

Abstract
Hydroxylation of aliphatic carbons by non-heme Fe(IV)-oxo (ferryl) complexes proceeds by hydrogen-atom (H•) transfer (HAT) to the ferryl and subsequent coupling between the carbon radical and Fe(III)-coordinated oxygen (termed rebound). Enzymes that use H•-abstracting ferryl complexes for other transformations must either suppress rebound or further process hydroxylated intermediates. For olefin-installing C–C desaturations, it has been proposed that a second HAT to the Fe(III)–OH complex from the carbon α to the radical preempts rebound. Deuterium (2H) at the second site should slow this step, potentially making rebound competitive. Desaturations mediated by two related L-arginine-modifying iron(II)- and 2-(oxo)glutarate-dependent (Fe/2OG) oxygenases behave oppositely in this key test, implicating different mechanisms. NapI, the L-Arg 4,5-desaturase from the naphthyridinomycin biosynthetic pathway, abstracts H• first from C5 but hydroxylates this site (leading to guanidine release) to the same modest extent whether C4 harbors 1H or 2H. By contrast, an unexpected 3,4-desaturation of L-homoarginine (L-hArg) by VioC, the L-Arg 3-hydroxylase from the viomycin biosynthetic pathway, is markedly disfavored relative to C4 hydroxylation when C3 (the second hydrogen donor) harbors 2H. Anchimeric assistance by N6 permits removal of the C4-H as a proton in the NapI reaction, but, with no such assistance possible in the VioC desaturation, a second HAT step (from C3) is required. The close proximity (≤ 3.5 Å) of both L-hArg carbons to the oxygen ligand in an x-ray crystal structure of VioC harboring a vanadium-based ferryl mimic supports and rationalizes the sequential-HAT mechanism. The results suggest that, although the sequential-HAT mechanism is feasible, its geometric requirements may make competing hydroxylation unavoidable, thus explaining the presence of α-heteroatoms in nearly all native substrates for Fe/2OG desaturases.
Graphical Abstract
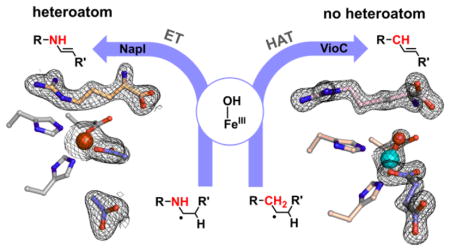
INTRODUCTION
Iron(II)- and 2-(oxo)glutarate-dependent (Fe/2OG) oxygenases hydroxylate, halogenate, epimerize, cyclize, and desaturate unactivated aliphatic carbon centers.1–3 The pathways to these diverse outcomes are thought to share common initial steps, which include addition of O2 to the non-heme Fe(II) cofactor, cleavage of 2OG to CO2 and succinate, formation of an Fe(IV)-oxo (ferryl) complex, and hydrogen atom transfer (HAT) from the substrate to the ferryl oxo.4–12 The resultant state, with Fe(III)–OH cofactor and substrate radical, is the likely branch point to the different outcomes.1 In the hydroxylation pathway, the carbon radical attacks the oxygen ligand to form a new C–O bond. This radical-coupling step, commonly referred to as oxygen rebound,13 regenerates the Fe(II) state of the cofactor for subsequent turnover. It is thought that rebound has a low activation barrier,14 consistent with the failure of the Fe(III)-OH/substrate-radical state to accumulate in reactions of Fe/2OG hydroxylases.6,8 Recent work has suggested, however, that this step can be suppressed to allow for different fates of the intermediate.1,15 For example, in stereoinversion of the bridgehead carbon (C5) of (3S,5S)-carbapenam-3-carboxylate by carbapenem synthase (CarC), HAT from C5 to the ferryl complex is followed by a butterfly-like flexion of the bicyclic substrate, which inverts the radical, moves it away from the Fe(III)-coordinated oxygen to disfavor rebound, and approximates its opposite face to a tyrosine residue that donates H• to complete the epimerization.12,16–18 In the Fe/2OG aliphatic halogenases, a chloro or bromo ligand cis to the oxygen couples to the carbon radical in preference to oxygen rebound.9,19,20 The halogenase SyrB2 directs this outcome by positioning the scissile C–H bond farther from the oxygen and closer to the halogen, trading proficiency in the HAT step for selectivity in the radical-coupling step.20,21 Whereas the required positioning might, in principle, have arisen by evolutionary relocation of the substrate-binding site within the conserved protein scaffold, recent analysis has suggested that the halogenases instead alter the configuration of the ferryl complex to achieve the required substrate-cofactor disposition.22–24 For both the carrier-protein-dependent SyrB2 and the recently discovered carrier-protein-independent halogenase, WelO5 (the first small-molecule halogenase identified), it has been proposed that the oxo of the ferryl locates trans to the proximal histidine ligand (found in the HXD coordination motif), essentially replacing the departing C1 carboxylate oxygen of 2OG.22,24 This hypothetical ferryl configuration has been termed offline, to distinguish it from the inline configuration with the oxo trans to the distal histidine. In both halogenases, the offline position of the oxo group should result in a perpendicular approach of the scissile C–H bond to the Fe=O unit. Frontier-orbital analysis of the SyrB2 reaction suggested that this disposition can rationalize both the unusually slow HAT step and the preference for C–Cl/Br over C–O coupling in the subsequent step.22,25,26 Enforcement of offline ferryl complexes could, in principle, be a general strategy to suppress rebound in other Fe/2OG enzymes that catalyze reactions other than hydroxylation.27
The mechanisms of known olefin-installing desaturations catalyzed by Fe/2OG enzymes remain poorly understood. Such transformations require, formally, removal of hydrogen atoms from two adjacent aliphatic carbons, perhaps requiring two successive H•-accepting intermediates. The modi operandi of other O2-activating iron enzymes, including those in the cytochrome P45028 and non-heme di-iron classes,29 appear well suited to this requirement (Scheme 1). In the former case, the Fe(IV)-oxo/porphyrin-radical intermediate known as compound I could serve as the first H• acceptor,30 leaving the Fe(IV)-OH complex, compound II (a strong oxidant in its own right),31 to remove the adjacent hydrogen (Scheme 1A). In the non-hemediiron case, an Fe2(IV) intermediate similar to Q from the soluble methane monooxygenase catalytic cycle32,33 could be the first HAT acceptor, leaving an Fe2(III/IV) complex, analogous to the tyrosine-oxidizing cluster X in the β subunit of class Ia ribonucleotide reductase,34 to cleave the adjacent C–H bond (Scheme 1B). These pathways can rationalize known desaturations at even completely unactivated aliphatic C–C bonds by heme-iron and non-heme di-iron oxidases in, for example, steroid and fatty acid metabolism, respectively.28,29
Scheme 1.
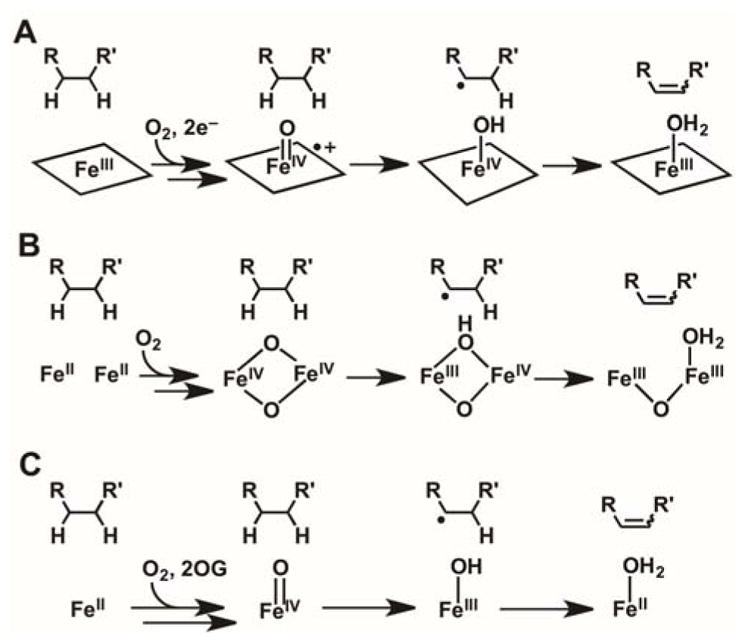
Hypothetical Pathways for C–C Desaturations Catalyzed by (A) Heme Cytochrome P450 (B) Non-heme Diiron, and (C) Fe/2OG Enzymes.
The catalytic manifold of Fe/2OG oxygenases seems less well suited for such a sequential-HAT mechanism (Scheme 1C): the Fe(III)-OH complex produced by HAT to the ferryl intermediate is expected to have modest oxidative potency. However, because the radical center weakens bonds to the adjacent (α) carbon, this mechanism has repeatedly been advanced in the literature.35–37 Two considerations motivate direct experimental scrutiny of the hypothesis. Firstly, in the absence of a very rapid change in the substrate-cofactor disposition between the first and second HAT steps, the imperative to suppress rebound could present a geometric conundrum. Perpendicular approach of the target C–H bond to the ferryl could, as proposed for the halogenases,22,24 disfavor subsequent C–O coupling, but the need for the adjacent carbon to approach the Fe(III)-OH species for the second HAT step would raise the question of why this carbon would not efficiently donate H• to the ferryl in the first place. Secondly, the vast majority of substrates for these enzymes have a lone-pair-harboring heteroatom (O or N) adjacent to one of the carbon atoms between which the olefin is installed.1 To our knowledge, only PrhA and BcmF among the known Fe/2OG desaturases are thought to transform a completely unactivated C–C unit,38,39 although an earlier example of mixed desaturation and hydroxylation of a non-native substrate by the Fe/2OG hydroxylase/oxacyclase/desaturase, clavaminate synthase, provides a notable second exception to this α-heteroatom rule.35 In the reactions conforming to the rule, the non-bonding electrons on the heteroatoms could enable alternative pathways via iminium or oxonium intermediates, from which the second hydrogen could be removed as a proton by a basic amino acid.
As a model system to address these mechanistic questions experimentally, we identified the L-arginine 4,5-desaturase, NapI, from the naphthyridinomycin bio-synthetic pathway (Scheme 2, top).40 In this selection, we viewed known L-Arg hydroxylases that are structurally similar to NapI (e.g., the 3-hydroxylase, VioC, from the viomycin pathway41,42 and the 3,4-dihydroxylase, OrfP, from the streptolidine pathway43) as important assets, because we anticipated that a comparative structural and mechanistic analysis might identify the crucial determinants of the divergent outcomes, an overarching goal of studies on this enzyme family.1 In the course of the mechanistic analysis, we fortuitously discovered that VioC can desaturate L-homoarginine (L-hArg, the L-Arg analog with an additional –CH2– unit in its side chain) between carbons 3 and 4 in competition with hydroxylation of either site (Scheme 2, bottom). As just the second demonstrated example (of which we are aware) of desaturation by an Fe/2OG oxygenase of a C–C unit lacking an activating α-heteroatom, this reaction presented a compelling case study to compare to the NapI reaction, which conforms to the α-heteroatom rule. We show here that the reactions actually proceed by different mechanisms. In the efficient, native 4,5-desaturation of L-Arg by NapI, anchimeric assistance (of either dehydration of a hydroxylated intermediate or electron transfer from the C5 radical) by N6 obviates the second HAT step in favor of removal of the C4 hydrogen as a proton. By contrast, with no such sequential, polar pathway for removal of the second hydrogen available in the absence of an α-heteroatom, the VioC reaction follows the oft-proposed, sequential-HAT pathway, but with marked diminution in selectivity. The results highlight a potentially general limitation of Fe/2OG enzymes for C–C desaturation reactions: effective suppression of rebound may be impossible for reactions that can proceed only by close approximation of adjacent C–H bonds to the cofactor, as is required for sequential HAT steps.
Scheme 2.

Naphthyridinomycin biosynthesis and the activities of NapI (top) and VioC (bottom).
RESULTS AND DISCUSSION
Kinetic Evidence for HAT from C5 of L-Arg to the Ferryl Intermediate in NapI
In the NapI desaturation reaction, the adjacent carbons from which hydrogens must be removed (C4 and C5) are distinguished by their β and α dispositions to the heteroatom, N6. To identify the site from which H• is abstracted by the presumptive ferryl intermediate, site-specifically deuterium-labeled substrates, 4,4-d2-L-Arg and 5,5-d2-L-Arg, were chemically synthesized (see Supporting Information) (Fig. S1) and used in single-turnover experiments monitoring formation and decay of the intermediate. Previous studies on multiple Fe/2OG hydroxylases, halogenases, and the epimerase/desaturase CarC established (1) that the conserved ferryl intermediate has an ultraviolet absorption feature at 320 nm and a Mössbauer quadrupole doublet with low isomer shift (~ 0.3 mm/s) that allow it to be monitored in time, and (2) that substitution of the abstracted hydrogen with deuterium markedly extends the lifetime of the intermediate, owing to the large, normal deuterium kinetic isotope effect (2H-KIE) on the HAT step.4,8,9,11,12,44,45 In accordance with these precedents, following rapid mixing of an anoxic solution of the NapI-•Fe(II)•2OG•L-Arg complex with air-saturated buffer, ΔA320 rises and falls (Fig. 1, black) in temporal correlation with the Mössbauer quadrupole doublet features diagnostic of the ferryl intermediate (Fig. S5A and C).45 In the reaction with 5,5-d2-L-Arg, the maximum absorbance is greater and the decay phase markedly delayed (Fig. 1, blue), reflecting a large 2H-KIE on decay of the ferryl complex (Fig. S5B). By contrast, deuterium substitution at C4 has only a modest effect on the kinetics of the ferryl complex (Fig. 1, red). Stabilization of the intermediate by deuteria on C5 but not C4 establishes that the ferryl complex in NapI abstracts H• from C5 to initiate the 4,5-desaturation.
Figure 1.
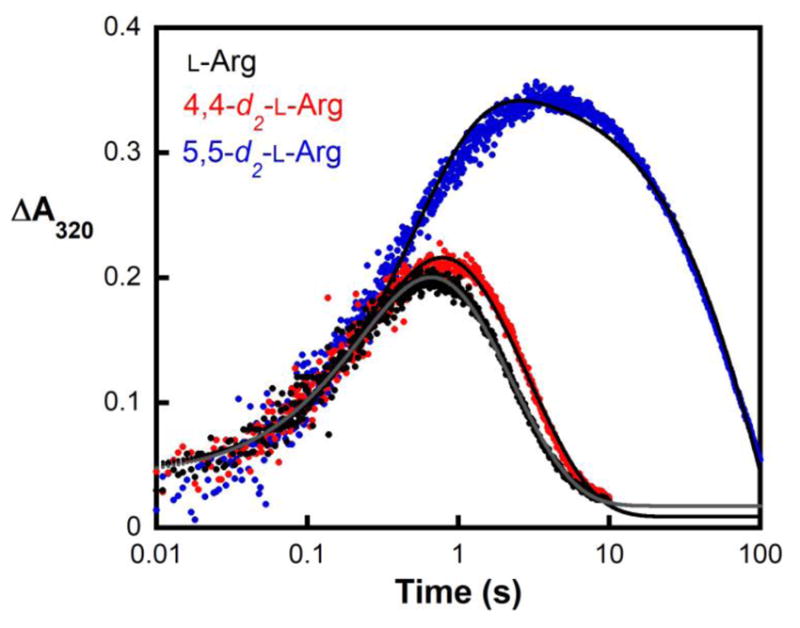
Kinetics of the ferryl complex in NapI monitored by its absorbance at 320 nm. An anoxic solution of NapI (1.2 mM), Fe(II) (1.1 mM), 2OG (6.0 mM) and 6.0 mM of either L-Arg (black), 4,4-d2-L-Arg (red), or 5,5-d2-L-Arg (blue) was mixed at 5 °C with an equal volume of air-saturated buffer (giving ~ 0.19 mM final [O2]). Regression fits to the data are shown as solid lines. The equation for fitting assumes absorbance from two consecutive, irreversible reactions, with isosbestic reactant and product and more intensely absorbing intermediate.
Product Yields with Deuterium-labeled Substrates Confirming Initial HAT from C5
Use of liquid chromatography and mass spectrometry (LC-MS) to compare the yields of the 4,5-dehydro-L-Arg product from the three substrate isotopologs provided additional confirmation of C5 targeting by the ferryl complex. At equivalent, limiting concentrations of 2OG, the L-Arg and 4,4-d2-L-Arg substrates give indistinguishable yields, whereas the C5-labeled substrate gives ~ 50% less desaturation (Figs. 2A and S6). As shown in previous work on TauD, SyrB2, and CarC, diminished yield of the primary product can reflect unproductive decay of the ferryl complex,12,46 its redirection to an adjacent site,20 or a combination of the two effects, caused by the large 2H-KIE on the initiating HAT step.4 Indeed, a complex of peaks at m/z = +16 relative to the substrate reflects stable hydroxylation occurring in competition with the predominant 4,5-desaturation. It has only modest intensity in reactions with the unlabeled L-Arg (Fig. 2C, black trace) but is considerably (4–5-fold) more intense upon use of the 5,5-d2-L-Arg isotopolog (blue traces), confirming that the large 2H-KIE can partially redirect the ferryl complex to another site and alter the outcome to stable hydroxylation. This complex of peaks is suppressed for the 4,4-d2-L-Arg isotopolog (red traces), implying that the hydroxylation side reaction occurs primarily at C4.
Figure 2.

Single-ion LC-MS chromatograms monitoring (A) desaturation (m/z = 173) (B) guanidine production (as a consequence of C5 hydroxylation), and (C) stable hydroxylation (m/z = 191) of substrates, L-Arg (black), 4,4-d2-L-Arg (red), and 5,5-d2-L-Arg (blue), by NapI. The green trace in B is the chromatogram of a 13C-guanidine standard. The lighter red and blue traces in C are traces for the m+15 ions (where m is the m/z value of that substrate) resulting from hydroxylation initiated by abstraction of deuterium. Peak areas relative to the normalized (independently for each panel) areas of the traces for unlabeled L-Arg are given to the left of each trace, with standard deviations from three trials in parentheses. Details of the analytical procedure are provided in the Supporting Information.
a, n.d. = not detected
Failure of C4 Deuteriation to Enhance C5 Hydroxylation Disfavoring Sequential-HAT Mechanism
Applied to NapI, the most widely cited mechanism for C–C desaturations would invoke a second HAT,35 from C4 to the Fe(III)-OH complex, to convert the ferryl-generated C5 radical directly to the 4,5-dehydro-L-Arg product (Scheme 3, pathway A). The precedent of the halogenases20 suggests that rebound to C5 might compete to some extent with the second HAT step. Competing rebound would generate an unstable hemiaminal-like species, which, if not directed to eliminate water on the normal desaturation pathway (Scheme 3, pathway B), would be expected to break down preferentially by elimination of guanidine (pKa of 12.5 for guanidinium versus 15.7 for water). Guanidinium (m/z = 60, green trace in Fig. 2B) is indeed produced in the reaction of the unlabeled substrate (black trace). The yield of the breakdown product is diminished by the presence of deuterium at C5 (blue trace) to the same extent as the yield of the primary 4,5-desaturation product, implying that both products form subsequent to HAT from C5. Under the assumption that the sequential-HAT mechanism (pathway A) is operant, guanidinium production would imply that the second HAT from C4 is preempted by rebound to C5 in a small fraction of events. In this scenario, because the second HAT step should also exhibit a sizable 2H-KIE, oxygen rebound to C5 should compete more effectively in the reaction of the 4,4-d2-L-Arg isotopolog, leading to increased guanidinium production and decreased desaturation. This expectation is not borne out, as neither yield is significantly altered by the presence of deuterium at C4 (Fig. 2A, B; compare red and black traces). The failure of this prediction of the sequential-HAT mechanism implies that rebound to C5 does not directly compete with HAT from C4 and, thus, that the reaction proceeds by a different mechanism.
Scheme 3.
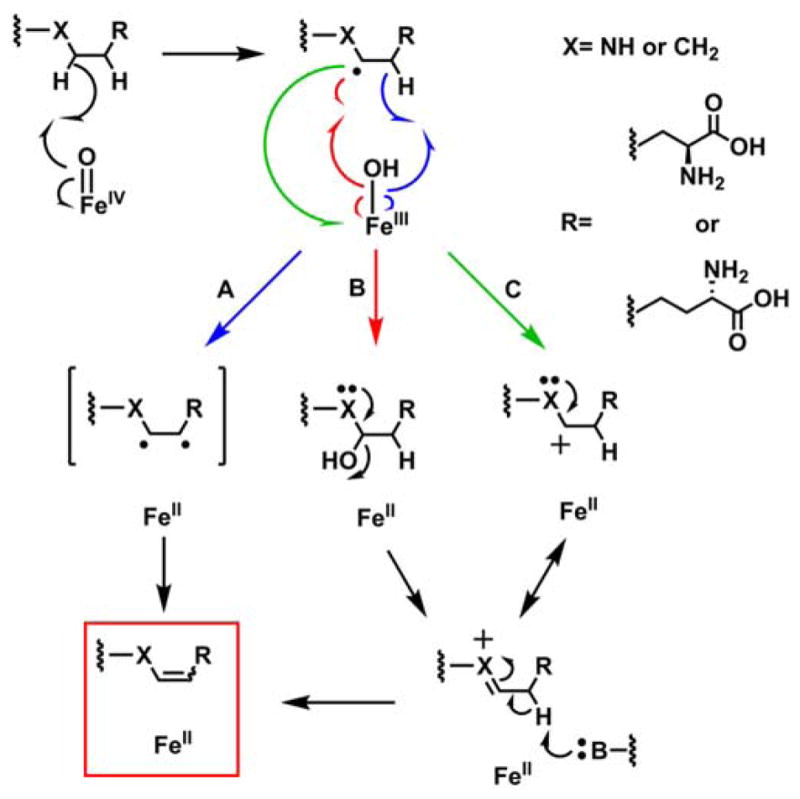
Possible pathways for C–C desaturation by an Fe/2OG oxygenase. (A) Sequential HAT (blue), (B) Intermediate Hydroxylation (red), and (C) Electron Transfer (green).
Inactivity of 6-Deaza L-Arg for Desaturation Suggesting Assistance of Polar C4-H Cleavage by N6
The two other most likely pathways for conversion of the C5 radical to 4,5-dehydro-L-Arg would involve rebound to C5 followed by dehydration and C4 deprotonation (Scheme 3, pathway B) or electron transfer from the radical to the Fe(III)-OH cofactor followed by C4 deprotonation (pathway C). Both of these pathways include an iminium-like intermediate involving the non-bonding electrons on N6, and neither is expected to be feasible in the absence of the α-heteroatom. To test this prediction, the 6-deaza analog of L-Arg, known as indospicine, was evaluated as a substrate for NapI. The analog is indeed a substrate but undergoes stable hydroxylation, as demonstrated by the prominent peaks at m/z +16 or +18 relative to the substrate in LC-MS chromatograms of the reactions carried out under air or 18O2, respectively (Fig. 3, black and red traces). Notably, no peak at m/z = –2 relative to indospicine develops (blue trace), confirming that N6 of L-Arg plays an essential role in the native 4,5-desaturation. These observations further validate the conclusion that the sequential-HAT mechanism (pathway A) is not operant for NapI.
Figure 3.
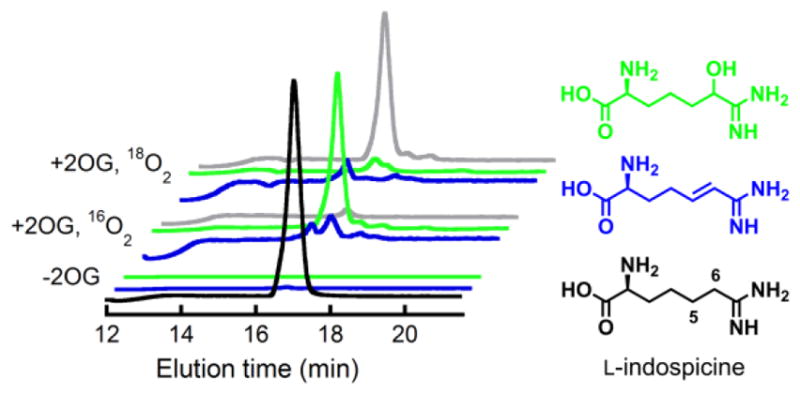
Positive-mode single-ion LC-MS chromatograms showing that NapI can hydroxylate but not desaturate the 6-deaza analogue of L-Arg, L-indospicine. The front three traces are from a control lacking the co-substrate, 2OG. The middle traces are from analysis of a reaction carried out under atmospheric O2. The rear traces are from a reaction under 18O2. Coloring coding of the traces is: black, m/z = 174 (indospicine); green, m/z = 190 (H16O-indospicine); gray, m/z = 192 (H18O-indospicine); blue, m/z = 172 (dehydroindospicine; not observed).
Kinetics of Reaction with 6-Deaza L-Arg Favoring Electron Transfer over Intermediate Hydroxylation
The distinction between pathways B and C is whether 5-hydroxy-L-Arg is (in B) or is not (in C) a precursor to the iminium intermediate that undergoes deprotonation of C4 to form 4,5-dehydro-L-Arg. Kinetic analysis of the reaction with L-indospicine suggests that pathway C is more likely. The hydroxylated product of the 6-deaza analog, 5-hydroxy-L-indospicine, would be expected to mimic the unstable hemiaminal-like intermediate of pathway B, but it would be foreign to pathway C. As unstable intermediates are generally held tightly within enzyme active sites, slow dissociation of the 5-hydroxy-L-indospicine product would be expected for the case that pathway B is operant, whereas rapid dissociation might be anticipated for a mechanism not involving a hydroxylated intermediate, such as pathway C. Previous work on TauD showed that product dissociation is ordered: succinate dissociation is the last and slowest step and is prevented by occupancy of the taurine site (by either substrate or product).6 Redevelopment of the 520-nm absorption band of the reactant complex in single-turnover reactions thus occurs only after release of both products. In single-turnover, stopped-flow experiments on NapI, the absorption feature of the reactant complex actually re-develops more rapidly in the L-indospicine reaction than in the reaction with the native substrate (Fig. S7). This result suggests that NapI readily releases the stably hydroxylated product, thus weighing against the intermediacy of its hemiaminal cognate in pathway B and in favor of pathway C.
X-ray Crystal Structure of NapI Rationalizing HAT from C5 to the Cofactor
NapI shares ~ 50% sequence identity with the structurally characterized L-Arg 3-hydroxylase, VioC, from the viomycin biosynthetic pathway. The two proteins are thus expected to have similar structures. The demonstration that the NapI ferryl intermediate abstracts H• from C5 (by contrast to abstraction from C3 in VioC) raises the intriguing question of how the shared substrate is differently positioned within the conserved protein architecture to permit different sites to be targeted for distinct outcomes. To answer this question, we solved an x-ray crystal structure of NapI in complex with Fe(II) and L-Arg in the absence of O2 (Fig. 4A, B). A superposition of the 2.1-Å-resolution NapI structure with the published structure of VioC (PDB accession code 6ALM) shows that the two proteins share an identical topology (rmsd of 1.13 Å over 314 Cα atoms), apart from a small difference in the fold of a dynamic lid loop (Fig. 5A).
Figure 4.
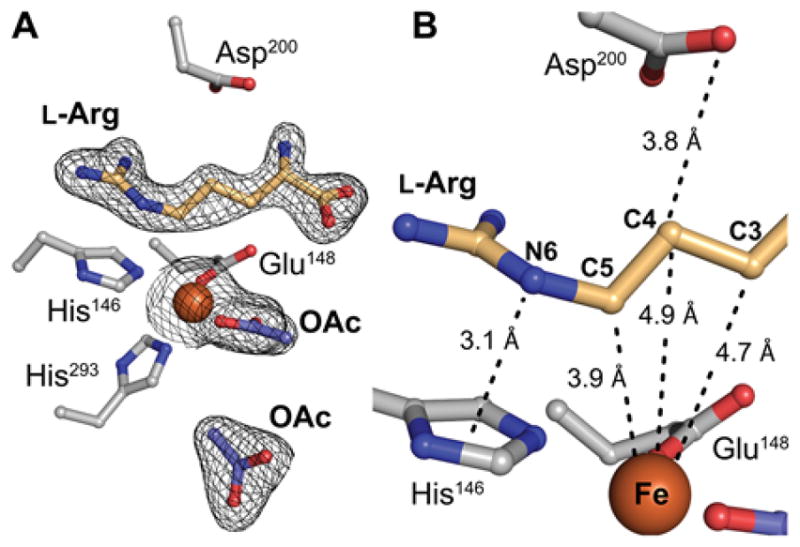
Structural analysis of NapI by x-ray crystallography. A, Active site model from a structure of L-Arg•FeII•NapI rationalizing initial HAT from C5. The enzyme exhibits a single conformation of L-Arg with continuous 2Fo-Fc electron density (black mesh) for the substrate when the map is contoured at 1.0 σ. B, Relative disposition of substrate and cofactor showing unusual proximity of C5 of L-Arg to the iron cofactor (orange sphere). Selected ligands and second-sphere side chains are shown in ball-and-stick format and colored by atom type. A pair of acetate ions (OAc; shown in purple) from the crystallization solution occupy the 2OG binding site.
Figure 5.

Comparison of the structures of VioC (PDB: 6ALM)44 and NapI. A, side-by-side comparison of the overall fold and conformation of the lid loop (inset). B and C, Comparison of the L-Arg side chain interactions in VioC and NapI, respectively, showing that their different sets of H-bonding partners position the common substrate uniquely within the active site. Selected side chains, backbone, and substrate molecules are shown in ball-and-stick format. Water molecules (red) and FeII cofactor (orange) are shown as spheres.
Two acetate ions from the crystallization solution occupy the 2OG binding site in the NapI structure; attempts to obtain high-quality crystals with the co-substrate bound were unsuccessful. However, the binding orientation of L-Arg, which is readily discernible in electron density maps of the complex (black mesh in Fig. 4A and Fig. S8), and the network of hydrogen-bonding contacts that anchor it in the active site (Fig. 5B–C) are unlikely to be affected by the absence of 2OG. Comparison of the VioC and NapI active site models (Figs. 5B–C and S9) shows that, despite their nearly identical folds, the two enzymes orient their common substrate using completely distinct contacts to the L-Arg amine, carboxylate, and guanidinium groups. Only a salt bridge between the substrate side chain and a second-sphere Asp (D245, NapI numbering) is conserved, but even this connection involves an alternate conformation of the substrate sidechain and different interacting atoms. Interestingly, NapI employs H-bonds between the terminal guanidine nitrogens and a pair of symmetrically disposed, bound water molecules to position C5 closest to the Fe(II) cofactor, just 3.9 Å away. C4 and C3 are more distant, at 4.9 Å and 4.7 Å, respectively (Fig. 4B). By contrast, in VioC, a single bound water molecule and an additional Asp residue (270) on the same side of the active site hydrogen bond with the terminal guanidine nitrogens, and these contacts are associated with a ~ 90° rotation of the L-Arg guanidine that serves to move C5 away from the cofactor and instead position C3 proximal to the Fe(II).
Active-site Metrics by HYSCORE Spectroscopy Rationalizing HAT from both C5 and C4
Analysis of hyperfine couplings between substrate hydrons and EPR-active (S = 3/2) iron-nitrosyl ({FeNO}7) cofactor adducts previously afforded active-site structural information for O2-activating non-heme-iron enzymes,47 including the Fe/2OG hydroxylase TauD48–50 and halogenase SyrB2.21 Orientation-selective HYSCORE spectra of the {FeNO}7 form of NapI in complex with either 5,5-d2-L-Arg or 4,4-d2-L-Arg reveal deuterons 3.3 ± 0.1 and 3.6 ± 0.2 Å from the iron, respectively (Fig. 6). For both substrates, the analysis yields angles between the Fe-2H and Fe-N(O) vectors of 55 ± 5° (Figs. S10 and S11). The HYSCORE data are thus also consistent with HAT to the ferryl complex primarily from C5. However, comparison of the metrics obtained by the two structural methods suggests that oxidation of the iron cofactor from +II in the crystal structure to (formally) +III in the complex probed by HYSCORE results in a leveling of the Fe–C5 and Fe–C4 distances. The implied movement of C4 toward the cofactor (Fig. 6), should it occur also upon conversion of the reactant complex to the ferryl intermediate, would explain the capacity of C4 to serve as H• donor, especially when HAT from C5 is slowed by the presence of deuterium.
Figure 6.

Analysis of substrate positioning in NapI by HYSCORE spectroscopy on its EPR-active, S = 3/2 iron-nitrosyl {FeNO}7 complex. A, Comparison of HYSCORE spectra (left) and simulations (right), collected at 330 mT on samples of NapI•{FeNO}7•2OG•5,5-d2-L-Arg (top) and NapI•{FeNO}7•2OG•4,4-d2-L-Arg (bottom). Spectra were collected with a microwave frequency of 9.77 GHz at a temperature of 4.0 K. Experimental conditions and simulation parameters are given in the legends to Figures S10 and S11. B, Comparison of distances obtained by x-ray crystallography (black) and HYSCORE spectroscopy (red), and structural models based on these distances. The transparent C3–C5 segment is based on the HYSCORE results. Hydrogens are shown as purple spheres.
Structural Features of NapI Potentially Promoting Polar C4-H Cleavage
The position of C5 in the substrate binding pocket, directly over the H146 ligand contributed by the HXD motif, is different from the positions of H• donors in other structurally characterized enzyme•Fe(II)•2OG•substrate complexes (Fig. S12). The binding mode allows for close interaction (3.1 Å) between the adjacent N6 and the imidazole ring of H146 (Fig. 4B), offering the possibility of cation-π stabilization51 of the full positive charge that would develop on N6 in pathways B and C of Scheme 3. Given the likelihood that the guanidine sidechain would already be protonated, formation of the iminium-like intermediate in pathway B or C could also involve coupled proton transfer from one of the terminal side-chain nitrogens to the salt-bridged Asp245 or an active-site water (Fig. 5C) to obviate formation of a dication intermediate. Both routes, B and C, would be completed by removal of a proton from C4, and the structural and HYSCORE data reveal two excellent candidates for the acceptor, the carboxylate side chain of D200 (which re-sides on a lid loop that closes over the substrate) and the Fe(II)-OH center itself, to act as the general base for this step. In the latter scenario, rotation of C4 to a position closer to the metal center (as suggested above) would have to occur. An additional possibility, suggested by the unusually short 3.9-Å distance between the H•-donating carbon and the Fe(II) cofactor (which, to our knowledge, is the shortest yet observed for any Fe/2OG oxygenase reactant complex3), is that the cofactor could function as a Lewis acid to promote elimination of hydroxide from C5 in pathway B of Scheme 3.
Discovery of Desaturation of L-homoarginine (L-hArg) by the L-Arg 3-Hydroxylase, VioC
Given the demonstrated importance of N6 in the 4,5-desaturation of L-Arg catalyzed by NapI, the discovery that the structurally similar (~50% identical) L-Arg 3-hydroxylase, VioC, from the viomycin biosynthetic pathway in Streptomyces vinaceus (Sv)41,52 can desaturate a C–C unit of L-hArg lacking an α-heteroatom (Scheme 2, bottom) presented an opportunity to probe experimentally how the requirement for anchimeric assistance is obviated. The discovery was made in an initial application of the LC-MS method used for NapI to the reaction of Sv VioC, Fe(II), 2OG, L-hArg and O2. Two major peaks, with Δm/z values of +16 and –2 relative to L-hArg, develop in this reaction; they reflect hydroxylation and desaturation, respectively, of the substrate analog (Fig. S13A). With varying, limiting quantities of 2OG, the intensities of both chromatographic peaks vary proportionally to [2OG] (Fig. S14), confirming that the species are authentic enzymatic products. Derivatization of the reaction products with the fluorenylmethyloxycarbonyl (Fmoc) protecting group and use of reverse-phase HPLC affords better chromatographic resolution of the products and reveals the presence of two hydroxylated species, for a total of three primary products (Fig. 7A, black trace). Use of 18O2 as a substrate results in a further +2 shift in Δm/z (to +18) for the two hydroxylated products, showing that they form by the canonical O-atom-insertion (HAT-rebound) mechanism. Not surprisingly, use of 18O2 does not change the Δm/z (−2) for the presumptively desaturated product. (Fig. S13B).
Figure 7.

LC-MS analysis of the transformations of L-hArg deuterium isotopologs by VioC, and interpretation of the results in terms of mechanistic pathways, 2H-KIE (kH/kD) values, and impacts on partition ratios. A, All-protium L-hArg. B, C3-labeled L-Arg (red), C4-labeled L-Arg (blue), and uniformly labeled L-Arg (purple). The red values are partition ratios for (i) the initial HAT to the ferryl complex from C3 or C4 (upper in A), (ii) the fate of the C3 radical, rebound or second HAT (middle in A), and (iii) overall products (lower in A and B). The red numbers in parentheses are the standard deviations of the values from three independent trials. The 2H-KIE values that rationalize the impacts of deuterium substitution on the partition ratios are shown in gray and black (in A). The arrows and dashes (in B) illustrate the qualitative impacts of the 2H-KIEs on the partitions between HAT from C3 or C4 to the ferryl complex (gray), between the two pathways for decay of the C3 radical (black), and among the three possible products (gray and black). Details of the analytical procedure are provided in the Supporting Information. The values resulting from product detection by A260 (from the Fmoc group) are compared in Table S3.
Characterization of the 3,4-Desaturated and 3/4-Hydroxylated Products from VioC/L-hArg Reaction
To determine the structure of the Δm/z = −2 product, the VioC/L-hArg reaction was carried out on a large scale, and the product was chromatographically purified and characterized by NMR spectroscopy (as summarized in the Experimental Procedures). Spectra from a suite of correlation experiments identify it as the Fmoc derivative of trans-3,4-dehydro-L-homoarginine (Fig S15–S19). Evidence summarized below confirms that the two hydroxylated species are 3S-hydroxy-L-homoarginine (3-OH) and 4-hydroxy-L-homoarginine (4-OH), implying that all three primary products form subsequent to HAT from either C3 or C4 to the ferryl intermediate.
Mechanistic Diagnosis of Partition Among 3,4-Desaturation and 3/4-Hydroxylation Outcomes
Chemical synthesis of 2,3,3-d3-L-hArg, 4,4-d2-L-hArg, and per-d9-L-hArg isotopologs (see Supporting Information, Fig. S2) afforded the tools to dissect the VioC/L-hArg desaturation reaction in the same manner as for NapI. Stopped-flow absorption experiments analogous to those on NapI were uninformative: although the ferryl intermediate was previously shown to accumulate substantially in the reaction of VioC with its native substrate (L-Arg),44 the complex with the non-native L-hArg substrate reacts with O2 too slowly to support measureable ferryl accumulation, even with uniform deuterium substitution to minimize its decay rate. Nevertheless, LC-MS analysis of the reactions with the series of isotopologs afforded significant mechanistic insight, revealing (1) that HAT from either labeled carbon to the ferryl complex is disfavored relative to HAT from the adjacent carbon lacking deuterium, thus changing the initial partition between C3 and C4 targeting, and (2) that the partition between C4 hydroxylation and 3,4-desaturation is shifted in favor of the former outcome by the presence of deuterium at C3. The measured product distributions across the series of L-hArg isotopologs could be rationalized by the two simple assumptions embodied in Fig. 7a. First, HAT to the ferryl intermediate from C3 (right pathway) commits the reaction to hydroxylation of this site, whereas HAT from C4 initiates either C4 hydroxylation (center) or 3,4-desaturation (left). Second, 2H-KIEs of ~ 10 on the HATs to the ferryl complex (from either C3 or C4) and the removal of the second hydrogen (exclusively from C3) impact the fates of the branch-point intermediates (the ferryl and Fe(III)-OH/substrate-radical states) to dictate the product distribution.
LC-MS Analysis of Reactions with Deuterium-labeled L-hArg Substrates to Assign Products
The first step in the analysis was to associate the two Δm/z = +16 chromatographic peaks with the appropriate hydroxylated products. The distinct impacts of C3 versus C4 deuterium substitution on the yields and Δm/z values of the two hydroxylation products enabled this assignment. The peak eluting at ~ 6 min was diminished and shifted to Δm/z = +15 (−2 for loss of 2H and +17 for gain of −OH) in the reaction with 2,3,3-d3-L-hArg, showing that it is associated with the 3-hydroxylated product (3-OH). Conversely, the peak eluting at ~ 5 min was diminished and shifted to Δm/z = +15 in the reaction with 4,4-d2-L-hArg, thus associating it with the 4-hydroxylated product (4-OH).
Effects of Deuterium at C3 and/or C4 on Partition Ratios: Evidence for Sequential HAT Steps
This assignment of the chromatographic peaks enabled determination of the relative yields of the three major products with each of the four L-hArg isotopologs (specifically labeled at either site, globally labeled, and unlabeled). Two independent methods of detection, mass spectrometry and UV absorption (at 260-nm, from the amine-appended Fmoc), gave mutually consistent results (Table S3). The anticipated redirecting effect of the large 2H-KIE on the initial HAT to the ferryl was confirmed by comparison of the chromatograms for the reactions of the unlabeled, 4,4-d2, and 2,3,3-d3-L-hArg isotopologs (Fig. 7). The partition ratio for HAT to the ferryl from carbons 3 and 4 (C3:C4) was diminished from ~ 1 (55:45) with the unlabeled substrate (panel A) to ~ 0.1 (8:92) with 2,3,3-d3-L-hArg (panel B, left), whereas it was increased to ~ 10 (94:6) with 4,4-d2-L-hArg (panel B, center). The effect of the 2H-KIE on the second hydrogen removal could be resolved by including the results for per-d9-L-hArg, because the very similar 2H-KIEs for HAT to the ferryl from carbons 3 and 4 resulted in a C3:C4 partition ratio identical to that for the unlabeled substrate. The ~ 10-fold increase in the 4-hydroxylation:3,4-desaturation partition ratio, from ~ 0.7 (18:27) with the unlabeled substrate to ~ 7 (43:6) with per-d9-L-hArg, thus reflected exclusively the 2H-KIE on removal of the second hydrogen from C3. Importantly, the 4-hydroxylation:3,4-desaturation partition ratio of ~ 7 (82:12) for 2,3,3-d3-L-hArg was almost identical to that for per-d9-L-hArg, thus establishing a consistent effect of C3 2H substitution, regardless of the value of the initial C3:C4 partition ratio for HAT to the ferryl complex. The existence and magnitude of this effect, not observed for the anchimerically-assisted desaturation mediated by NapI, are most consistent with the oft-proposed sequential-HAT mechanism (Scheme 3, pathway A).35
Structure of VioC•Fe(II)•2OG•L-hArg Complex Rationalizing HAT to Ferryl from C3
To underpin mechanistic interpretation of the product distribution and impact of specific deuterium labeling of the substrate thereupon, we solved the structure of the VioC•Fe(II)•2OG•L-hArg complex to 1.7 Å resolution. Continuous composite and difference electron density is visible for the iron cofactor, its ligands, and L-hArg (Fig. 8A and Fig. S20A). The extended (4-guanidinobutyl) side chain is readily accommodated in the active site, and the analog forms the same hydrogen-bonding interactions with second-sphere residues as the native L-Arg substrate does in the previously published structure.44 As in the prior structure, hydrogen bonding of the substrate α-amine to iron ligand E170 and interactions of both D268 and D270 with the guanidine moiety combine to position C3 closest to the Fe(II) center (4.0 Å), explaining its preferential targeting by the ferryl complex (Fig. 9A and Fig. S21A).
Figure 8.

X-ray crystal structures of VioC complexes revealing changes in the conformation of the substrate/product side chain through the reaction cycle. A, Reactant complex with Fe(II) (orange), 2OG (yellow), and L-hArg (pink). B, Complex with vanadium [cyan; (hydr)oxo shown in red], succinate (purple), and L-hArg (pink). C, Complex after exposure of crystals of the reactant complex to O2 showing electron density consistent with formation of (3S)-OH-L-hArg (green) (color coding of other ligands as in A). In all structures, a 2Fo-Fc electron density map for selected active site components is shown in black mesh and contoured to 1.5 σ.
Figure 9.
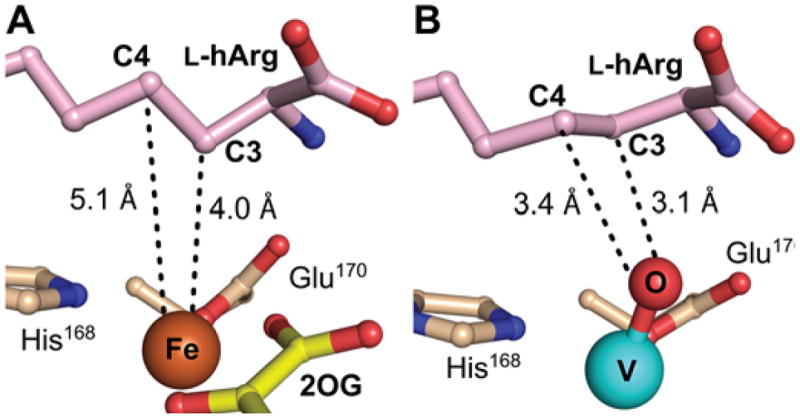
Views of the active site models for the VioC reactant and vanadium complexes showing substrate side chain conformational changes potentially associated with ferryl formation. A, Reactant complex (from Figure 8A), in which C3 is proximal (4.0 Å) to the metal center but C4 is too far removed (5.1 Å) and poorly oriented for HAT to the cofactor. B, Vanadium complex showing C3 and C4 comparably well poised (3.1 Å and 3.4 Å, respectively) for HAT, consistent with the ability of both carbons to be targeted by the ferryl intermediate.
Structure of Product Complex Revealing C3 Hydroxylation and its Stereochemistry
Exposure of the crystals of the VioC•Fe(II)•2OG•L-hArg complex to O2 and subsequent freezing allowed for solution of the structure of a product complex at 1.95-Å resolution (Fig. 8C). The composite and difference electron density clearly reveal conversion of L-hArg to the C3-hydroxylated product (3-OH) with S stereochemistry (Fig. S20C). The apparent accumulation of only the 3-OH product in crystallo could be a consequence of restraints on the active site that disfavor the 4-hydroxylation and 3,4-desaturation pathways (vide infra). Alternatively, the 3-OH product could selectively accumulate, owing to its slow dissociation, tight binding, or both. As in several previously reported structures of product complexes of Fe/2OG hydroxylases,44,53 the newly installed oxygen coordinates the Fe(II) cofactor, an interaction that could contribute to its predominance in the active site.
Structural Analysis of a Ferryl-Mimicking Vanadium Complex Rationalizing Ambiguous C3/C4 Targeting and Desaturation by Sequential HATs
Although the structure of the VioC•Fe(II)•2OG•L-hArg reactant complex is consistent with HAT from C3 to either the ferryl complex (to initiate 3-hydroxylation) or the Fe(III)–OH intermediate (to complete 3,4-desaturation), the position of C4 seems inconsistent with HAT from this carbon to the ferryl complex (to initiate either 4-hydroxylation or 3,4-desaturation). C4 is 5.1 Å from the iron with its hydrogens directed away from the Fe(II) center, not suitably poised for abstraction by either of the oxidized intermediates. The structure thus implies that the conformation of the substrate side-chain must change significantly during the reaction to permit C4 to donate H•, as deduced above for the NapI/L-Arg case. To obtain evidence for such substrate dynamics, we determined the structure of VioC in complex with a mimic of the ferryl center. Recent studies have shown that the stable V(IV)-oxo (vanadyl) dication can bind in the active sites of Fe/2OG oxygenases, including VioC.44,50 Specific relocations of activesite residues in the VioC vanadyl complex, matching V–ligand and Fe–ligand distances determined by extended X-ray absorption fine structure (EXAFS) spectroscopy, accuracy of ferryl Mössbauer parameters calculated from the geometry of the vanadyl complex, and a marked stabilization of the otherwise dynamic interaction between the halogenase, SyrB2, and its carrier-protein substrate, SyrB1, in both its ferryl and vanadyl forms provided evidence for faithful “metallomimicry” of the reactive states. A 1.70-Å-resolution structure obtained by x-ray-diffraction analysis of crystals of the VioC•VO•succinate•L-hArg complex (Fig. 8B) provides even stronger evidence. Although the refined V–O distance of 1.86 Å is consistent with the previously reported photo-reduction of the vanadyl ion in the x-ray beam, details of the structure suggest that this process does not abrogate the ferryl-mimicking features of the original (unreduced) complex.44 The observed interactions of the protein with the substrate are nearly identical to those in the reactant complex, but, importantly, C4 is much closer to the metal center (Fig. 9B). Subtle changes in the conformation of the L-hArg sidechain relative to that in the reactant complex bring carbons 3 and 4 into nearly equal proximity to the vanadyl oxo (3.1 versus 3.4 Å, respectively). In this configuration, the two carbons would be similarly well-poised to serve as HAT donors, thus rationalizing the observed distribution of products.
HYSCORE analysis of the vanadyl complex itself (not photo-reduced) provides support for the conclusions from the x-ray crystal structure. Hyperfine coupling of the vanadyl center to deuterium is observed in the complex with either 3,3-d2-L-hArg or 4,4-d2-L-hArg bound. Analysis of the orientation-selective HYSCORE spectra gives distances of 3.4 ± 0.2 Å and 3.8 ± 0.4 Å between the vanadium ion and the C3 and C4 deuterons, respectively (Figs. S22 and S23). The magnitude and rank order of these V–2H distances, and the difference between them, match well to those indicated by the crystallographic structural model (3.6 Å for the pro-S C3-2H and 4.4 Å for the pro-R C4-2H). These comparisons imply that, other than the low-barrier elongation of the V–O and other metal-ligand bonds, the cryogenic photo-reduction of the vanadyl unit in the x-ray beam does not grossly alter the chemically relevant dispositions of atoms in the active site, in agreement with the conclusions from the earlier study.44 The implications are: (i) that crucial dispositions of substrate atoms may change in these enzymes in progression through the catalytic cycle, (ii) that reactant complexes may, therefore, fail to recapitulate reactivity-determining features of the ferryl complexes, and (iii) that the vanadyl complex may provide a better basis for rationalizing and perhaps even predicting reaction outcomes.
CONCLUSIONS
The selective, natural desaturation of L-Arg by NapI and the relatively unselective, non-native desaturation of L-hArg by VioC appear to proceed by different mechanisms, as judged by the divergent impact of deuterium substitution of the second hydrogen donor on the extent of competing oxygen rebound to the substrate radical formed by the first HAT. Facilitation of polar cleavage of the second C–H bond by the α-heteroatom, present in the NapI reaction but absent in the VioC/L-hArg desaturation, is most likely responsible and can rationalize the presence of α-heteroatoms in the majority of known, natural Fe/2OG-desaturase substrates.1 Given the clear evidence for divergent mechanisms, continuing study of desaturations of substrates lacking a heteroatom to define the limitations of the Fe/2OG mechanistic manifold seem warranted. For example, the previously reported non-native 3–4-desaturation of deoxyproclavaminate by clavaminate synthase is reportedly more selective (90% desaturation versus 10% hydroxylation) than the VioC/L-hArg desaturation.35 Similarly, reactions tentatively attributed to PrhA and BcmF in the paraherquonin and bicyclomycin biosynthetic pathways, respectively, would be examples of desaturations without the possibility of heteroatom assistance;38,39 these native desaturations are likely to be relatively efficient. Given that Fe/2OG oxygenases appear to present an outstanding set of targets for development of new biocatalytic activities and that olefin insertion between unactivated carbons would be an immensely useful synthetic capability, defining the inherent limitations of the mechanistic motif for this type of reactivity is a worthy goal.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM119707 to A.K.B; GM113106 and GM118812 to J.M.B. and C.K.) and the National Science Foundation Graduate Research Fellowship Program (Grant No. DGE1255832 to R.J.M.). We thank Professor Scott A. Showalter for assistance with collection of NMR spectra. We gratefully acknowledge the resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). The Eiger 16M detector was funded by an NIH-Office of Research Infrastructure Programs, High-End Instrumentation Grant (1S10OD012289-01A1).
Footnotes
ASSOCIATED CONTENT
Experimental section, Figures S1–S23, and Tables S1–S3. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Bollinger JM, Jr, Chang W-c, Matthews ML, Martinie RJ, Boal AK, Krebs C. In: 2-oxoglutarate-dependent oxygenases. Hausinger RP, Schofield CJ, editors. The Royal Society of Chemistry; London: 2015. p. 95. [Google Scholar]
- 2.Martinez S, Hausinger RP. J Biol Chem. 2015;290:20702. doi: 10.1074/jbc.R115.648691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aik W, McDonough MA, Thalhammer A, Chowdhury R, Schofield CJ. Curr Opin Struct Biol. 2012;22:691. doi: 10.1016/j.sbi.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Price JC, Barr EW, Glass TE, Krebs C, Bollinger JM., Jr J Am Chem Soc. 2003;125:13008. doi: 10.1021/ja037400h. [DOI] [PubMed] [Google Scholar]
- 5.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 6.Price JC, Barr EW, Hoffart LM, Krebs C, Bollinger JM., Jr Biochemistry. 2005;44:8138. doi: 10.1021/bi050227c. [DOI] [PubMed] [Google Scholar]
- 7.Riggs-Gelasco PJ, Price JC, Guyer RB, Brehm JH, Barr EW, Bollinger JM, Jr, Krebs C. J Am Chem Soc. 2004;126:8108. doi: 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- 8.Hoffart LM, Barr EW, Guyer RB, Bollinger JM, Jr, Krebs C. Proc Natl Acad Sci U S A. 2006;103:14738. doi: 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galonic DP, Barr EW, Walsh CT, Bollinger JM, Jr, Krebs C. Nat Chem Biol. 2007;3:113. doi: 10.1038/nchembio856. [DOI] [PubMed] [Google Scholar]
- 10.Fujimori DG, Barr EW, Matthews ML, Koch GM, Yonce JR, Walsh CT, Bollinger JM, Jr, Krebs C, Riggs-Gelasco PJ. J Am Chem Soc. 2007;129:13408. doi: 10.1021/ja076454e. [DOI] [PubMed] [Google Scholar]
- 11.Matthews ML, Krest CM, Barr EW, Vaillancourt FH, Walsh CT, Green MT, Krebs C, Bollinger JM., Jr Biochemistry. 2009;48:4331. doi: 10.1021/bi900109z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang W-c, Guo Y, Wang C, Butch SE, Rosenzweig AC, Boal AK, Krebs C, Bollinger JM., Jr Science. 2014;343:1140. doi: 10.1126/science.1248000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groves JT. J Chem Ed. 1985;62:928. [Google Scholar]
- 14.Ye S, Riplinger C, Hansen A, Krebs C, Bollinger JM, Jr, Neese F. Chemistry. 2012;18:6555. doi: 10.1002/chem.201102829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matthews ML, Chang WC, Layne AP, Miles LA, Krebs C, Bollinger JM., Jr Nat Chem Biol. 2014;10:209. doi: 10.1038/nchembio.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma G, Zhu W, Su H, Cheng N, Liu Y. ACS Catal. 2015;5:5556. [Google Scholar]
- 17.Topf M, Sandala GM, Smith DM, Schofield CJ, Easton CJ, Radom L. J Am Chem Soc. 2004;126:9932. doi: 10.1021/ja047899v. [DOI] [PubMed] [Google Scholar]
- 18.Borowski T, Broclawik E, Schofield CJ, Siegbahn PE. J Comput Chem. 2006;27:740. doi: 10.1002/jcc.20384. [DOI] [PubMed] [Google Scholar]
- 19.Blasiak LC, Vaillancourt FH, Walsh CT, Drennan CL. Nature. 2006;440:368. doi: 10.1038/nature04544. [DOI] [PubMed] [Google Scholar]
- 20.Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, Bollinger JM., Jr Proc Natl Acad Sci U S A. 2009;106:17723. doi: 10.1073/pnas.0909649106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinie RJ, Livada J, Chang WC, Green MT, Krebs C, Bollinger JM, Jr, Silakov A. J Am Chem Soc. 2015;137:6912. doi: 10.1021/jacs.5b03370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong SD, Srnec M, Matthews ML, Liu LV, Kwak Y, Park K, Bell CB, 3rd, Alp EE, Zhao J, Yoda Y, Kitao S, Seto M, Krebs C, Bollinger JM, Jr, Solomon EI. Nature. 2013;499:320. doi: 10.1038/nature12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinie RJ, Livada J, Chang W-c, Green MT, Krebs C, Bollinger JM, Jr, Silakov A. J Am Chem Soc. 2015;137:6912. doi: 10.1021/jacs.5b03370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitchell AJ, Zhu Q, Maggiolo AO, Ananth NR, Hillwig ML, Liu X, Boal AK. Nat Chem Biol. 2016;12:636. doi: 10.1038/nchembio.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srnec M, Solomon EI. J Am Chem Soc. 2017;139:2396. doi: 10.1021/jacs.6b11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Srnec M, Wong SD, Matthews ML, Krebs C, Bollinger JM, Jr, Solomon EI. J Am Chem Soc. 2016;138:5110. doi: 10.1021/jacs.6b01151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boal AK, Bollinger JM, Jr, Chang WC. Proc Natl Acad Sci U S A. 2015;112:11989. doi: 10.1073/pnas.1514689112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 29.Shanklin J, Guy JE, Mishra G, Lindqvist Y. J Biol Chem. 2009;284:18559. doi: 10.1074/jbc.R900009200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rittle J, Green MT. Science. 2010;330:933. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 31.Green MT, Dawson JH, Gray HB. Science. 2004;304:1653. doi: 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- 32.Lee SK, Nesheim JC, Lipscomb JD. J Biol Chem. 1993;268:21569. [PubMed] [Google Scholar]
- 33.Shu L, Nesheim JC, Kauffmann K, Munck E, Lipscomb JD, Que L., Jr Science. 1997;275:515. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- 34.Bollinger JM, Jr, Edmondson DE, Huynh BH, Filley J, Norton JR, Stubbe J. Science. 1991;253:292. doi: 10.1126/science.1650033. [DOI] [PubMed] [Google Scholar]
- 35.Zhou J, Kelly WL, Bachmann BO, Gunsior M, Townsend CA, Solomon EI. J Am Chem Soc. 2001;123:7388. doi: 10.1021/ja004025+. [DOI] [PubMed] [Google Scholar]
- 36.Ishikawa N, Tanaka H, Koyama F, Noguchi H, Wang CC, Hotta K, Watanabe K. Angew Chem Int Ed Engl. 2014;53:12880. doi: 10.1002/anie.201407920. [DOI] [PubMed] [Google Scholar]
- 37.Brauer A, Beck P, Hintermann L, Groll M. Angew Chem Int Ed. 2016;55:422. doi: 10.1002/anie.201507835. [DOI] [PubMed] [Google Scholar]
- 38.Matsuda Y, Iwabuchi T, Fujimoto T, Awakawa T, Nakashima Y, Mori T, Zhang H, Hayashi F, Abe I. J Am Chem Soc. 2016;138:12671. doi: 10.1021/jacs.6b08424. [DOI] [PubMed] [Google Scholar]
- 39.Meng S, Han W, Zhao J, Jian XH, Pan HX, Tang GL. Angew Chem Int Ed Engl. 2018;57:719. doi: 10.1002/anie.201710529. [DOI] [PubMed] [Google Scholar]
- 40.Hiratsuka T, Koketsu K, Minami A, Kaneko S, Yamazaki C, Watanabe K, Oguri H, Oikawa H. Chem Biol. 2013;20:1523. doi: 10.1016/j.chembiol.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 41.Yin X, Zabriskie TM. Chembiochem. 2004;5:1274. doi: 10.1002/cbic.200400082. [DOI] [PubMed] [Google Scholar]
- 42.Helmetag V, Samel SA, Thomas MG, Marahiel MA, Essen LO. FEBS J. 2009;276:3669. doi: 10.1111/j.1742-4658.2009.07085.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang CY, Lyu SY, Liu YC, Hsu NS, Wu CC, Tang CF, Lin KH, Ho JY, Wu CJ, Tsai MD, Li TL. Angew Chem Int Ed Engl. 2014;53:1943. doi: 10.1002/anie.201307989. [DOI] [PubMed] [Google Scholar]
- 44.Mitchell AJ, Dunham NP, Martinie RJ, Bergman JA, Pollock CJ, Hu K, Allen BD, Chang WC, Silakov A, Bollinger JM, Jr, Krebs C, Boal AK. J Am Chem Soc. 2017;139:13830. doi: 10.1021/jacs.7b07374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujimori DG, Krebs C, Walsh CT, Bollinger JM., Jr Acc Chem Res. 2007;40:484. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bollinger JM, Jr, Krebs C. J Inorg Biochem. 2006;100:586. doi: 10.1016/j.jinorgbio.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 47.Brown CD, Neidig ML, Neibergall MB, Lipscomb JD, Solomon EI. J Am Chem Soc. 2007;129:7427. doi: 10.1021/ja071364v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muthukumaran RB, Grzyska PK, Hausinger RP, McCracken J. Biochemistry. 2007;46:5951. doi: 10.1021/bi700562t. [DOI] [PubMed] [Google Scholar]
- 49.Casey TM, Grzyska PK, Hausinger RP, McCracken J. J Phys Chem B. 2013;117:10384. doi: 10.1021/jp404743d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martinie RJ, Pollock CJ, Matthews ML, Bollinger JM, Krebs C, Silakov A. Inorg Chem. 2017;56:13382. doi: 10.1021/acs.inorgchem.7b02113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gallivan JP, Dougherty DA. Proc Natl Acad Sci U S A. 1999;96:9459. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ju J, Ozanick SG, Shen B, Thomas MG. Chembiochem. 2004;5:1281. doi: 10.1002/cbic.200400136. [DOI] [PubMed] [Google Scholar]
- 53.Strieker M, Kopp F, Mahlert C, Essen LO, Marahiel MA. ACS Chem Biol. 2007;2:187. doi: 10.1021/cb700012y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.