

Supplemental Digital Content is available in the text.
Keywords: inflammatory, isosteviol sodium, nuclear factor kappa-light-chain-enhancer of activated B cells, toll-like receptors, traumatic brain injury
Abstract
Previous studies have shown that isosteviol sodium (STVNa) protects against permanent cerebral ischemia injury by inhibition of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-mediated inflammatory responses. Overwhelming evidence shows that toll-like receptors (TLRs) are the upstream regulators of NF-κB. On the basis of the similarity of the pathology caused by traumatic brain injury (TBI) and stroke, we speculated that STVNa may have a therapeutic effect against TBI through regulation of the TLRs/NF-κB signaling-mediated inflammatory response. Thus, we studied the potential therapeutic effects of STVNa and the underlying mechanisms. Male rats, subjected to controlled cortical impact (CCI) injury, were injected intraperitoneally with STVNa (5, 10, 20, 40, and 80 mg/kg, daily for 3 or 7 days) after trauma. Neurobehavioral scores, relative numbers of cortical lesions, and histology were examined. We also measured the mRNA and protein expression levels of TLRs/NF-κB signaling pathway-related genes including TLR2, TLR4, and NF-κB by quantitative real-time-PCR and western blotting, respectively, and concentrations of tumor necrosis factor-α and interleukin-1β by an enzyme-linked immunosorbent assay. The results indicated that STVNa (20 mg/kg) showed significant neuroprotective effects 3 and 7 days after TBI, including the reduction of cortical lesions, improvement of the neurological severity score, significantly increased number of restored neurons, decreased number of astrocytes, and lower concentrations of tumor necrosis factor-α and interleukin-1β. Results from quantitative real-time-PCR and western blotting also show that the mRNA and protein expression levels of TLR2, TLR4, and NF-κB were significantly lower in STVNa-treated rats compared with the vehicle-treated rats. The administration of STVNa attenuates the TLR/NF-κB signaling pathway-mediated inflammatory responses in the injured rat brain, and this may be the mechanism by which STVNa improves the outcome following TBI.
Introduction
Traumatic brain injury (TBI) is the leading cause of severe disability and human deaths worldwide, especially in children and young adults 1,2. Approximately 10 million individuals are hospitalized and 1.7 million individuals die every year in the USA owing to TBI 3. This exceedingly complex disorder is caused by both primary and secondary brain injury, which results from metabolic, cellular, and delayed neurochemical changes, and evolves over hours to days following the initial traumatic insult, and causes progressive white and gray matter damage. A complex series of sterile inflammatory responses have been reported to play an important role in secondary brain injury following TBI 4,5. Although the mechanisms regulating this process are not well understood, several studies have reported the inhibition of proinflammatory mediators to offer a potentially effective therapeutic approach against TBI 6,7.
Toll-like receptors (TLRs), which are involved in the recognition of pathogen-associated molecular patterns present on bacteria, are transmembrane proteins 8. Among these, TLR2 and TLR4, which are expressed widely in the brain, detect endogenous agonists, intracellular components of ruptured cells, and products of genes that are activated by inflammation 9. Moreover, TLR2 and TLR4 have been shown to play a major role in initiating the cerebral inflammation related to ischemia, Parkinson’s disease, and Alzheimer’s disease 10. It is well known that TLRs activate a common signaling pathway that culminates in the activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). As one of the most important downstream molecules in the TLR signaling pathway, NF-κB activation is required for the transcription of genes of several inflammatory mediators, such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) 11.
Stevioside is a natural sweet-tasting glycoside isolated from the herb Stevia rebaudiana, which is composed of steviol, a diterpenic carboxylic alcohol with three glucose molecules. It has a sweetening potency 300 times that of sucrose and has long been used as a food sweetener in many countries for decades 12. Previous studies have shown that stevioside can lower blood sugar 13 and blood pressure 14, which may exert beneficial effects in cerebral ischemia. Isosteviol is obtained by the acid hydrolysis of stevioside, a process that maintains the desirable pharmacological activities of stevioside. A previous study reported that pretreatment with isosteviol inhibits NF-κB expression, thus reducing inflammation and apoptosis in a rat model of stroke 15. However, isosteviol is a beyerene diterpene and is therefore not suitable as an aqueous injection to treat ischemic diseases because it has poor solubility and low bioavailability. An injectable formulation of isosteviol sodium salt (STVNa) possesses much greater solubility and bioavailability, and has the potential to be applied widely as an emergency treatment 16. Our group had previously reported the neuroprotective effects of STVNa against acute focal cerebral ischemia in rats 17 and further reported that it protects against permanent cerebral ischemia injury in mice by inhibition of NF-κB-mediated inflammatory and apoptotic responses. However, whether STVNa exerts protective effects against TBI injury remains completely unknown.
The aim of the present study was to investigate whether STVNa attenuates the TBI-induced activation of the TLRs/NF-κB signaling pathway in the penumbra field of rats with acute focal cerebral ischemia. We speculate that modulation of the TLR/NF-κB signaling pathway is the mechanism by which STVNa confers neuroprotection by inhibition of astrocyte proliferation, increased neuronal survival rate, and reduced swelling of the hippocampus, promoting behavioral recovery after TBI.
Materials and methods
Experimental animals
Male Sprague-Dawley rats (8–10 weeks of age), weighing 250–300 g, were purchased from the Animal Research Centre of Guangzhou University of Chinese Medicine (Guangzhou, China). The rats were housed in a temperature-controlled environment (25±2°C), with a 12-h light–dark cycle, and allowed free access to food and water. All efforts were made to minimize animal suffering and reduce the number of animals used. The experimental procedures were approved by the Institutional Animal Care and Use Committee of Guangdong Pharmaceutical University.
Controlled cortical impact of the TBI model
The experimental TBI method used in this study was performed as described previously, with minor modifications 18. Rats were anesthetized with 2% isoflurane and placed on a stereotaxic frame. After a midline skin incision, a 3.5 mm circular craniotomy was performed midway between the lambda and the bregma, 2.0 mm to the right of the central suture, using an electric drill. During the craniotomy, rats were excluded from the study if the dura mater was breached. The controlled cortical impact (CCI) was induced with an electric impact device using an impact tip. We used a PCI3000 precision cortical impactor (Hatteras Instruments, Cary, North Carolina, USA) and a 2.0 mm diameter flat fact tip with a slightly rounded edge (velocity=1.5 m/s, depth=5 mm, and contact time=120 ms). The temperature was monitored in all groups using a rectal thermometer, and maintained at 37±0.5°C during surgery, using a heating pad controlled by a homeothermic blanket control unit (Harvard Apparatus, Holliston, Massachusetts, USA). After the injury, the skin was sutured and rats were allowed to recover. Animals in the sham and TBI groups were injected with vehicle after TBI and their body temperatures were maintained at 37°C in a humidity-controlled incubator for up to 6 h after TBI. In the other groups, animals were administered STVNa injections and the temperature was not maintained artificially.
STVNa administration
STVNa was obtained from the Chemical Development Laboratories of Key Biological Pharmaceutical Company (Dongguan, China). The drug was administered intraperitoneally immediately after the CCI injury. To assess dose–response, rats were distributed randomly into seven groups: sham (n=6), vehicle (n=6), and STVNa (5, 10, 20, 40, and 80 mg/kg, n=6/group). All the groups were injected as mentioned above. The injection volume was maintained around 1.2 ml. To investigate the mechanism, STVNa treatment was performed at a dose of 20 mg/kg. To explore the effects of STVNa treatment on astrogliosis and neuron survival in the phases of TBI, rats were randomly injected intraperitoneally with vehicle (n=3) or 20 mg/kg STVNa (n=3) immediately after TBI and then injected consecutively for 3 or 7 days until the end of the experiment. Anesthesia was administered only during the TBI procedure; animals recovered from isoflurane anesthesia and received an intraperitoneal injection of STVNa without further sedation.
Neurological deficits analysis
After 3 and 7 days of TBI, the rats were subjected to behavioral evaluation. Rats were tested for neurological deficits according to the Modified Neurological Severity Score (mNSS) 19,20. Neurological function was graded on a scale of 0–18 (normal score, 0; maximal deficit score, 18). The mNSS is a composite of motor, sensory, reflex, and balance tests. During scoring of the severity of injury, a score of 1 was assigned for inability to perform the test or lack of the tested reflex. The scores indicate three levels of neurological damage: mild damage (1–6), moderate damage (7–12), and severe damage (13–18). To evaluate the sensory functions of animals more intuitively and clearly, we used the Bederson test 21, which assesses motor deficits and circling behavior. Motor and behavioral changes were assessed using a five-point scale as follows: 0=no observable neurological deficit, 1=failure to extend left forepaw, 2=decreased resistance to lateral push towards the paretic site (and forelimb flexion) without circling, 3=same behavior as grade 2 with the addition of circling, and 4=loss of spontaneous walking, and a depressed level of consciousness.
Evaluation of contusion volume
To measure the contusion volumes, Nissl staining was performed 3 days after TBI. Freshly frozen 20 μm-thick brain sections containing target lesions were collected and fixed with a 1 : 1 mixture of 10% formalin and acetic acid for 10 min. After washing with distilled water for 5 min, slices were stained with a solution containing a buffer (0.1 M acetate acid and 0.1 M sodium acetate at a ratio of 94 : 6) and cresyl violet acetate at a ratio of 5 : 1. The sections were then dehydrated in 100% ethyl alcohol and mounted.
After Nissl staining, the sections were imaged using ImageJ software (National Institutes of Health, Bethesda, Maryland, USA) and the areas of the contusion in the two hemispheres were quantified by an investigator blinded to the treatment of the animals. Contusion volume was assessed on the basis of the Cavalieri method of stereology 22. The number of sections and the section thickness were multiplied by the mean area of the remaining cortex. Contusion volumes were calculated on the basis of the contusion areas (C) obtained from 10 to 12 sections as follows: C1×D+0.2 (C1+C2+C3+…C12), with D being the distance between two sections. In addition, to consider the effects of swelling or edema, hemispheric tissue loss was measured as a percentage calculated by: [(contralateral hemispheric volume−ipsilateral hemispheric volume)/(contralateral hemispheric volume)×100%]. The result was reported as the ratio of lesion volume against the corrected hemispheric volume.
Immunohistochemical assays
Immunohistochemistry for glial fibrillary acidic protein (GFAP) and neuronal nuclei (NeuN) were performed using polyclonal rabbit anti-GFAP (1 : 3000 dilution; Dako, Carpinteria, California, USA) and anti-NeuN (1 : 2000 dilution; Abcam, Cambridge, UK) primary antibodies, respectively. Sections were then washed in PBS and incubated for 40 min at 37°C with the secondary antibody (anti-mouse IgG antibody EnVision+System; HRP, Dako, Glostrup, Denmark), and after a final wash in PBS, bound antibodies were visualized with 3,3′-diaminobenzidine using a 3,3′-diaminobenzidine-enhanced liquid substrate system (Dako). All incubations were performed under humidified conditions. Finally, sections were counter-stained in hematoxylin. To confirm the specificity of the immunostaining, a negative control test was performed using mouse (G3A1) mAb IgG1 Isotype Control (Cell Signaling Technology Inc., Danvers, Massachusetts, USA) in place of the primary antibodies. NeuN-positive or GFAP-positive cells were observed and counted using a fluorescence microscope (Leica DM4000 B LED; Leica, Wetzlar, Germany).
RNA extraction and qRT-PCR
Total RNA was extracted using TriPure Reagent (Roche Diagnostics, Indianapolis, Indiana, USA) according to the manufacturer’s instructions. The RNA (500 ng) from each sample was reverse-transcribed into cDNA using the PrimeScript RT reagent kit (Takara Bio Inc., Kusatsu, Japan). RT-PCR was performed using the 7500 real-time-PCR system (Applied Biosystems, Foster City, California, USA) with AceQ qPCR SYBR Green Master Mix (Q111-02; Vazyme Biotech Co. Ltd., Nanjing City, China). RT-PCR primers were as follows: TLR2, 5′CTCTTCAGCAAACGCTGTTCT-3′ and 5′-GGCGTCTCCCTCTATTGTATTG-3′; TLR4, 5′ATGGCATGGCTTACACCACC-3′ and 5′-GAGGCCAATTTTGTCTCCACA-3′; NF-κB, 5′-TGCGATTCCGCTATAAATGCG-3′ and 5′-ACAAGTTCATGTGGATGAGGC-3′; Gapdh, 5′-TCAACAGCAACTCCCACTCTTCCA-3′ and 5′-ACCCTGTTGCTGTAGCCGTATTCA-3′. The obtained data were normalized to Gapdh expression levels for each sample.
Enzyme-linked immunosorbent assay
The levels of inflammatory mediators were quantified using specific ELISA kits for rats according to the manufacturers’ instructions (TNF-α; Diaclone Research, France; IL-1β; BioSource Europe SA, Belgium) and previous studies 23,24. Results were expressed as ng/g protein.
Western blotting
Seventy-two hours after surgery, 12 mice (three from the sham group, three from the TBI group, and three from the TBI+20 mg/kg STVNa group) were euthanized, perfused with ice-cold PBS through the ascending aorta until the perfusion buffer flowing from the right atrium was clear, and then pericontusional brain tissue (brain area within 3 mm of the epicenter of the injury) was harvested. Western blotting was performed as described previously 25. Briefly, total proteins were extracted and the protein concentration was determined using a BCA kit (Solarbio, Beijing, China). The samples were subjected to SDS-polyacrylamide gel electrophoresis. Separated proteins on the gel were subsequently transferred onto polyvinylidene fluoride membranes (Roche Diagnostics GmbH, Mannheim, Germany) by a transfer apparatus at 300 mA for 90 min. The blots were then blocked with 5% fat-free dry milk for 2 h at room temperature. Subsequently, the blots were incubated with the indicated primary antibodies overnight at 4°C, including mouse anti-TLR2 monoclonal antibody (dilution 1 : 500), mouse anti-TLR4 monoclonal antibody (dilution 1 : 500), mouse anti‑NF-κB p65 polyclonal antibody (dilution 1 : 500), and mouse anti-β-actin monoclonal antibody (both from Abcam, Cambridge, Massachusetts, USA). They were then incubated with horseradish peroxidase-conjugated anti-mouse IgG (1 : 2000 dilution) (all from Abcam) secondary antibodies for 2 h at room temperature. The immunoblot on the membrane was then visible following development with an enhanced chemiluminescence detection system and the densitometric signals were quantified using an ImageJ software (National Institutes of Health). The intensity of the immunoreactive bands was normalized to the intensity of the corresponding bands for β-actin. The results were analyzed using the ImageJ software (National Institutes of Health).
Statistical analysis
Data were analyzed, and measurements were expressed as mean±SEM, using the one-way analysis of variance with Tukey’s multiple-comparison test to evaluate the statistical significance of the differences among means. A P value of less than 0.05 was considered statistically significant.
Results
Dose–response study
To investigate the effects of CCI, the treated groups were compared with sham-operated groups, which had only been subjected to craniotomy. On the basis of the previous studies indicating that STVNa provides neuroprotection against ischemic stroke 26, and that similar mechanisms of action limit anatomical brain damage and neurological deficits following both experimental ischemic stroke and TBI 27,28, we hypothesized that STVNa may exert a protective effect against TBI.
To study the dose–response of STVNa, we used Nissl staining to calculate the relative cortical lesion after 3 days (Fig. 1). The result showed that the lesion size of the vehicle group (12.25±1.59%) was considerably large. A significant decrease in the lesion size was observed at 10 (10.55±0.77%), 20, or 40 mg/kg, and the most significant decrease in the lesion were at 20 and 40 mg/kg (8.68±0.85 and 8.76±0.89%) doses. From the data, the 5 mg/kg dose (12.31±1.11%) was ineffective, and no significant reduction in the lesion size was observed with the 40 mg/kg dose compared with the 20 mg/kg dose (Fig. 1a).
Fig. 1.
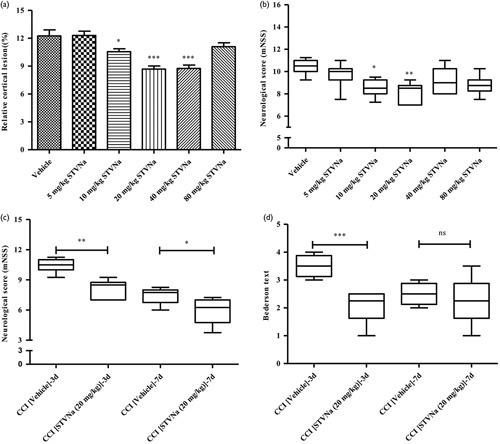
Results of the dose–response relationship of isosteviol sodium (STVNa) were measured 3 and 7 days after traumatic brain injury (TBI) in rats. (a) The distribution of percent area of infarct in serial brain sections stained with cresyl violet-stain 3 days after TBI in vehicle-treated and STVNa-treated rats. (b) Neurological scores of Modified Neurological Severity Score (mNSS) 3 days after TBI in the study of the dose–response relationship of STVNa. (c) Neurological scores of mNSS at 3 and 7 days after TBI. (d) Neurological scores of the Bederson test at 3 and 7 days after TBI. Data were expressed as the mean±SEM (n=8/group). *P<0.05, **P<0.01 and ***P<0.001 versus the vehicle group by one-way analysis of variance with Tukey’s multiple-comparison test.
Furthermore, 3 days after treatment, we evaluated functional improvements in STVNa-treated animals using the mNSS. Figure 1b shows that the rats in the sham group showed no neurological deficits, whereas rats in the vehicle group displayed considerable neurological deficits after CCI. The 10 and 20 mg/kg doses of STVNa decreased the mNSS values compared with the vehicle group (Fig. 1b), with the latter showing the maximal effect. Therefore, 20 mg/kg of STVNa treatment significantly reduced cortical lesion and improved neurological function, and was selected for subsequent experiments.
Effect of STVNa on neurobehavioral outcomes
A neurobehavioral assessment was performed 3 and 7 days after CCI. Marked neurological impairment was observed in vehicle-treated CCI rats, as reflected in the increased neurological score, whereas no or only marginal neurological symptoms were observed in the sham-operated animals. Figure 1c and d show significant neurological impairment following CCI at the 3-day and 7-day time-points compared with sham-operated animals. Our data indicate that treatment with STVNa at a dose of 20 mg/kg significantly improves the neurological score after CCI at both the time-points tested.
Effect of STVNa on anatomical outcomes
To determine whether STVNa could improve short-term anatomical outcomes following CCI, brain sections were analyzed 3 and 7 days after surgery. Rats were assigned randomly to three groups: sham, CCI vehicle, and 20 mg/kg STVNa-treated group. At the mentioned time-points, CCI caused complex cortical lesions, including hematoma, a decrease in the cellular density of the surrounding areas, and loss of cortical tissue, referred to as cavitation. In addition to cortical injury, significant hippocampal swelling (146.84±17.22% of the contralateral side) was observed in all vehicle-treated CCI animals compared with the sham group (P<0.05, n=6). Treatment with 20 mg/kg STVNa reduced CCI-induced hippocampal swelling to levels significantly different from those of the vehicle group.
At the later time-point in the CCI group, the lesions were characterized by structurally defined cortical cavitation (Fig. 2a) and significant hippocampal swelling was still present, although less prominent compared with the 3-day time-point. Seven days after injury, hippocampal swelling in the CCI group was measured to be 127.46±8.42% (n=8) of the contralateral side (Fig. 2b and c), and was significantly lower than the value obtained at the 3-day time-point (P<0.05, Student’s independent t-test between the two CCI groups). Treatment with STVNa reduced CCI-induced hippocampal swelling to a level significantly lower than that of the vehicle group.
Fig. 2.
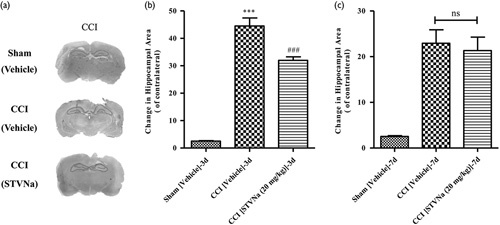
Brain pathology after controlled cortical impact (CCI). (a) Representative photographs of cresyl-violet-stained brain sections from sham and CCI-injured wild type (WT) mice showing cortical cavitation and remaining hippocampal swelling in the areas 3 days after injury. (b, c) Quantitative analysis of relative hippocampal areas normalized to the contralateral side in saline-treated and isosteviol sodium (STVNa) (20 mg/kg)-treated rats at 3 days (b) and 7 days (c) after CCI or sham. Data are presented as mean±SEM, ***P<0.001, versus the saline-treated sham group, and ###P<0.001, versus the saline-treated CCI group, one-way analysis of variance, followed by Tukey’s multiple-comparison test (n=7–9).
Effects of STVNa on astrogliosis and neuronal survival following CCI
Considering the important role of astrocytes and neuronal survival in TBI and the neuroprotective effects of STVNa 26, we carried out immunohistochemical studies to detect changes in the levels of astrocytic and neuronal markers in rats 3 and 7 days after injury (Fig. 3a). At this time-point, CCI caused apparent astrogliosis, detected as increased GFAP immunoreactivity in the surrounding areas of cortical injury, referred to as penumbra, as well as in the ipsilateral hippocampal regions within lateral dorsal and posterior nuclei, compared with the corresponding contralateral areas. In the CCI-injured animals, increased GFAP immunoreactivity was accompanied by morphological changes in the immunopositive cells. Figure 3b shows representative images of the morphological changes in the hippocampal GFAP-positive cells, suggesting astrogliosis in the ipsilateral side compared with the contralateral side. A slight increase was observed in the GFAP levels in the ipsilateral cortical area surrounding the craniotomy site, but not in the other brain regions, and was also observed in sham-operated animals. However, the changes in the GFAP levels in sham-operated animals were markedly lower than that in the cortical penumbra of the animals from the CCI group. In the STVNa-treated animals, the morphological changes in the microglia were less pronounced than those in the vehicle-treated ones. Three days after injury, GFAP immunoreactivity increased significantly in the cortical penumbra of the STVNa-treated CCI animals compared with the vehicle-treated CCI group, whereas in the hippocampus, GFAP immunoreactivity decreased to the control level at this time-point.
Fig. 3.
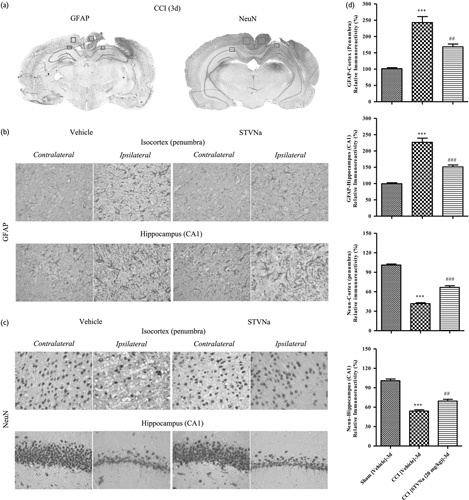
Effects of isosteviol sodium (STVNa) on astrogliosis and neuron survival following controlled cortical impact (CCI). (a) Representative brain sections and microphotographs of selected brain regions from the same corresponding sections used for immunohistochemistry quantification 3 days after traumatic brain injury (TBI). (b) Effects of STVNa on glial fibrillary acidic protein (GFAP) immunoreactivity in TBI (n=3/group). Scale bar=50 m. (c) Effect of STVNa on neuronal immunoreactivity in the rats’ contusive brain (n=3/group). Scale bar=50 m (d) Quantitative analyses of GFAP and neuronal nuclei (NeuN) immunoreactivity in the ipsilateral brain regions. Data are represented as mean±SEM (n=3; ***P<0.001 vs. the sham group; ##P<0.01 and ###P<0.001 vs. the CCI/vehicle group).
To study neuronal survival, we used NeuN immunoreactivity. Similar to GFAP levels, the NeuN levels increased considerably in the cortical penumbra as well as in the ipsilateral hippocampal regions compared with the corresponding contralateral areas in the CCI-treated animals at both time-points. A large decrease in the NeuN levels was observed in the cortical area surrounding the craniotomy site in the sham-operated animals. Significant differences in the NeuN immunoreactivities between the ipsilateral and the contralateral hippocampal regions were observed in the CCI-injured and STVNa-treated rats (Fig. 3c). In the cortical penumbra and hippocampus, increased neuronal survival was observed 3 days after injury. Similarly, STVNa treatment for 7 days also significantly inhibited the proliferation of astrocytes and the loss of neurons caused by CCI injury compared with the vehicle group (Supplementary Fig. 1, Supplemental digital content 1, http://links.lww.com/WNR/A468).
STVNa inhibits the TLRs/NF-κB signaling pathway following TBI
Many studies reported that the pathological processes including inflammatory response, apoptosis, and loss of neurons usually reached the maximum peak at 3 days after TBI injury 29–34. To further explore the mechanism underlying the STVNa-mediated neuroprotective effects, the levels of TLR2, TLR4, and NF-κB were measured after the administration of STVNa for 3 days following TBI (Fig. 4). These genes play a crucial role in the TLR/NF-κB signaling pathway, and are activated by TBI. The expression of NF-κB was higher in the vehicle-treated group than in the sham-operated group, confirming an observation from our previous study 26. However, treatment with 20 mg/kg STVNa 3 days after TBI effectively reduced the activation of NF-κB (Fig. 4d). As expected, the levels of TLR2 and TLR4 increased significantly 3 days after TBI in the vehicle group versus the sham-operated group (P<0.05), as observed previously. The administration of STVNa significantly reduced the levels of TLR2 and TLR4 compared with the vehicle group (P<0.05) (Fig. 4c and Fig. 5).
Fig. 4.
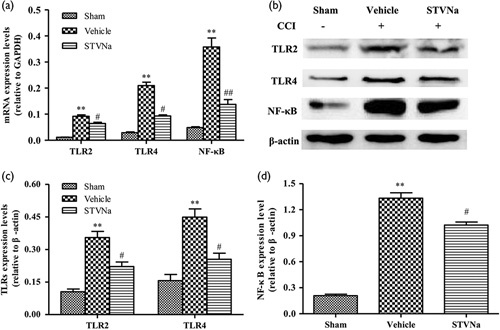
Isosteviol sodium (STVNa) inhibits the toll-like receptors (TLRs)/nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway after traumatic brain injury (TBI) in vivo. (a) The mRNA expression levels of TLR2, TLR4, and NF-ΚB in the injured tissue were detected by RT-PCR at 3 days after trauma. (b) The protein expression levels of TLR2, TLR4, and NF-κB were detected by western blotting at 3 days after trauma. (c, d) Relative expression levels of TLR2, TLR4 (c), and NF-κB (d) were calculated by normalizing to that of GAPDH, respectively. Data are represented as mean±SEM (n=3; **P<0.01 vs. the sham group; #P<0.05 and ##P<0.01 vs. the vehicle group).
Fig. 5.
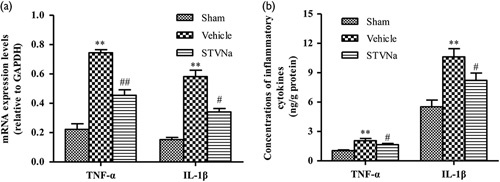
Isosteviol sodium (STVNa) treatment significantly downregulates the expression of inflammatory cytokines in the brain. (a, b) The expression levels of tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) in the injured brains were determined by RT-PCR (a) and ELISA (b). Data are represented as mean±SEM (n=3; **P<0.01 vs. the sham group; #P<0.05 and ##P<0.01 vs. the vehicle group).
STVNa treatment significantly reduces the expression of inflammatory cytokines in the brain
To investigate the downstream inflammatory mechanism induced by the TLR/NF-κB signaling pathway further, ELISA was performed to examine the expression of inflammatory cytokines in the hippocampus. The results showed that the expression levels of TNF-α and IL-1β were significantly upregulated in the hippocampal tissues of rats in the TBI group compared with the sham-operated group 3 days after TBI, and both were significantly reduced by STVNa. These results indicate that STVNa significantly reduces the production of inflammatory factors in the hippocampus of a rat model of TBI.
Discussion
TBI is the deadliest and most disabling form of acute brain trauma, accompanied by inflammatory responses and neuron apoptosis 35. Unfortunately, there is no effective treatment for TBI so far. Our previous studies have shown that STVNa protects against permanent cerebral ischemia injury by inhibition of NF-κB-mediated inflammatory responses 26. On the basis of the similarity of the pathology caused by TBI and stroke, in this study, we investigated whether STVNa has any therapeutic effect against TBI and its underlying mechanisms.
Our results in the present study indicated that: (a) 20 mg/kg STVNa treatment significantly reduces infarct volumes, improves neurological function, and reduces CCI-induced hippocampal swelling; (b) the astrocytic hyperplasia and neuronal loss because of TBI can be inhibited by STVNa; (c) the cortical levels of molecules involved in the TLR/NF-κB signaling pathway, such as TLR2, TLR4, and NF-κB, are suppressed by treatment with STVNa; and (d) following the administration of STVNa, the expression of TNF-α and IL-1β in the cortical regions surrounding the injured site is significantly decreased. These findings suggest that STVNa attenuates the TBI-induced activation of the TLRs/NF-κB signaling pathway, which may lead to secondary brain damage following primary trauma in the rat model of TBI.
The TLRs are a family of transmembrane proteins and act as signal transduction molecules 36. They play a major role in controlling the innate immune response to a wide variety of pathogen-associated molecules 37. Some TLRs have been authenticated in animal brains and human cells. Several studies have shown that TLR2 and TLR4 are the key to lipopolysaccharide-induced injury in the central nervous system. Thus, TLR2 and TLR4 are well positioned in the central nervous system and may initiate inflammation following TBI.
Although the results from this study indicate that the expression levels of TLR2 and TLR4 in the contused brain increase following TBI, and can be suppressed by the administration of STVNa, the mechanism underlying the initial effect of the TLR signaling pathway following TBI remains unclear. As mentioned earlier, certain endogenous stimuli of TLR2 and TLR4, such as heat-shock proteins 60 and 70, fibrinogens, and fibronectin, have been reported to enter or increase in level in the cerebrospinal fluid and brain after TBI 38,39. However, the principal TLR ligands in the brain and the mechanisms responsible for the beneficial effects of STVNa against TBI warrant further research.
The functional importance of NF-κB in acute inflammation is based on its ability to regulate the promoters of a variety of genes whose products, such as TNF-α, IL-1β, and acute-phase proteins, are critical to inflammatory processes 40. The inhibition of NF-κB activation by corticosteroid hormones, antioxidants, protease inhibitors, and other compounds may provide a pharmacological basis for interference with the pathological inflammatory conditions 41. Pettus et al. 42 suggested that progesterone administered after TBI reduced the initial cytotoxic surge of inflammatory factors, including complement factor C3, GFAP, and NF-κB. A previous study showed that NF-κB-binding activity is significantly reduced at 5 days after injury after the administration of progesterone, which is in agreement with the results of the study by Pettus et al. 42, in which western blotting was used to analyze NF-κB expression relative to total protein from brain samples.
Activation of TLRs recruits downstream signaling proteins, leading to transcription of genes encoding inflammation-associated molecules and cytokines. For example, combined with specific ligands, the activation of TLR2 and TLR4, the first mammalian TLRs recognized, dissociates the inhibitor from NF-κB, following which it translocates into the nucleus, and activates and regulates the transcription of genes related to inflammatory responses, such as TNF-α and IL-1β 43,44. Accumulating evidence indicates that TLR/NF-κB signaling contributes toward inflammatory responses, and thereby induces neuronal apoptosis during TBI 45–48.
Although the mechanisms of STVNa against TBI still need to be further explored in vivo and in vitro, our results, for the first time, show the protective effects of STVNa on the brain after TBI by inhibiting TLR/NF-κB signaling pathway. We found that TBI upregulates mRNA and protein expression of TLR2, TLR4, and NF-κB, and the levels of TNF-α and IL-1β in the brain regions surrounding the injured site, which are inhibited markedly by the administration of STVNa. These results suggest that TBI induces the activation of the TLR/NF-κB signaling pathway in rats, and this activation plays a central role in the inflammatory response that leads to secondary insults following TBI. The therapeutic effects of STVNa injection after TBI are probably through inhibiting TLRs/NF-κB-dependent inflammatory responses.
Supplementary Material
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website, www.neuroreport.com.
Acknowledgements
This study was supported in part by the Science and Technology Planning Project of Guangdong Province (no. 2015B010109004), the 2017 PhD Start-up Fund of the Natural Science Foundation of Guangdong Province of China (2017A030310404), the China Postdoctoral Science Foundation (no. 2017M622642), the Science and Technology Planning Project of Guangzhou (no.123000068), and the National Natural Science Foundation of China (31601089).
The authors are grateful to Key Biopharmaceutical Co. Ltd for supplying STVNa.
Conflicts of interest
There are no conflicts of interest.
Footnotes
Jie Zan and Hao Zhang contributed equally to the writing of this article.
References
- 1.Runyan DK. The challenges of assessing the incidence of inflicted traumatic brain injury: a world perspective. Am J Prev Med 2008; 34:S112–S115. [DOI] [PubMed] [Google Scholar]
- 2.Coronado VG, Xu L, Basavaraju SV, McGuire LC, Wald MM, Faul MD, et al. Surveillance for traumatic brain injury-related deaths – United States, 1997–2007. Morbidity and mortality weekly report. MMWR Surveill Summ 2011; 60:1–32. [PubMed] [Google Scholar]
- 3.Mcgarry LJ, Thompson D, Millham FH, Cowell L, Snyder PJ, Lenderking WR, Weinstein MC. Outcomes and costs of acute treatment of traumatic brain injury. J Trauma 2002; 53:1152. [DOI] [PubMed] [Google Scholar]
- 4.Helmy A, De Simoni MG, Guilfoyle MR, Carpenter KL, Hutchinson PJ. Cytokines and innate inflammation in the pathogenesis of human traumatic brain injury. Prog Neurobiol 2011; 95:352–372. [DOI] [PubMed] [Google Scholar]
- 5.Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics 2010; 7:22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chakrabarti M, Das A, Samantaray S, Smith JA, Banik NL, Haque A, et al. Molecular mechanisms of estrogen for neuroprotection in spinal cord injury and traumatic brain injury. Rev Neurosci 2015; 27:271–281. [DOI] [PubMed] [Google Scholar]
- 7.Jain KK. Neuroprotection in traumatic brain injury. The handbook of neuroprotection: Humana Press; 2011. 1082–1089. [Google Scholar]
- 8.Kopp E, Medzhitov R. Recognition of microbial infection by toll-like receptors. Curr Opin Immunol 2003; 15:396–401. [DOI] [PubMed] [Google Scholar]
- 9.Johnson GB, Brunn GJ, Platt JL. Activation of mammalian Toll-like receptors by endogenous agonists. Crit Rev Immunol 2003; 23:15. [DOI] [PubMed] [Google Scholar]
- 10.Lee SJ, Lee S. Toll-like receptors and inflammation in the CNS. Curr Drug Targets Inflamm Allergy 2002; 1:181. [DOI] [PubMed] [Google Scholar]
- 11.Takeda K, Akira S. TLR signaling pathways. Semin Immunol 2004; 16:3–9. [DOI] [PubMed] [Google Scholar]
- 12.Soejarto DD, Kinghorn AD, Farnsworth NR. Potential sweetening agents of plant origin. III. Organoleptic evaluation of Stevia leaf herbarium samples for sweetness. J Nat Prod 1982; 45:590. [DOI] [PubMed] [Google Scholar]
- 13.Gregersen S, Jeppesen PB, Holst JJ, Hermansen K. Antihyperglycemic effects of stevioside in type 2 diabetic subjects. Metabolism 2004; 53:73–76. [DOI] [PubMed] [Google Scholar]
- 14.Chan P, Xu DY, Liu JC, Chen YJ, Tomlinson B, Huang WP, Cheng JT. The effect of stevioside on blood pressure and plasma catecholamines in spontaneously hypertensive rats. Life Sci 1998; 63:1679–1684. [DOI] [PubMed] [Google Scholar]
- 15.Xu D, Du W, Zhao L, Davey AK, Wang J. The neuroprotective effects of isosteviol against focal cerebral ischemia injury induced by middle cerebral artery occlusion in rats. Planta Med 2008; 74:816–821. [DOI] [PubMed] [Google Scholar]
- 16.Lai W, Kang Q, Zou C, Li Q, Sun H, Tan W. Development of a liquid formulation of poorly water-soluble isosteviol sodium using the co-solvent technology. Pharm Dev Technol 2017; 22:275–282. [DOI] [PubMed] [Google Scholar]
- 17.Hu H, Sun XO, Tian F, Zhang H, Liu Q, Tan W. Neuroprotective effects of isosteviol sodium injection on acute focal cerebral ischemia in rats. Oxid Med Cell Longev 2016; 2016:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prostaglandin F2alpha FP receptor antagonist improves outcomes after experimental traumatic brain injury. [DOI] [PMC free article] [PubMed]
- 19.Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, Chopp M. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke 2001; 32:1005–1011. [DOI] [PubMed] [Google Scholar]
- 20.Chen Y, Constantini S, Trembovler V, Weinstock M, Shohami E. An experimental model of closed head injury in mice: pathophysiology, histopathology, and cognitive deficits. J Neurotrauma 1996; 13:557–568. [DOI] [PubMed] [Google Scholar]
- 21.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke 1986; 17:472. [DOI] [PubMed] [Google Scholar]
- 22.Ahmad AS, Saleem S, Ahmad M, Doré S. Prostaglandin EP1 receptor contributes to excitotoxicity and focal ischemic brain damage. Toxicol Sci 2006; 89:265–270. [DOI] [PubMed] [Google Scholar]
- 23.Bagge A, Clausen TR, Larsen S, Ladefoged M, Rosenstierne MW, Larsen L, et al. MicroRNA-29a is up-regulated in beta-cells by glucose and decreases glucose-stimulated insulin secretion. Biochem Biophys Res Commun 2012; 426:266. [DOI] [PubMed] [Google Scholar]
- 24.Sun J, Li F, Chen J, Xu J. Effect of ketamine on NF-kappa B activity and TNF-alpha production in endotoxin-treated rats. Ann Clin Lab Sci 2004; 34:181. [PubMed] [Google Scholar]
- 25.Hua F, Ma J, Ha T, Kelley JL, Kao RL, Schweitzer JB, et al. Differential roles of TLR2 and TLR4 in acute focal cerebral ischemia/reperfusion injury in mice. Brain Res 2009; 1262:100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang H, Sun X, Xie Y, Zan J, Tan W. Isosteviol sodium protects against permanent cerebral ischemia injury in mice via inhibition of NF-κB mediated inflammatory and apoptotic responses. J Stroke Cerebrovasc Dis 2017; 26:2603–2614. [DOI] [PubMed] [Google Scholar]
- 27.Elting JW, de Jager AEJ, Teelken AW, Schaaf MJ, Maurits NM, van der Naalt J, et al. Comparison of serum S-100 protein levels following stroke and traumatic brain injury. J Neurol Sci 2000; 181:104–110. [DOI] [PubMed] [Google Scholar]
- 28.Fandiñorivera J. Traumatic brain injury and ischemic stroke: a delayed sequela? Rev Neurol 2004; 38:912. [PubMed] [Google Scholar]
- 29.Itoh T, Imano M, Nishida S, Tsubaki M, Nakayama T, Mizuguchi N, et al. Appearance of neural stem cells around the damaged area following traumatic brain injury in aged rats. J Neural Transm (Vienna) 2013; 120:361–374. [DOI] [PubMed] [Google Scholar]
- 30.Itoh T, Satou T, Hashimoto S, Ito H. Isolation of neural stem cells from damaged rat cerebral cortex after traumatic brain injury. Neuroreport 2005; 16:1687–1691. [DOI] [PubMed] [Google Scholar]
- 31.Itoh T, Satou T, Nishida S, Hashimoto S, Ito H. Cultured rat astrocytes give rise to neural stem cells. Neurochem Res 2006; 31:1381. [DOI] [PubMed] [Google Scholar]
- 32.Chen S, Pickard JD, Harris NG. Time course of cellular pathology after controlled cortical impact injury. Exp Neurol 2003; 182:87–102. [DOI] [PubMed] [Google Scholar]
- 33.Douen AG, Dong L, Vanance S, Munger R, Hogan MJ, Thompson CS, Hakim AM. Regulation of nestin expression after cortical ablation in adult rat brain. Brain Res 2004; 1008:139–146. [DOI] [PubMed] [Google Scholar]
- 34.Itoh T, Imano M, Nishida S, Tsubaki M, Hashimoto S, Ito A, Satou T. Exercise increases neural stem cell proliferation surrounding the area of damage following rat traumatic brain injury. J Neural Transm 2011; 118:193–202. [DOI] [PubMed] [Google Scholar]
- 35.Yu WH, Dong XQ, Hu YY, Huang M, Zhang ZY. Ginkgolide B reduces neuronal cell apoptosis in the traumatic rat brain: possible involvement of toll-like receptor 4 and nuclear factor kappa B pathway. Phytother Res 2012; 26:1838–1844. [DOI] [PubMed] [Google Scholar]
- 36.Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature 2000; 406:782. [DOI] [PubMed] [Google Scholar]
- 37.Medzhitov R. Toll-like receptors and innate immunity. Adv Immunol 2001; 78:1. [DOI] [PubMed] [Google Scholar]
- 38.Truettner J, Schmidt-Kastner R, Busto R, Alonso OF, Loor JY, Dietrich WD, Ginsberg MD. Expression of brain-derived neurotrophic factor, nerve growth factor, and heat shock protein HSP70 following fluid percussion brain injury in rats. J Neurotrauma 1999; 16:471–486. [DOI] [PubMed] [Google Scholar]
- 39.Lai YL, Stange C, Wisniewski SR, Adelson PD, Janesko-Feldman KL, Brown DS, et al. Mitochondrial heat shock protein 60 is increased in cerebrospinal fluid following pediatric traumatic brain injury. Dev Neurosci 2006; 28:336–341. [DOI] [PubMed] [Google Scholar]
- 40.Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell 1996; 87:13. [DOI] [PubMed] [Google Scholar]
- 41.Beauparlant P, Hiscott J. Biological and biochemical inhibitors of the NF-kappa B/Rel proteins and cytokine synthesis. Cytokine Growth Factor Rev 1996; 7:175–190. [DOI] [PubMed] [Google Scholar]
- 42.Pettus EH, Wright DW, Stein DG, Hoffman SW. Progesterone treatment inhibits the inflammatory agents that accompany traumatic brain injury. Brain Res 2005; 1049:112–119. [DOI] [PubMed] [Google Scholar]
- 43.Anderson KV. Toll signaling pathways in the innate immune response. Curr Opin Immunol 2000; 12:13. [DOI] [PubMed] [Google Scholar]
- 44.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2001; 2:675. [DOI] [PubMed] [Google Scholar]
- 45.Chen G, Shi J, Jin W, Wang L, Xie W, Sun J, Hang C. Progesterone administration modulates TLRs/NF-kappaB signaling pathway in rat brain after cortical contusion. Ann Clin Lab Sci 2008; 38:65–74. [PubMed] [Google Scholar]
- 46.Morganti-Kossmann MC, Rancan M, Otto VI, Stahel PF, Kossmann T. Role of cerebral inflammation after traumatic brain injury: a revisited concept. Shock 2001; 16:165–177. [DOI] [PubMed] [Google Scholar]
- 47.Chen G, Zhang S, Shi J, Ai J, Qi M, Hang C. Simvastatin reduces secondary brain injury caused by cortical contusion in rats: possible involvement of TLR4/NF-kappaB pathway. Exp Neurol 2009; 216:398–406. [DOI] [PubMed] [Google Scholar]
- 48.Lu J, Goh SJ, Tng PY, Deng YY, Ling EA, Moochhala S. Systemic inflammatory response following acute traumatic brain injury. Front Biosci (Landmark Ed) 2009; 14:3795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s website, www.neuroreport.com.