

Abstract
Opioids alleviate pain, but adverse effects severely limit their usefulness. To solve this problem, biased ligands favoring 1 signaling pathway downstream of the μ-opioid receptor over another are being developed. In the target article, the authors synthesize compounds that preferentially activate G-protein or β-arrestin signaling. They find that increased bias towards G-protein signaling produces better antinociception with minimal side effects in mice models. G-protein–biased opioids may provide a safer treatment strategy.
Keywords: Functional selectivity, Bias, Opioid, Signaling
Commentary on: Schmid CL, Kennedy NM, Ross NC, Lovell KM, Yue Z, Morgenweck J, Cameron MD, Bannister TD, Bohn LM. Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 2017;171:1165–1175.
1. Introduction
Opioids are among the most effective treatments for acute and chronic pain, yet their use is limited by unpleasant and dangerous side effects (eg, constipation, addiction, and life-threatening respiratory depression). Furthermore, the United States is crippled by the “opioid epidemic” where overdose drug deaths have surpassed car accidents as the number one cause of accidental death. Thus, detailed understanding of opioid receptors and their signaling pathways will undoubtedly help create safe and effective treatments for chronic pain.
Clinically effective analgesic opioids act at the μ-opioid receptor, a 7-transmembrane domain protein that couples to intracellular effectors including G-protein and β-arrestin.10 An emerging paradigm in opioid-based analgesic drug discovery is biased agonism. Biased agonism describes the preferential activation of 1 signaling partner downstream of receptor activation over another (ie, G-protein activation vs β-arrestin recruitment).4,5 The rationale for this stemmed from experiments in β-arrestin knockout mice, where μ-opioid receptor activation produced analgesia, but significantly less side effects associated with opioid administration including constipation, respiratory depression, and tolerance.2,8 Therefore, significant efforts have been placed on designing ligands biased toward G-protein activation, a requirement for analgesic efficacy, while avoiding interaction with β-arrestin.
TRV130 (OLINVO; oliceridine, Chesterbrook, PA), a G-protein–biased opioid agonist, was developed by Trevena, Inc. and recently completed phase 3 clinical trials. Despite extremely promising preclinical in vivo data, TRV130 had underwhelming results in clinical trials, demonstrating only a trend toward reduced side effects, which were not significantly different from morphine.3 This has led to much debate about the promise of biased agonism at the μ-opioid receptor and the numerous reports of new biased agonists in the literature. A common, yet often incorrect, approach is to first define bias in an in vitro system between 2 ligands and later ascribe the observed in vivo differences to observed in vitro bias. This approach ignores alternative pharmacokinetic and pharmacodynamic explanations. Ultimately, these problems have truly muddied the waters surrounding biased agonism.
In an effort to solve this problem, Schmid et al.9 detail a variety of novel μ-opioid receptor agonists that show a range of bias for G-protein vs β-arrestin recruitment in vitro and find a strong correlation with a larger therapeutic window for G-protein–biased ligands (eg, SR-17018; Fig. 1). The authors compare these findings with calculated in vivo therapeutic indices, derived using data from tests of antinociception and respiratory depression, taking into account brain penetration of the compounds. The authors highlight a strong correlation between degree of G-protein bias and a wider therapeutic window, thereby building a foundation for G-protein bias-based drug discovery efforts for the treatment of pain.
Figure 1.
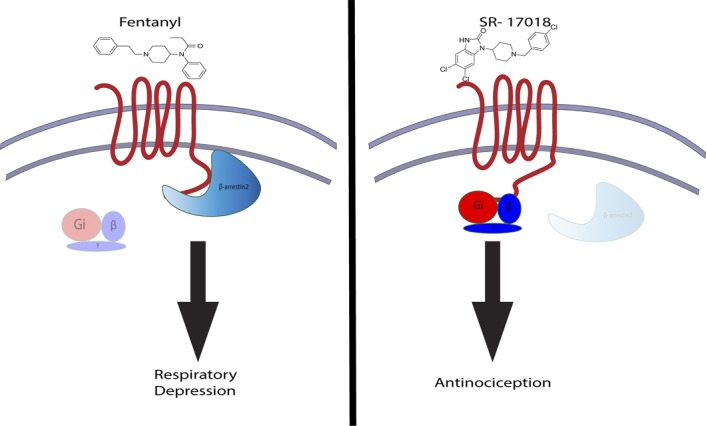
G-protein–biased compounds produce antinociception with reduced side effects. Fentanyl is a clinically used opioid analgesic that preferentially recruits β-arrestin downstream of the μ-opioid receptor (left). Consequently, fentanyl produces significant respiratory depression. In the target article, Schmid et al.9 synthesize SR-17018, an opioid that preferentially engages G-protein signaling (right). The authors demonstrate that SR-17018 produces antinociception with minimal respiratory depression relative to fentanyl.
2. Novel scaffold development produces ligand-biased signaling in vitro
A major difficulty with the development of biased ligands is correlating the magnitude of bias with observed in vivo effects. The authors overcome this difficulty by using a medicinal chemistry approach to provide a series of compounds on a single scaffold and describe structure–activity relationship regarding both G-protein and β-arrestin engagement, giving a range of bias. In particular, they demonstrate the extent to which halogen substitutions significantly impact the observed signaling bias between G-protein and β-arrestin. Given the context-dependent nature of bias calculations, generating a wide range of biases within a single scaffold is particularly useful in translating in vitro findings to in vivo validation. This approach also has the potential to rapidly generate and validate novel opioid-based analgesics.
Schmid et al.9 quantify bias using various readouts for G-protein activation (direct measure of G-protein and measure of its downstream effector, adenylyl cyclase) and β-arrestin recruitment to the receptor using heterologous cell systems that express the human or mouse μ-opioid receptor, highlighting the conservation of rank order of bias in their series of compounds. The authors provide a comprehensive evaluation of in vitro bias; however, there is 1 puzzling finding. Given that inhibition of adenylyl cyclase is directly downstream of G-protein activation, there should be no difference in bias rank order when using either of these 2 readouts. However, the authors note that 2 opioids, fentanyl and sufentanil, switch from β-arrestin biased to G-protein biased when adenylyl cyclase inhibition is used as a measure of G-protein activation. The reason for this is unclear although one potential explanation may simply be the way bias is measured. Bias calculations in the literature are quite variable, even when the same cell type and calculation method are used. Thus, this finding raises the important question of how reliable certain bias calculations really are, an issue that has recently gained much attention.7 Nonetheless, the wealth of in vitro evidence provided by Schmid et al.9 provides support for the structure–activity relationship of their series of biased compounds.
3. G-protein–biased compounds look promising in vivo
An important part of the analgesic drug discovery process is to ensure that putative analgesics can produce antinociception in the absence of side effects in preclinical models. To that end, Schmid et al.9 used a multitude of measures to demonstrate that their compounds: (1) penetrate the brain through systemic delivery; (2) produce antinociception; and (3) produce less respiratory depression compared with fentanyl. Simultaneous collection of these data provides a nearly comprehensive analysis of pharmacokinetics, antinociception, and side effects.
Importantly, the authors acknowledge that their analysis is incomplete. Exploring whether bias factors correlate with other opioid-induced side effects (eg, constipation and tolerance) remains unknown. A previous study demonstrated that repeated administration of TRV130 produced unwanted constipating and abuse-related behavioral effects, despite its bias for G-protein signaling.1 In fact, the behavioral effects of chronic TRV130 treatment resemble those of morphine.1 Evaluating the effects of chronic administration of biased agonists is crucial if these compounds are to be considered possible analgesics.
4. Future directions and concluding remarks
The data presented by Schmid et al.9 certainly build a firm foundation for the development of biased opioid ligands for the treatment of pain. However, as with any groundbreaking and important study, there are numerous interesting questions that could be addressed in future lines of study.
Ideally, the trio of in vivo analyses (pharmacokinetics, antinociception, and side effects) should be used to characterize all new potential analgesics. However, a logical next step would be to conduct these same studies using mouse models of persistent pain such as inflammatory pain, neuropathic pain, and assess more sophisticated pain-related behaviors including pain-depressed behaviors.6
The authors mention the story of TRV130, a G-protein–biased compound that displayed similarly exciting animal data, and attribute its pitfalls to the relatively small separation between G-protein activation and β-arrestin recruitment. Given the context-dependent nature of observed bias, a direct comparison of the newer ligands with TRV130, a biased opioid ligand tested in humans, would have provided valuable insight on the potential success of these compounds and highlight whether the observed correlation is maintained with alternate G-protein–biased scaffolds.
Collectively, Schmid et al.9 provide compelling evidence correlating in vitro biased G-protein activation of the μ-opioid receptor with an improved therapeutic index in vivo. In a tour de force, their techniques range from scaffold development to preclinical models of respiratory depression and antinociception. This study addresses many of the concerns and pitfalls associated with previous observations of ligand-biased signaling. In conclusion, the data presented by Schmid et al.9 are a strong positive step forward in the development of biased opioid ligands for the treatment of pain.
Disclosures
The authors have no conflict of interest to declare.
M.A. Stanczyk is a doctoral candidate supported by T32 GM007767. R. Kandasamy is a postdoctoral fellow supported by T32 DA007268 and R37 DA039997.
Acknowledgments
The authors thank Dr. John R. Traynor for his helpful comments on the article.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
References
- [1].Altarifi AA, David B, Muchhala KH, Blough BE, Akbarali H, Negus SS. Effects of acute and repeated treatment with the biased mu opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J Psychopharmacol 2017;31:730–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 1999;286:2495–8. [DOI] [PubMed] [Google Scholar]
- [3].DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther 2013;344:708–17. [DOI] [PubMed] [Google Scholar]
- [4].Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 2013;12:205. [DOI] [PubMed] [Google Scholar]
- [5].Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci 2012;3:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Negus SS, Vanderah TW, Brandt MR, Bilsky EJ, Becerra L, Borsook D. Preclinical assessment of candidate analgesic drugs: recent advances and future challenges. J Pharmacol Exp Ther 2006;319:507–14. [DOI] [PubMed] [Google Scholar]
- [7].Onaran HO, Ambrosio C, Uğur Ö, Koncz EM, Grò MC, Vezzi V, Rajagopal S, Costa T. Systematic errors in detecting biased agonism: analysis of current methods and development of a new model-free approach. Scientific Rep 2017;7:44247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Raehal KM, Walker JK, Bohn LM. Morphine side effects in β-arrestin 2 knockout mice. J Pharmacol Exp Ther 2005;314:1195–201. [DOI] [PubMed] [Google Scholar]
- [9].Schmid CL, Kennedy NM, Ross NC, Lovell KM, Yue Z, Morgenweck J, Cameron MD, Bannister TD, Bohn LM. Bias factor and therapeutic window correlate to Predict safer opioid analgesics. Cell 2017;171:1165–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Traynor J. μ-Opioid receptors and regulators of G protein signaling (RGS) proteins: from a symposium on new concepts in mu-opioid pharmacology. Drug Alcohol Depend 2012;121:173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]