

Abstract
Melanoma is a lethal form of skin cancer. Skin pigmentation, which is regulated by the melanocortin 1 receptor (MC1R), is an effective protection against melanoma. However, the endogenous MC1R agonists lack selectivity for the MC1R and thus can have side effects. The use of noncanonical amino acids in previous MC1R ligand development raises safety concerns. Here we report the development of the first potent and selective hMC1R agonist with only canonical amino acids. Using γ-MSH as a template, we developed a peptide, [Leu3, Leu7, Phe8]-γ-MSH-NH2 (compound 5), which is 16-fold selective for the hMC1R (EC50 = 4.5 nM) versus other melanocortin receptors. Conformational studies revealed a constrained conformation for this linear peptide. Molecular docking demonstrated a hydrophobic binding pocket for the melanocortin 1 receptor. In vivo pigmentation study shows high potency and short duration. [Leu3, Leu7, Phe8]-γ-MSH-NH2 is ideal for inducing short-term skin pigmentation without sun for melanoma prevention.
Graphical abstract

INTRODUCTION
Melanoma is the most dangerous form of skin cancer. In 2016, there is an estimated 73 870 new cases of melanoma, with an estimated 10 130 deaths in the U.S. alone.1 Exposure to UV radiation is the primary risk factor for melanoma.2 Skin pigmentation, on the other hand, is an effective protection against melanoma. Melanocytes can produce melanin, which is able to dissipate over 99.9% of UV radiation.3 The strong correlation between skin pigmentation and melanoma risk is also evidenced by the fact that in the U.S., Caucasians have 20–30 times higher chances of getting melanoma than Asians and Blacks.4 Current efforts seek to prevent UV damage to human skin, which in many cases leads to melanoma and other skin cancers. As a result, inducing skin pigmentation without UV exposure is considered an effective way to prevent UV induced melanoma.
Skin pigmentation is regulated by the melanocortin 1 receptor (MC1R) on melanocytes.5 Activation of MC1R by melanocyte stimulating hormones (MSH) leads to cyclic adenosine monophosphate (cAMP) production, which eventually leads to transcription of multiple pigment synthesis genes and melanin production.6–22 The natural MSHs consist of three peptides: α-melanocyte-stimulating hormone (α-MSH), β-melanocyte-stimulating hormone (β-MSH), and γ-melanocyte-stimulating hormone (γ-MSH). All MSHs share the same pharmacophore of His-Phe-Arg-Trp. The MSHs not only activate MC1R but also are able to activate the melanocortin 3 receptor (MC3R), melanocortin 4 receptor (MC4R), and melanocortin 5 receptor (MC5R), which regulate distinct physiological processes such as feeding behavior, energy homeostasis, sexual function, immune responses, and sebaceous gland secretion.23 Thus, improving selectivity to MC1R is of critical importance for MSHs to trigger skin pigmentation for melanoma prevention without interfering with other physiological functions.
The major challenge is that sunscreen lotion is viewed as both a cosmetic and a drug in the United States and thus must follow regulations from the U.S. Food and Drug Administration (FDA).24 Since 2014, the α-MSH analogue [Nle4, D-Phe7]α-MSH (NDP-α-MSH, brand name Scenesse) has been shown to effectively induce skin pigmentation25 and has been approved in Europe for treating erythropoietic protoporphyria (EPP). However, the FDA approval for its use in the U.S. is still pending. Two major aspects on NDP-α-MSH can be improved to pose less health concerns and better fit FDA regulations. One is that NDP-α-MSH is a universal agonist for all melanocortin receptors. The other is that it uses noncanonical amino acids that may not be metabolized the same way as canonical amino acids. Our goal is to develop MC1R selective peptides with canonical amino acids so that it will be more safe and easy degraded into natural building block amino acids in the body.
Design of Selective MC1R γ-MSH Analogues
Our earlier research led to a potent drug NDP-α-MSH (MT-I) or “afamelanotide” for the treatment of congenital erythropoietic porphyria in Europe and skin color disorders in Australia. However, MT-I does not have high selectivity for any one of the melanocortin receptor subtypes, which can lead to unexpected side effects such as headache and nausea.25 One of our previous studies of a γ-MSH analogue (compound 1, Table 1) had improved potency and selectivity for the MC1R. Efforts have been made to move compound 1 to be a melanoma prevention agent, but the process of drug development has been very slow due to the un-natural amino acids that are involved.
Table 1.
Sequence of γ-MSH Analogues
| no. | peptide sequence |
|---|---|
| 1. Nle3, DNal(2′)6, DTrp8-γ-MSH-NH2 | H-Tyr-Val-Nle-Gly-His-D-Nal(2)-Arg-D-Trp-Asp-Arg-Phe-Gly-NH2 |
| 2. Leu3, Pro5, γ-MSH-NH2 | H-Tyr-Val-Leu-Gly-Pro-Phe-Arg-Trp-Asp-Arg-Phe-Gly-NH2 |
| 3. Leu3, Pro5, Phe8 γ-MSH-NH2 | H-Tyr-Val-Leu-Gly-Pro-Phe-Arg-Phe-Asp-Arg-Phe-Gly-NH2 |
| 4. Leu3, Leu7, γ-MSH-NH2 | H-Tyr-Val-Leu-Gly-His-Phe-Leu-Trp-Asp-Arg-Phe-Gly-NH2 |
| 5. Leu3, Leu7, Phe8 γ-MSH-NH2 | H-Tyr-Val-Leu-Gly-His-Phe-Leu-Phe-Asp-Arg-Phe-Gly-NH2 |
| 6. Leu3, Pro5, Le u7 γ-MSH-NH2 | H-Tyr-Val-Leu-Gly-Pro-Phe-Leu-Trp-Asp-Arg-Phe-Gly-NH2 |
| 7. Leu3, Pro5, Leu7, Phe8 γ-MSH-NH2 | H-Tyr-Val-Leu-Gly-Pro-Phe-Leu-Phe-Asp-Arg-Phe-Gly-NH2 |
| 8. Pro5 γ-MSH-NH2 | H-Tyr-Val-Met-Gly-Pro-Phe-Arg-Trp-Asp-Arg-Phe-Gly-NH2 |
| 9. Leu3, γ-MSH | H-Tyr-Val-Leu-Gly-His-Phe-Arg-Trp-Asp-Arg-Phe-Gly-OH |
| 10. Leu3, γ-MSH-NH2 | H-Tyr-Val-Leu-Gly-His-Phe-Arg-Trp-Asp-Arg-Phe-Gly-NH2 |
| 11. γ-MSH | H-Tyr1-Val2-Met3-Gly4-His5-Phe6-Arg7-Trp8−Asp9-Arg10-Phe11-Gly12-OH |
| 12. γ-MSH-NH2 | H-Tyr1-Val2-Met3-Gly4-His5-Phe6-Arg7-Trp8–Asp9-Arg10-Phe11-Gly12-NH2 |
| 13. Ac-NDP-γ-MSH-NH2 | Ac-Tyr1-Val2-Nle3-Gly4-His5-D-Phe6-Arg7-Trp8-Asp9-Arg10-Phe11-Gly12-NH2 |
| 14. α-MSH | Ac-Ser-Tyr-Ser-Met-Glu-His-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2 |
| 15. NDP-α-MSH | Ac-Ser-Tyr-Ser-Nle-Glu-His-D-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2 |
This motivated us to design a selective MC1R ligand with only canonical amino acids starting from this γ-MSH analogue. We introduced modifications on five different sites of γ-MSH: (1) Introducing the C-terminal amide group to γ-MSH and its analogues was shown to increase their binding affinities to all melanocortin receptor subtypes but MC3R.26 (2) To keep the similarity and stability of compound 1, we substitute Nle3 with Leu3 since natural γ-MSH has Met3 which is easily oxidized. (3) His5 was substituted to proline, which was previously shown to increase potency and selectivity to MC1R.26 (4) Trp8 was substituted to phenylalanine, which was also shown to improve MC1R selectivity.27 (5) Most importantly, our chimeric receptor studies demonstrated that the electrostatic interaction, Arg(L)-Asp(R), between the Arg8 of the NDP-α-MSH and the Asp122, Asp126 of the hMC4R is of critical importance to achieve binding and receptor activation, as Asp126Asn mutation on the MC4R caused more than a 400-fold increase in the EC50 value.28 Similarly, a key interaction between the Arg8 of NDP-α-MSH and the Asp154 as well as the Asp158 of the MC3R is necessary, as Asp158Ala mutation on MC3R caused more than 350-fold increase for the EC50 value.29 In contrast, mutation of either Asp117 or Asp121 to alanine on MC1R only had around a 10-fold influence on the IC50 and EC50 of NDP-α-MSH, suggesting a smaller role of ionic interactions between MC1R and its agonists.30 Finally, our latest docking studies of hMC1R with selective hMC1R ligand revealed that there is a very hydrophobic binding pocket for the hMC1R. This suggested that increasing hydrophobicity of the hMC1R ligand can improve the selectivity (data in the process of publication). Therefore, our hypothesis is switching the arginine in the γ-MSH analogue to a neutrally charged amino acid, in particular leucine should reduce binding toward the hMC3R and the hMC4R. We envision that enhanced selectivity toward the MC1R can be reached with reduced electrostatic interaction between the Arg(L)-Asp(R) of the γ-MSH analogues and the respective aspartic acids on the MC3R and MC4R receptors. Herein, a series of Leu3, Pro5, Leu7, Phe8 γ- MSH-NH2 analogues are designed and synthesized to test these hypotheses (Table 1).
RESULTS
Bioassay Results
Competitive binding assays with [125I]NDP-α-MSH and adenylate cyclase assays were performed with HEK293 cells stably expressing human MC1R (hMC1R), human MC3R (hMC3R), human MC4R (hMC4R), and human MC5R (hMC5R) on the γ-MSH analogues (Table 1, Table 2). γ-MSH (compound 11) is a universal agonist to melanocortin receptors with 1.8-fold selectivity to hMC1R (EC50 = 300 nM). By addition of the C-terminal amide group to γ-MSH, compound 12 loses its selectivity to hMC1R, as the potency on hMC1R (EC50 = 70 nM) is in the same range as the potency on hMC4R (EC50 = 65 nM). However, the potency to hMC1R is enhanced 3-fold. Compounds 9 and 10 with methionine substituted by leucine both have enhanced potency and selectivity to the hMC1R. Compound 9 has a 10-fold increase in potency (EC50= 31 nM) to the hMC1R compared to γ-MSH with 13-fold selectivity. Compound 10 has better potency and selectivity with EC50 of 8 nM on the hMC1R and a selectivity of over 22-fold. The methionine to leucine substitution also caused compounds 9 and 10 to have less binding efficiency and no activation on the hMC5R, which is consistent with the D-amino acid scan showing that Met3 in γ-MSH is important for hMC5R activation.31 Thus, we focused our further design using Leu3 γ-MSH-NH2 (compound 10) as a template.
Table 2.
Competitive Binding Assay and cAMP Assay Results of γ-MSH Analoguesa
| no. | hMC1R
|
hMC3R
|
hMC4R
|
hMC5R
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50, nM | % BE | EC50, nM | % max effect | IC50, nM | % BE | EC50, nM | % max effect | IC50, nM | % BE | EC50, nM | % max effect | IC50, nM | % BE | EC50, nM | % max effect | |
| 1 | 0.30 ± 0.02 | 100 | 3.0 ± 0.2 | 70 | 5.0 ± 0.6 | 100 | 300 ± 6 | 6 | 6.0 ± 1 | 100 | 630 ± 70 | 18 | 3.5 ± 0.5 | 100 | NA | 6 |
| 2 | 82 ± 7 | 100 | 4.6 ± 0.9 | 100 | 4.1 ± 0.4 | 100 | 5.0 ± 0.4 | 100 | 5.0 ± 0.4 | 100 | 23 ± 3 | 90 | 18 ± 3 | 100 | 16 ± 1 | 82 |
| 3 | NB | 0 | >1000 | 100 | 18 ± 2 | 33 | NA | 0 | NB | 0 | NA | 0 | NB | 0 | NA | 0 |
| 4 | 137 ± 11 | 100 | 604 ± 74 | 100 | 17 ± 1 | 26 | NA | 0 | >1000 | 47 | NA | 0 | NB | 0 | NA | 0 |
| 5 | 24 ± 3 | 100 | 4.5 ± 0.4 | 100 | >1000 | 45 | >1000 | 50 | 210 ± 5 | 58 | 71 ± 8 | 54 | NB | 0 | NA | 0 |
| 6 | 16 ± 2 | 29 | NA | 0 | 117 ± 24 | 32 | NA | 0 | NB | 0 | NA | 0 | 0.42 ± 0.07 | 32 | NA | 0 |
| 7 | 1.1 ± 0.1 | 32 | >1000 | 100 | >1000 | 47 | >1000 | 100 | 6.7 ± 0.9 | 36 | 200 ± 25 | 100 | NB | 0 | NA | 0 |
| 8 | >1000 | 52 | 40 ± 6 | 100 | 118 ± 14 | 73 | 521 ± 72 | 100 | 360 ± 42 | 62 | 128 ± 13 | 78 | 36 ± 1 | 47 | >1000 | 100 |
| 9 | 23 ± 2 | 100 | 31 ± 6 | 100 | 56 ± 3 | 69 | >1000 | 100 | 20 ± 1 | 86 | 444 ± 69 | 100 | 254 ± 41 | 38 | NA | 0 |
| 10 | 42 ± 7 | 100 | 8 ± 1.8 | 100 | >1000 | 53 | >1000 | 100 | 68 ± 11 | 100 | 178 ± 23 | 100 | >1000 | 60 | NA | 0 |
| 11 | 1000 ± 20 | 100 | 300 ± 30 | 100 | 71 ± 10 | 100 | 700 ± 89 | 100 | 760 ± 80 | 100 | 710 ± 70 | 100 | 2200 ± 200 | 100 | 550 ± 60 | 100 |
| 12 | 1.2 ± 0.2 | 100 | 70 ± 10 | 100 | 40 ± 10 | 100 | 520 ± 50 | 38 | 45 ± 5 | 100 | 65 ± 12 | 76 | 330 ± 57 | 100 | 390 ± 25 | 73 |
| 13 | 0.5 ± 0.01 | 100 | 1.5 ± 0.1 | 100 | 2.0 ± 0.02 | 100 | 2.0 ± 0.2 | 100 | 1.2 ± 0.2 | 100 | 1.4 ± 0.1 | 100 | 2.4 ± 0.3 | 100 | 1.9 ± 0.2 | 100 |
| 14 | 0.4 ± 0.01 | 100 | 0.7 ± 0.01 | 100 | 30 ± 3.9 | 100 | 6.7 ± 1 | 100 | 5 ± 1 | 100 | 2.1 ± 0.6 | 100 | 18 ± 2 | 100 | 8.1 ± 1.5 | 100 |
| 15 | 0.01 | 100 | 0.01 | 100 | 3.3 ± 0.3 | 100 | 0.8 ± 0.1 | 100 | 0.4 ± 0.02 | 100 | 0.2 ± 0.04 | 100 | 2.2 ± 0.5 | 100 | 1 ± 0.3 | 100 |
| a IC | is the | peptide at 50% | fic | (N = 4). | % BE | the | of [125 | α-MSH | at 10 μM. | means | that 0% of [125 | -MSH | ||||
IC50 is the concentration of peptide at 50% specific binding (N = 4). % BE is the percentage of [125I]NDP-α-MSH displacement at 10 μM. NB means that 0% of [125I]NDP-α-MSH displacement was observed at 10 μM. EC50 is the effective concentration of peptide that could generate 50% maximal intracellular cAMP accumulation (N = 4). % max effect is the percentage of cAMP produced at a ligand concentration of 10 μM, in relation to MT-II. NA indicates 0% cAMP accumulation observed at 10 μM. The peptides were tested over a range of concentrations from 10−10 to 10−5 M. Compounds 1 and 11–15 were previously published data and are included for comparison.
To improve selectivity on the hMC1R, we did modifications on the melanotropin pharmacophore His5-Phe6-Arg7-Trp8. Peptides were designed with a combination of mutations on His5, Arg7, and Trp8. As expected, compound 2 with the proline substitution had improved potency, but it lost all selectivity as it activates hMC1R (EC50 = 4.6 nM) and hMC3R (EC50 = 5.0 nM) with almost the same potency. The leucine substitution on compound 4 did affect its binding to hMC3R and hMC4R, as they only showed weak binding and no activity on these receptors. However, the potency to hMC1R (EC50 = 604 nM) is also decreased by 76-fold. A double replacement of His5 to Pro5 and Arg7 to Leu7 (compound 6) did not show synergistic effects, as compound 6 lost all activities for any melanocortin receptor. The loss of potency is also seen in the triple replacements of His5 to Pro5, Arg7 to Leu7, and Trp8 to Phe8 (compound 7). The replacements of His5 to Pro5 and Trp6 to Phe6 (compound 3) also showed loss of activities at all melanocortin receptors. The purpose of our design was achieved with a double replacement of Arg7 to Leu7 and Trp8 to Phe8 (compound 5, [Leu3, Leu7, Phe8]-γ-MSH-NH2). Compound 5 ([Leu3, Leu7, Phe8]-γ-MSH-NH2) has 1.8-fold enhanced potency for the hMC1R (EC50 = 4.5 nM) than the template compound (compound 10). The selectivity to hMC1R is over 16-fold. Furthermore, compound 5 ([Leu3, Leu7, Phe8]-γ-MSH-NH2) has only enhanced potency for the hMC1R (EC50= 4.5 nM) compared with the template compound (compound 10). The selectivity to hMC1R is over 16-fold. Furthermore, compound 5 ([Leu3, Leu7, Phe8]-γ-MSH-NH2) is only a partial agonist with 54% maximal activation on the hMC4R. The template compound (compound 10), on the other hand, is a full agonist on the hMC4R. These results suggest that [Leu3, Leu7, Phe8]-γ-MSH-NH2 is an ideal peptide with strong potency and selectivity for the hMC1R.
Human Serum Stability
To examine the stability of [Leu3, Leu7, Phe8]-γ-MSH-NH2 in comparison to γ-MSH and NDP-MSH, serum stability assays were performed. The endogenous ligand γ-MSH has the shortest half-life of around 5 min. Compared to γ-MSH, compound 5 has an improved serum stability of 17.5 min. The elongated half-life is possibly due to the replacement of methionine on the γ-MSH template. NDP-MSH, which contains D-Phe, has the longest half-life in human serum of around 30 min.
In Vivo Study Results
Pigmentation studies of [Leu3, Leu7, Phe8]-γ-MSH-NH2 in the Anolis carolinensis show it is a superpotent agonist in vivo. The black color appeared in 1 min after injection (ip, 2 μg/g) (Figure 1b). This is exactly the same response of injection of the same dose of NDP-α-MSH (Figure 1a). In comparison, intraperitoneal injection of the vehicle did not produce any pigmentation effect (data not shown). The green color was able to resume in less than 24 h after [Leu3, Leu7, Phe8]-γ-MSH-NH2 injection but not for the NDP-α-MSH. It took 2 weeks to resume the green color for the NDP-α-MSH (Figure 1a). The much longer half-life of the NDP-α-MSH in vivo is probably due to two un-natural amino acids, Nle and D-Phe, which are not recognized by proteases in vivo, thus resulting in biological stability. However, [Leu3, Leu7, Phe8]-γ-MSH-NH2 is composed of all of natural amino acids. This indicates that [Leu3, Leu7, Phe8]-γ-MSH-NH2 will be ideal to be used in a skin care product to induce skin pigmentation and protect against melanoma during sun exposure. The natural color can be resumed in 1 day with a lower dosage. Furthermore, [Leu3, Leu7, Phe8]-γ-MSH-NH2 is highly selective for hMC1R and therefore avoids possible side effects due to activation of the other hMCRs subtypes.
Figure 1.

(a) NDP-α-MSH induced pigmentation on the lizard: (left) before injection; (right) after injection in 1 min. (b) [Leu3, Leu7, Phe8]-γ-MSH-NH2 induced pigmentation on the lizard: (left) before injection; (right) after injection in 1 min.
Conformational Study of [Leu3, Leu7, Phe8]-γ-MSH-NH2
The lowest energy conformation search via MacroModel (Schrodinger) revealed a very constrained sphere-like structure for the [Leu3, Leu7, Phe8]-γ-MSH-NH2 (Figure 2a). The major force for the constraint is hydrophobicity due to the presence of dominant hydrophobic amino acids in this peptide. The Ramachandran plot shows that all of the amino acids are in the region of a 310 helix (Figure 2b) except for the Gly 4, Phe6, and Asp9. These three amino acids are in the turn region.
Figure 2.
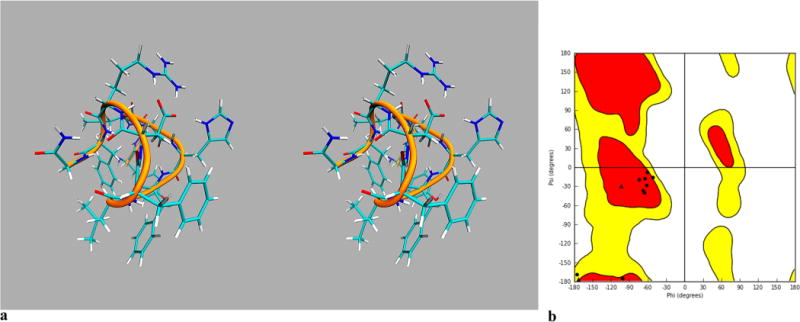
Conformational studies of [Leu3, Leu7, Phe8]-γ-MSH-NH2: (a) stereoview of lowest energy conformation search derived structure of [Leu3, Leu7, Phe8]-γ-MSH-NH2; (b) Ramachandran plot of lowest energy conformation search derived structure of [Leu3, Leu7, Phe8]-γ-MSH-NH2, showing that most of the amino acids are in the 310 helix region except for the Gly4, Phe6, and Asp9, which are in the loop area.
Molecular Docking Studies
The initial hMC1R structure applied for this study is from previous published work from the Mosberg lab.32 First we searched for a potential binding site with Maestro SiteMap and identified the one with the highest druggability score of 1.06 (Figure 3). We then performed a flexible docking simulation with Maestro Glide targeting that pocket.
Figure 3.
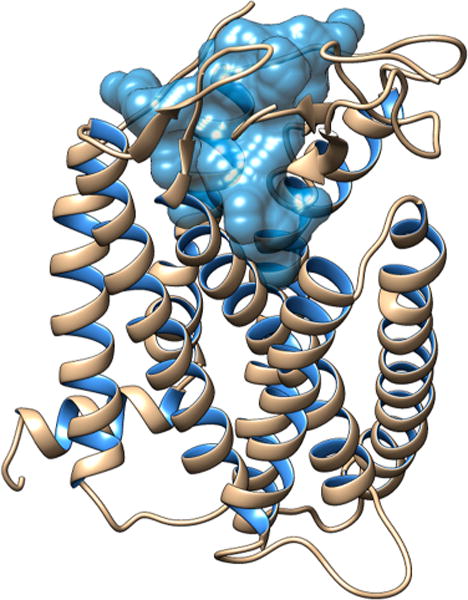
Binding site of hMC1R for the [Leu3, Leu7, Phe8]-γ-MSH-NH2.
Molecule docking revealed a very hydrophobic binding pocket for the hMC1R. A docking score of −11.32 kcal/mol was achieved, and key interactions between [Leu3, Leu7, Phe8]-γ-MSH-NH2 and the hMC1R were identified (Figure 4). [Leu3, Leu7, Phe8]-γ-MSH-NH2 showed an antiparallel β-sheet conformation with the modified pharmacophore, His5-Phe6-Leu7-Phe8, at the turn region (Figure 4, Table 3). His5 and Phe6 interact with the transmembrane segment 6 (TM6) of the hMC1R. His5 is stabilized by π–π stacking interactions with Thr262 and His260 inside the hMC1R. Phe6 has π–π stacking interactions with both Trp254 and Phe257. Both His260 and Phe257 have been shown by previous mutagenesis studies to be important for melanocortin receptor activation.28,30 Phe8 interacts with Cys125 on the TM3 domain. Other residues on [Leu3, Leu7, Phe8]-γ-MSH-NH2 interact with the TM1 and TM3. The relative movement on the TM6 was shown to be critical for G protein coupled receptor activation.33 Our results indicated that the pharmacophore of [Leu3, Leu7, Phe8]-γ-MSH-NH2 interacts with TM6 to generate its movement while other residues on [Leu3, Leu7, Phe8]-γ-MSH-NH2 anchor to other transmembrane segments on hMC1R. It has to be noted that this peptide maintains all the important interactions as reported from previous studies. The key for its high affinity and selectivity lies in the fact that it introduces new types of interactions. It also has many hydrogen bond interactions, indicative of stronger binding.
Figure 4.
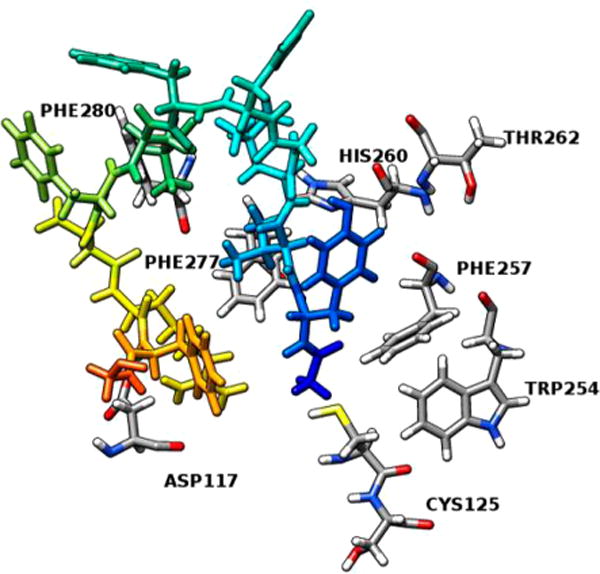
Molecular interactions between the modified pharmacophore -His-Phe-Leu-Phe- of [Leu3, Leu7, Phe8]-γ-MSH-NH2 and the hMC1R.
Table 3.
Molecular Interactions between [Leu3, Leu7, Phe8]-γ-MSH-NH2 and hMC1R
| interacting residue on 5 | interacting residue on hMC1R | distance, Å | type of interaction |
|---|---|---|---|
| His5 NE2 | Thr262 | 1.14 | H-bond |
| His5 | His260 | 5.12 | π–π stacking |
| Phe6 | Trp254 | 3.68 | π–π stacking |
| Phe6 | Phe257 | 3.16 | π–π stacking |
| Tyr1 | Phe277 | 5.32 | π–π stacking |
| Tyr1 | Phe280 | 3.99 | π–π stacking |
| Leu7 N | Cys125 | 2.79 | H-bond |
| Asp9 OD1 | Ser126 | 2.62 | H-bond |
| Arg10 NH2 | Asp117 | 1.79 | salt bridge |
| Phe11 | Phe45 | 3.55 | π–π stacking |
We also emphasize that the evaluation of the binding site where our ligand was bound led to an increased druggability score of 1.21, suggesting that the peptide targeted a highly druggable part of the binding site.
Furthermore, we tested compound 1 for comparison purposes, and after having it docked inside the receptor cavity and analyzing its interactions, we note that compound 1 has a slightly higher and therefore worse docking score (−11.08 kcal/ mol) compared to [Leu3, Leu7, Phe8]-γ-MSH-NH2. It also presents less interactions with the receptor, although most of them are hydrogen bonds as shown in Table 4. Surprisingly, only the pharmacophore of compound 1 (His-D-NaI(2′)-Arg-Trp) interacts with hMC1R (Figure 5), whereas in [Leu3, Leu7, Phe8]-γ-MSH-NH2 the majority of its amino acids are participating in interactions. In [Leu3, Leu7, Phe8]-γ-MSH-NH2 we see more different types of interactions which can partially explain its enhanced selectivity toward the hMC1R.
Table 4.
Molecular Interactions between Compound 1 and hMC1R
| interacting residue on 1 | interacting residue on hMC1R | distance, Å | type of interaction |
|---|---|---|---|
| His N | Ile276 | 2.30 | H-bond |
| Arg NE | Glu94 | 1.93 | H-bond |
| Arg NH2 | Glu94 | 2.27 | H-bond |
| Arg NH2 | Thr124 | 2.31 | H-bond |
| Arg NH1 | Asp121 | 2.72 | H-bond |
| Trp | Phe179 | 5.02 | π–π stacking |
| Trp N | Asp121 | 1.90 | H-bond |
Figure 5.
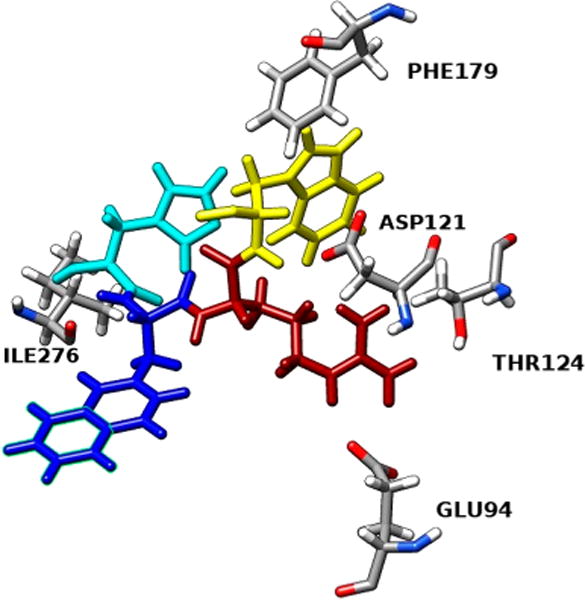
Molecular interactions between the pharmacophore -His-D-NaI(2′)-Arg-Trp- of compound 1 and the hMC1R.
DISCUSSION AND CONCLUSIONS
Seeking effective treatments of melanoma is very hot in the current era of science and technology. However, prevention of melanoma has been neglected. We are providing a very useful natural AA made peptide here, and it will be more applicable and beneficial for most people. In this study, we first successfully developed a natural aa made peptide, [Leu3, Leu7, Phe8]-γ-MSH-NH2, which is a potent selective hMC1R agonist. The composition of canonical amino acids ensures that the peptide can be easily degraded into natural building block amino acids. The use of only natural amino acids versus unnatural can alleviate most safety concerns. The high selectivity of the [Leu3, Leu7, Phe8]-γ-MSH-NH2 for the hMC1R and shorter half-life provide a safer and reduced-side-effect agent. As peptides have been widely used in cosmeceuticals as active ingredients and can be applied transdermally,34 [Leu3, Leu7, Phe8]-γ-MSH-NH2 is suitable as a safe skin care product for the prevention of melanoma skin cancer.
Compared to our previously developed peptide (compound 1), [Leu3, Leu7, Phe8]-γ-MSH-NH2 seems to have less selectivity to hMC1R in terms of EC50. However, compound 1 is also an antagonist at the hMC3R, hMC4R, and hMC5R as suggested by its strong binding affinity to those MCRs. The binding affinity of compound 1 to the hMC1R (IC50 = 0.3 nM) is only 12-fold stronger than hMC5R (IC50 = 3.5 nM). As hMC5R regulates sebum production in the skin,35 compound 1 may cause hMC5R antagonism and decreased production of sebum. On the other hand, [Leu3, Leu7, Phe8]-γ-MSH-NH2 does not have detectable binding to hMC5R in our biological assays. Binding of [Leu3, Leu7, Phe8]-γ-MSH-NH2 to the hMC3R and hMC4R is also weak as evidenced by high IC50 and low % binding. More importantly, MC4R is exclusively expressed in the central nervous system.36 Long linear peptides such as NDP-α-MSH cannot pass through the blood–brain barrier to reach the brain.37 With similar length and sequence as NDP-α-MSH, compound 5 is unlikely to reach the brain and interfere with MC4R’s function. As a result, compound 5 is selective to hMC1R and less likely to interfere with physiological processes regulated by other MCRs.
The serum stability assay showed a half-life of 17.5 min for [Leu3, Leu7, Phe8]-γ-MSH-NH2 in human serum. It is worth noting that the skin pigmentation effect lasts longer than the peptide’s half-life in serum. Even though the half-life of NDP-α-MSH in human serum is 30 min, the European standard is to have NDP-α-MSH in a controlled-release implant and subcutaneously administrated to EPP patients once every 2 months.38 It was further confirmed with our in vivo assay that the pigmentation effect would last for less than 24 h. It is worth mentioning that even though pigmentation effect in lizards is mostly regulated by melanosome dispersion rather than melanogenesis, the melanosome dispersion process in coldblooded animals is also regulated by the MC1R/cAMP pathway.39 [Leu3, Leu7, Phe8]-γ-MSH-NH2 is expected to effectively induce short period skin pigmentation in human and thus can be used in skin care products for short-term melanoma prevention.
In our in silico study, we discovered that hMC1R has a very hydrophobic binding pocket. The modified pharmacophore of [Leu3, Leu7, Phe8]-γ-MSH-NH2, His-Phe-Leu-Phe, has enhanced hydrophobicity compared to the natural MSHs pharmacophore (His-Phe-Arg-Phe) which contributes to hMC1R selectivity. Computational chemistry combined with chimeric receptors studies verifies our hypothesis that enhanced hydrophobicity of the MSHs pharmacophore can increase the hMC1R selectivity. The newly designed hMC1R selective agonist [Leu3, Leu7, Phe8]-γ-MSH-NH2 fits perfectly within the hydrophobic binding pocket of hMC1R and provides new insights for future drug design for the hMC1R.
Molecular docking is a very useful tool in structural biology and computer aided drug design, especially for GPCR based drug design owing to the limited crystal structures available (none of the melanocortin receptors) and the dynamic environment of the membrane protein. The major goal of ligand–receptor docking is to evaluate the feasible binding geometries of a putative ligand with a target protein of known three-dimensional structure. Normally, docking calculations can help obtain an idea about the binding affinity of the ligand. It also provides useful insights regarding the interactions between ligand and receptors and their roles. Here we study the [Leu3, Leu7, Phe8]-γ-MSH-NH2 and hMC1R interaction by conjugating the study of biological information from multiple mutagenesis, chimeric receptor studies of the hMC1R, and molecular docking.
Finally, structure-based drug design has become a useful approach for current drug discovery. In our long-term peptide-based drug development, peptide truncation and amino acid scans have been used to discover the major pharmacophore. Conformational constraints were applied to produce numerous stable and selective melanotropins, and the three-dimensional structures of ligands using NMR spectroscopy combined with computational based drug design have led to several selective compounds. Peptide mimetic studies also led to selectivity and potency. However, few of these strategies led to a drug. In this study, we first used a strategy by applying useful information from multiple mutagenesis and chimeric receptor studies along with computational chemistry to rationally design a hMC1R peptide ligand using canonical amino acids. The economic favorability of canonical amino acids and straightforward synthesis strategy can provide lower cost for mass production. Our strategy represents the rational design to fulfill specific requirements for developing skin care products for short-term skin pigmentation and melanoma prevention.
EXPERIMENTAL SECTION
Synthesis
Nα-Fmoc-amino acids were obtained from Bachem, NovaBiochem, and Advanced ChemTech. The side chain protecting groups were Boc and tBu [Fmoc-Asp(tBu)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Arg(pbf)-OH, Fmoc-His(trt)-OH, Fmoc-Ser(tBu)-OH, and Fmoc-Tyr(tBu)-OH]. Fmoc-Rink amide resin was purchased from Polymer Laboratories. Organic solvents and reagents were purchased from Aldrich and used without further purification. All peptides were synthesized by the N-Fmoc solid-phase peptide strategy using DIEA and HCTU as the coupling reagents. Rink amide resin (0.65 mmol/g) or 2-chlorotrityl resin (1 mmol/g) was placed into a 5 mL polypropylene syringe with a frit on the bottom and swollen in DCM (2 mL) and DMF (2 mL) for 1 h. The Fmoc protecting group on the Rink linker was removed by 20% piperidine in DMF. After 20 min the solution of piperidine was removed and the resin was washed with DMF (2 mL, 4 times) and DCM (2 mL, 4 times). N-Fmoc amino acid (3 equiv) and HCTU (3 equiv) were dissolved in DMF, and then DIEA (3 equiv) was added. The coupling mixture was transferred into the syringe with the resin and shaken for 30 min. Coupling completion was monitored with a Kaiser test. The coupling mixture was removed, and the resin was washed with DMF (2 mL, 4 times) and DCM (2 mL, 4 times). N-Fmoc groups were removed with 20% piperidine in DMF in 20 min. Each coupling and deprotection step was repeated until a linear peptide was assembled. The final wash of the resin was done with DMF (2 mL, four times) and DCM (2 mL, four times). The product was cleaved from the resin with a mixture of 95% TFA, 2.5% TIPS, and 2.5% water during 3 h. Side chain protecting groups were removed during the cleavage step as well. The cleaved mixture was evaporated on a rotary evaporator, and the crude peptide was dissolved in H2O/methanol and purified by HPLC.
The peptide was lyophilized and purified by preparative RP-HPLC on a C18 bonded silica column (Vydac 218TP152022, 250_22 mm, 15–20 μm, 300 Å). A C18 analytical column (YMC-Pack ODS-AM 150_4.6 mm, S-3 μm, 120 Å) was used to analyze the purity, eluted with a linear gradient of acetonitrile (gradient, 2–80% B in A over 30 min, flow rate 0.8 mL/min). System 1: solvent A, 0.1% TFA in water; solvent B, 0.08% TFA in acetonitrile. System 2: solvent A, 1% formic acid in water; solvent B, 1% formic acid in methanol) and aqueous 0.1% TFA (v/v). The major peak of all compounds accounted for ≥95% of the combined total peak area monitored by a UV detector at 254 nM.
Biological Assays
Competitive binding assays with [125I]NDP-MSH and adenylate cyclase assays were performed using previously described protocols.26,41–43
Pigmentation Study
Lizards Anolis carolinensis were purchased from Carolina online. Peptide samples were dissolved in saline at the concentration of 1 mM. Lizards were anesthetized by diethyl ether before injection. The total amount of peptide was through ip injection with 3 μg/g of each lizard. The strategy follows previous publications.44–48
Serum Stability Assay
Compounds 5 and 15 were dissolved at 10 mg/mL as stock solution. An amount of 1 mL of RPMI supplemented with 25% of human serum was allocated into a 1.5 mL tube and warmed up to 37 °C before adding 5 μL of peptide stock solution. An amount of 100 μL of the reaction solution is removed from times 0, 10, 20, 30, and 40 min. An amount of 200 μL of 96% ethanol was added for precipitation of serum proteins. The reaction sample was cooled (4 °C) for 15 min and then spun at 18 000g (Eppendorf centrifuge) for 2 min. The reaction supernatant was then analyzed using RP-HPLC on a 5 μm, 25 cm × 0.4 cm Vydac C-18 column. A linear gradient from 100% buffer A to 50–50% of buffer A and buffer B is used over 30 min. The absorbance was detected at 280 nm. Area under the expected peak is expected to be proportional to the amount of remaining peptide. The half-life was estimated by fitting the data into an exponential decay curve.
Molecular Modeling
Molecular modeling experiments employed MacroModel version 10.5 equipped with Maestro 10.5 graphical interface (Schrödinger, LLC, New York, NY, 2016) installed on a Linux Red Hat 11 system and were performed as previously described. Peptide structures were built into extended structures with standard bond lengths and angles, and they were minimized using the OPLS3 force field and the Polak–Ribier conjugate gradient (PRCG). Optimizations were converged to a gradient rmsd of less than 0.05 kJ/Å mol or continued until a limit of 50 000 iterations was reached. Aqueous solution conditions were simulated using the continuum dielectric water solvent model (GB/SA). Extended cutoff distances were defined at 8 Å for van der Waals, 20 Å for electrostatics, and 4 Å for H-bonds.
Conformational profiles of the cyclic peptides were investigated by the hybrid Monte Carlo/low frequency mode (MCMM/LMCS) procedure as implemented in MacroModel using the energy minimization parameters as described above. MCMM torsional variations and low mode parameters were set up automatically within Maestro graphical user interface. A total of 20 000 search steps were performed, and the conformations with energy difference of 50 kJ/mol from the global minimum were saved. Interatomic dihedral angles were measured for each peptide analogue using the Maestro graphical user interface.
Molecular Docking
Molecular docking studies used the Glide programs (version 7.0, Schrodinger, LLC, New York, 2016). To analyze the docking results and execute the protocol, the Maestro user interface (version 10.5, Schrodinger, LLC, New York, 2016) was employed. Docking was performed using the SP (standard precision mode) protocol. This includes the following. (1) Preparation of protein: The protein was subjected to energy minimization using Schrodinger implementation of OPLS3 force field. (2) Preparation of ligand: The ligand was prepared using the LigPrep 3.7 module of the Schrodinger suite using the standard protocol with OPLS3 force field. (3) Active site prediction: We employed Sitemap (version 3.8) to search for potential binding sites. Sitemap applies theoretical methods and predicts the most accurate binding site. Again, we used Sitemap after we had docked our ligand to evaluate the binding site. (4) Grid generation–docking calculation: Glide used a series of hierarchical filters to search for possible locations for the ligand in the active site region of the receptor. For the grid-based ligand docking, the receptor grid generation process was used. A grid box of 30 × 30 × 30 Å3 with a default inner box (10 × 10 × 10 Å3) was centered on the corresponding ligand. The receptor grid was defined as an enclosed box at the centroid of the ligand. Lastly, we performed a flexible docking calculation using the “standard precision” Glide algorithm and after the postdocking minimization we kept the pose with the best docking score.
Preparation of Protein and Ligand
The hMC1R model was obtained from Chai et al.,32 imported and prepared by a multistep process through the Protein Preparation Wizard of Maestro. The protocol was especially used to obtain the optimized and minimized energy conformation of the protein. First, we assigned bond orders and added hydrogen atoms. Water molecules that did not participate in interactions were removed. Following the above steps of preparation, the protein was subjected to energy minimization using Schrodinger implementation of OPLS3 force field. The structure for compound 5 and compound 1 was built in 3D coordinates and minimized in MacroModel to obtain the lowest energy conformation, and retaining the geometry was then prepared using the LigPrep 3.7 module of the Schrodinger suite using the standard protocol with OPLS3 force field.
Active Site Prediction
We employed Sitemap (version 3.8) to search for potential binding sites. Sitemap applies theoretical methods and predicts the most accurate binding site. The OPLS3 force field generates site points, possible for ligand interaction within the protein. Sitemap searches for positions favorable for a donor, acceptor, and hydrophobic group to be present in the receptor.
We used Sitemap to identify the top ranked potential receptor binding sites, keeping all parameters as default. Five sites with different site scores were obtained as output, and the site with the highest score (SiteScore and Dscore) was selected.
We again used Sitemap after we had docked our ligand to evaluate the binding site based on the druggability score (Dscore). Druggability is a term used in drug discovery to describe a biological target that is known to bind or is predicted to bind with high affinity to a drug. If that score is higher than 0.75, the target is considered to be druggable.
Molecular Docking Studies
We performed molecular docking studies using the Glide program (version 7.0, Schrödinger, LLC, New York, 2016). To analyze the docking results and execute the protocol, the Maestro user interface (version 10.5, Schrödinger, LLC, New York, 2016) was employed.
We used the set of site points from the proposed binding site to generate a grid box for the docking calculation. The receptor grid was defined as an enclosing box at the centroid of the ligand. Then we performed a flexible docking calculation using the “standard precision” Glide algorithm, and after the postdocking minimization we kept the pose with the best docking score.
Supplementary Material
Acknowledgments
These studies were supported in part by grants from the U.S. Public Health Service, National Institutes of Health, Grants DK017420 and GM 108040, and Proof of Concept grant from the University of Arizona Tech Launch Transfer.
ABBREVIATIONS USED
- AAA
amino acid analysis
- Boc
tert-butyloxycarbonyl
- Fmoc
fluorenylmethoxycarbonyl
- Fmo
fluorenylmethyl
- Bzl
benzyl
- tBu
tert-butyl
- CH3CN
acetonitrile
- DCM
dichloromethane
- DIEA
diisopropylethylamine
- DMF
N,N-dimethylformamide
- DIC
diisopropylcarbodiimide
- HOBt
N-hydroxybenzotriazole
- Nal(2)
naphthylalanine
- TFA
trifluoroacetic acid
- TIPS
triisopropylsilyl
- SPPS
solid-phase peptide synthesis
- RP-HPLC
reverse phase high-performance liquid chromatography
- hMC1R
human melanocortin 1 receptor
- MSH
melanocyte-stimulating hormone
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b01295.
Physiochemical and spectral data (1H NMR, mass spectrometry), HPLC chromatograms, hMC1R binding site prediction, and overall conformation of compound 5 bound to hMC1R (PDF)
Molecular formula strings and some data (CSV)
ORCID
Minying Cai: 0000-0001-9504-2091
Notes
The authors declare no competing financial interest.
ADDITIONAL NOTE
Abbreviations used for amino acids and designation of peptides follow the rules of the IUPAC-IUB Commission of Biochemical Nomenclature in J. Biol. Chem.40
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. Ca-Cancer J Clin. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Kanavy HE, Gerstenblith MR. Ultraviolet radiation and melanoma. Semin Cutaneous Med Surg. 2011;30(4):222–228. doi: 10.1016/j.sder.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 3.Meredith P, Riesz J. Radiative relaxation quantum yields for synthetic eumelanin. Photochem Photobiol. 2004;79(2):211–216. doi: 10.1562/0031-8655(2004)079<0211:rcrqyf>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 4.Ward-Peterson M, Acuña JM, Alkhalifah MK, Nasiri AM, Al-Akeel ES, Alkhaldi TM, Dawari SA, Aldaham SA. Association between race/ethnicity and survival of melanoma patients in the United States over 3 decades: a secondary analysis of SEER data. Medicine (Philadelphia, PA, U. S.) 2016;95(17):e3315. doi: 10.1097/MD.0000000000003315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Engel J, Schlesinger-Raab A, Emeny R, Holzel D, Schubert-Fritschle G. Quality of life in women with localised breast cancer or malignant melanoma 2 years after initial treatment: a comparison. Int J Behav Med. 2014;21(3):478–486. doi: 10.1007/s12529-013-9334-x. [DOI] [PubMed] [Google Scholar]
- 6.Lin JY, Fisher DE. Melanocyte biology and skin pigmentation. Nature. 2007;445(7130):843–850. doi: 10.1038/nature05660. [DOI] [PubMed] [Google Scholar]
- 7.Zmijewski MA, Slominski AT. Is Mc1r an important regulator of non-pigmentary responses to UV radiation? Exp Dermatol. 2013;22(12):790–791. doi: 10.1111/exd.12129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolnicka-Glubisz A, De Fabo E, Noonan F. Functional melanocortin 1 receptor Mc1r is not necessary for an inflammatory response to UV radiation in adult mouse skin. Exp Dermatol. 2013;22(3):226–228. doi: 10.1111/exd.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao J, Wan L, Hacker E, Dai X, Lenna S, Jimenez-Cervantes C, Wang Y, Leslie NR, Xu GX, Widlund HR, Ryu B, Alani RM, Dutton-Regester K, Goding CR, Hayward NK, Wei W, Cui R. MC1R is a potent regulator of PTEN after UV exposure in melanocytes. Mol Cell. 2013;51(4):409–422. doi: 10.1016/j.molcel.2013.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadekaro AL, Chen J, Yang J, Chen S, Jameson J, Swope VB, Cheng T, Kadakia M, Abdel-Malek Z. Alpha-melanocyte-stimulating hormone suppresses oxidative stress through a p53-mediated signaling pathway in human melanocytes. Mol Cancer Res. 2012;10(6):778–786. doi: 10.1158/1541-7786.MCR-11-0436. [DOI] [PubMed] [Google Scholar]
- 11.Abdel-Malek Z, Suzuki I, Tada A, Im S, Akcali C. The melanocortin-1 receptor and human pigmentation. Ann N Y Acad Sci. 1999;885:117–133. doi: 10.1111/j.1749-6632.1999.tb08669.x. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki I, Im S, Tada A, Scott C, Akcali C, Davis MB, Barsh G, Hearing V, Abdel-Malek Z. Participation of the melanocortin-1 receptor in the UV control of pigmentation. J Invest Dermatol Symp Proc. 1999;4(1):29–34. doi: 10.1038/sj.jidsp.5640177. [DOI] [PubMed] [Google Scholar]
- 13.Scholzen TE, Brzoska T, Kalden DH, Hartmeyer M, Fastrich M, Luger TA, Armstrong CA, Ansel JC. Expression of functional melanocortin receptors and proopiomelanocortin peptides by human dermal microvascular endothelial cells. Ann N Y Acad Sci. 1999;885:239–253. doi: 10.1111/j.1749-6632.1999.tb08681.x. [DOI] [PubMed] [Google Scholar]
- 14.Pawelek JM. Approaches to increasing skin melanin with MSH analogs and synthetic melanins. Pigm Cell Res. 2001;14(3):155–160. doi: 10.1034/j.1600-0749.2001.140304.x. [DOI] [PubMed] [Google Scholar]
- 15.Scott MC, Wakamatsu K, Ito S, Kadekaro AL, Kobayashi N, Groden J, Kavanagh R, Takakuwa T, Virador V, Hearing VJ, Abdel-Malek ZA. Human melanocortin 1 receptor variants, receptor function and melanocyte response to UV radiation. J Cell Sci. 2002;115(11):2349–2355. doi: 10.1242/jcs.115.11.2349. [DOI] [PubMed] [Google Scholar]
- 16.Kadekaro AL, Kanto H, Kavanagh R, Abdel-Malek Z. Significance of the melanocortin 1 receptor in regulating human melanocyte pigmentation, proliferation, and survival. Ann N Y Acad Sci. 2003;994:359–365. doi: 10.1111/j.1749-6632.2003.tb03200.x. [DOI] [PubMed] [Google Scholar]
- 17.Kadekaro AL, Kavanagh R, Kanto H, Terzieva S, Hauser J, Kobayashi N, Schwemberger S, Cornelius J, Babcock G, Shertzer HG, Scott G, Abdel-Malek ZA. Alpha-melanocortin and endothelin-1 activate antiapoptotic pathways and reduce DNA damage in human melanocytes. Cancer Res. 2005;65(10):4292–4299. doi: 10.1158/0008-5472.CAN-04-4535. [DOI] [PubMed] [Google Scholar]
- 18.Abdel-Malek ZA, Knittel J, Kadekaro AL, Swope VB, Starner R. The melanocortin 1 receptor and the UV response of human melanocytes–a shift in paradigm. Photochem Photobiol. 2008;84(2):501–508. doi: 10.1111/j.1751-1097.2008.00294.x. [DOI] [PubMed] [Google Scholar]
- 19.Abdel-Malek ZA, Ruwe A, Kavanagh-Starner R, Kadekaro AL, Swope V, Haskell-Luevano C, Koikov L, Knittel JJ. Alpha-MSH tripeptide analogs activate the melanocortin 1 receptor and reduce UV-induced DNA damage in human melanocytes. Pigm Cell Melanoma Res. 2009;22(5):635–644. doi: 10.1111/j.1755-148X.2009.00598.x. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Borron JC, Abdel-Malek Z, Jimenez-Cervantes C. MC1R, the cAMP pathway, and the response to solar UV: extending the horizon beyond pigmentation. Pigm Cell Melanoma Res. 2014;27(5):699–720. doi: 10.1111/pcmr.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nasti TH, Timares L. MC1R, eumelanin and pheomelanin: their role in determining the susceptibility to skin cancer. Photochem Photobiol. 2015;91(1):188–200. doi: 10.1111/php.12335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin K, Sturm RA, Smith AG. MC1R and NR4A receptors in cellular stress and DNA repair: implications for UVR protection. Exp Dermatol. 2014;23(7):449–452. doi: 10.1111/exd.12420. [DOI] [PubMed] [Google Scholar]
- 23.Cai M, Hruby VJ. The melanocortin receptor system: a target for multiple degenerative diseases. Curr Protein Pept Sci. 2016;17(5):488–496. doi: 10.2174/1389203717666160226145330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biba E. Protection: the sunscreen pill. Nature. 2014;515(7527):S124–125. doi: 10.1038/515S124a. [DOI] [PubMed] [Google Scholar]
- 25.Langendonk JG, Balwani M, Anderson KE, Bonkovsky HL, Anstey AV, Bissell DM, Bloomer J, Edwards C, Neumann NJ, Parker C, Phillips JD, Lim HW, Hamzavi I, Deybach JC, Kauppinen R, Rhodes LE, Frank J, Murphy GM, Karstens FP, Sijbrands EJ, de Rooij FW, Lebwohl M, Naik H, Goding CR, Wilson JH, Desnick RJ. Afamelanotide for erythropoietic protoporphyria. N Engl J Med. 2015;373(1):48–59. doi: 10.1056/NEJMoa1411481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cai M, Mayorov AV, Cabello C, Stankova M, Trivedi D, Hruby VJ. Novel 3D pharmacophore of alpha-MSH/gamma-MSH hybrids leads to selective human MC1R and MC3R analogues. J Med Chem. 2005;48(6):1839–1848. doi: 10.1021/jm049579s. [DOI] [PubMed] [Google Scholar]
- 27.Holder JR, Xiang Z, Bauzo RM, Haskell-Luevano C. Structure-activity relationships of the melanocortin tetrapeptide Ac-His-D-Phe-Arg-Trp-NH2 at the mouse melanocortin receptors. 4. modifications at the Trp position. J Med Chem. 2002;45(26):5736–5744. doi: 10.1021/jm020296e. [DOI] [PubMed] [Google Scholar]
- 28.Chen M, Cai M, Aprahamian CJ, Georgeson KE, Hruby V, Harmon CM, Yang Y. Contribution of the conserved amino acids of the melanocortin-4 receptor in D-[Nle4, Phe7]-alpha-melanocyte-stimulating hormone binding and signaling. J Biol Chem. 2007;282(30):21712–21719. doi: 10.1074/jbc.M702285200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen M, Aprahamian CJ, Celik A, Georgeson KE, Garvey WT, Harmon CM, Yang Y. Molecular characterization of human melanocortin-3 receptor ligand-receptor interaction. Biochemistry. 2006;45(4):1128–1137. doi: 10.1021/bi0521792. [DOI] [PubMed] [Google Scholar]
- 30.Yang Y, Dickinson C, Haskell-Luevano C, Gantz I. Molecular basis for the interaction of [Nle4,D-Phe7]melanocyte stimulating hormone with the human melanocortin-1 receptor. J Biol Chem. 1997;272(37):23000–23010. doi: 10.1074/jbc.272.37.23000. [DOI] [PubMed] [Google Scholar]
- 31.Grieco P, Balse PM, Weinberg D, MacNeil T, Hruby VJ. D-Amino acid scan of gamma-melanocyte-stimulating hormone: importance of Trp(8) on human MC3 receptor selectivity. J Med Chem. 2000;43(26):4998–5002. doi: 10.1021/jm000211e. [DOI] [PubMed] [Google Scholar]
- 32.Chai BX, Pogozheva ID, Lai YM, Li JY, Neubig RR, Mosberg HI, Gantz I. Receptor-antagonist interactions in the complexes of agouti and agouti-related protein with human melanocortin 1 and 4 receptors. Biochemistry. 2005;44(9):3418–3431. doi: 10.1021/bi0478704. [DOI] [PubMed] [Google Scholar]
- 33.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477(7366):549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malerich S, Berson D. Next generation cosmeceuticals: the latest in peptides, growth factors, cytokines, and stem cells. Dermatol Clin. 2014;32(1):13–21. doi: 10.1016/j.det.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Thiboutot D, Sivarajah A, Gilliland K, Cong Z, Clawson G. The melanocortin 5 receptor is expressed in human sebaceous glands and rat preputial cells. J Invest Dermatol. 2000;115(4):614–619. doi: 10.1046/j.1523-1747.2000.00094.x. [DOI] [PubMed] [Google Scholar]
- 36.Kishi T, Aschkenasi CJ, Lee CE, Mountjoy KG, Saper CB, Elmquist JK. Expression of melanocortin 4 receptor mRNA in the central nervous system of the rat. J Comp Neurol. 2003;457(3):213–235. doi: 10.1002/cne.10454. [DOI] [PubMed] [Google Scholar]
- 37.Vaidyanathan G, Zalutsky MR. Fluorine-18-labeled [Nle4,D-Phe7]-alpha-MSH, an alpha-melanocyte stimulating hormone analogue. Nucl Med Biol. 1997;24(2):171–178. doi: 10.1016/s0969-8051(96)00211-9. [DOI] [PubMed] [Google Scholar]
- 38.Kim ES, Garnock-Jones KP. Afamelanotide: a review in erythropoietic protoporphyria. Am J Clin Dermatol. 2016;17(2):179–185. doi: 10.1007/s40257-016-0184-6. [DOI] [PubMed] [Google Scholar]
- 39.Richardson J, Lundegaard PR, Reynolds NL, Dorin JR, Porteous DJ, Jackson IJ, Patton EE. Mc1r pathway regulation of zebrafish melanosome dispersion. Zebrafish. 2008;5(4):289–295. doi: 10.1089/zeb.2008.0541. [DOI] [PubMed] [Google Scholar]
- 40.IUPAC-IUB Commission on Biochemical Nomenclature. Symbols for amino-acid derivatives and peptides. Recommendations (1971) J Biol Chem. 1972;247(4):977–983. [PubMed] [Google Scholar]
- 41.Cai M, Cai C, Mayorov AV, Xiong C, Cabello CM, Soloshonok VA, Swift JR, Trivedi D, Hruby VJ. Biological and conformational study of beta-substituted prolines in MT-II template: steric effects leading to human MC5 receptor selectivity. J Pept Res. 2004;63(2):116–131. doi: 10.1111/j.1399-3011.2003.00105.x. [DOI] [PubMed] [Google Scholar]
- 42.Cai M, Stankova M, Pond SJ, Mayorov AV, Perry JW, Yamamura HI, Trivedi D, Hruby VJ. Real time differentiation of G-protein coupled receptor (GPCR) agonist and antagonist by two photon fluorescence laser microscopy. J Am Chem Soc. 2004;126(23):7160–7161. doi: 10.1021/ja049473m. [DOI] [PubMed] [Google Scholar]
- 43.Cai M, Mayorov AV, Ying J, Stankova M, Trivedi D, Cabello C, Hruby VJ. Design of novel melanotropin agonists and antagonists with high potency and selectivity for human melanocortin receptors. Peptides. 2005;26(8):1481–1485. doi: 10.1016/j.peptides.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 44.Sawyer TK, Hruby VJ, Darman PS, Hadley ME. [half-Cys4,half-Cys10]-alpha-Melanocyte-stimulating hormone: a cyclic alpha-melanotropin exhibiting superagonist biological activity. Proc Natl Acad Sci U. S. A. 1982;79(6):1751–1755. doi: 10.1073/pnas.79.6.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. 4-Norleucine, 7-D-phenylalanine-alpha-melanocyte-stimulating hormone: a highly potent alpha-melanotropin with ultralong biological activity. Proc Natl Acad Sci U. S. A. 1980;77(10):5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Castrucci AM, Hadley ME, Hruby VJ. Melanotropin bioassays: in vitro and in vivo comparisons. Gen Comp Endocrinol. 1984;55(1):104–111. doi: 10.1016/0016-6480(84)90134-5. [DOI] [PubMed] [Google Scholar]
- 47.Tanaka S, Park MK, Takikawa H, Wakabayashi K. Comparative studies on the electric nature of amphibian gonadotropin. Gen Comp Endocrinol. 1985;59(1):110–119. doi: 10.1016/0016-6480(85)90425-3. [DOI] [PubMed] [Google Scholar]
- 48.Sugg EE, De Castrucci LAM, Hadley ME, van Binst G, Hruby VJ. Cyclic lactam analogues of Ac-[Nle4]alpha-MSH4–11-NH2. Biochemistry. 1988;27(21):8181–8188. doi: 10.1021/bi00421a029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.