

Abstract
Objective(s):
Aberrant expression of CCL5 has been found in several kinds of inflammatory diseases, and the roles of CCL5 in these diseases have also been reported. However, the role of CCL5 in infantile pneumonia is still unclear. Thus, the function and acting mechanism of CCL5 in the in vitro model of infantile pneumonia were researched in this study.
Materials and Methods:
Human fetal lung fibroblast WI-38 cells were subjected with lipopolysaccharide (LPS) to mimic an in vitro model of pneumonia. CCL5 was silenced by transfection with CCL5-targeted siRNA, and then cell viability, apoptosis, and the expressions of apoptosis-associated factors were respectively assessed by CCK-8 assay, flow cytometry and Western blot. Besides, expressions of CCL5 and pro-inflammatory factors were analyzed by qRT-PCR and Western blot. The secretions of pro-inflammatory factors were measured by ELISA. Finally, the expressions of main factors in JNK and NF-κB pathways were detected.
Results:
LPS treatment suppressed cell viability, promoted cell apoptosis, and enhanced the secretion of IL-6, MCP-1, and TNF-α. Overexpression of CCL5 was found in LPS-treated cells. CCL5 silence protected WI-38 cells from LPS-induced inflammatory damage, with increasing cell viability, inhibiting cell apoptosis, and reducing the production of pro-inflammatory cytokines. Besides, CCL5 silence inhibited LPS-induced activations of JNK and NF-κB pathways.
Conclusion:
Down-regulation of CCL5 could protect WI-38 cells from LPS-induced inflammatory damage via inactivating JNK and NF-κB pathways.
Keywords: CCL5, Infantile pneumonia, JNK pathway, Lipopolysaccharide (LPS), NF-κB pathway, Pro-inflammatory cytokines
Introduction
Pneumonia is an important cause of mortality and morbidity in children aged < 5 years (1, 2). Among children, infants are especially susceptible to this disease, probably because their immunity and respiratory anatomies are still under-developed (2). The main symptoms of infantile pneumonia are cough, fever, shortness of breath, and breathing difficulty. The severe patients are easily accompanied with high fever, possibly leading to febrile convulsions, even respiratory failure and heart failure (3). The infantile pneumonia is usually induced by viral and bacterial co-infection. Accordingly, antiviral drugs and antibiotics are main therapies. However, due to some common problems, such as serious adverse reactions of the chemical drugs and overuse of antibiotics, the efficacy of the conventional treatment with western medicines is decreased to some extent (4). Therefore, seeking more active and effective therapeutic strategies for infantile pneumonia is very important.
Accumulating evidences have indicated the important role of chemokines and their respective receptors in the regulation of local inflammation (5). Regulated upon activation normal T cell expressed and secreted (RANTES), also known as CCL5 and originally recognized as a product of activated T cells, now is widely established as an inflammatory chemokine and can mediate chemotactic activity in T cells, monocytes, natural killer cells, etc. (6, 7). CCL5 belongs to the CC chemokine family, and is recognized by CCR1, CCR3 and CCR5 receptors (8). CCL5 is associated with some chronic inflammatory diseases, such as rheumatoid arthritis and inflammatory bowel disease (9, 10). CCL5 plays an important role in some types of cancers and inflammatory diseases. It can modulate cell migration and invasion in some cancer cells (7, 11, 12). At present, an association between CCL5 expression and cancers has been widely reported. However, the specific effect of CCL5 on inflammatory diseases is rarely reported.
The present work aimed to delineate the functional effects of CCL5 on lipopolysaccharide (LPS)-induced inflammatory damage in human WI-38 fibroblasts, and to reveal the potential of CCL5 targeting on infantile pneumonia treatment (13). We first found that CCL5 was highly expressed in response to LPS stimulation. Thus, we speculated CCL5 was involved in the development of infantile pneumonia. The expression of CCL5 in WI-38 cells was suppressed by transfection with CCL5-targeted siRNA. The effect of CCL5 knockdown on LPS-induced inflammatory damage was analyzed. In order to further reveal the underlying mechanisms, the effects of CCL5 knockdown on JNK and NF-κB pathways were studied. Our findings will give a new direction for treating infantile pneumonia.
Materials and Methods
Cell culture and LPS treatment
Human WI-38 fibroblasts, obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA), were incubated in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Carlsbad, CA, USA), supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco), 100 μg/ml streptomycin, and 100 U/ml penicillin (Sigma, St Louis, Missouri, USA) in a humidified atmosphere of 5% CO2 at 37 °C. Once the cells achieved a sub-confluence of 80-90%, they were dissociated through treatment with 0.2% trypsin/0.02% ethylenediamine tetraacetic acid for 5 min. Subsequently, cells were collected by centrifugation at 300 x g for 2 min at 25°C. The cells were plated in 24-well plates at a density of 2×104 cells per well and maintained for 48 hr to reach optimal confluence. LPS used in this study was purchased from Sigma, and the following LPS concentrations were used for the experiment 5, 10, and 20 μg/ml (14, 15). WI-38 cells may respond to LPS when under serum-free conditions. Thus, the serum-free culture medium was applied for LPS-treated cells. The experiments were conducted 24 hr later in the WI-38 cells.
siRNA preparation and transfection
The siRNA of CCL5 and the corresponding negative control were designed and synthesized by GenePharma (Shanghai, China). They were referred as to si-CCL5 and si-NC. The cell transfection was performed using Lipofectamine 3000 (Invitrogen Life Technologies, Carlsbad, CA, USA) following the manufacturer’s protocol. After 48 hr of transfection, cells were collected for forthcoming analyses.
Cell viability assay
Cell viability was assessed by a Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Gaithersburg, MD). Briefly, cells were seeded in 96-well plate with 5000 cells/well. After stimulation, 20 μl CCK-8 solution was added to the wells, and the cultures were then incubated for 1 hr at 37 °C in humidified 95% air and 5% CO2. The absorbance was measured at 450 nm using a Microplate Reader (Bio-Rad, Hercules, CA).
Apoptosis assay
Apoptosis assay was performed using Annexin V-FITC/PI apoptosis detection kit (Beijing Biosea Biotechnology, Beijing, China). The WI-38 cells were seeded in 6-well plate with 100,000 cells/well. Treated cells were washed twice and resuspended in 200 μl binding buffer containing 10 μl Annexin V-FITC. After incubation at room temperature in the dark for 30 min, 5 μl propidium iodide (PI) and 300 μl phosphate buffered saline (PBS) were added, and the samples were analyzed with the cytometer (Beckman Coulter, USA).
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated using Trizol reagent (Life Technologies Corporation, Carlsbad, CA, USA). First-strand cDNA was synthesized from total mRNA using Multiscribe Reverse Transcriptase kit (Applied Biosystems, CA, USA) and qRT-PCR was performed using SYBR® Premix Ex TaqTM II (TaKaRa Dalian, China). The mRNA level of each gene was normalized to GAPDH with 2-ΔΔCt method using Bio-Rad CFX Manager V1.1.308.1111 software (16).
Enzyme-linked immunosorbent assay (ELISA)
Culture supernatant was collected from 24-well plates and the productions of pro-inflammatory factors were determined by specific ELISA kits as instructed and normalized to cell protein concentrations. Human ELISA kits for IL-6 (ab46042), MCP-1 (ab100586), and TNF-α (ab181421) were purchased from Abcam (Cambridge, MA).
Western blot
The cellular lysates were prepared as described previously (17). Proteins were resolved on SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The blots were blocked with 4% bovine serum albumin for 1 hr at room temperature and then probed with primary antibodies against IL-6 (ab6672), MCP-1 (ab151538), TNF-α (ab1793), CCL5 (ab9679), Bcl-2 (ab32124), Bax (ab32503), pro caspase-3 (ab4051), cleaved caspase-3 (ab32042), pro caspase-9 (ab32539), cleaved caspase-9 (ab32539), JNK (ab179461), p-JNK (ab124956), c-Jun (ab32137), p-c-Jun (ab32385), p65 (ab16502), p-p65 (ab86299), IkBα (ab7217), p-IkBα (ab133462), and GADPH (ab125247, 1:1000 dilution; Abcam, Cambridge, MA) for 1 hr at 4°C. After rinsing, the blots were subsequently incubated with appropriate peroxidase-conjugated secondary antibody (1:1,000 dilution, Abcam) for 1 hr at room temperature. The blots were visualized by enhanced chemiluminescence method using Kodak XOMAT LS film (Eastman Kodak, Rochester, NY, USA).
Statistical analysis
All experiments were repeated three times. The data were expressed as the mean±SD, and statistical evaluation was performed using one-way analysis of variance (ANOVA). Statistical significance between groups was calculated using Graphpad 6.0 statistical software (GraphPad Software Inc., San Diego, CA). Values of P<0.05 were considered statistically significant and marked with * or #.
Results
LPS induced inflammatory damage in WI-38 cells
To investigate the effect of LPS on the viability of WI-38 cells, the cells were treated with various doses (5, 10, and 20 μg/ml) of LPS for 24 hr. As shown in Figure 1A, cell survival rate was decreased in a dose dependent manner, with a viability decrease of about 60% by 10 μg/ml LPS treatment (P<0.05) and about 20% by 20 μg/ml LPS treatment (P<0.01) relative to control group (0 μg/ml). Considering that 10 μg/ml LPS resulted in a significant decrease in cell viability, 10 μg/ml was selected as a LPS-stimulating condition for use in the following experiments. Subsequently, the effect of 10 μg/ml LPS on the secretion of pro-inflammatory cytokines was analyzed. Compared with control group, the mRNA levels of IL-6, MCP-1 and TNF-α were all significantly increased by LPS according to qRT-PCR analysis (all P<0.001, Figure 1B). The levels of these pro-inflammatory cytokines in cell culture supernatant were quantitatively measured by ELISA, and they were dramatically increased by LPS stimulation (all P<0.001, Figure 1C). Similar results were displayed in Western blot assay and expressions of all IL-6, MCP-1 and TNF-α were significantly decreased (all P<0.001, Figure 1D). Furthermore, LPS with 10 μg/ml significantly promoted the apoptosis of WI-38 cells (P<0.01, Figure 1E). LPS down-regulated anti-apoptotic protein Bcl-2, up-regulated pro-apoptotic protein Bax, activated caspase-3, and caspase-9 (P<0.01, P<0.001, P<0.001, and P<0.001, Figure 1F). These data indicated that LPS induced inflammatory damage in WI-38 cells.
Figure 1.
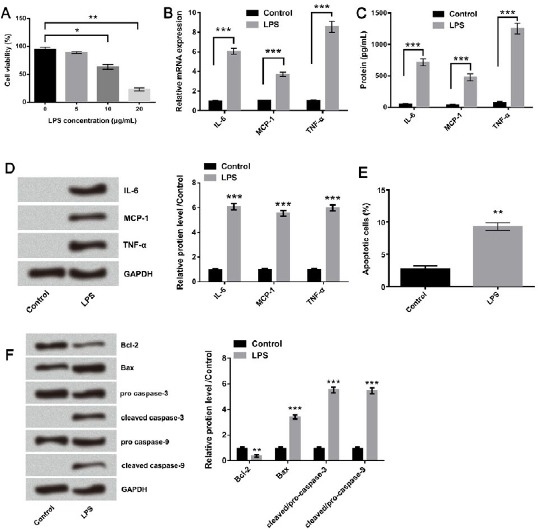
Lipopolysaccharide (LPS) induced inflammatory damage in WI-38 cells. (A) The effect of LPS with different concentrations (5, 10, and 20 μg/ml) on cell viability was analyzed by CCK-8 assay. The expression levels of IL-6, MCP-1, and TNF-α were analyzed by (B) qRT-PCR, (C) ELISA, and (D) Western blot after treatment with 10 μg/ml LPS. The effect of LPS with 10 μg/ml on cell apoptosis was evaluated by (E) Annexin V-FITC/PI double staining method and (F) Western blot. Means ± SD were shown. * P<0.05, ** P<0.01 *** P<0.001 (ANOVA)
CCL5 expression was up-regulated in LPS-treated WI-38 cells
The mRNA expression levels of CCL5 in WI-38 cells treated with 10 μg/ml and 20 μg/ml LPS were both significantly higher than un-treated group (P<0.01, Figure 2A). Besides, the protein expressions of CCL5 were also enhanced at different degrees after LPS treatments at various levels (Figure 2B). CCL5 overexpression was found in WI-38 cells with LPS treatment, indicating an association between CCL5 expression and LPS-induced inflammatory response.
Figure 2.
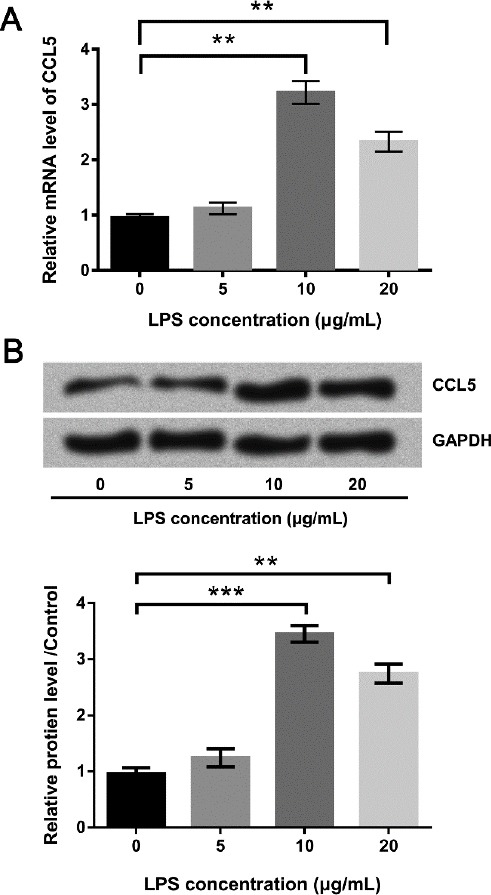
CCL5 was up-regulated in lipopolysaccharide (LPS)-treated WI-38 cells. (A) The mRNA and (B) protein expression levels of CCL5 were respectively detected by qRT-PCR and Western blot when cells were treated with various doses of LPS. Means±SD were shown. ** P<0.01 (ANOVA)
Effect of CCL5 silence on LPS-induced cell growth impairment
The si-CCL5 was transfected into WI-38 cells to explore whether CCL5 was implicated in the cell growth-inhibiting effect of LPS. Firstly, the transfection efficiency was verified by detecting the expression of CCL5. Results from qRT-PCR and Western blot (Figure 3A and B) showed that the level of CCL5 was decreased after si-CCL5 transfection when compared with si-NC group (both P<0.01). Results from CCK-8 assay (Figure 3C) showed that cell viability was much higher in CCL5-suppressing cells (P<0.05) compared to si-NC group. Flowcytometry detection results given in Figure 3D displayed that the cell apoptotic rate was decreased by CCL5 silence (P<0.05) compared to si-NC. Western blot results in Figure 3E showed that up-regulation of Bcl-2, down-regulation of Bax, cleaved caspase-3, and cleaved capsase-9 were observed in LPS+si-CCL5 group (all P <0.001). These data revealed that CCL5-knockdown attenuated the inhibitory impact of LPS on WI-38 cell growth.
Figure 3.
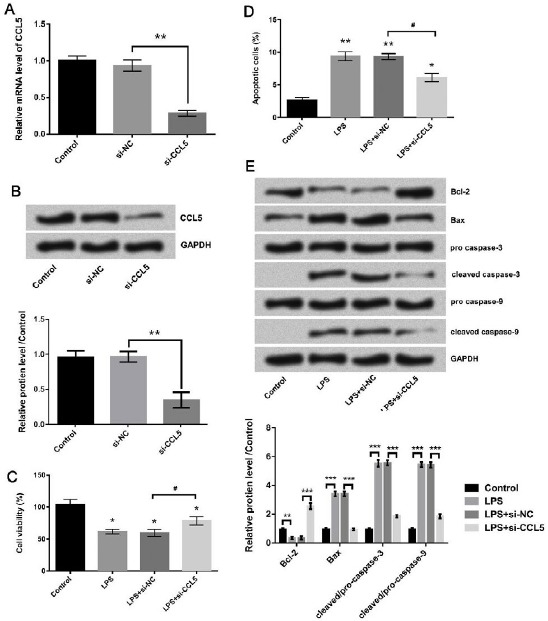
The viability was enhanced and the apoptosis was inhibited in lipopolysaccharide (LPS)-treated cells after CCL5 knockdown. Transfection efficiency was verified by (A) qRT-PCR and (B) Western blot. (C) The effect of CCL5 knockdown on viability of LPS-treated cells was evaluated by CCK-8 assay. The effect of CCL5 knockdown on apoptosis of LPS-treated cells was evaluated by (D) Annexin V-FITC/PI double staining method and (E) Western blot. #/* P<0.05, *** P<0.001 (ANOVA)
Effect of CCL5 silence on LPS-induced pro-inflammatory cytokine productions in WI-38 cells
To investigate the effect of CCL5 on the LPS-induced pro-inflammatory cytokine productions, the si-CCL5 was transfected into LPS-treated WI-38 cells, and qRT-PCR, ELISA, and Western blot were all performed to observe the changes of pro-inflammatory factor expressions (Figure 4). As shown in Figure 4A, in LPS-treated WI-38 cells, CCL5 knockdown significantly decreased the mRNA expression levels of IL-6 (P<0.01), MCP-1 (P<0.01), MCP-1 (P<0.01), and TNF-α (P<0.001). According to results of ELISA, CCL5 silence significantly inhibited the productions of IL-6 and TNF-α (both P<0.001), but it had no remarkable influence on MCP-1 (Figure 4B). The Western blot analysis displayed that all of pro-inflammatory cytokines including IL-6, MCP-1, and TNF-α were remarkably down-regulated after CCL5 silencing (all P<0.01, Figure 4C). These data further verified that CCL5 knockdown protected cells from LPS-induced inflammatory damage.
Figure 4.
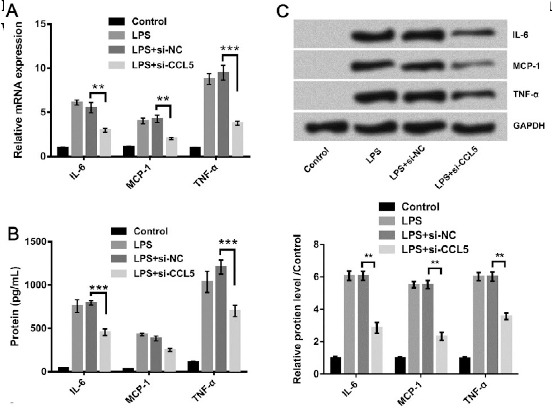
Pro-inflammatory factors were down-regulated by CCL5 knockdown in lipopolysaccharide (LPS)-treated cells. The expression levels of IL-6, MCP-1, and TNF-α were detected by (A) qRT-PCR, (B) ELISA, and (C) Western blot. ** P<0.01, *** P<0.001 (ANOVA)
Effect of CCL5 silence on LPS-stimulated JNK and NF-κB pathways
JNK and NF-κB pathways can be activated in pneumonia, and activations of these pathways are associated with the up-regulations of some pro-inflammatory cytokines (18, 19). Thus, we detected the expression of main factors in both pathways to identify whether JNK and NF-κB pathways were implicated in CCL5-mediated cytoprotection. As shown in Figure 5A, p-JNK and p-c-Jun were up-regulated after LPS treatment (both P<0.001), while they were remarkably down-regulated when CCL5 was silenced (both P <0.001). The regulation of CCL5 involved in NF-κB pathway was displayed in Figure 5B; p-p65 and p-IkBα were up-regulated in LPS-treated cells (both P<0.001), while they were remarkably down-regulated after CCL5 knockdown (both P<0.001). These data provided evidence that JNK and NF-κB pathways might be closely related to the role of CCL5 in inflammatory damage in WI-38 cells.
Figure 5.
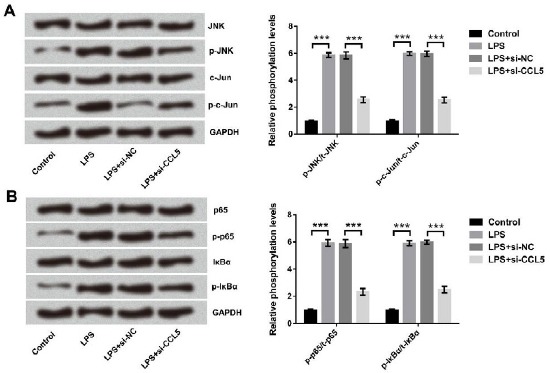
The activations of (A) JNK and (B) NF-κB pathways induced by lipopolysaccharide (LPS) were inhibited after CCL5 silence. WI-38 cells were transfected with CCL5 targeted siRNA after LPS treatment. The expressions of the proteins were detected by Western blot
Discussion
The infantile pneumonia is one of the common respiratory tract diseases in infancy. Providing new insights into therapeutic target for treating infantile pneumonia is urgent due to high mortality and morbidity of this disease. In our study, WI-38 cells were used and treated with prototypic pathogen-derived pro-inflammatory stimulant LPS. After LPS treatment, some pro-inflammatory cytokines, including IL-6, MCP-1, and TNF-α were activated. IL-6 and TNF-α are involved in the pathogenesis of mycoplasma pneumonia and are correlated with disease severity in children (20). The levels of IL-6 and TNF-α can be used as indexes for evaluating the prognosis and treatment of pneumonia (20). In addition, MCP-1 may involve in activating monocytes / macrophages and T lymphocyte and the imbalance of Thl / Th2 in the infection of mycoplasma pneumonia (21). Thus, IL-6, MCP-1, and TNF-α are reliable signals, which reflect the grades of lung inflammation.
As the results showed, LPS induced overproductions of IL-6, MCP-1, and TNF-α, inhibited the viability, and promoted apoptosis of WI-38 cells, indicating that the in vitro model of pneumonia was successfully established. Meanwhile, CCL5 was dramatically up-regulated in LPS-treated cells, and its expression was positively correlated with the concentration of LPS. In human monocytes, CCL5 transcription can be up-regulated rapidly and transiently in response to LPS (22). Castellani et al. revealed that expression and secretion of CCL5 were enhanced in granulomatous calcified tissue of the lung after LPS treatment in vivo (23). Together with these previous studies, we therefore briefly concluded that CCL5 expression might be closely related to LPS-induced inflammatory damage.
CCL5 is secreted by macrophages, epithelial cells, platelets, fibroblasts, and activated T cells (24). As a number of CC chemokine family, it is known to regulate T cell differentiation and polarize Th1 >> Th2 subtypes as well as numerous physiological functions of leukocytes including migration (25, 26). T cell differentiation and the pro-inflammatory Th1 and Th2 cytokine levels are reported to be associated with pulmonary inflammation and lung injury (27). Therefore, CCL5 may play an important role in infantile pneumonia.
As the data indicated in the present study, the growth of WI-38 cells was inhibited and the secretion of pro-inflammatory cytokines was enhanced after LPS treatment, while the cell growth impairment and the pro-inflammatory cytokine overproduction were inhibited after CCL5 knockdown. The abnormal expression of CCL5 is associated with many pathological inflammatory states, such as glomerulonephritis, arthritis, and cancer (28). Increased levels of CCL5 were found in body of Lewis rat suffered with rheumatoid arthritis; and blocking of CCL5 by specific antibodies ameliorated symptoms as much as indomethacin (29). Thus, inhibiting CCL5 might be helpful to ease inflammation.
NF-κB is an indispensable transcription factor in the LPS-TLR4 signaling pathway for regulating inflammatory processes (30, 31). The activation of MAPKs is critical in mediating some cellular responses, including cell proliferation and differentiation, transcription factor activation, and cytokine gene expression and production (32, 33). JNK is one of the important MAPK family members. Thus, inhibitors of NF-κB and JNK have been used as therapeutic drugs for inflammation-associated human diseases in clinical applications (33). Here, we showed that CCL5 knockdown potently suppressed LPS-induced activations of NF-κB and JNK pathways. Accordingly, it can be inferred that regulating CCL5 can protect cells from LPS-induced inflammatory damage via modulation of NF-κB and JNK pathways, which may provide a novel therapy for inflammatory diseases, including infantile pneumonia.
Conclusions
In summary, our study revealed that augmented expression of CCL5 might be associated with infantile pneumonia, and down-regulating CCL5 would inhibit the cell injury induced by LPS. CCL5 silence reduced the effects of LPS on cell viability, apoptosis, as well as the secretion of pro-inflammatory cytokines. Furthermore, CCL5 silence suppressed the activity of JNK and NF-κB pathways. This study will provide a better understanding of CCL5 in infantile pneumonia, and suggest its potential application in the treatment of this disease.
Conflict of interest
The authors declare that no conflict of interest exists.
References
- 1.Wardlaw T, Salama P, Johansson EW, Mason E. Pneumonia:the leading killer of children. Lancet. 2006;368:1048–1050. doi: 10.1016/S0140-6736(06)69334-3. [DOI] [PubMed] [Google Scholar]
- 2.Chen CH, Wen HJ, Chen PC, Lin SJ, Chiang TL, Hsieh IC, et al. Prenatal and postnatal risk factors for infantile pneumonia in a representative birth cohort. Epidemiol Infect. 2012;140:1277–1285. doi: 10.1017/S0950268811001890. [DOI] [PubMed] [Google Scholar]
- 3.Yao YQ, Wang ZW, Ding YX, Yu Y, Jiang WX, Liu XH, et al. Effect of Zhifei mixture combined western drugs on symptoms and signs of children with mycoplasma pneumonia. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2014;34:522–525. [PubMed] [Google Scholar]
- 4.Hu HF, Cao LJ. Treatment of pediatric pneumonia. J Applie Clin Pediatr. 2011;4:3. [Google Scholar]
- 5.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schall TJ, Jongstra J, Dyer BJ, Jorgensen J, Clayberger C, Davis MM, et al. A human T cell-specific molecule is a member of a new gene family. J Immunol. 1988;141:1018–1025. [PubMed] [Google Scholar]
- 7.Huang CY, Fong YC, Lee CY, Chen MY, Tsai HC, Hsu HC, et al. CCL5 increases lung cancer migration via PI3K, Akt and NF-kappaB pathways. Biochem Pharmacol. 2009;77:794–803. doi: 10.1016/j.bcp.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 8.Singh S, Sadanandam A, Singh RK. Chemokines in tumor angiogenesis and metastasis. Cancer Metastasis Rev. 2007;26:453–467. doi: 10.1007/s10555-007-9068-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mccormack G, Moriarty D, O'Donoghue DP, Mccormick PA, Sheahan K, Baird AW. Tissue cytokine and chemokine expression in inflammatory bowel disease. Inflamm Res. 2001;50:491–495. doi: 10.1007/PL00000223. [DOI] [PubMed] [Google Scholar]
- 10.Ben-Baruch A. Inflammation-associated immune suppression in cancer:the roles played by cytokines, chemokines and additional mediators. Semin Cancer Biol. 2006;16:38–52. doi: 10.1016/j.semcancer.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 11.Pinilla S, Alt E, Abdul Khalek FJ, Jotzu C, Muehlberg F, Beckmann C, et al. Tissue resident stem cells produce CCL5 under the influence of cancer cells and thereby promote breast cancer cell invasion. Cancer Lett. 2009;284:80–85. doi: 10.1016/j.canlet.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 12.Chuang JY, Yang WH, Chen HT, Huang CY, Tan TW, Lin YT, et al. CCL5/CCR5 axis promotes the motility of human oral cancer cells. J Cell Physiol. 2009;220:418–426. doi: 10.1002/jcp.21783. [DOI] [PubMed] [Google Scholar]
- 13.Smeding L, Kuiper JW, Plotz FB, Kneyber MC, Groeneveld AJ. Aggravation of myocardial dysfunction by injurious mechanical ventilation in LPS-induced pneumonia in rats. Respir Res. 2013;14:92. doi: 10.1186/1465-9921-14-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silswal N, Singh AK, Aruna B, Mukhopadhyay S, Ghosh S, Ehtesham NZ. Human resistin stimulates the pro-inflammatory cytokines TNF-αand IL-12 in macrophages by NF-κB-dependent pathway. Biochem Biophys Res Commun. 2005;334:1092–1101. doi: 10.1016/j.bbrc.2005.06.202. [DOI] [PubMed] [Google Scholar]
- 15.Zeidler D, Zahringer U, Gerber I, Dubery I, Hartung T, Bors W, et al. Innate immunity in Arabidopsis thaliana:lipopolysaccharides activate nitric oxide synthase (NOS) and induce defense genes. Proc Natl Acad Sci U S A. 2004;101:15811–15816. doi: 10.1073/pnas.0404536101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Wang Y, Dong H, Zhu M, Ou Y, Zhang J, Luo H, et al. Icariin exterts negative effects on human gastric cancer cell invasion and migration by vasodilator-stimulated phosphoprotein via Rac1 pathway. Eur J Pharmacol. 2010;635:40–48. doi: 10.1016/j.ejphar.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 18.Jiang YZ. Pseudomonas aeruginosa colonization increased the ventilator-associated pneumonia in mice through the TNF-αand JNK signaling pathway. Biol Sci. 2014;14:66. [Google Scholar]
- 19.Hwang MH, Damte D, Lee JS, Gebru E, Chang ZQ, Cheng H, et al. Mycoplasma hyopneumoniae induces pro-inflammatory cytokine and nitric oxide production through NFκB and MAPK pathways in RAW264.7 cells. Vet Res Commun. 2011;35:21–34. doi: 10.1007/s11259-010-9447-5. [DOI] [PubMed] [Google Scholar]
- 20.Shou-Jun LI, Zhang Y, Neng-Shun WU, Sun JH. Detection of serum IL-6, IL-10 and TNF-αin children with mycoplasma pneumonia and their relationship with disease severity. J Nanchang Univ. 2012;4:432. [Google Scholar]
- 21.Wang AG. Significance of serum levels of MCP-1 and interleukin-12P40 in children infected with mycoplasma pneumonia. Modern Prev Med. 2009;36:3582–3585. [Google Scholar]
- 22.Fessele S, Boehlk S, Mojaat A, Miyamoto NG, Werner T, Nelson EL, et al. Molecular and in silico characterization of a promoter module and C/EBP element that mediate LPS-induced RANTES/CCL5 expression in monocytic cells. FASEB J. 2001;15:577–579. doi: 10.1096/fj.00-0459fje. [DOI] [PubMed] [Google Scholar]
- 23.Castellani ML, Shanmugham LN, Petrarca C, Simeonidou I, Frydas S, Colli MD, et al. Expression and secretion of RANTES (CCL5) in granulomatous calcified tissue before and after lipopolysaccharide treatment In Vivo. Calcif Tissue Int. 2007;80:60–67. doi: 10.1007/s00223-006-0115-2. [DOI] [PubMed] [Google Scholar]
- 24.Appay V, Rowland-Jones SL. RANTES:a versatile and controversial chemokine. Trends Immunol. 2001;22:83–87. doi: 10.1016/s1471-4906(00)01812-3. [DOI] [PubMed] [Google Scholar]
- 25.Gerdes N, Zhu L, Ersoy M, Hermansson A, Hjemdahl P, Hu H, et al. Platelets regulate CD4- T-cell differentiation via multiple chemokines in humans. Thromb Haemost. 2011;106:353–362. doi: 10.1160/TH11-01-0020. [DOI] [PubMed] [Google Scholar]
- 26.Sakthivel SK, Singh UP, Singh S, Taub DD, Igietseme JU, Lillard JW., Jr CCL5 regulation of mucosal chlamydial immunity and infection. BMC Microbiol. 2008;8:136. doi: 10.1186/1471-2180-8-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mckinley L, Logar AJ, Mcallister F, Zheng M, Steele C, Kolls JK. Regulatory T cells dampen pulmonary inflammation and lung injury in an animal model of pneumocystis pneumonia. J Immunol. 2006;177:6215–6226. doi: 10.4049/jimmunol.177.9.6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marques RE, Guabiraba R, Russo RC, Teixeira MM. Targeting CCL5 in inflammation. Expert Opin Ther Targets. 2013;17:1439–1460. doi: 10.1517/14728222.2013.837886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barnes DA, Tse J, Kaufhold M, Owen M, Hesselgesser J, Strieter R, et al. Polyclonal antibody directed against human RANTES ameliorates disease in the Lewis rat adjuvant-induced arthritis model. J Clin Invest. 1998;101:2910–2919. doi: 10.1172/JCI2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ling M, Li Y, Xu Y, Pang Y, Shen L, Jiang R, et al. Regulation of miRNA-21 by reactive oxygen species-activated ERK/NF-κB in arsenite-induced cell transformation. Free Radic Biol Med. 2012;52:1508–1518. doi: 10.1016/j.freeradbiomed.2012.02.020. [DOI] [PubMed] [Google Scholar]
- 31.Han DW, Lee MH, Kim HH, Hyon SH, Park JC. Epigallocatechin-3-gallate regulates cell growth, cell cycle and phosphorylated nuclear factor-[kappa]B in human dermal fibroblasts. Acta Pharmacol Sin. 2011;32:637–646. doi: 10.1038/aps.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 33.Liu D, Cao G, Han L, Ye Y, Sima Y, Ge W. Flavonoids from radix tetrastigmae inhibit TLR4/MD-2 mediated JNK and NF-κB pathway with anti-inflammatory properties. Cytokine. 2016;84:29–36. doi: 10.1016/j.cyto.2015.08.003. [DOI] [PubMed] [Google Scholar]