of tracking was set to 1 cm along the path of movement. These data were recorded and analyzed in 10-min bins. For thigmotaxis assessment, the amount of time spent in each of three predefined zones, termed here as wall, neutral, and center zones, in the open field was measured. The wall zone was set to begin at the edge of the circular arena and was set to 5 cm wide. The center zone was set to 15 cm in diameter. The neutral zone was the width of the space between the wall and center zones, set to 20 cm wide. The amount of time that the center point of the mouse was in each zone was recorded and analyzed. The number of movement initiations was measured by analyzing the frequency that the mouse moved at a set speed of 2.0 cm/s or greater.
Rotorod
An automated 4-lane rotarod apparatus (Economex, Columbus Instruments, USA) with a mouse rod (3.5 cm diameter) was used. The training session comprised of two stages: 1) teach the task: mice were placed on the rotarod at 4 revolutions per min (rpm) for 5 min; 2) habituation to handling: place the mice on the rod at 4 rpm, take them off, repeat 5 times. Animals were trained for two sessions before the experiment, one session per day. For rotarod testing, the rod was set to accelerate from 4 to 34 rpm over 5 min (0.1 accel), and the time to fall was recorded. Mice received 4 trials (5 min between trials) and the results were averaged to obtain a single value for each animal; the averaged value was used for statistical analyses.
DigiGait
Gait assessment using video recordings was performed by the DigiGait imaging apparatus (DigiGait, Mouse Specifics, Inc., Boston, MA). Mice were placed on a motor-driven treadmill within a plexiglass compartment (~25 cm long and ~5 cm wide). A high-speed digital video camera was mounted underneath the transparent treadmill belt to visualize paw contacts. The treadmill was set at a fixed speed of 20 cm/s at which most animals that tested here were able to run continuously. The videos (~5 s) were analyzed by the DigiGait software to calculate stride duration and stride length for each limb.
Morris Water Maze
Spatial memory performance was assessed using the Morris Water Maze task as previously described (Morris, 1984; Mannix et al., 2011). A white pool [41.5 cm (radius) × 60 cm (depth)] was filled with water to 29 cm depth and maintained at a temperature of 24° C. The platform was positioned 1 cm below water surface. Across all trials, mice were positioned at each of one of 4 different starting locations. For hidden platform training, mice were exposed to 2 trials per day for 2 consecutive days and a 5th trial on the following day. Mice were given a maximum of 90 s to locate the submerged platform. Mice that did not locate the platform within 90 s were placed on the platform by the experimenter where they were allowed to remain for 10 s. The probe trial occurred ~1 h following the hidden platform training trial on day 3. For the probe trial, the platform was removed, and the time that the animal swam in the target quadrant was recorded (maximum 60 s). During hidden platform training, time to reach the platform was recorded and analyzed. During the probe trial, percent time in the target quadrant was recorded and analyzed. Across all trials, times were recorded directly by an experimenter blind to the genotype of the mouse being evaluated.
Fear Conditioning
Fear conditioning to cues and context was assessed according to the standard procedure (Saxe et al., 2006). Conditioning was conducted in Any-Maze fear conditioning chambers [17 cm (length) × 17 cm (width) × 15 cm (height)] (Stolting, Wood Dale, IL). Each chamber was located inside a larger sound and light attenuating chamber that prevented entry of outside noise and light. Each chamber was equipped with a camera, cue light, and speaker as well as an electrified grid floor. The detection of freezing was automated, and based on video analysis, using Any-Maze software. Freezing was defined as the complete absence of motion, for a minimum of 0.5 s.
Fear conditioning occurred over three consecutive days and each mouse was exposed to one session/day. On day 1 (training), each mouse was placed in a fear conditioning chamber and allowed to habituate for 5 min. Mice were then presented with 2 tone cues alone (1500 Hz), followed by 3 tone cues that co-terminated with the delivery of a 2 s footshock (0.4 mA). Each tone lasted 30 s and the duration of the training session was 8 min. Freezing behavior was scored before and after the end of each tone/footshock. On day 2 (tone cue test), each mouse was placed into the same chamber with a different contextual background (black and white striped wall inserts vs. no wall inserts on day 1). Mice were allowed to habituate to the chambers for 5 min and were subsequently presented with 5 (30 s) tones without footshock. The tone cue session lasted 8 min. Freezing behavior was scored before and after delivery of each tone. On day 3 (context test), each mouse was placed into the same chamber containing the original context from day 1 (chamber only, no wall inserts) and freezing was scored for the duration of the test (8 min). The percentage of freezing time was used for statistical analysis.
Novel Object Recognition
Novel Object Recognition was conducted similarly to the methods established by (Ennaceur and Delacour, 1988). On days 1–3, mice were habituated to the same open field arenas mentioned above for Open Field testing. Each habituation session lasted 15 min and during this time no objects were present in the arena. Mice were similarly habituated to arenas on day 4, but immediately following the 15 min period, two identical objects (white cylinders) were placed on opposite sides of the arena and the mouse was left in the arena for 15 min to explore both objects. Immediately following the 15 min, mice were placed back in their home cages for one hour. After one hour had elapsed, mice were placed back in the arena which then contained one of the original objects (white cylinder) and one novel object (blue cylinder) and mice were given 15 min to explore both objects. The familiar object (white cylinder) was always placed on the same side of the arena for a given mouse. However, between mice, the side that the white cylinder was placed on was counterbalanced. Percent time exploring the objects for both phases was recorded and analyzed. Times were recorded directly by an experimenter blind to the genotype of the mouse being evaluated. Mice had to be actively investigating the object (e.g., sniffing, licking, rearing, etc) in order for the time to be recorded and reported as “exploration of the novel object”.
Prepulse Inhibition of Startle (PPI)
PPI was performed similarly to Paylor and Crawley (1997) and was measured using the Hamilton Kinder Scientific Startle Reflex Behavioral Monitoring System (Kinder Scientific, Cridersville, OH). Mice were placed in the startle chambers for a 10 minute habituation period with a constant background noise of 60 dB. Following habituation, mice were presented with 7 trials which included: a 120 dB pulse alone, and prepulses of 66, 68, 70, 72, 74, and 76 dB above background noise followed by a 120 dB pulse. The prepulse (20 ms duration) preceded the 120 dB pulse (40 ms duration) by 100 ms. The test session included 10 rounds of the 7 trials (15 s between each trial). Each of the 6 prepulses followed by the 120 dB pulse were presented in a counterbalanced manner. The Startle Monitoring system recorded the average startle response (amplitude) for 150 ms following each of the pulse alone trials and prepulse + pulse trials. The percent startle response to each of the prepulse intensities was calculated as follows: 1-[prepulse+pulse startle response/pulse startle response alone]*100.
Social Interaction
Social Interaction was conducted as previously described (Crawley, 2004). Mice were habituated to the 3-chambered test apparatus (Noldus, Leesburg, VA) for 10 min whereby they had free access to all 3 chambers. Following habituation, a novel (stranger) mouse was placed in a tubular, wired cage placed in one of the outer chambers. The opposite outer chamber contained an empty tubular, wired cage (no mouse inside). The mouse was given free access to all 3 chambers for 10 min. The outer chamber that contained the novel mouse was counterbalanced between mice. The time spent in all 3 chambers was recorded directly by an experimenter blind to the genotype of the mouse being evaluated.
AMP Cumulative Dosing
Separate groups of experimental and control mice were tested for their acute response to AMP using a cumulative dosing paradigm (Yap and Miczek, 2007; Jung et al., 2013; Jedynak et al., 2016). Over 3 consecutive days all mice were habituated to custom-made open field arenas [20 cm (length) × 20 cm (width) × 15 cm (height)] for 45 min following an intraperitoneal (ip) saline injection. On the cumulative dosing experiment day, locomotor activity was recorded for 15 min in response to a saline injection (ip). Following the initial 15 min, locomotor activity was recorded for a total of 120 min. Episodes of 30 min, 30 min and 60 min were recorded following AMP cumulative doses of 0.56 mg/kg, 1.78 mg/kg (cumulative 2.34 mg/kg), and 3.0 mg/kg (cumulative 5.34 mg/kg), (ip), respectively. Locomotor activity in mice was tracked by Ethovision XT v10 video tracking software. Minimum distance of tracking was set to 1 cm along the path of movement and analyzed in 5-min bins. Stereotyped behavior was defined as repetitive head bobbing, rearing, and grooming in the absence of locomotor activity as previously described (McNamara et al., 2006). These behaviors were scored from videotape recordings for 60-s periods starting 5 min after each AMP injection, and continuing every 5 min for 1 h. The total amount of time spent head bobbing, rearing and grooming was recorded and analyzed.
Drugs
D-amphetamine hemisulfate (Sigma-Aldrich) was dissolved in physiological (0.9%) saline solution and injected i.p. in a body volume of 10 ml/kg.
Microdialysis experiments
Surgery
Mice were anesthetized with a mixture of ketamine and xylazine (120 and 16 mg/kg, respectively; ip) and positioned into a mouse stereotaxic apparatus (Stoelting Co., Wood Dale, IL, USA). An incision was made in the scalp to expose the skull. Next, a small hole approximately 2-mm in diameter was drilled into the skull to expose the dura. Mice were then implanted randomly in the right or left side of the brain with a concentric diaylsis probe (see below). During surgical implantation, the dialysis probe was secured in a CMA/10 clip (CMA/Microdialysis AB, Solna, Sweden) that was mounted on a stereotaxic holder; the probe was continuously perfused with saline. The probe was positioned to target the NAc shell, according to Paxinos and Franklin (Paxinos and Franklin, 2001) and previously published reports [anterior: +1.5; lateral: ± 0.6; ventral: −5.1; coordinates are uncorrected in millimeters from bregma; (Tanda et al., 2009; Mereu et al., 2015)]. The implanted probe was secured to the skull with dental cement (GlasIonomer Cement CX-Plus, Henry Schein, Melville, NY). Following surgery, mice were allowed to recover overnight in hemispherical CMA-120 cages (CMA/Microdialysis AB) that were equipped with overhead fluid swivels (Instech Laboratories Inc, Plymouth, PA) for connection to the dialysis probes. All microdialysis studies were conducted in these cages.
In Vivo Microdialysis
Microdialysis was performed following a previously published procedure (Desai et al., 2010). Concentric dialysis probes for mice were constructed using AN69 dialyzing membranes (Hospal Dasco, Lyon, France) as previously described (Tanda et al., 2005; Tanda et al., 2009). Briefly, the probe materials consisted of two (inlet and outlet) 4-cm pieces of silica fused tubing that were inserted into a 22-gauge stainless steel needle. A small drop of glue was used to fix the inlet and outlet tubes at 6- and 5-mm, respectively, from the end of the needle. Next, the inlet and outlet tubes were inserted into a 4-mm dialyzing fiber with the other end closed by a drop of glue; the glue tip of the dialyzing membrane was shaped into a cone. The inlet was set at approximately 0.1-mm away from the closed end of the fiber, and the outlet was 1-mm away from the tip of the inlet. After inserting tubes into the dialyzing membrane, using a thin layer of glue, the fiber was fixed to the remaining portion of silica fused tubing and the end of the 22-gauge needle, leaving only the dialyzing surface of the membrane, i.e, membrane not covered with glue, to the lowest 1-mm portion of the probe. The total length of the probes was approximately 24-mm.
The day after implanting probes, microdialysis studies were performed on freely moving mice in the same CMA-120 cages in which they recovered overnight following surgical procedures. Using a 2.5-ml syringe (Hamilton Co., Reno, NV) attached to a CMA 402 Syringe Pump (CMA/Microdialysis AB), Ringer’s solution (147.0 mM NaCl, 2.2 mM CaCl2, and 4.0 mM KCl) was delivered at a constant flow rate of 1 μl/min through the dialysis probes. Dialysate sampling began 20–30 min after the pump was started, and samples were collected every 10 min thereafter. DA was analyzed immediately, as described below. Injections of saline and AMP were given after three consecutive baseline samples had DA values with less than 10% variability (typically after 1-h). AMP was administered at doses of 0.3, 0.7 (cumulative 1.0 mg/kg), 2.0 (cumulative 3 mg/kg), and 7.0 mg/kg (cumulative 10 mg/kg), respectively, every 30-min following the saline injection. Samples continued to be taken every 10-min throughout the experiment, and ended 300-min after the first injection, i.e., 180-min after the last dose of AMP was administered.
Analytical Procedure
Midbrain, striatum, nucleus accumbens (NAc; core and shell combined) and medial prefrontal cortex (mPFC) were dissected from AMP naïve animals and used for assay of DA total tissue content. The tissue was assayed on the same day as dissection. Dialysate samples (10 μl) were injected, without purification, into a high-performance liquid chromatography system consisting of an MD 150 × 3.2 mm chromatographic column (particle size 3.0 μm; ESA Inc., Chelmsford, MA), and a coulometric detector (Coulochem III; ESA, Dionex, Sunnyvale, CA) to quantify DA. Oxidation and reduction electrode potentials of the analytical cell (5014B; ESA Inc.), respectively, were set at +125 mV and −175 mV. The mobile phase consisting of 100 mM NaH2PO4, 0.1 mM Na2EDTA, 0.5 mM n-octyl sulfate, and 18% (v/v) methanol (pH adjusted to 5.5 with Na2HPO4) was pumped at 0.6 ml/min by a solvent delivery module (584; ESA Inc.). DA assay sensitivity was 2 fmol/sample.
Histology
At the end of the in vivo microdialysis experiment, mice were euthanized with an overdose of pentobarbital, and their brains were removed and fixed in 4% formaldehyde in saline solution. Brains were then cut into 30–50 μm serial coronal sections oriented according to (Paxinos and Franklin, 2001) using a microtome (Microm HM 650 V; Microm International, Thermo Scientific, Waldorf, Germany). Brain slices were then examined under a microscope to identify the location of the probes. The position of the probe was verified in all mice, and the dialyzing portion of all probes implanted in the NAc shell is depicted in Fig. 10 c; rectangles show the boundaries of probe tracks that were inside the shell and were considered appropriate probe placements. Data from mice with probe placements outside these boundaries (i.e., outside the NAc shell) were excluded from the study. While the probes were implanted randomly in both hemispheres, all probe placements are shown on only one side of the brain for simplicity; anterior coordinates measured from bregma are shown in brain sections based on drawings from Paxinos and Franklin (Paxinos and Franklin, 2001).
Fast scan cyclic voltammetry
Coronal striatal brain slices (250 μm) were prepared from adult mice using a vibratome (Leica VT1200; Nussloch, Germany) and ice-cold cutting solution containing (in mM): 125.2 NaCl, 26 NaHCO3, 10 glucose, 2.5 KCl, 2.7 MgSO4, 0.3 KH2PO4 and 0.3 NaH2PO4. Slices were allowed to recover in the solution for 30 min at 34 °C, and then transferred to recording ACSF containing (in mM): 125.2 NaCl, 26 NaHCO3, 10 glucose, 2.4 CaCl2, 2.5 KCl, 1.3 MgSO4, 0.3 KH2PO4 and 0.3 NaH2PO4. The recording chamber temperature was maintained at 32°C (± 2°C). Electrochemical recordings were performed with carbon fiber electrodes (5 μm diameter) with a freshly cut surface that were placed into the desired region (dorsolateral striatum, core, and medial shell) ∼50 μm below the exposed surface. For cyclic voltammetry, a triangular voltage wave (−400 to +900 mV at 280 V/s vs Ag/AgCl) was applied to the electrode every 100 ms. Currents were evoked by local electrical stimulation with tungsten electrode (World Precision Instruments, Sarasota, FL) and stimuli (100–400 μA, 100 μs duration) were delivered every 2 min by ISO-flex stimulus isolator (AMPI, Jerusalem, Israel) and Master-8 pulse generator (AMPI). Evoked currents were recorded with an Axopatch 200B amplifier with a low-pass Bessel filter setting at 5 kHz, digitized at 25 kHz (ITC-18 board; InstruTech, Great Neck, NY). Triangular wave generation and data acquisition were controlled by a computer running a locally written macro (Dr. E. Mosharov, Columbia University, New York, NY) in IGOR Pro (WaveMetrics, Lake Oswego, OR). Carbon fiber electrodes were calibrated with 1 μM DA before and after recording. Data are presented as mean ± SEM.
Statistical Analyses
GraphPad Prism v6.0b software was used for statistical analyses. Group data were calculated as mean ± SEM. For open field analysis, total distance traveled data were expressed as area under the curve (AUC) and for thigmotaxis, the cumulative time spent in each of the three zones was analyzed; both measures were analyzed via two-way ANOVA followed by Sidak’s post hoc comparisons. Movement initiations were assessed by the frequency that the mouse moved at a set speed of 2.0 cm/s or greater and analyzed via unpaired t-test. Rotorod data were assessed by taking the averaged value amongst the 4 trials and were analyzed via unpaired t-test. Digigate data were expressed as AUC for both fore- and hindlimbs and were analyzed via two-way ANOVA with Sidak’s post hoc comparisons. Fear conditioning data, both training and cued-tone test, were analyzed by two-way repeated measures ANOVA with Sidak’s post hoc comparisons. Context fear conditioning data were assessed by percent of total freeze time and were analyzed via unpaired t-test. For Morris Water Maze, training data were analyzed via two-way repeated measures ANOVA with Sidak’s post hoc comparisons and probe trial data were assessed by percent time in the target quadrant and analyzed via unpaired t-test. Novel Object Recognition data (expressed as percent time investigating the novel object) were analyzed by two-way ANOVA with Sidak’s post hoc comparisons. Social Interaction, and PPI were analyzed by two-way ANOVA with Sidak’s post hoc comparisons. AMP acute dosing data were expressed as area under the curve and were analyzed by two-way ANOVA with Sidak’s post hoc comparisons. Data from in vivo microdialysis experiments are expressed as a percentage of basal levels of DA. Basal levels were calculated as the mean of values from three consecutive samples that were taken immediately before the first injection and displayed <10% variability. All results are presented as group means (+SEM), and data were analyzed using a two-way repeated measures ANOVA; overall changes from basal levels were subjected to Sidak’s post hoc analyses. The effects of each cumulative dose of a drug were determined by averaging the three samples after its injection. Cyclic voltammetry and total tissue content of DA were analyzed by two-way repeated measures ANOVA with Sidak’s post hoc comparisons.
Results
Behavioral characterization of synGLT-1 KO mice
Baseline locomotor activity
Previous studies revealed no abnormalities in the synGLT-1 KO mice in a basic behavioral screening battery of tests (SmithKline Beecham, Harwell, Imperial College and Royal London Hospital phenotype assessment; SHIRPA) (Brooks and Dunnett, 2009; Petr et al., 2015). Our goal here was to continue the behavioral phenotyping of the synGLT-1 KO mouse line. To determine whether knockout of neuronal GLT-1 alters basal locomotor activity and associated patterns of behavior, we assessed locomotor activity using the open field test in synGLT-1 KO (N=8) and WT littermate control (N=8) mice (Fig. 1). There was no difference in total distance traveled over 60 min by synGLT-1 KO mice compared to WT littermate controls (no main effect of genotype F(1,14) = 1.188, p = 0.2942). Both synGLT-1 KO and WT littermate controls displayed a time-dependent decrease in locomotion (i.e. habituation) (main effect of time F(5,70) = 11.52, p < 0.0001; no genotype × time interaction F(5,70) = 0.4293, p = 0.8267) (Fig. 1 a). Movement initiations is a measure of basal activity in the open field that provides an indication of general arousal level (Roth and Katz, 1979; Forster et al., 1996), and is altered by disruptions in DA signaling (Eilam et al., 1989). There was no difference in the number of movement initiations between genotypes (t = 1.090, df = 14, p = 0.2942) (Fig. 1 b). Thigmotaxis, or the tendency to remain close to the outer edges or walls of the arena, is a commonly used open field measure of anxiety in mice (Simon et al., 1994). For this test, the amount of time spent in each of three pre-defined zones, an outer zone referred to as the ‘wall zone’, a ‘center zone’, and a zone in between the outer and center zones referred to as the ‘neutral zone’ was assessed. There was no difference in the amount of time spent in each of the three zones between genotypes F(1, 14) = 2.229, p = 0.1576) (Fig. 1 c).
Fig. 1. synGLT-1 KO mice showed normal baseline locomotor activity.

In an open field, synGLT-1 KO mice (N = 8) displayed no differences in total distance traveled (a), number of movement initiations (b) or thigmotaxis (c) relative to littermate controls (N = 8) over 60 min. Error bars indicate SEM
Motor Function
In addition to measuring basic locomotor activity in the open field, we wanted to more comprehensively evaluate motor function. Digigate measures the gait patterns of both fore- and hindlimbs and is widely used in genetically altered mice. We found no significant differences between synGLT-1 KO (N=17) and WT (N=16) mice in either stride duration F(1, 62) = 0.06303, p = 0.8026, or stride length F(1, 62) = 0.05472, p = 0.8158 (Fig. 2 a, b). Similarly, the accelerating rotarod is used to analyze motor coordination and balance of genetically altered mice. We found no significant differences in the latency to fall between synGLT-1 KO (N=26) and WT (N=23) mice (t = 1.325, df = 47, p = 0.1917) (Fig. 2 c).
Fig. 2. synGLT-1 KO mice showed no abnormalities in gate or rotorod performance.
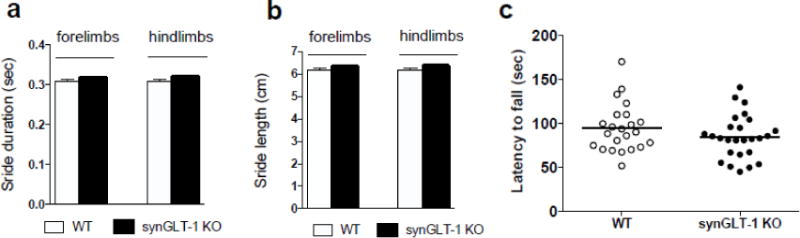
Digigate analysis revealed no differences between synGLT-1 KO (N=17) and WT littermate controls (N=16) in stride duration (a) or stride length (b) (speed of the treadmill=20cm/s). No motor deficit on rotarod (c) was found in synGLT-1 KO mice (N=26; closed circles) compared to WT mice (N=23; open circles). Error bars indicate SEM
Aversive Learning
GLT-1 is relatively highly expressed in CA3 neurons of the hippocampus (Berger and Hediger, 1998; Berger et al., 2005), and in axon terminals both in the CA1 and CA3 regions of the hippocampus (Chen et al., 2004; Furness et al., 2008). Interestingly, a recent report demonstrated that blocking GLT-1 function results in a reduction of the acquisition of fear conditioning learning (John et al., 2015). Therefore, we predicted that fear conditioning may be hindered in the synGLT-1 KO mice. However, the results revealed no difference in freezing behavior by synGLT-1 KO mice (N=8) compared to WT littermate controls (N=8) in response to repeated CS-US pairings during fear conditioning training (no main effect of genotype F(1,36) = 0.01221, p = 0.9126). Both synGLT-1 KO and WT littermate controls displayed an increase in freezing behavior across successive CS-US trials (main effect of CS-US trial F(2,36) = 40.77, p < 0.0001; no genotype × CS-US trial interaction F(2,36) = 0.2333, p = 0.7931) (Fig. 3 a1). There was no difference in freezing behavior between genotypes in response to the tone during the tone cue test (no main effect of genotype F(1,12) = 1.230, p = 0.2891) (Fig. 3 a2). Both genotypes displayed an increase in freezing behavior during the tone period relative to the pre-tone period (main effect of trial period F(1,12) = 91.74, p < 0.0001; no genotype × trial period interaction F(1,12) = 0.002174, p = 0.9636) (Fig. 3 a2). During the context test, there was no difference in freezing behavior between genotypes (t = 1.003, df = 12, p = 0.3357) (Fig. 3 a3).
Fig. 3. synGLT-1 KO mice performed similar to controls in learning and memory tasks.
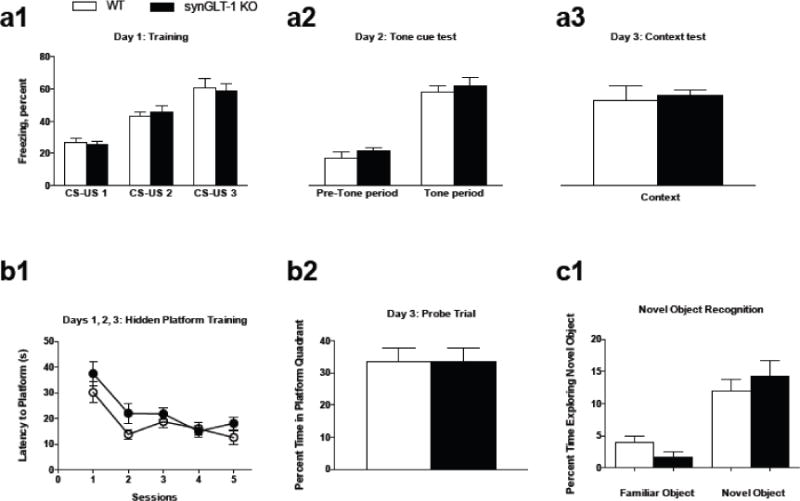
a1, During fear conditioning training, both synGLT-1 KO mice (N=8) and WT littermate controls (N=6) displayed increased freezing in response to repeated CS-US pairings. a2, During the tone cue test, both genotypes froze significantly more during the tone period compared to the pre-tone period. a3, synGLT-1 KO and WT littermates displayed similar freezing behavior during the context test.
b1, SynGLT-1 KO (N=11) and WT littermate controls (N=12) displayed similar acquisition behavior (i.e. decreased amount of time to reach the platform) over 5 training sessions in the Morris Water Maze test. b2, During the probe trial, no differences in percent time spent in the target quadrant between genotypes were detected.
c1, SynGLT-1 KO (N=12) and WT littermate controls (N=11) showed an increased amount of time exploring the novel object compared to the familiar one; there were no differences between genotypes. Error bars indicate SEM
Learning and Memory
Disrupting glutamate transport by loss of GLT-1 protein and function has been shown to hinder performance in tests of learning and memory, including the acquisition and retention trials of the Morris Water Maze task (Bechtholt-Gompf et al., 2010; Mookherjee et al., 2011) as well as memory for the novel object in the novel object recognition task (Takahashi et al., 2015). Interestingly, when GLT-1 expression was restored, novel object recognition behavior was enhanced (Takahashi et al., 2015). In light of these reports, we predicted that learning and memory during these tasks in our synGLT-1 KO would be compromised. Thus, we first assessed the spatial memory performance of synGLT-1 KO (N=11) and WT littermate control (N=12) mice in the Morris Water Maze. There was no difference in latency time to platform between synGLT-1 KO and WT mice during the hidden platform training of the Morris Water Maze test (no main effect of genotype F(1,21) = 3.303, p = 0.0834) (Fig. 3 b1). Both genotypes displayed a decrease in latency time to platform across successive hidden platform training trials (main effect of training trial F(4,84) = 15.30, p < 0.0001; no genotype × training trial interaction F(4,84) = 0.8809, p = 0.4790) (Fig. 3 b1). During the probe test, there was no difference in the percent time spent in the platform quadrant between genotypes (t = 0.1294, df = 10, p = 0.8996) (Fig. 3 b2). Both synGLT-1 KO and WT mice performed above chance [defined as 20/60 s (Mannix et al., 2011)] in time spent swimming in the platform quadrant. Next, we tested a separate group of mice for novel object recognition. During this task, both synGLT-1 KO (N=6) and WT (N=8) littermate controls displayed a significant increase in the percent time exploring the novel object (main effect of time F(2,30) = 69.02, p = 0.0010; no differences in the amount of time exploring the novel object occurred between genotypes (no genotype × time interaction F(1,5) = 0.029, p = 0.8709 (Fig. 3 c1).
Social Interaction
In addition to testing for cognitive deficits in the synGLT-1 KO, we also wanted to determine if other behaviors including sociability and sensorimotor gating were altered. Alteration in glutamate signaling by blockade of NMDA receptors was found to reduce performance during both the social interaction test and the prepulse inhibition of startle task (Kinney et al., 2003; Morales and Spear, 2014). In order to determine if social behavior is reduced in the synGLT-1 KO, we ran the social interaction test. During this test, both synGLT-1 KO (N=7) and WT (N=5) littermate controls displayed a significant increase in the amount of time in the chamber with the novel mouse when compared to the other two chambers (main effect of time F(1,5) = 47.32, p < 0.0001). Additionally, there were no differences between genotypes in the amount of time spent with the novel mouse (no genotype × time interaction F(2,30) = 3.037, p = 0.0629 (Fig. 4 a).
Fig. 4. synGLT-1 KO mice performed similar to controls during measures of sociability and sensorimotor gating.
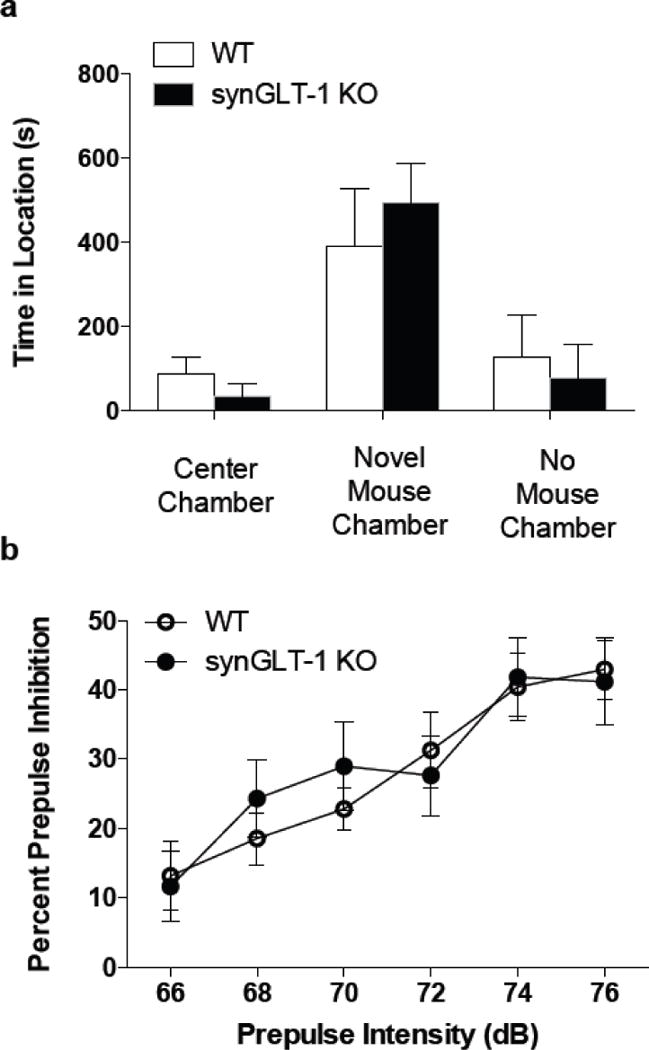
a, synGLT-1 KO and WT littermate controls spent a greater amount of time in the chamber with the novel mouse. There were no differences between genotypes in the amount of time spent in any of the chambers.
b, synGLT-1 KO and WT littermate controls exhibited a progressive increase PPI with increasing prepulse intensities. No differences between genotypes occurred. Error bars indicate SEM
Sensorimotor Gating
The prepulse inhibition of startle (PPI) task is a measure of sensorimotor gating and refers to the ability of a weak prepulse stimulus to inhibit the response to a subsequent stronger stimulus. We found that while the PPI effect was enhanced with increasing prepulse intensity, there were no differences between synGLT-1 KO (N=15) and WT (N=11) mice in PPI at any of the 6 prepulse intensities used (main effect of prepulse intensity F(5,120) = 34.67, p < 0.0001; no main effect of genotype × prepulse intensity interaction F(5,120) = 1.122, p = 0.3526 (Fig. 4 b).
AMP-induced hyperlocomotion
Alterations in glutamatergic signaling can modulate the behavioral effects of AMP (Burns et al., 1994; Wolf, 1998; David and Abraini, 2003). Specifically, intra-accumbal infusions of glutamate agonists and antagonists at the NMDA, AMPA, and group 1 metabotropic receptor (mGluR) subtypes increased and decreased the locomotor response to AMP, respectively (Burns et al., 1994; David and Abraini, 2003). Interestingly, the reduced locomotor response to AMP by the group 1 mGluR antagonist was blocked by infusion of a group 2 mGluR agonist. Group 2 mGluRs negatively regulate glutamate release (Baker et al., 2002) and have also been shown to be in close proximity to GLT-1 (Bellesi and Conti, 2010). Based on such findings, we predicted that the defect in glutamate homeostasis produced by deletion of GLT-1 in neurons, which would presumably increase local extracellular glutamate levels, might diminish the behavioral effects of AMP, conceivably via increased activation of group 2 mGluRs. Using a cumulative dosing procedure (Yap and Miczek, 2007) to study the acute effects of AMP, we found that the AMP-induced increase in locomotor activity following the injection of 3 mg/kg in synGLT-1 KO mice (N=16) was significantly lower than in WT mice (N=21) (main effect of time and genotype F(20,735) = 2.313, p = 0.0010. Sidak’s multiple comparisons showed a significant difference during the last three time bins following the 3 mg/kg AMP dose during the last three time bins (min. 95, p = 0.0078; min. 100, p = 0.0006; min. 105, p < 0.0001) (Fig. 5 a). Minimal stereotyped behavior was observed (grooming, rearing, head bobbing) and was not different between genotypes (data not shown).
Fig. 5. The locomotor response to acute AMP administration was blunted in synGLT-1 KO mice.
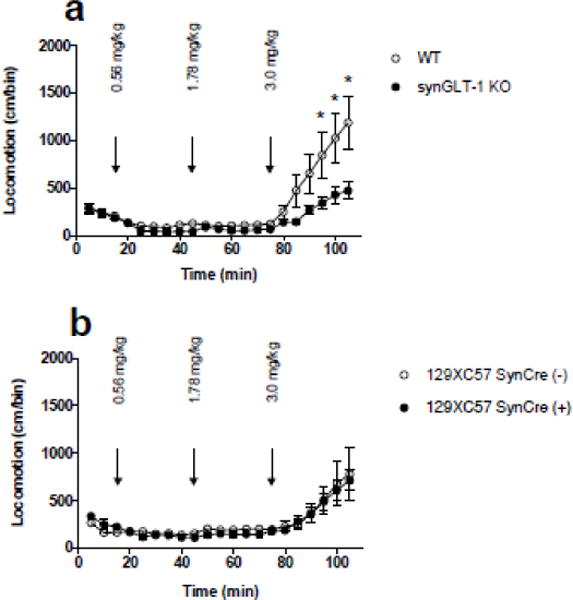
a, Locomotor activity was recorded in response to saline and 3 increasing doses of AMP (exact doses: 0.56, 1.78, and 3.00 mg/kg, ip; cumulative doses: 0.56, 2.34, 5.34 mg/kg). SynGLT-1 KO mice (N=16) displayed a blunted locomotor response following the highest dose (3.0 mg/kg) of AMP compared to WT littermate control mice (N=21) (p = 0.045). b, SynCre(+) mice (N=13) on the same background as the mice tested above (mixed 129XC57) showed no differences in their locomotor response to AMP compared to SynCre(−) mice (N=9) on the same background. Arrows indicate time of injection. Error bars indicate SEM
In experiments involving genetic alterations via cre-recombinase, it is important to conduct studies with the appropriate cre control groups to exclude the possibility that the observed phenotype is not produced solely by cre-recombinase (Harno et al., 2013). Here, we examined control mice on a mixed 129XC57 background by comparing the effects of AMP on synapsin-Cre+ and synapsin-Cre− mice. To produce the 129XC57BL/6J mouse line for this experiment, we obtained the 129 mouse line from Jackson Labs that was derived from the same mouse line that served as the source of the mouse embryonic fibroblasts used to produce the conditional knockout line. No differences between SynCre(+) (N=13) and SynCre(−) (N=9) in their acute locomotor response to AMP were observed (no main effect of genotype F(1,20) = 0.2527, p = 0.6206) (Fig. 5 b).
AMP-stimulated DA release was similar in synGLT-1 KO and WT mice
The major effect of psychostimulants such as AMP is to increase extracellular concentrations of DA at dopaminergic synapses (Kuczenski et al., 1995), specifically within NAc shell (Heidbreder and Feldon, 1998; Desai et al., 2010; Sulzer, 2011). One possible explanation for the decreased response to AMP in synGLT-1 KO mice is decreased AMP-evoked DA release. We used in vivo microdialysis techniques to measure AMP-stimulated DA release within the NAc shell using a cumulative dosing procedure. Surprisingly, there was no difference between the baseline DA values for the two genotypes; synGLT-1 KO (N=8) baseline values were 14 ± 3.1 (SEM) fmol/sample and WT (N=6) were 13.1 ± 3.7 fmol/sample (Fig. 6 a). Administration of saline did not significantly alter extracellular levels of DA in the NAc shell in either synGLT-1 KO or WT mice (ts ≤ 0.03682, ps > 0.9999). Cumulatively administered AMP dose-dependently increased extracellular levels of DA in the dialysate samples from the NAc shell in both genotypes (F(5,60) = 27.22, p < 0.0001). While increases in DA were approximately 500% of basal DA levels following administration of a cumulative dose of 3 mg/kg AMP in both synGLT-1 KO and WT, only the highest cumulative dose (10 mg/kg) produced a significant increase in extracellular DA, compared to baseline levels (0.3 – 3.0 mg/kg synGLT-1 KO and WT: Sidak’s post-test ts ≤ 2.137, ps ≥ 0.1705; 10 mg/kg synGLT-1 KO and WT: Sidak’s post-test ts ≥ 5.542, ps< 0.001). Additionally, there were no differences between the two genotypes at any of the time points (F(1,12) = 0.4972 p = 0.4942) (Fig. 6 b). Only data from animals in which probe placements corresponded to the medial NAc shell were used (Fig. 6 c).
Fig. 6. Microdialysis revealed no change in AMP stimulated extracellular DA release in the medial shell of the NAc.
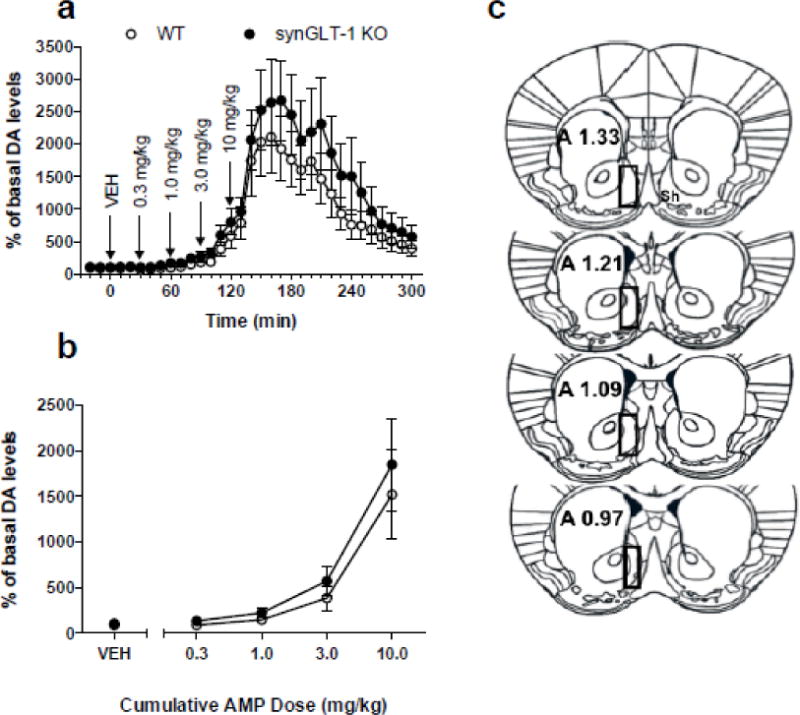
a, Time course of the effects of cumulative administration of AMP (0.3–10 mg/kg) on the extracellular levels of DA in synGLT-1 KO (N=8) and WT (N=6) mice. Dialysate samples were taken from the NAc shell every ten min. Each arrow indicates time points at which incremental cumulative injections of AMP were administered. Ordinates; percentage of basal DA level; abscissae, time in min after injection. Each point indicates the mean (±SEM) effect shown as of percentage of basal DA levels; levels were uncorrected for probe recovery. There were no differences that occurred between the synGLT-1 KO and WT genotypes (F1,384 = 2.47, p = 0.14); however, a significant effect of time (F32,384 = 19.26, p < 0.0001) and a time × genotype interaction (F32,384 = 1.921, p = 0.0024) was observed. b, Cumulative dose-response. Changes in extracellular levels of DA in the NAc shell of synGLT-1 KO and WT mice after administration of AMP are shown. Ordinates, percentage of basal DA levels; abscissae cumulative drug dose in milligrams per kilogram. Each data point represents the mean (±SEM) of three samples taken in the thirty min following administration of each cumulative dose.
C, Drawings of the forebrain sections based on Paxinos and Franklin (2001) with superimposed rectangles that show the confines within which the microdialysis probe tracks were considered to be in the NAc shell. Data were only included from all subjects with probe tracts within the rectangles. The anterior coordinate (measured from the bregma) is located on each section
Fast scan cyclic voltammetry on brain slices revealed no effect of neuronal GLT-1 knockout on DA release
To directly investigate whether GLT-1 deficiency altered DA release, we measured evoked transmitter release from coronal brain slices of dorsal striatum (dSTR) and NAc (core and shell) of synGLT-1 KO (N=4) and WT (N=3) mice. DA release was stimulated via application of a single electrical pulse (100 μs duration) and monitored at a proximal carbon fiber electrode in cyclic voltammetric mode of detection (see Methods). Local electrical stimulation induced a rapid and transient increase in extracellular DA (Fig. 7 a) that was not different between control (Cre-) and GLT-1 knockout (Cre+) slices in all three striatal regions (Fig. 7 b, c).
Fig. 7. Deletion of GLT-1 does not change evoked and AMP-induced DA release.
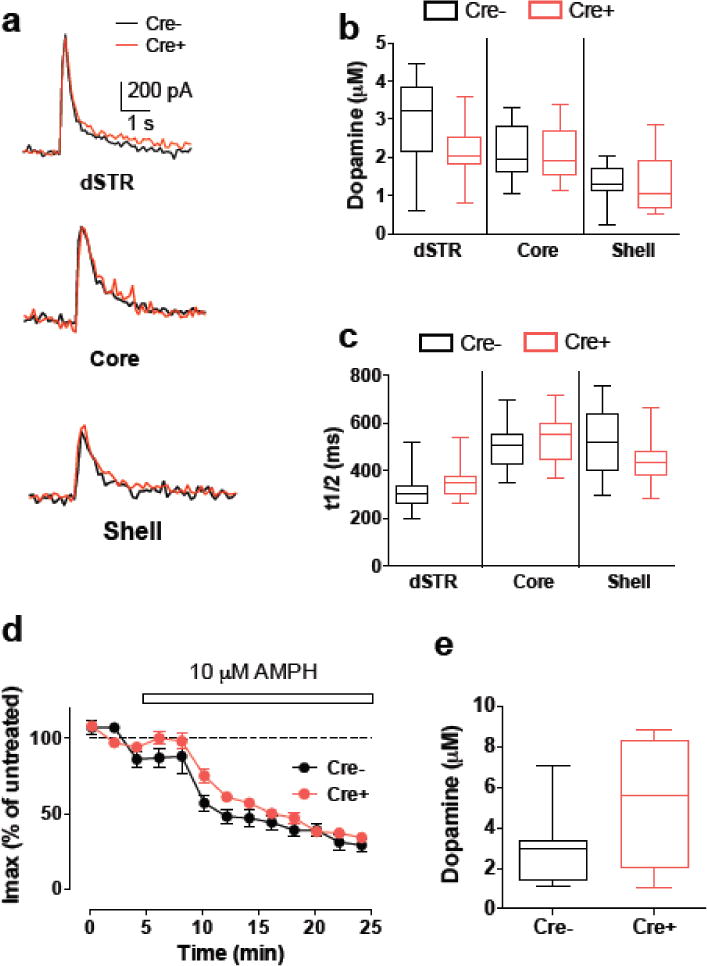
(a) Representative recordings of evoked DA release measured by cyclic voltammetry in response to single-pulse electrical stimulation from wild-type (Cre−) and GLT-1 KO (Cre+) acute striatal slices. (b, c) GLT-1 KO did not change maximal amplitude (b) or width at half-height (t1/2, c) of evoked DA release in dSTR and NAc (core and shell). (d) Time course of AMP-induced inhibition of evoked DA release in the NAc shell was not different between control and GLT-1 KO striatal slices. (e) No difference in the amplitude of AMP-induced DA efflux between control and GLT-1 KO striatal slices. No significant difference was observed between any of the data pairs on all panels
We next examined whether deletion of GLT-1 had any effect on AMP-induced changes in DA release and DAT-mediated efflux. Consistent with previous reports (Jones et al., 1998; Schmitz et al., 2001), perfusion of 10 μM AMP for 20 min induced gradual decrease in evoked DA release (Fig. 7 d) and an increase in DA overflow (not shown). The kinetics of the changes in the amplitude of evoked DA peaks was not different between the groups (Fig. 7 d). Similarly, the magnitude of AMP-induced DA efflux was similar in control and GLT-1 KO slices (Fig. 7 e; F(1,2), p = 0.8404). Overall, our data indicate that GLT-1 KO had no significant effect on stimulation-dependent DA release or AMP-induced changes in striatal DA terminals in the dSTR and NAc.
Tissue content of DA
To investigate the possibility that neuronal GLT-1 knockout might alter DA signaling we assayed DA tissue content in tissue samples from the midbrain, dSTR, NAc, and (mPFC) (Fig. 8). We found no change in DA content in any of these regions between synGLT-1 KO (N=5) and WT (N=3) mice (no main effect of genotype F(1, 25) = 0.0147, p = 0.9045) (Fig. 8).
Fig. 8. Pan-neuronal knockout does not change tissue DA content.
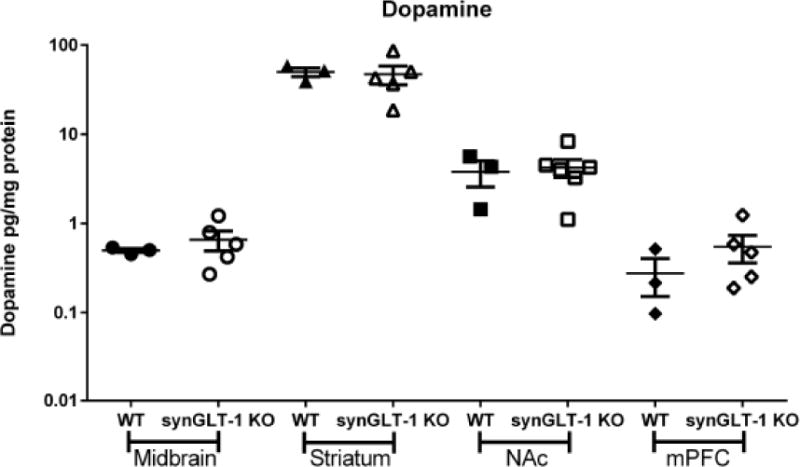
HPLC-EC measurement of DA in midbrain, dSTR, NAc and medial pre-frontal cortex tissue of WT (N=3) and synGLT-1 KO (N=5) mice. No differences in tissue content of DA from these regions were detected
Discussion
We have utilized a conditional GLT-1 knockout mouse described previously (Petr et al., 2015) to investigate the behavioral phenotype of mice in which the GLT-1 gene is inactivated in neurons by the expression of Cre under the synapsin 1 promoter. In the Petr et al., 2015 paper, the efficacy of the synapsin-Cre driven knockout of GLT-in neurons in the hippocampus was established by electron microscopic immunocytochemistry: in the knockout there was a 87.5% reduction in labeling of axon terminals in the CA1 region without significant change in the GLT-1 labeling of astrocytic processes in the same region. Another demonstration of the efficacy of the synapsin-Cre driven knockout was its effect on synaptosomal glutamate uptake. Using crude synaptosomes prepared from the whole forebrain, we found a 40% decrease in uptake in the synGLT-1 KO compared with littermate controls, and, surprisingly, no significant effect on uptake in the astrocyte GLT-1 knockout. These results imply that the knockout of GLT-1 in neurons is widespread throughout the forebrain. In addition, we have more recently assayed synaptosomal glutamate uptake in synaptosomes from specific regions of the brain, e.g. cortex, hippocampus, and striatum, and find results very similar to those we obtained using synaptosomes from the whole forebrain (unpublished).
In the present study, we found that pan-neuronal knockout of GLT-1 resulted in no alterations in locomotor activity in open field, rotorod and DigiGait tests. Because GLT-1 mRNA and protein are highly expressed in neurons and axon terminals of the hippocampus (Berger and Hediger, 1998; Berger et al., 2005), we expected diminished learning and memory function during tests dependent upon hippocampal function in the synGLT-1 KO mice. Surprisingly, performance in the Morris Water Maze, cue and context dependent fear conditioning, and novel object recognition tests was unaltered in the synGLT-1 KO mice. In addition, we found no alteration in sensorimotor gating and social interaction in these mice. Other reports show diminished learning and memory behavior following GLT-1 loss of expression/function (Bechtholt-Gompf et al., 2010). However our imposed loss of GLT-1 is specific to neurons, which may explain the discrepancy in our findings compared with others. One possible explanation for this is that GLT-1 expressed in neurons does not contribute significantly to glutamate clearance at the hippocampal synapses involved in learning and memory, and in fact, it has been demonstrated that astrocytic transporters are the major contributors to glutamate uptake at Schaffer collateral synapses in the hippocampus (Bergles and Jahr, 1998). When tested for their response to AMP, we found synGLT-1 mice to exhibit a significantly blunted locomotor response to acute administration of AMP. Control studies testing for an effect of expression of Cre-recombinase per se under the synapsin promoter showed no differences in locomotor responses to AMP between mice expressing Cre-recombinase and littermate controls on the same 129XC57 genetic background as the synGLT-1 KO mice. The diminished locomotor responses to AMP in the synGLT-1 KO cannot be attributed to a general decrease in basal locomotor activity in the synGLT-1 KO mice or to an increase in AMP-induced stereotyped behaviors.
The present findings raise important questions regarding the mechanisms underlying the modulation of responsiveness to AMP by GLT-1 expression. Considerable evidence suggests that changes in glutamate homeostasis play a key role in the pathophysiology underlying responses to repeated exposure to psychostimulants and withdrawal. GLT-1 in particular, always assumed to be expressed in astrocytes, has been specifically implicated in phenomena related to withdrawal, specifically reinstatement (Kalivas, 2009; Roberts-Wolfe and Kalivas, 2015). GLT-1 has never previously been shown to affect the acute responses to AMP. However, the neuronal glutamate transporter EAAC1/EAAT3 also appears to play a role in modulating acute responses to AMP (Underhill et al., 2014). In that study, Underhill et al. reported that AMP-induced internalization of DAT is associated with internalization of EAAT3, and consequent potentiation of excitatory current responses measured in identified substantia nigra pars compacta neurons, possibly due to decreased clearance of extracellular glutamate. The role of EAAT3 in the behavioral responses to AMP remains unclear.
How could deletion of GLT-1 specifically from neurons result in a blunted locomotor response to AMP? Broadly considered, there are two major classes of roles that GLT-1 expressed in neurons might assume, one related to DA signaling and one related to glutamate signaling. The effects of AMP are due to its producing an increase in extracellular DA in and around dopaminergic synapses though a variety of actions, including a weak base effect to compromise the pH gradient across dopaminergic synaptic vesicles, inhibition of monoamine oxidase, and inhibition of DA uptake (Sulzer, 2011). The most obvious explanation, therefore, for the modulatory effects of GLT-1 deletion in neuron on response to AMP is that loss of GLT-1 from some population of axon terminals, presumably glutamatergic, in some way reduces the amount of DA that accumulates extracellularly following AMP administration. This type of effect might be achieved by a number of mechanisms.
Developmental
It is conceivable that the loss of GLT-1 in neurons has a developmental effect resulting, for example, in decreased number or projections of DA neurons. It has been previously reported that NMDA receptor activation has a trophic effect on DA neurons suggesting that abnormalities of glutamate homeostasis might compromise dopaminergic neuron survival (Fortin et al., 2012). In this case, a 30% decrease in DA terminal number and a 37% decrease in evoked DA release in the NAc shell were reported in the DAT-driven vGLUT2 KO in which glutamatergic signaling by dopaminergic neurons was downregulated by loss of VGLUT2 in these neurons. (Fortin et al., 2012).
Metabotropic receptors
Another possibility is that altered glutamate homeostasis in proximity of dopaminergic terminals may alter the activation of metabotropic receptors, specifically mGluR2/3, known to be expressed in dopaminergic axons, where they negatively regulate DA release (Chaki et al., 2006; Karasawa et al., 2006; Lane et al., 2013; Fujioka et al., 2014). This regulation can be detected by its effects on basal and stimulated extracellular DA concentration and intracellular DA content.
Vesicular synergy
Yet another possibility is suggested by previous work providing evidence that vesicular synergy, the enhancement of packaging of one transmitter into synaptic vesicles by the accumulation of a second transmitter into the same vesicles, is an important phenomenon in dopaminergic neurons (Hnasko and Edwards, 2012). Abnormalities in glutamate homeostasis produced by deletion of neuronal GLT-1 might in some way compromise vesicular synergy in dopaminergic neurons. It would be easy to imaging this scenario if GLT-1 were expressed in DA neurons. If vesicular synergy were an important phenomenon (Trudeau et al., 2014), then intra-terminal concentration of glutamate would be expected to be important, and GLT-1 expressed in the plasma membrane of dopaminergic terminals might provide this glutamate. If so, its absence could nullify vesicular synergy, because a mechanism to maintain glutamate levels in dopaminergic terminals were lacking. Compromise of vesicular synergy has been shown to result in a decrease in stimulated release of DA into the extracellular space in mice with conditional deletion of VGLUT2 expression restricted to DA neurons (Hnasko et al., 2010).
Localization of the effect of neuronal knockout of GLT-1 on response to AMP
Although the region(s) and neuronal populations responsible for the effect of neuronal knockout of GLT-1 on the locomotor response to AMP is unknown, the NAc is a likely candidate given its role in AMP-induced locomotor hyperactivity (Sellings and Clarke, 2003). The NAc can be divided into two subregions, the core and shell, both of which are different functionally and structurally (Zahm and Brog, 1992; Weiner et al., 1996). It has been shown that both electrolytic and DA lesions of the NAc core result in a reduction of the acute locomotor response to AMP (Weiner et al., 1996; Sellings and Clarke, 2003). However, in the Sellings and Clark report, it was shown that DA lesions in the shell also resulted in a reduction in the acute locomotor response to AMP (Sellings and Clarke, 2003). Thus, it is conceivable that deletion of GLT-1 specifically from NAc core and/or shell neurons diminishes the dopaminergic signal in that region and is responsible for the blunted locomotor response to AMP. However, if this were the case, we would have likely observed evidence of diminished dopaminergic signalling, which was not the case. Although in the assay of total tissue content of dopamine, core and shell tissue were combined, we were able to use cyclic voltammetry in brain slices to assay DA release from the core and shell regions separately (please refer to Figure 7 A–C), as well as in the dorsal striatum. No differences were detected in electrically evoked dopamine release in knockout and wildtype littermates from any of these regions.
The fact that we observed no changes in tissue DA content, AMP stimulated changes in extracellular DA concentration detected by microdialysis, or AMP or electrically stimulated DA release detected by cyclic voltammetry, makes the three explanations noted above unlikely, though not entirely excluded. For example, it is possible that in one or more regions others than those sampled by microdialysis or voltammetry there is a critical mGluR2/3 mediated effect of GLT-1 deletion in neurons. However, this possibility seems unlikely given the wide range of regions sampled for the tissue content studies reported above. It is also possible that such an effect is taking place in a specific circuit but the changes in that circuit are being diluted and rendered undetectable by the surrounding tissue in which no changes are taking place. With these caveats in mind, it seems likely that the effect of neuronal deletion of GLT-1 to decrease response to AMP is not due to a change in DA dynamics.
An alternative possibility is that the effect of deletion of GLT-1 in neurons is due to a compromise of a glutamatergic signal. It is well-established that excitatory signalling plays a critical role in the behavioral responses to psychostimulant drugs, but, generally, changes in excitatory signaling induced by exposure to these drugs have been thought to underlie their long-lasting effects such as sensitization and reward conditioning, rather than acute behavioral responses (Wolf, 1998; Ungless et al., 2001; Luscher and Malenka, 2011; Wolf and Tseng, 2012; Ikemoto and Bonci, 2014). However, increases in extracellular glutamate have been detected following acute AMP administration in the NAc, ventral tegmental area, and PFC (Xue et al., 1996; Wolf et al., 2000)(Ash et al., 2014). These observable increases in glutamate efflux in the NAc and ventral tegmental area following acute AMP administration occurred with too long of a delay to mediate the acute locomotor response to AMP, as been noted (Xue et al., 1996). Stimulation of glutamate release by AMP administration may result in changes in glutamate receptor expression or distribution in the ventral tegmental area and NAc that may contribute to the induction of AMP sensitization, which occurs within a time scale of hours and days. However, there is evidence that implicates mGluR5 receptors in the acute locomotor stimulant effects of AMP and cocaine (McGeehan et al., 2004; Gill et al., 2012). In either case, removal of a glutamate clearance mechanism would be expected to enhance excitatory signaling, so, if anything, potentiate the effects of psychostimulants rather than diminish their effects, as we see here. As an example, Underhill and colleagues have shown that DAT internalization caused by exposure to AMP induces internalization of EAAC1, a glutamate transporter expressed in dopaminergic neurons, resulting in potentiation of excitatory inputs onto midbrain dopaminergic neurons (Underhill et al., 2014). Our findings imply that there is a glutamate signal that augments response to AMP that is dependent upon GLT-1 expression in neurons and that is diminished, not increased, by removal of GLT-1 from neurons. What exactly is the nature of that signal is unclear. At present, we can only say that the signal emanates from some population of neurons that expresses GLT-1. Undoubtedly, our understanding of the underlying mechanisms will be greatly increased by identifying the specific neuronal population or populations involved.
Acknowledgments
The authors wish to acknowledge Nick Andrews and Georgia Gunner of the Neurodevelopmental Behavioral Core at Boston Children’s Hospital for technical assistance with behavioral experiments. The authors would also like to acknowledge Kush Kapur for statistical consultation. This work was supported by NIH grants NS066019, NS075222, MH104318, DA031734, HD018655, and by the Tommy Fuss Fund. KF was supported by T32 NS007473.
Abbreviations
- AMP
amphetamine
- SynCre
synapsin 1-cre
- AUC
area under the curve
- PPI
prepulse inhibition of startle
- NAc
nucleus accumbens
- dSTR
dorsal striatum
- DA
dopamine
Footnotes
Conflicts of Interest: The authors declare no competing financial interests.
References
- Ash ES, Heal DJ, Clare Stanford S. Contrasting changes in extracellular dopamine and glutamate along the rostrocaudal axis of the anterior cingulate cortex of the rat following an acute d-amphetamine or dopamine challenge. Neuropharmacology. 2014;87:180–187. doi: 10.1016/j.neuropharm.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechtholt-Gompf AJ, Walther HV, Adams MA, Carlezon WA, Jr, Ongur D, Cohen BM. Blockade of astrocytic glutamate uptake in rats induces signs of anhedonia and impaired spatial memory. Neuropsychopharmacology. 2010;35:2049–2059. doi: 10.1038/npp.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellesi M, Conti F. The mGluR2/3 agonist LY379268 blocks the effects of GLT-1 upregulation on prepulse inhibition of the startle reflex in adult rats. Neuropsychopharmacology. 2010;35:1253–1260. doi: 10.1038/npp.2009.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger UV, Hediger MA. Comparative analysis of glutamate transporter expression in rat brain using differential double in situ hybridization. Anat Embryol. 1998;198:13–30. doi: 10.1007/s004290050161. [DOI] [PubMed] [Google Scholar]
- Berger UV, DeSilva TM, Chen W, Rosenberg PA. Cellular and subcellular mRNA localization of glutamate transporter isoforms GLT1a and GLT1b in rat brain by in situ hybridization. J Comp Neurol. 2005;492:78–89. doi: 10.1002/cne.20737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE. Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. J Neurosci. 1998;18:7709–7716. doi: 10.1523/JNEUROSCI.18-19-07709.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks SP, Dunnett SB. Tests to assess motor phenotype in mice: a user’s guide. Nature reviews Neuroscience. 2009;10:519–529. doi: 10.1038/nrn2652. [DOI] [PubMed] [Google Scholar]
- Burns LH, Everitt BJ, Kelley AE, Robbins TW. Glutamate-dopamine interactions in the ventral striatum: role in locomotor activity and responding with conditioned reinforcement. Psychopharmacology (Berl) 1994;115:516–528. doi: 10.1007/BF02245576. [DOI] [PubMed] [Google Scholar]
- Chaki S, Yoshikawa R, Okuyama S. Group II metabotropic glutamate receptor-mediated regulation of dopamine release from slices of rat nucleus accumbens. Neurosci Lett. 2006;404:182–186. doi: 10.1016/j.neulet.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Chen W, Mahadomrongkul V, Berger UV, Bassan M, DeSilva T, Tanaka K, Irwin N, Aoki C, Rosenberg PA. The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci. 2004;24:1136–1148. doi: 10.1523/JNEUROSCI.1586-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN. Designing mouse behavioral tasks relevant to autistic-like behaviors. Ment Retard Dev Disabil Res Rev. 2004;10:248–258. doi: 10.1002/mrdd.20039. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Danbolt NC, Furness DN, Zhou Y. Neuronal vs glial glutamate uptake: Resolving the conundrum. Neurochem Int. 2016;98:29–45. doi: 10.1016/j.neuint.2016.05.009. [DOI] [PubMed] [Google Scholar]
- David HN, Abraini JH. Blockade of the locomotor stimulant effects of amphetamine by group I, group II, and group III metabotropic glutamate receptor ligands in the rat nucleus accumbens: possible interactions with dopamine receptors. Neuropharmacology. 2003;44:717–727. doi: 10.1016/s0028-3908(03)00052-2. [DOI] [PubMed] [Google Scholar]
- Desai RI, Paronis CA, Martin J, Desai R, Bergman J. Monoaminergic psychomotor stimulants: discriminative stimulus effects and dopamine efflux. J Pharmacol Exp Ther. 2010;333:834–843. doi: 10.1124/jpet.110.165746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilam D, Golani I, Szechtman H. D2-agonist quinpirole induces perseveration of routes and hyperactivity but no perseveration of movements. Brain Res. 1989;490:255–267. doi: 10.1016/0006-8993(89)90243-6. [DOI] [PubMed] [Google Scholar]
- Ennaceur A, Delacour J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav Brain Res. 1988;31:47–59. doi: 10.1016/0166-4328(88)90157-x. [DOI] [PubMed] [Google Scholar]
- Forster MJ, Dubey A, Dawson KM, Stutts WA, Lal H, Sohal RS. Age-related losses of cognitive function and motor skills in mice are associated with oxidative protein damage in the brain. Proc Natl Acad Sci U S A. 1996;93:4765–4769. doi: 10.1073/pnas.93.10.4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin GM, Bourque MJ, Mendez JA, Leo D, Nordenankar K, Birgner C, Arvidsson E, Rymar VV, Berube-Carriere N, Claveau AM, Descarries L, Sadikot AF, Wallen-Mackenzie A, Trudeau LE. Glutamate corelease promotes growth and survival of midbrain dopamine neurons. J Neurosci. 2012;32:17477–17491. doi: 10.1523/JNEUROSCI.1939-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka R, Nii T, Iwaki A, Shibata A, Ito I, Kitaichi K, Nomura M, Hattori S, Takao K, Miyakawa T, Fukumaki Y. Comprehensive behavioral study of mGluR3 knockout mice: implication in schizophrenia related endophenotypes. Molecular brain. 2014;7:31. doi: 10.1186/1756-6606-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness DN, Dehnes Y, Akhtar AQ, Rossi DJ, Hamann M, Grutle NJ, Gundersen V, Holmseth S, Lehre KP, Ullensvang K, Wojewodzic M, Zhou Y, Attwell D, Danbolt NC. A quantitative assessment of glutamate uptake into hippocampal synaptic terminals and astrocytes: new insights into a neuronal role for excitatory amino acid transporter 2 (EAAT2) Neuroscience. 2008;157:80–94. doi: 10.1016/j.neuroscience.2008.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill MJ, Arnold JC, Cain ME. Impact of mGluR5 during amphetamine-induced hyperactivity and conditioned hyperactivity in differentially reared rats. Psychopharmacology (Berl) 2012;221:227–237. doi: 10.1007/s00213-011-2565-0. [DOI] [PubMed] [Google Scholar]
- Harno E, Cottrell EC, White A. Metabolic pitfalls of CNS Cre-based technology. Cell metabolism. 2013;18:21–28. doi: 10.1016/j.cmet.2013.05.019. [DOI] [PubMed] [Google Scholar]
- Heidbreder C, Feldon J. Amphetamine-induced neurochemical and locomotor responses are expressed differentially across the anteroposterior axis of the core and shell subterritories of the nucleus accumbens. Synapse. 1998;29:310–322. doi: 10.1002/(SICI)1098-2396(199808)29:4<310::AID-SYN3>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Hnasko TS, Edwards RH. Neurotransmitter corelease: mechanism and physiological role. Annu Rev Physiol. 2012;74:225–243. doi: 10.1146/annurev-physiol-020911-153315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS, Chuhma N, Zhang H, Goh GY, Sulzer D, Palmiter RD, Rayport S, Edwards RH. Vesicular glutamate transport promotes dopamine storage and glutamate corelease in vivo. Neuron. 2010;65:643–656. doi: 10.1016/j.neuron.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemoto S, Bonci A. Neurocircuitry of drug reward. Neuropharmacology. 2014;76(Pt B):329–341. doi: 10.1016/j.neuropharm.2013.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedynak J, Hearing M, Ingebretson A, Ebner SR, Kelly M, Fischer RA, Kourrich S, Thomas MJ. Cocaine and Amphetamine Induce Overlapping but Distinct Patterns of AMPAR Plasticity in Nucleus Accumbens Medium Spiny Neurons. Neuropsychopharmacology. 2016;41:464–476. doi: 10.1038/npp.2015.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John CS, Sypek EI, Carlezon WA, Cohen BM, Ongur D, Bechtholt AJ. Blockade of the GLT-1 Transporter in the Central Nucleus of the Amygdala Induces both Anxiety and Depressive-Like Symptoms. Neuropsychopharmacology. 2015;40:1700–1708. doi: 10.1038/npp.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung ES, Lee HJ, Sim HR, Baik JH. Cocaine-induced behavioral sensitization in mice: effects of microinjection of dopamine d2 receptor antagonist into the nucleus accumbens. Experimental neurobiology. 2013;22:224–231. doi: 10.5607/en.2013.22.3.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nature reviews Neuroscience. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Karasawa J, Yoshimizu T, Chaki S. A metabotropic glutamate 2/3 receptor antagonist, MGS0039, increases extracellular dopamine levels in the nucleus accumbens shell. Neurosci Lett. 2006;393:127–130. doi: 10.1016/j.neulet.2005.09.058. [DOI] [PubMed] [Google Scholar]
- Kinney GG, Burno M, Campbell UC, Hernandez LM, Rodriguez D, Bristow LJ, Conn PJ. Metabotropic glutamate subtype 5 receptors modulate locomotor activity and sensorimotor gating in rodents. J Pharmacol Exp Ther. 2003;306:116–123. doi: 10.1124/jpet.103.048702. [DOI] [PubMed] [Google Scholar]
- Kuczenski R, Segal DS, Cho AK, Melega W. Hippocampus norepinephrine, caudate dopamine and serotonin, and behavioral responses to the stereoisomers of amphetamine and methamphetamine. J Neurosci. 1995;15:1308–1317. doi: 10.1523/JNEUROSCI.15-02-01308.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane TA, Boerner T, Bannerman DM, Kew JN, Tunbridge EM, Sharp T, Harrison PJ. Decreased striatal dopamine in group II metabotropic glutamate receptor (mGlu2/mGlu3) double knockout mice. BMC neuroscience. 2013;14:102. doi: 10.1186/1471-2202-14-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannix RC, Zhang J, Park J, Zhang X, Bilal K, Walker K, Tanzi RE, Tesco G, Whalen MJ. Age-dependent effect of apolipoprotein E4 on functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2011;31:351–361. doi: 10.1038/jcbfm.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeehan AJ, Janak PH, Olive MF. Effect of the mGluR5 antagonist 6-methyl-2-(phenylethynyl)pyridine (MPEP) on the acute locomotor stimulant properties of cocaine, D-amphetamine, and the dopamine reuptake inhibitor GBR12909 in mice. Psychopharmacology (Berl) 2004;174:266–273. doi: 10.1007/s00213-003-1733-2. [DOI] [PubMed] [Google Scholar]
- McNamara RK, Logue A, Stanford K, Xu M, Zhang J, Richtand NM. Dose-response analysis of locomotor activity and stereotypy in dopamine D3 receptor mutant mice following acute amphetamine. Synapse. 2006;60:399–405. doi: 10.1002/syn.20315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereu M, Tronci V, Chun LE, Thomas AM, Green JL, Katz JL, Tanda G. Cocaine-induced endocannabinoid release modulates behavioral and neurochemical sensitization in mice. Addiction biology. 2015;20:91–103. doi: 10.1111/adb.12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingote S, Chuhma N, Kalmbach A, Thomsen GM, Wang Y, Mihali A, Sferrazza CE, Zucker-Scharff I, Siena AC, Welch MG, Lizardi-Ortiz J, Sulzer D, Moore H, Gaisler-Salomon I, Rayport S. Dopamine neuron dependent behaviorsmediated by glutamate cotransmission. eLife. 2017:6. doi: 10.7554/eLife.27566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mookherjee P, Green PS, Watson GS, Marques MA, Tanaka K, Meeker KD, Meabon JS, Li N, Zhu P, Olson VG, Cook DG. GLT-1 loss accelerates cognitive deficit onset in an Alzheimer’s disease animal model. Journal of Alzheimer’s disease: JAD. 2011;26:447–455. doi: 10.3233/JAD-2011-110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, Spear LP. The effects of an acute challenge with the NMDA receptor antagonists, MK-801, PEAQX, and ifenprodil, on social inhibition in adolescent and adult male rats. Psychopharmacology (Berl) 2014;231:1797–1807. doi: 10.1007/s00213-013-3278-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods. 1984;11:47–60. doi: 10.1016/0165-0270(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The Mouse Brain in Sterotaxic Coordinates. 2nd. Academic Press; 2001. [Google Scholar]
- Paylor R, Crawley JN. Inbred strain differences in prepulse inhibition of the mouse startle response. Psychopharmacology (Berl) 1997;132:169–180. doi: 10.1007/s002130050333. [DOI] [PubMed] [Google Scholar]
- Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA, Fischer KD, Armsen W, Wang J, Danbolt NC, Rotenberg A, Aoki CJ, Rosenberg PA. Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci. 2015;35:5187–5201. doi: 10.1523/JNEUROSCI.4255-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempe D, Vangeison G, Hamilton J, Li Y, Jepson M, Federoff HJ. Synapsin I Cre transgene expression in male mice produces germline recombination in progeny. Genesis. 2006;44:44–49. doi: 10.1002/gene.20183. [DOI] [PubMed] [Google Scholar]
- Rimmele TS, Rosenberg PA. GLT-1: The elusive presynaptic glutamate transporter. Neurochem Int. 2016;98:19–28. doi: 10.1016/j.neuint.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts-Wolfe DJ, Kalivas PW. Glutamate Transporter GLT-1 as a Therapeutic Target for Substance Use Disorders. CNS & neurological disorders drug targets. 2015;14:745–756. doi: 10.2174/1871527314666150529144655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth KA, Katz RJ. Stress, behavioral arousal, and open field activity–a reexamination of emotionality in the rat. Neurosci Biobehav Rev. 1979;3:247–263. doi: 10.1016/0149-7634(79)90012-5. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Saxe MD, Battaglia F, Wang JW, Malleret G, David DJ, Monckton JE, Garcia AD, Sofroniew MV, Kandel ER, Santarelli L, Hen R, Drew MR. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci U S A. 2006;103:17501–17506. doi: 10.1073/pnas.0607207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield MD, Heinsbroek JA, Gipson CD, Kupchik YM, Spencer S, Smith AC, Roberts-Wolfe D, Kalivas PW. The Nucleus Accumbens: Mechanisms of Addiction across Drug Classes Reflect the Importance of Glutamate Homeostasis. Pharmacol Rev. 2016;68:816–871. doi: 10.1124/pr.116.012484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellings LH, Clarke PB. Segregation of amphetamine reward and locomotor stimulation between nucleus accumbens medial shell and core. J Neurosci. 2003;23:6295–6303. doi: 10.1523/JNEUROSCI.23-15-06295.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon P, Dupuis R, Costentin J. Thigmotaxis as an index of anxiety in mice. Influence of dopaminergic transmissions. Behav Brain Res. 1994;61:59–64. doi: 10.1016/0166-4328(94)90008-6. [DOI] [PubMed] [Google Scholar]
- Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron. 2011;69:628–649. doi: 10.1016/j.neuron.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Kong Q, Lin Y, Stouffer N, Schulte DA, Lai L, Liu Q, Chang LC, Dominguez S, Xing X, Cuny GD, Hodgetts KJ, Glicksman MA, Lin CL. Restored glial glutamate transporter EAAT2 function as a potential therapeutic approach for Alzheimer’s disease. J Exp Med. 2015;212:319–332. doi: 10.1084/jem.20140413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Tanda G, Ebbs A, Newman AH, Katz JL. Effects of 4′-chloro-3 alpha-(diphenylmethoxy)-tropane on mesostriatal, mesocortical, and mesolimbic dopamine transmission: comparison with effects of cocaine. J Pharmacol Exp Ther. 2005;313:613–620. doi: 10.1124/jpet.104.080465. [DOI] [PubMed] [Google Scholar]
- Tanda G, Newman AH, Ebbs AL, Tronci V, Green JL, Tallarida RJ, Katz JL. Combinations of cocaine with other dopamine uptake inhibitors: assessment of additivity. J Pharmacol Exp Ther. 2009;330:802–809. doi: 10.1124/jpet.109.154302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau LE, Hnasko TS, Wallen-Mackenzie A, Morales M, Rayport S, Sulzer D. The multilingual nature of dopamine neurons. Prog Brain Res. 2014;211:141–164. doi: 10.1016/B978-0-444-63425-2.00006-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill SM, Wheeler DS, Li M, Watts SD, Ingram SL, Amara SG. Amphetamine modulates excitatory neurotransmission through endocytosis of the glutamate transporter EAAT3 in dopamine neurons. Neuron. 2014;83:404–416. doi: 10.1016/j.neuron.2014.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Weiner I, Gal G, Rawlins JN, Feldon J. Differential involvement of the shell and core subterritories of the nucleus accumbens in latent inhibition and amphetamine-induced activity. Behav Brain Res. 1996;81:123–133. doi: 10.1016/s0166-4328(96)00051-4. [DOI] [PubMed] [Google Scholar]
- Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/s0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Tseng KY. Calcium-permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why? Frontiers in molecular neuroscience. 2012;5:72. doi: 10.3389/fnmol.2012.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME, Xue CJ, Li Y, Wavak D. Amphetamine increases glutamate efflux in the rat ventral tegmental area by a mechanism involving glutamate transporters and reactive oxygen species. J Neurochem. 2000;75:1634–1644. doi: 10.1046/j.1471-4159.2000.0751634.x. [DOI] [PubMed] [Google Scholar]
- Xue CJ, Ng JP, Li Y, Wolf ME. Acute and repeated systemic amphetamine administration: effects on extracellular glutamate, aspartate, and serine levels in rat ventral tegmental area and nucleus accumbens. J Neurochem. 1996;67:352–363. doi: 10.1046/j.1471-4159.1996.67010352.x. [DOI] [PubMed] [Google Scholar]
- Yap JJ, Miczek KA. Social defeat stress, sensitization, and intravenous cocaine self-administration in mice. Psychopharmacology (Berl) 2007;192:261–273. doi: 10.1007/s00213-007-0712-4. [DOI] [PubMed] [Google Scholar]
- Zahm DS, Brog JS. On the significance of subterritories in the “accumbens” part of the rat ventral striatum. Neuroscience. 1992;50:751–767. doi: 10.1016/0306-4522(92)90202-d. [DOI] [PubMed] [Google Scholar]