

Abstract
Introduction:
Hereditary multiple intestinal atresia associated with severe combined immunodeficiency (MIA-SCID) is a very rare disease caused by deleterious mutations in the tetratricopeptide repeat domain-containing protein 7A gene TTC7A. It is characterized by intestinal obstruction, sepsis, and a poor prognosis. Insights into phenotype–genotype correlations could help to guide genetic counseling and increase our knowledge of the natural history of this disease.
Case presentation:
We report the case of a newborn in which his fetal magnetic resonance imaging showed jejunal atresia and microcolon and an abdominal x-ray at birth confirmed intestinal obstruction. The clinical course was complicated by multiple episodes of sepsis, and laboratory investigations showed SCID. The genetic analysis identified a homozygous c.53344_53347 mutation in the TTC7A gene compatible with MIA-SCID syndrome. The patient required 3 operations because of new intestinal atresias in the first months of life. She underwent bone marrow transplantation at 8 months of age but died of liver failure secondary to graft-versus-host disease.
Conclusion:
Immunologic assessment and genetic screening for TTC7A mutations are important in patients with MIA. Greater knowledge of the functions of the TTC7A protein will have important therapeutic implications for patients with MIA-SCID syndrome.
Keywords: exome, gastrointestinal tract, neonatal sepsis, parenteral nutrition
1. Introduction
Hereditary multiple intestinal atresia (MIA) (OMIM #243150) is a severe, very rare, congenital disease that involves multiples atretic injuries in diverse segments of the gastrointestinal tract, from the stomach to the rectum.[1,2] Approximately 10% to 15% of cases occur in association with severe combined immunodeficiency (SCID) and recurrent sepsis (MIA-SCID syndrome,).[3,4] According to a PubMed search, just 21 cases of MIA-SCID have been published[4–13] since the first case was described by Moreno et al[5] in 1990. All the cases to date have involved disruption of the epithelial barrier along the gastrointestinal tract, inversion of apicobasal polarity of the epithelial cells, and signs of intestinal epithelial apoptosis. Total parenteral nutrition (TPN) dependence and repeated episodes of sepsis are associated with a very poor prognosis and death occurs before the age of 2 years in 70% of patients.[4–13]
In 2013, Samuels et al[6] identified the causative gene of MIA, the tetratricopeptide repeat domain-containing protein 7A gene TTC7A, through whole-exome sequencing. Since then, 20 mutations have been identified: skipping of exons 7, 12, and 18, p.L823P, p.Y105fs, p.K254fs, p.L823P, p.S678X, p.Q712X, p.L399P, p.Q277X, p.A832X, p.S539L, p.Y336X, p.L493fsX13, p.A558GfsX7, p.E71K, p.Q526X, and p.A832T.[7–13] However, the relation between TTC7A mutations and phenotype in patients with MIA-SCID syndrome remains to be clarified.
We present a complicated case of MIA-SCID in an infant girl. The girl's parents provided informed consent to the publication of this case. There were no other ethical requirements.
2. Case report
A preterm female infant was born by vaginal delivery to healthy nonconsanguineous parents at 32 weeks of gestation. This was the first pregnancy of the mother, aged 35 years. The antenatal ultrasound at 6 months of gestation had shown a cystic malformation of the pelvis (25 × 94 cm) suggestive of dilated colon. Subsequent magnetic resonance imaging of the fetus confirmed jejunal atresia and microcolon. Chorionic villus sampling showed a normal female karyotype (46 XX).
The birth weight was 1610 grams and the 10-minute Apgar score was 9. On admission, the patient had stable respiratory and hemodynamic status. Her abdomen was soft and nondistended and palpation did not elicit pain. There were no palpable masses. An abdominal x-ray at birth confirmed intestinal obstruction associated with bilious nasogastric tube drainage (Fig. 1). The first surgical intervention, performed on day 1 of life, showed a dilated stomach and duodenum in addition to multiple jejunal and ileal atresias and intestinal malrotation. Jejunostomy and ileostomy were performed with resection through 20 cm of cecum (Fig. 2). Histopathological examination showed the classic aspect of intestinal lumen fibrosis but normal innervation. TPN and antibiotic therapy were established in the postoperative period.
Figure 1.
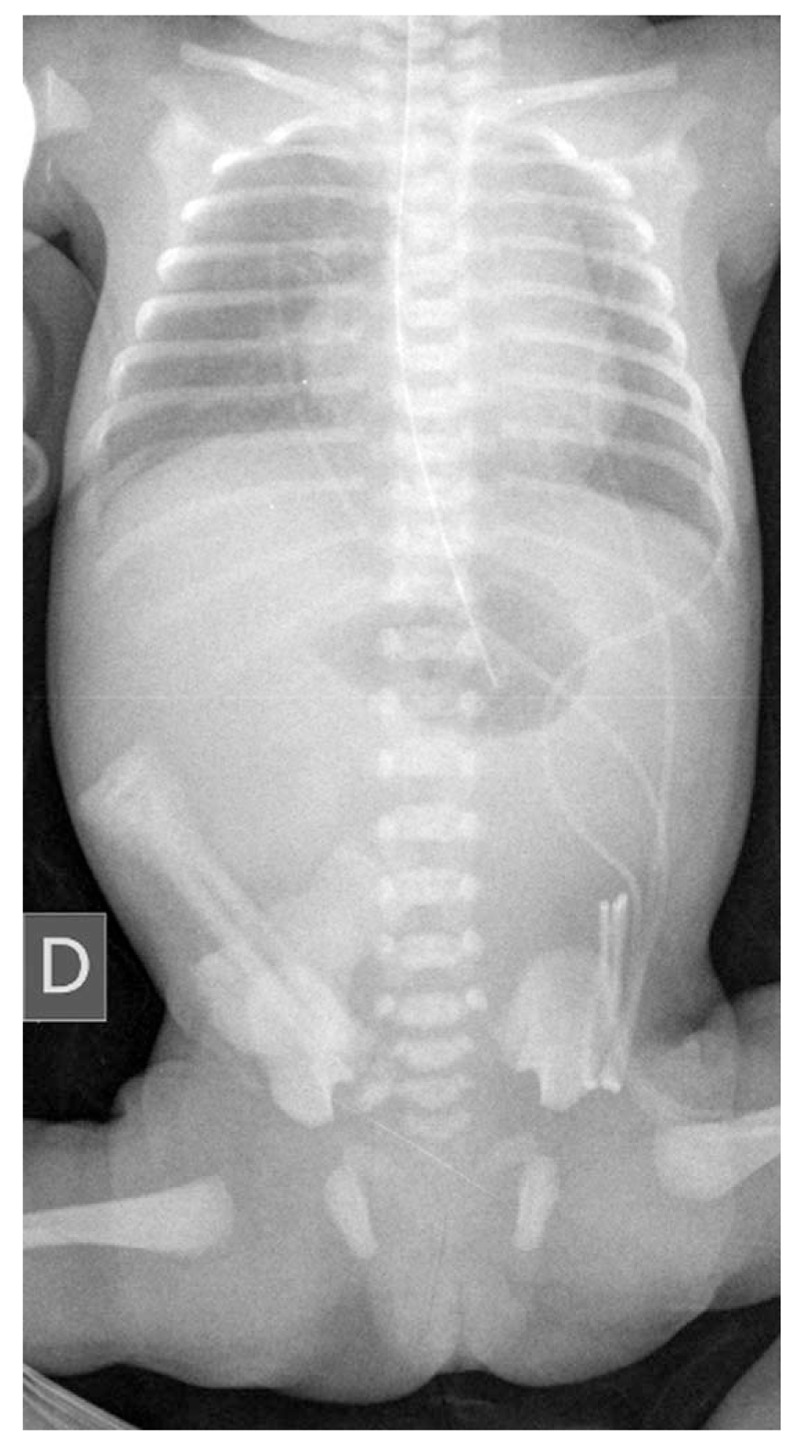
Abdominal x-ray at birth confirming intestinal obstruction.
Figure 2.
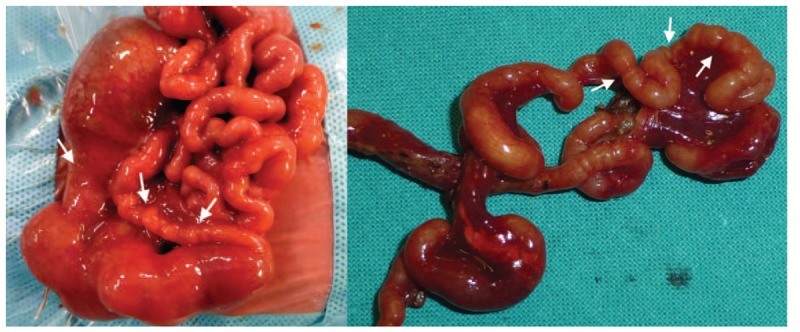
Multiple intestinal atresias observed in the macroscopic examination during the first surgical intervention. (A) Preresection; (B) postresection.
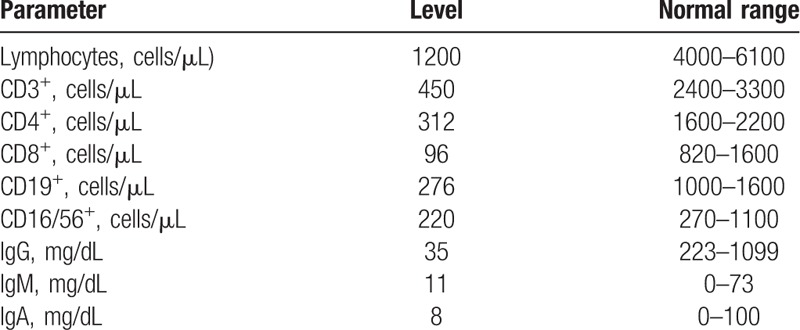
At 1 month of life, after a failed attempt to start trophic enteral feeding, a new ileal atresia with secondary microcolon was detected by lower gastrointestinal tract radiography (Fig. 3). A second surgical intervention identified additional atretic lesions in the ileal segment requiring ileocecal valve resection and another anastomosis. The remaining small bowel was about 25 to 30 cm in length. A skin abrasion around the stoma was detected after this second operation. The wound worsened despite topical antibiotics, antifungals, and steroids, and at 3 months of life, the gastrointestinal tract was reconstructed and the ostomies were closed by end-to-end anastomosis. Bilious nasogastric tube drainage continued to be observed, even with TPN. Additional evidence of intestinal atresia was detected by contrast radiography. The patient's clinical course was complicated by multiple episodes of sepsis caused by central lines or intestinal bacteria, such as Enterobacter cloacae and Pseudomona aeruginosa. Laboratory investigations at the age of 3 months showed severe T-cell lymphopenia and profound hypogammaglobulinemia (Table 1), confirming the diagnosis of SCID. Genetic analysis confirmed a homozygous c.53344_53347 mutation in the TTC7A gene, compatible with MIA-SCID syndrome. At 4 months of life, the patient developed moderate hepatic impairment, with laboratory results showing a maximum conjugated bilirubin level of 3.3 mg/dL, GOT 130 UI/L, GPT 120 UI/L, and prolonged prothrombin time. The patient underwent bone marrow transplantation at 8 months of life but died of liver failure secondary to graft-versus-host disease.
Figure 3.
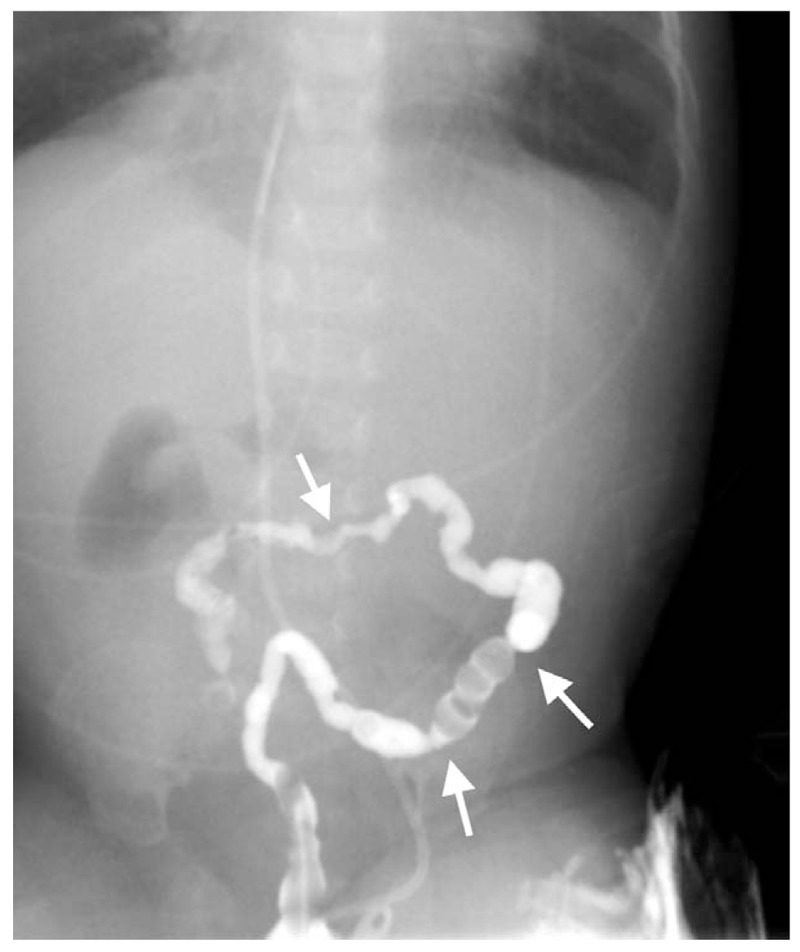
Ileal atresias detected by lower gastrointestinal tract radiography in the first month of life, after the first operation.
Table 1.
Laboratory results for patient with multiple intestinal atresia-severe combined immunodeficiency syndrome at 3 months of life.
3. Discussion
We have presented a new case of MIA-SCID with a homozygous c.53344_53347 mutation in the TTC7A gene. Clinical manifestations of hereditary MIA develop in the first 3 months of life, but in most cases, they are present from birth.[3–8] Atresias in MIA can occur in any segment of the gastrointestinal tract, but they are most common in the small bowel. Treatment consists of early surgical intervention to restore the continuity of the gastrointestinal tract through resections and anastomoses. Surgery and short-bowel syndrome, however, are an indication for TPN, which is associated with an increased risk of cholestasis, liver cirrhosis, and hepatic failure.[13,14] Patients with MIA also have altered immune system development and function. Only 6 living patients with MIA-SCID have been described in the literature. The oldest of these, presented by Bigorgne et al,[8] was 8 years old and had severe T-cell and B-cell and mild natural killer cell lymphopenia. Transplantation was contraindicated due to severe bronchiectasis.
There is limited experience with attempts to restore the immune system in patients with MIA-SCID syndrome. Gilroy et al[15] described donor immune reconstitution after liver-small bowel transplantation in a 16-month-old boy with MIA-SCID. The boy achieved complete enteral autonomy and normal liver function and showed no evidence of allograft rejection 2 years post-transplant. Three of the cases of living patients with MIA-SCID were described by Chen et al[7] in 2013. The patients were 12, 22, and 24 months old and had multiple episodes of sepsis except the 22-month-old patient achieved reconstitution of T-cell immunity after bone marrow transplantation at 3 months of life. The last known living patient was described by Agarwal et al[10] in March 2014. At the time of writing, the infant was 5 months old and had undergone multiple surgical interventions and 4 episodes of sepsis.
Our patient died at 8 months of life following bone marrow transplantation. The cause of death was liver failure secondary to graft-versus-host disease, a high-risk complication of transplantation.[16,17] She had a homozygous c.53344_53347 mutation in the TTC7A gene. This homozygous mutation had been described in 4 other patients with a similar phenotype, and they all died within the first year of life.[6,7,9] Other mutations identified in MIA-SCID syndrome have been associated with significant phenotypic variations. A recent study showed TTC7A mutations in 5 children from 3 unrelated families with very early onset inflammatory bowel disease associated with apoptotic enterocolitis.[11] None of them, however, had MIA-SCID syndrome, suggesting genotype-phenotype correlations in this disease.
In conclusion, TTC7A mutations should be investigated in patients with multiple intestinal atresias with or without SCID. Certain homozygous mutations may suggest a more common phenotype. Surgery is currently the treatment of choice for MIA, but it is associated with multiple complications. Particular attention should be paid to intestinal sepsis, which is the main cause of death in these patients. Further clarification of the role of the TTC7A protein in both the immune system and intestinal mucosa could improve the prognosis of patients with hereditary MIA and SCID.
Author contributions
Conceptualization: Alejandro Pérez-Muñuzuri, Olalla López-Suárez, Carolina López-Sanguos.
Data curation: Natalia Mandiá, Alejandro Pérez-Muñuzuri.
Formal analysis: Alejandro Pérez-Muñuzuri, Olalla López-Suárez, Carolina López-Sanguos.
Methodology: Natalia Mandiá, Carolina López-Sanguos, Adolfo Bautista-Casasnovas.
Validation: Olalla López-Suárez.
Writing – original draft: Adolfo Bautista-Casasnovas, María-Luz Couce.
Writing – review & editing: María-Luz Couce.
Footnotes
Abbreviations: MIA = multiple intestinal atresia, TPN = total parenteral nutrition, SCID = combined immunodeficiency.
Source of Support: Article-processing charge was covered by the Fundación IDIS-C012.
The authors have no conflicts of interest to disclose.
References
- [1].Mishalany HG, Der Kaloustian VM. Familial multiple-level intestinal atresias: report of two siblings. J Pediatr 1971;79:124–5. [DOI] [PubMed] [Google Scholar]
- [2].Guttman FM, Braun P, Garance PH, et al. Multiple atresias and a new syndrome of hereditary multiple atresias involving the gastrointestinal tract from stomach to rectum. J Pediatr Surg 1973;8:633–40. [DOI] [PubMed] [Google Scholar]
- [3].Lambrecht W, Kluth D. Hereditary multiple atresias of the gastrointestinal tract: report of a case and review of the literature. J Pediatr Surg 1998;33:794–7. [DOI] [PubMed] [Google Scholar]
- [4].Cole C, Freitas A, Clifton MS, et al. Hereditary multiple intestinal atresias: 2 new cases and review of the literature. J Pediatr Surg 2010;45:E21–4. [DOI] [PubMed] [Google Scholar]
- [5].Moreno LA, Gottrand F, Turck D, et al. Severe combined immunodeficiency syndrome associated with autosomal recessive familial multiple gastrointestinal atresias: study of a family. AmJ Med Genet 1990;37:143–6. [DOI] [PubMed] [Google Scholar]
- [6].Samuels ME, Majewski J, Alirezaie N, et al. Exome sequencing identifies mutations in the gene TTC7A in French–Canadian cases with hereditary multiple intestinal atresia. J Med Genet 2013;50:324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen R, Giliani S, Lanzi G, et al. Whole-exome sequencing identifies tetratricopeptide repeat domain 7A (TTC7A) mutations for combined immunodeficiency with intestinal atresias. J Allergy Clin Immunol 2013;132:656–64. e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bigorgne AE, Farin HF, Lemoine R, et al. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. J Clin Invest 2014;124:328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Avitzur Y, Guo C, Mastropaolo L, et al. Mutations in tetratricopeptide repeat domain 7A results in a severe form of very early onset inflammatory bowel disease. Gastroenterology 2014;146:1028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Agarwal NS, Northrop L, Anyane-Yeboa K, et al. Tetratricopeptide repeat domain 7A (TTC7A) mutation in a newborn with multiple intestinal atresia and combined immunodeficiency. J Clin Immunol 2014;34:607–10. [DOI] [PubMed] [Google Scholar]
- [11].Bigorgne AM, Farin HF, Lemoine R, et al. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. J Clin Invest 2014;124:328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yang W, Lee PPW, Thong MK, et al. Compound heterozygous mutations in TTC7A cause familial multiple intestinal atresias and severe combined immunodeficiency. Clin Genet 2015;88:542–9. [DOI] [PubMed] [Google Scholar]
- [13].Guanà R, Garofalo S, Teruzzi E. The complex surgical management of the first case of severe combined immunodeficiency and multiple intestinal atresias surviving after the fourth year of life. Pediatr Gastroenterol Hepatol Nutr 2014;17:257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ali YA, Rahman S, Bhat V, et al. Hereditary multiple intestinal atresia with severe combined immunodeficiency: a case report of two siblings and review of the literature on MIA, HMIA, and HMIA with immunodeficiency over the last 50 years. BMJ Case Rep 2011;2011:bcr0520103031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gilroy RK, Coccia PF, Talmadge JE, et al. Donor immune reconstitution after liver-small bowel transplantation for multiple intestinal atresia with immunodeficiency. Blood 2004;103:1171. [DOI] [PubMed] [Google Scholar]
- [16].Walker MW, Lovell MA, Kelly TE, et al. Multiple areas of intestinal atresia associated with immunodeficiency and post transfusion graft-versus-host disease. J Pediatr 1993;123:93–5. [DOI] [PubMed] [Google Scholar]
- [17].Fischer RT, Friend B, Talmon GA, et al. Intestinal transplantation in children with multiple intestinal atresias and immunodeficiency. Pediatr Transplant 2014;18:190–6. [DOI] [PubMed] [Google Scholar]