

Abstract
Background:
Rheumatoid arthritis (RA) is the most common inflammatory arthritis and is a major cause of disability. The nuclear factor-kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway has been reported to be involved in the pathogenesis of RA with unclear mechanisms. Therefore, this study aims to explore the effect of NF-κB pathway on proliferation, apoptosis, and angiogenesis of human fibroblast-like synovial cells (HFLS) in RA.
Methods:
Normal HFLS and RA-HFLS were selected as the normal and control groups, respectively. RA-HFLS were treated by BAY11-7082 (an inhibitor of NF-κB) in different concentrations, namely 2.5 μmol/L BAY11-7082, 5 μmol/LBAY11-7082 and 10 μmol/L BAY11-7082. MTT assay was employed to detect cell proliferation. Cell apoptosis was determined by flow cytometry at 24, 48, and 72 hours after culture. Western blot analysis was employed to detect the expressions of NF-κB, angiogenesis-related factors (VEGF, Ang1, and Ang2).
Results:
Initially, we found that BAY11-7082 inhibited NF-κB expression in a concentration-dependent manner. According to the findings of MTT assay and flow cytometry, we understood that RA-HFLS treated by BAY11-7082 (an inhibitor of NF-κB), the inhibition of NF-κB pathway, suppressed RA-HFLS proliferation and induced RA-HFLS apoptosis in a concentration and time-dependent manner. Furthermore, RA-HFLS treated by BAY11-7082 presented decreased VEGF, Ang1 and Ang2 expressions in a concentration-dependent manner.
Conclusion:
The study concluded that inhibition of NF-κB pathway induced cell apoptosis and suppressed proliferation and angiogenesis of RA-HFLS, which could serve as a novel target in the treatment of RA.
Keywords: angiogenesis, apoptosis, human fibroblast-like synovial cells, NF-κB pathway, proliferation, rheumatoid arthritis
1. Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease, characterized by inflammation of multiple joints,[1] which can result in articular cartilage and bone destruction, leading to significant disability.[1] Currently, about 1% of adult population worldwide has been diagnosed with RA, which results in their poor quality of life.[2] The underlying hypothesis in the pathogenesis of RA is alleged to be the proliferation of human fibroblast-like synovial cells (HFLS) in response to inflammatory cytokines.[1] RA progression is primarily characterized by dysregulated cell proliferation in the synovial lining, such as HFLS, resulting in hyperplasia, pannus formation, and destruction of associated joint cartilage.[3] HFLS display surprisingly tumor-like behavior, including increasing in number, mediating inflammation, and acquiring aggressive phenotypes, all of which contribute to joint cartilage erosion.[4] Inflammatory responses, including low-grade fever, weight loss, and fatigue, are crucial for RA development along the progression of HFLS proliferation.[5] HFLS cells obtained from synovia of patients with RA could retain their aggressive phenotype without in vitro stimulation for several passages.[6] In RA, excessive migration of circulating leukocytes into the inflamed joint compels the formation of new blood vessels to provide adequate nutrients and oxygen to the hypertrophic joint.[7] Angiogenesis has now been considered as a significant event in the maintenance and formation of the pannus in RA.[8] Although the relationship between several genes and RA has already been highlighted the genetic regulation beneficial for RA patients in the progression of the disease still remains unclear.[9]
Nuclear factor-kappa-light-chain-enhancer of activated B cells (NF-κB) is one of the numerous transcription factors, which has the ability to result in severe autoimmune diseases under abnormal activity, including arthritis.[10] NF-κB has the ability to control the expression of gene products which affect various cellular responses, such as inflammation, immunity, cell proliferation, and apoptosis.[2] NF-κB is ubiquitously expressed in the cytoplasm of virtually all cell types.[11,12] The NF-κB pathway comprises of a core I kappa B kinase (IKK) complex, inhibitor I kappa B (IκB) protein and transcription factor NF-κB.[12] Additionally, NF-κB pathway is critical to epithelial immune defense against invading microbial pathogens and has been involved in secretion of antimicrobial peptides, activation of adaptive immunity, and release of cytokines or chemokines to regulate immune effector cells.[13] Activation of the NF-κB pathway participates in chronic inflammatory disorders of pathogenesis, including RA and inflammatory bowel disease.[9] In the present study, we investigated the effects of the NF-κB pathway on proliferation, apoptosis, and angiogenesis of HFLSCs in RA, which could provide a novel target to the treatment of RA.
2. Materials and methods
2.1. Ethics statement
The ethical approval is not necessary for the reason that it is a research on cells.
2.2. Cell culture and grouping
HFLS and RA-HFLS cells were obtained from the Cell Applications INC (San Diego). Cells were cultured in Dulbecco's modified Eagle's Medium (DMEM) supplemented with 2 mM glutamine, 100 units/mL penicillin, 80 units/mL streptomycin, and 10% fetal bovine serum (FBS) (Hyclone, Logan, UT) and were incubated in a 5% CO2 incubator at 37°C. The experimental cells from the 4th to 8th generation in order to ensure the cells are in good condition.
The cells were classified into 5 groups: the normal group (Normal-HFLS), the control group (RA-HFLS), the 2.5 μmol/L BAY11-7082 group (treated with 2.5 μmol/L BAY11-7082 [an inhibitor of NF-κB]), the 5 μmol/L BAY11-7082 (treated with 5 μmol/L BAY11-7082), and the 10 μmol/L BAY11-7082 group (treated with 10 μmol/L BAY11-7082). BAY11-7082 was dissolved in dimethyl sulfoxide (DMSO). In order to avoid the interference effect from the solvent, same amount of DMSO (0.1%, v/v) was dissolved in each group.[14]
2.3. 3-[4,5-dimethylthiazol-2-yl] 2,5-diphenyltetrazolium bromide (MTT) assay
Cell viability was evaluated using MTT assay. Briefly, after the cells were digested by trypsin, cells in the logarithmic growth phase in each group were combined and prepared into a single cell suspension with the density of 1.5 × 105 cells/mL. Single cell suspension was cultured in 96-well plates (200 μL/well), and was then treated with 20 μL of MTT solution (5 mg/mL) at 24, 48 and 72 hours after culture. Next, after further incubation for 4 hours, the medium was removed, 150 μL of DMSO was added into each well and shocked at a low-speed for 10 min, when the water-insoluble formazan crystals were fully dissolved. Optical density (OD) value of each well was measured with an automatic enzyme reader (Bio-Rad, La Jolla, CA) at a wavelength of 490 nm, after which the cell viability (%) was calculated. This test was repeated for 6 wells.
2.4. Flow cytometry
The apoptotic rate was detected by using the Annexin V-FITC apoptosis detection kit (APOAF-50TST, Sigma-Aldrich, St. Louis, MO). The cells among the 5 groups were cultured for 48 hours, and then were adjusted to 1 × 106 cells/mL. Cells were washed twice with cold phosphate buffer saline (PBS), followed by addition of 10 × binding buffer (pH 7.5, 100 mM HEPES/NaOH, 1.4 M NaCl, and 25 mM CaCl2), Annexin-V-FITC and PI to the cells, respectively. After the completing mixing the solution, cells were incubated in conditions devoid of light for 15 minutes at room temperature and were analyzed by flow cytometry.
2.5. Western blot analysis
Cells in the logarithmic growth phase in each group were harvested in precooled 3 to 5 times volume lysis buffer (containing: 150 mM NaCl (S5886, Sigma-Aldrich, St. Louis, MO), 1% Triton X-100 (X100, Sigma-Aldrich, St. Louis, MO), 50 mM Tris–HCl (93363, Sigma-Aldrich, St. Louis, MO) and 1 SigmaFASTä protease inhibition tablet (S8820, Sigma-Aldrich, St. Louis, MO) per 100 mL) and made into homogenate. After lysis, the cell lysates were centrifuged for 1 h at 12000 g at 4°C and the supernatant was collected. The protein concentration was quantified by Bradford method, and samples were modulated to the same concentration. The protein samples (20 μg) were mixed with gel loading buffer (5 × sodium dodecyl sulfate [SDS]) and were boiled in water for 5 minutes. The proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane after separation by 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE). PVDF membrane was sheared at about 60 kDa. After the membrane was blocked with 5% non-fat milk for 1 hour at 37°C, and the primary rabbit anti-human antibodies NF-κB, VEGF, Ang1, Ang2 and β-actin (1: 1000; Cell Signaling Technology) were added. Next, the membranes were incubated with the aforementioned antibodies overnight at 4°C and were washed with tris-buffered saline Tween 20 (TBST) 3 times for 5 minutes each time, then were incubated with the corresponding horseradish peroxidase labelled secondary antibodies (1: 2000; Cell Signaling Technology) at room temperature for 2 hours. The membranes were treated with electrochemiluminescence (ECL) followed by analysis with by gel documentation system (GDS), and then an image was taken with an enhanced chemiluminescence western detection system (Perkin-Elmer Life Sciences, Boston, MA), after which the protein levels were normalized to the expression of β-actin.
2.6. Statistical analysis
SPSS 21.0 statistical software (IBM Corp. Armonk, NY) was employed for statistical analysis. The measurement data were presented using mean ± standard deviation (SD). The t-test was adopted to compare the average value of 2 samples. The one-way analysis of variance (ANOVA) method was adopted to compare among multiple groups. A probability value of P < .05 indicated the difference was statistically significant.
3. Results
3.1. BAY11-7082 inhibited NF-κB expression in a concentration-dependent manner
The results of western blotting (Fig. 1) revealed an obvious increase in NF-κB expression in the control group compared with the normal group (P < .05), thereby suggesting that the NF-κB expression of RA-HFLS was higher than the normal HFLS. The NF-κB expression in the 2.5 μmol/L BAY11-7082, 5 μmol/L BAY11-7082 and 10 μmol/L BAY11-7082 groups were between the normal group and the control group. The increased concentration of BAY11-7082, consequently lead to a significant decrease in the NF-κB expression in the 2.5 μmol/L BAY11-7082, 5 μmol/L BAY11-7082 and 10 μmol/L BAY11-7082 groups (all P < .05). In comparison with the normal group and control group, the NF-κB expressions in the 2.5 μmol/L BAY11-7082 and 5 μmol/L BAY11-7082 groups had notable differences (all P < .05). The NF-κB expression in the 10 μmol/L BAY11-7082 group did not exhibit a significant difference compared to the normal group (P > .05). Results were indicative of the fact that BAY11-7082 could down-regulate NF-κB expression in a concentration-dependent manner.
Figure 1.
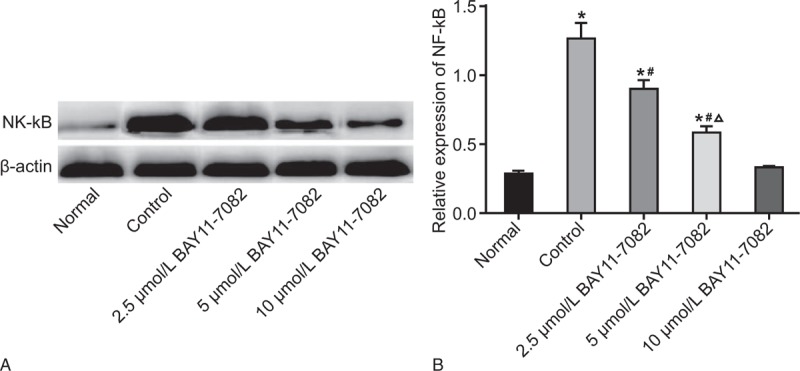
According to the results of western blot analysis, BAY11-7082 inhibits NF-κB expression in a concentration-dependent manner. A, protein bands of NF-κB expression among 5 groups; B, NF-κB expression among 5 groups detected by western blot analysis; ∗P < .05 compared to the normal group; #P < .05 compared to the control group; Δ, P < .05 compared to the 2.5 μmol/L BAY11-7082 group. NF-κB = nuclear factor-kappa-light-chain-enhancer of activated B cells.
3.2. BAY11-7082 suppressed proliferation of RA-HFLS in the concentration and time-dependent manner
As shown in Figure 2, over time, the cell viability increased. No significant difference was observed in cell viability at any specific time point in the normal and 10 μmol/L BAY11-7082 groups (P > .05). In the control and 2.5 μmol/L BAY11-7082 groups, the cell viability at 48 h and 72 hours was significantly different compared with the cell viability recorded at the previous time point (P < .05). In the 5 μmol/L BAY11-7082 group, the cell viability at 72 hours was markedly higher than that at 48 hours (P < .05). No significant difference was observed in cell viability at 0 and 24 hours among the 5 groups (P > .05). At 48 hours, the cell viability evidently increased in the control and 2.5 μmol/L BAY11-7082 groups compared with the normal group (P < .05), whereas cell viability was higher in the control group compared to the 2.5 μmol/L BAY11-7082 group (P < .05). Among the 5 μmol/L BAY11-7082 and 10 μmol/L BAY11-7082 groups, no marked difference was observed in cell viability compared to the normal group (P > .05). At 72 hours, the cell viability greatly ascended in the control, 2.5 μmol/L BAY11-7082 and 5 μmol/L BAY11-7082 groups compared to the normal group, whereas the cell viability decreased among the control, 2.5 μmol/L BAY11-7082 and 5 μmol/L BAY11-7082 groups and there were statistically significant differences in pairwise comparisons (P < .05). In the 10 μmol/L BAY11-7082 group, cell viability did not exhibit any marked difference compared to the normal group (P > .05). To summarize, the control, 2.5 μmol/L BAY11-7082 and 5 μmol/L BAY11-7082 groups showed higher cell proliferation than the normal group, whereas cell proliferation declined in the 2.5 μmol/L BAY11-7082, 5 μmol/L BAY11-7082 and 10 μmol/L BAY11-7082 groups compared to the control group. The results revealed that BAY11-7082 may inhibit RA-HFLS proliferation in the concentration and in a time-dependent manner.
Figure 2.
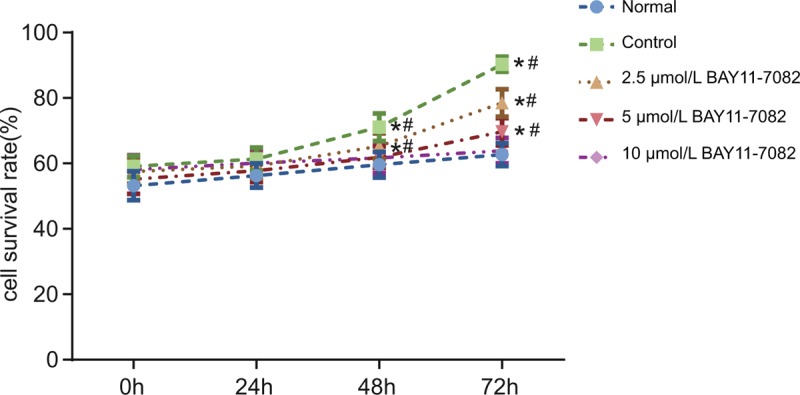
MTT assay findings suggest that BAY11-7082 suppresses RA-HFLS proliferation in a concentration and time-dependent manner. ∗P < .05 compared to the previous time point in the same group; #P < .05 compared to the other groups at the same time point. HFLS = fibroblast-like synovial cells, MTT = 3-[4 = 5-dimethylthiazol-2-yl] 2 = 5-diphenyltetrazolium bromide, RA = rheumatoid arthritis.
3.3. BAY11-7082 promoted RA-HFLS apoptosis in a concentration-dependent manner
The results of flow cytometry (Fig. 3) showed that the apoptosis rate significantly decreased in the control, 2.5 μmol/L BAY11-7082, and 5 μmol/L BAY11-7082 groups compared to the normal group (P < .05), while the apoptosis rate elevated successively in the control, 2.5 μmol/L BAY11-7082, and 5 μmol/L BAY11-7082 groups (P < .05). In the 10 μmol/L BAY11-7082 group, no significant difference was observed in apoptosis rate compared to the normal group (P > .05). The results indicated that BAY11-7082 could stimulate RA-HFLS apoptosis in a concentration-dependent manner.
Figure 3.
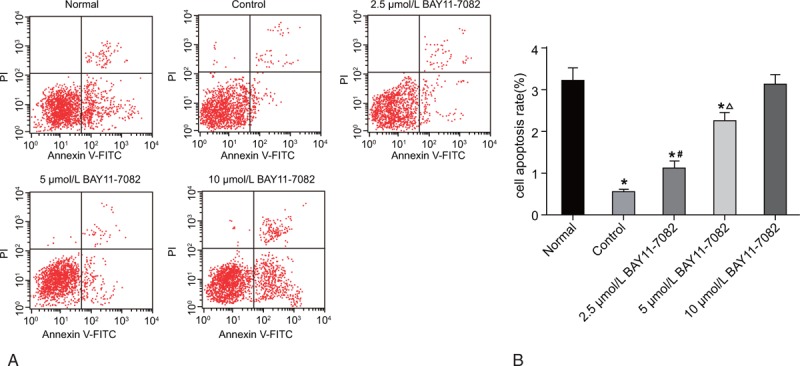
According to the results of flow cytometry, BAY11-7082 promotes RA-HFLS apoptosis in a concentration-dependent manner. (A) The apoptosis of RA-HFLS among 5 groups determined by flow cytometry; (B) the apoptosis rate of RA-HFLS among 5 groups; compared with the Normal group P < .05; #, compared with the Control group P < .05; Δ, compared with the 2.5 μmol/L NF-κB group P < .05. HFLS = fibroblast-like synovial cells, RA = rheumatoid arthritis.
3.4. BAY11-7082 decreased VEGF, Ang1 and Ang2 expressions in a concentration-dependent manner
As revealed in Figure 4, the expressions of VEGF, Ang1, and Ang2 were upregulated in the control, 2.5 μmol/L BAY11-7082 and 5 μmol/L BAY11-7082 groups compared with the normal group, whereas the expressions of VEGF, Ang1, and Ang2 were downregulated successively in the control, 2.5 μmol/L BAY11-7082, and 5 μmol/L BAY11-7082 groups (P < .05). In the 10 μmol/L BAY11-7082 group, no significant difference was observed in levels of VEGF, Ang1 and Ang2 compared with the normal group (P > .05). The results indicated that BAY11-7082 might inhibit the expressions of RA-HFLS angiogenesis-related factors in a concentration-dependent manner.
Figure 4.
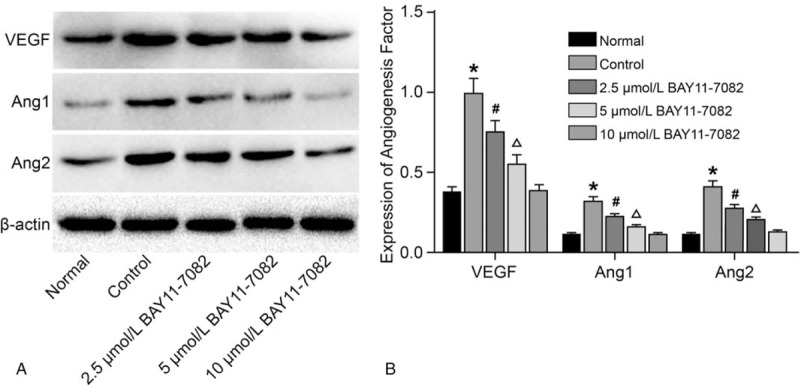
According to the results of western blot analysis, BAY11-7082 decreases the expressions angiogenesis-related factors (VEGF, Ang1, and Ang2) in a concentration-dependent manner. A, protein bands of angiogenesis-related factors; B, angiogenesis-related factors expression; ∗P < .05 compared to the normal group; #P < .05 compared to the control group; Δ, P < .05 compared to the 2.5 μmol/L BAY11-7082 group; VEGF, Vascular endothelial growth factor; Ang1, Angiotension1; Ang2, Angiotension2. Ang = angiopoietins.
4. Discussion
RA occurs in 0.5% to 1.0% of the adult population worldwide, characterized principally by inflammatory polyarthritis with local inflammation in joint synovial tissue as well as progressive bone and cartilage degradation.[15] The complex interactions among immunologic, genetic, and environmental factors (family history, air, or food pollution) are of high clinical significance in RA development.[15] NF-κB pathway is a major participant in general inflammatory responses and especially along the pathogenesis of RA.[11] HFLS plays an indispensable role in the formation of RA pannus and in pannus invasion and destruction of cartilage and bone.[16] This study was performed with the underlying aim to explore the effect of NF-κB pathway on the proliferation, apoptosis and angiogenesis-related factors of RA-HFLS in RA patients. From the results, we concluded that inhibition of NF-κB pathway induced cell apoptosis and suppressed proliferation and angiogenesis of RA-HFLS.
For clear comparisons, our study comprised of 5 groups, including the normal, control, 2.5 mmol/L BAY11-7082, 5 μmol/L BAY11-7082, and 10 μmol/L BAY11-7082 groups. The comparative results show that the NF-κB expression of RA-HFLS was higher compared to the expression recorded in the normal HFLS, while the NF-κB expression decreased with an increase in BAY11-7082 concentration. The results exhibited that BAY11-7082 inhibited NF-κB expression, showing a time-dependent manner. NF-κB activation promotes synovial hyperplasia via accelerating proliferation and inhibiting apoptosis of HFLS.[1] A significant increase in NF-κB was observed in PGC-1β overexpressed RA-HFLS through its inhibitory role in cell growth.[17] Besides, BAY11-7082 (an inhibitor of NF-κB activation) is reported to reduce the IL-8 production and NF-κB binding activity via blocking p38 or STAT1.[18] The expression of ASCC1 reduced NF-κB transcriptional activity in a dose-dependent manner.[9]
This study also showed that cell viability increased in the control and the 2.5 μmol/L BAY11-7082 groups compared to the normal group. A notable increase in cell viability was observed in at 72 hours in the control, 2.5 μmol/L BAY11-7082 and 5 μmol/L BAY11-7082 groups compared with the normal group. Thus, we propose that downregulation of NF-κB might play an important role in enhancing cell viability of RA-HFLS. NF-κB plays a crucial role in anti-apoptosis, and a previous study has highlighted the role of NF-κB inhibitor in inducing cell apoptosis.[19] Results also show that apoptosis induced by the suppression of NF-κB was significantly decreased in the control, 2.5 μmol/L BAY11-7082, 5 μmol/L BAY11-7082 groups compared with the normal group. In accordance with our results, NF-κB inhibitor BAY11-7082 has been demonstrated to induce cell apoptosis of human uveal melanoma cells.[20] For the previously studied various types of transcription factors, NF-κB is indispensable in the production of cytokines and proteases by RA-HFLS, along with its role in the regulating of the resistance of RA-HFLS to cell apoptosis.[1] Autophagy induction in primary RA-HFLS can negatively regulate inflammatory response.[19] Therefore, it has the ability to balance the influence of inflammation in RA development and HFLS apoptosis.[1] A previous study demonstrated that the product of lipid peroxidation might induce cell death in RA-HFLS due to synovial inflammations by activating NF-κB pathways.[1]
Additionally, the present study also revealed that the expressions of VEGF, Ang1 and Ang2 increased in the control, 2.5 μmol/L BAY11-7082 and 5 μmol/L BAY11-7082 groups compared to the normal group, while the expression of VEGF declined with an increase in BAY11-7082 concentration. It revealed that BAY11-7082 inhibited the expressions of various angiogenesis-related factors (soluble endoglin, placental growth factor (PlGF), and transforming growth factor-beta1 (TGF-beta1), in RA-HFLS in a concentration-dependent manner. A previous study revealed that the expression of VEGF declined in genetically diabetic mice after treatment with BAY 11–7082.[21] Growth factor release has been associated with enhanced proliferation of synoviocytes, along with angiogenesis in synovial tissue.[3] The release of VEGF and other growth factors enhanced angiogenesis in inflamed synovial tissue, and thus elevated serum growth factor levels might serve as an indicator of RA severity.[3] VEGF was effective principally in restricting primary tumor growth, while the Ang/Tie2 pathway-targeted agents (anti-Tie2, anti-Ang2, BowAng1) were relatively ineffective at controlling the growth of primary tumor.[22] Numerous studies have reported that the dependence of cytoprotection on VEGF was abolished by pretreatment with BAY11–7085 (a specific inhibitor of NF-κB).[23]
Angiopoietins (Ang) are angiogenic factors essential for vascular development, maturation, and inflammation.[24] Angiopoietin-1 (Ang1) and its context-dependent antagonist angiopoietin-2 (Ang2) are secreted by endothelial growth factors (VEGF121, VEGF145, and VEGF165), which bind to the extracellular domain of the tyrosine kinase receptor Tie2 expressed predominantly in endothelial cells.[24] Ang1 contributes to stabilizing newly formed vessels, while Ang2 disrupts pre-existing vasculature.[25,26] Ang2 expression is induced to compensate for weak Ang1 signaling by restoring Tie2 activity and promoting the survival or proliferation of tumor endothelial cells.[27] The concentration of Ang1 was significantly lower in the serum of cirrhotic patients compared the concentration in the non-cirrhotic ones, and Ang2 serum levels were higher in patients with cirrhosis.[28] Zhao et al[29] demonstrated that Ang2 activated the AT1R/PI3K/Akt pathway, which further activated NF-κB, consequently leading to an increased expression of matrix metalloproteinase (MMP)-2 and MMP-9 and migration in breast cancer cells. In consistent with our findings, activated NF-κB pathway was attributed to HFLS acceleration, thus facilitating the progress of RA.
Collectively, the key findings of the study revealed that NF-κB pathway is upregulated in RA, and its inhibition could induce cell apoptosis and prevent from proliferation and angiogenesis of RA-HFLS, which may potentially be a clinically viable target in the treatment of RA.
5. Conclusion
In conclusion, these data could serve as an explanation that inhibition of NF-κB pathway could induce cell apoptosis and suppress proliferation angiogenesis of RA-HFLS, which contributed to the development of potential therapeutic strategies for treating RA. However, more prospective studies are needed for deeper investigation in order to confirm the effects of NF-κB pathway on proliferation, apoptosis and angiogenesis of RA-HFLS, and to move forward towards the goal of developing novel and promising therapeutic strategies for the treatment of RA.
Acknowledgments
We would like to give our sincere appreciation to the reviewers for their helpful comments on this article.
Author contributions
Conceptualization: Zhong-Bin Xia, Guo-Qing Li, Fan-Bo Feng.
Data curation: Fan-Ru Meng, Xia Wu, Ying Liu.
Formal analysis: Zhong-Bin Xia, Fan-Ru Meng, Chun-Wang Zhang, Dan Liu.
Funding acquisition: Guo-Qing Li.
Investigation: Zhong-Bin Xia, Yu-Xuan Fang, Xia Wu.
Methodology: Yu-Xuan Fang, Chun-Wang Zhang.
Resources: Dan Liu, Fan-Bo Feng.
Supervision: Guo-Qing Li.
Validation: Xia Wu, Hai-Yang Qiu.
Visualization: Ying Liu.
Writing – original draft: Fan-Ru Meng, Yu-Xuan Fang, Fan-Bo Feng, Hai-Yang Qiu.
Writing – review & editing: Zhong-Bin Xia, Fan-Ru Meng, Yu-Xuan Fang, Xia Wu, Chun-Wang Zhang, Ying Liu, Dan Liu, Guo-Qing Li, Fan-Bo Feng, Hai-Yang Qiu.
Footnotes
Abbreviations: Ang = angiopoietins, ANOVA = analysis of variance, DMEM = dulbecco's modified Eagle's Medium, DMSO = dimethyl sulfoxide, ELC = electrochemiluminescence, FBS = fetal bovine serum, GDS = gel documentation system, HFLS = fibroblast-like synovial cells, IκB = inhibitor I kappa B, IKK = I kappa B kinase, MMP = matrix metalloproteinase, MTT = 3-[4 = 5-dimethylthiazol-2-yl] 2 = 5-diphenyltetrazolium bromide, NF-κB = nuclear factor-kappa-light-chain-enhancer of activated B cells, OD = optical density, PBS = phosphate buffer saline, RA = rheumatoid arthritis, SD = standard deviation, SDS-PAGE = sodium dodecyl sulfate -polyacrylamide gel electrophoresis, TBST = tris-buffered saline Tween 20.
ZBX and FRM are regarded as the co-first authors.
This study was supported by grants from National Natural Science Foundation of China (No. 81402936 and 81302576), Jiangsu Provincial Natural Science Foundation of China (No. BK20131234), Six Talent Peak Research Project in Jiangsu Province (No. 2015-WSN-105), the 333 Project of Jiangsu Province, Jiangsu Province Youth Medical Talent Project, as well as by Jiangsu Province Clinical Medical Science and Technology Project (No. BL2013034).
Competing interests: The authors have declared that no competing interests exist.
References
- [1].Yin G, Wang Y, Cen XM, et al. Lipid peroxidation-mediated inflammation promotes cell apoptosis through activation of NF-kappaB pathway in rheumatoid arthritis synovial cells. Mediators Inflamm 2015;2015:460310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wang HZ, Wang HH, Huang SS, et al. Inhibitory effect of baicalin on collagen-induced arthritis in rats through the nuclear factor-kappaB pathway. J Pharmacol Exp Ther 2014;350:435–43. [DOI] [PubMed] [Google Scholar]
- [3].Lahoti TS, Hughes JM, Kusnadi A, et al. Aryl hydrocarbon receptor antagonism attenuates growth factor expression, proliferation, and migration in fibroblast-like synoviocytes from patients with rheumatoid arthritis. J Pharmacol Exp Ther 2014;348:236–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kono M, Yasuda S, Stevens RL, et al. Ras guanine nucleotide-releasing protein 4 is aberrantly expressed in the fibroblast-like synoviocytes of patients with rheumatoid arthritis and controls their proliferation. Arthritis Rheumatol 2015;67:396–407. [DOI] [PubMed] [Google Scholar]
- [5].Yin G, Li Y, Yang M, et al. Pim-2/mTORC1 pathway shapes inflammatory capacity in rheumatoid arthritis synovial cells exposed to lipid peroxidations. Biomed Res Int 2015;2015:240210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol 2013;9:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Elshabrawy HA, Chen Z, Volin MV, et al. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 2015;18:433–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yang X, Qiu P, Chen B, et al. KIAA1199 as a potential diagnostic biomarker of rheumatoid arthritis related to angiogenesis. Arthritis Res Ther 2015;17:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Torices S, Alvarez-Rodriguez L, Grande L, et al. A truncated variant of ASCC1, a novel inhibitor of NF-kappaB, is associated with disease severity in patients with rheumatoid arthritis. J Immunol 2015;195:5415–20. [DOI] [PubMed] [Google Scholar]
- [10].Swierkot J, Nowak B, Czarny A, et al. The activity of JAK/STAT and NF-kappaB in patients with rheumatoid arthritis. Adv Clin Exp Med 2016;25:709–17. [DOI] [PubMed] [Google Scholar]
- [11].Noort AR, Tak PP, Tas SW. Non-canonical NF-kappaB signaling in rheumatoid arthritis: Dr Jekyll and Mr Hyde? Arthritis Res Ther 2015;17:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sun J, Xu G, Wang Z, et al. The effect of NF-kappaB signalling pathway on expression and regulation of nacrein in pearl oyster, pinctada fucata. PLoS One 2015;10:e0131711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhou R, Gong AY, Chen D, et al. Histone deacetylases and NF-kB signaling coordinate expression of CX3CL1 in epithelial cells in response to microbial challenge by suppressing miR-424 and miR-503. PLoS One 2013;8:e65153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chow JY, Wong CK, Cheung PF, et al. Intracellular signaling mechanisms regulating the activation of human eosinophils by the novel Th2 cytokine IL-33: implications for allergic inflammation. Cell Mol Immunol 2010;7:26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Citro A, Scrivo R, Martini H, et al. CD8+ T cells specific to apoptosis-associated antigens predict the response to tumor necrosis factor inhibitor therapy in rheumatoid arthritis. PLoS One 2015;10:e0128607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang H, Jia XZ, Sui CJ, et al. Effects of thapsigargin on the proliferation and survival of human rheumatoid arthritis synovial cells. ScientificWorldJournal 2014;2014:605416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhou JJ, Ma JD, Mo YQ, et al. Down-regulating peroxisome proliferator-activated receptor-gamma coactivator-1 beta alleviates the proinflammatory effect of rheumatoid arthritis fibroblast-like synoviocytes through inhibiting extracellular signal-regulated kinase, p38 and nuclear factor-kappaB activation. Arthritis Res Ther 2014;16:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cong W, Ruan D, Xuan Y, et al. Cardiac-specific overexpression of catalase prevents diabetes-induced pathological changes by inhibiting NF-kappaB signaling activation in the heart. J Mol Cell Cardiol 2015;89:314–25. [DOI] [PubMed] [Google Scholar]
- [19].Lee H, Kang SW, Byun HS, et al. Brazilin limits inflammatory responses through induction of prosurvival autophagy in rheumatoid fibroblast-like synoviocytes. PLoS One 2015;10:e0136122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hu S, Luo Q, Cun B, et al. The pharmacological NF-kappaB inhibitor BAY11-7082 induces cell apoptosis and inhibits the migration of human uveal melanoma cells. Int J Mol Sci 2012;13:15653–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bitto A, Altavilla D, Pizzino G, et al. Inhibition of inflammasome activation improves the impaired pattern of healing in genetically diabetic mice. Br J Pharmacol 2014;171:2300–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wu FT, Paez-Ribes M, Xu P, et al. Aflibercept and Ang1 supplementation improve neoadjuvant or adjuvant chemotherapy in a preclinical model of resectable breast cancer. Sci Rep 2016;6:36694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mo SJ, Hong J, Chen X, et al. VEGF-mediated NF-kappaB activation protects PC12 cells from damage induced by hypoxia. Neurosci Lett 2016;610:54–9. [DOI] [PubMed] [Google Scholar]
- [24].Robinson-Cohen C, Katz R, Price BL, et al. Association of markers of endothelial dysregulation Ang1 and Ang2 with acute kidney injury in critically ill patients. Crit Care 2016;20:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dumas E, Neagoe PE, McDonald PP, et al. New insights into the pro-inflammatory activities of Ang1 on neutrophils: induction of MIP-1beta synthesis and release. PLoS One 2016;11:e0163140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nambu H, Nambu R, Oshima Y, et al. Angiopoietin 1 inhibits ocular neovascularization and breakdown of the blood-retinal barrier. Gene Ther 2004;11:865–73. [DOI] [PubMed] [Google Scholar]
- [27].Daly C, Eichten A, Castanaro C, et al. Angiopoietin-2 functions as a Tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer Res 2013;73:108–18. [DOI] [PubMed] [Google Scholar]
- [28].Hernandez-Bartolome A, Lopez-Rodriguez R, Borque MJ, et al. Angiopoietin-2/angiopoietin-1 as non-invasive biomarker of cirrhosis in chronic hepatitis C. World J Gastroenterol 2016;22:9744–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhao Y, Wang H, Li X, et al. Ang II-AT1R increases cell migration through PI3K/AKT and NF-kappa B pathways in breast cancer. J Cell Physiol 2014;229:1855–62. [DOI] [PubMed] [Google Scholar]