

Abstract
Pheochromocytomas are rare tumors arising from chromaffin cells of the adrenal medulla and extramedullary sympathetic ganglia. The incidence of asymptomatic disease is rising due to increased detection rates from widespread use of computed tomography. We describe a case of one of the largest documented pheochromocytomas resected in the United States, an 18-cm tumor in a patient who presented with exertional dyspnea, abdominal pain, constipation, weight loss, and intermittent hypertension. After biochemical and appropriate imaging workup, the patient underwent an open resection of the mass.
Keywords: Pheochromocytoma, paraganglionoma, adrenal, kidney, endocrine
Pheochromocytomas are rare tumors arising from chromaffin cells of the adrenal medulla and extramedullary sympathetic ganglia. They are a cause of hypertension in adults in fewer than 0.2% of cases and present in the fourth and fifth decades of life (1). Most cases are sporadic, but they may be associated with multiple endocrine neoplasia type 2 (MEN2),vonHippel-Lindau syndrome (VHL), and less commonly neurofibromatosis type 1 (NF-1) (2,3). Patients classically present with the paroxysmal triad of headache, diaphoresis, and tachycardia; hypertension is variably present (1). The incidence of asymptomatic disease is rising due to increased detection rates from widespread use of computed tomography (CT). Approximately 10% of pheochromocytomas are extramedullary (i.e. they are catecholamine-secreting para-ganglionomas), 10% are bilateral, and 10% are malignant (4).
The approach to the workup of a suspected pheochromocytoma includes biochemical studies as well as imaging. Plasma free metanephrine testing may be performed first. Due to the low specificity of this test, a positive result should prompt confirmation with measurement of 24-hour urinary catecholamines and their metabolites (5). Imaging studies include CT or magnetic resonance imaging (MRI), with or without additional nuclear studies such as iodine-123 meta-iodobenzylguanidine (MIBG) scintigraphy and fluorodeoxyglucose positron-emission tomography (1,6). Genetic testing may also be performed if personal or family histories suggest the presence of an inherited syndrome.
The only curative option for non-metastatic disease is surgical resection after medical optimization (7). Patients require preoperative volume expansion and sympathetic control with alpha- followed by beta-adrenergic blockade (7). The choice of approach includes both laparoscopic and open surgery, depending on tumor characteristics and patient factors.
We describe a case of one of the largest documented pheochromocytomas resected in the United States, an 18-cm tumor in a patient who presented with exertional dyspnea, abdominal pain, constipation, weight loss, and intermittent hypertension.
Case Report
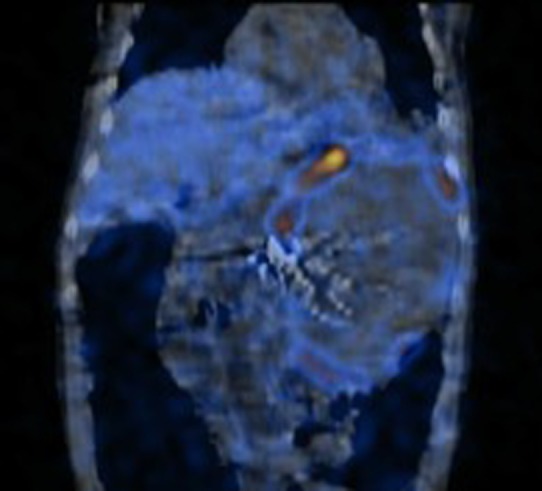
The patient was a 57-year-old male with past medical history of hypertension who presented with symptoms of abdominal pain and increasing distention. The patient was in overall good health and did not regularly see a physician. He underwent a CT scan, which demonstrated a large, vascular tumor with dimensions of 19×13×12 cm (Figure 1). Interpretation of the CT imaging was of concern for malignancy, as the mass had significant central necrosis and high vascularity. MIBG scan showed a large left-sided mass with no other lesions (Figure 2).
Figure 1. Computed tomography revealed a large left-sided retroperitoneal mass with highly vascular features .

Figure 2. 123I-Meta-iodobenzylguanidine scan showing a large leftsided mass.
Biochemical workup included measurement of serum cortisol, normetanephrine and metanephrines. Urinary metanephrine, noremetanephrine and vanillylmandelic acid (VMA) were also measured. A significantly elevated level of normetanephrine (41.5 nmol/l) was found. Urinaryno-rmetanephrines were elevated at 20000 μg/g urinary creatinine (Crt) and urinary normetanephine at 11000 μg/l. Urinary VMA was found to be 41 mg/g Crt.
With the biochemical workup complete, it was concluded that this mass was likely a pheochromocytoma. The patient was started on an alpha-blocker (terazosin) which was continued for 2 weeks. The patient initially had some orthostatic hypotension and was treated with intravenous fluid hydration. He was discharged to complete his preoperative preparation. A beta-blocker was started 2 days prior to the operation. The patient’s blood pressure was well controlled with the treatment regimen. The patient was readmitted prior to the operation and received addition fluid hydration as well as a bowel prep.
Operative course. The patient was placed supine and intubated by the anesthesia team. An internal jugular central line catheter and radial arterial line were placed. A midline incision with a left lateral extension was fashioned and a self-retaining retractor was utilized allowing for optimal visualization. The mass protruded through the mesentery of the transverse colon (Figure 3). The mass compressed the kidney but was not adherent to it. A large adrenal vein arose from the mass and was ligated. Once the medial and lateral attachments were taken down, the mass was resected en bloc (Figure 4).
Figure 3. Initial view upon entering the abdomen .

Figure 4. Resected mass, with greatest dimension of 18 cm.

During dissection of the mass, the patient had several episodes of hypertension that were corrected. After resection, fluids and vasopressors were required to maintain blood pressure. These were titrated off shortly after the operation and the patient was extubated later that evening.
Postoperative course. Postoperatively the patient did very well. He maintained a normal blood pressure. He was discharged on postoperative day 7.
Pathological evaluation. The specimen weighed 1729 g and measured 18.0×12.5×9.5 cm. The entire specimen appeared to be a well-circumscribed, encapsulated mass. A thin rim of normal appearing adrenal tissue was identified on one edge. Sectioning revealed necrotic and hemorrhagic cut surfaces.
On microscopic evaluation, the tumor was found to be composed of small nests of tumor cells surrounded by sustentacular cells and capillaries (Figure 5). The neoplastic cells had hyperchromatic nuclei containing finely stippled chromatin (Figure 6). Lymphovascular invasion was present. Large areas of necrosis were identified. Rare mitoses (approximately 4/50 high-power fields)were identified. On immunohistochemical analysis, tumor cells were strongly immunoreactiveforchromogranin (Figure 7).
Figure 5. Microscopic evaluation of the tumor revealed small nests of tumor cells surrounded by capillaries (H&E stain; a: ×100; b: ×200).
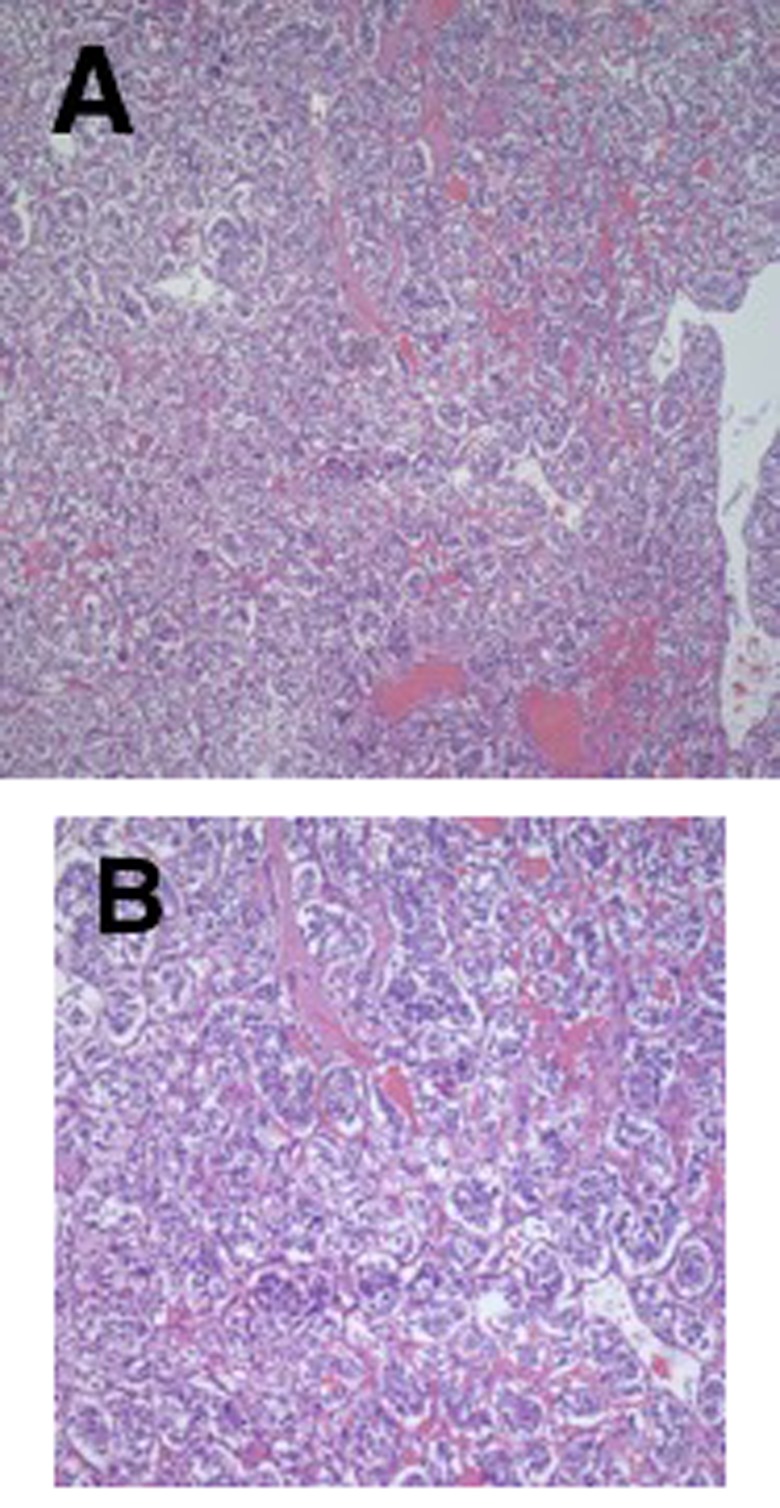
Figure 6. Microscopic evaluation of the tumor revealed neoplastic cells with hyperchromatic nuclei containing finely stippled chromatin (H&E stain;×400).

Figure 7. Neoplastic cells were found to be immunoreactive for chromogranin (×20).

Discussion
Surgical resection of a pheochromocytoma remains the mainstay treatment for this disease. Laparoscopic surgery has emerged as a viable option for removal of most of these tumors. Factors that the surgeon must take into account when determining whether to perform the operation laparoscopically are the size and vascularity of the tumor, signs of malignancy, presence of local invasion, and degree of expertise in laparoscopic approach (8). Our patient had an 18-cm mass on imaging with signs of malignancy, including high vascularity and necrosis. For this reason, an open operation was selected.
Optimal exposure permits increased visualization and less manipulation of the mass. The surgeon should minimize manipulation of the mass. Furthermore, great care should be taken to avoid entering or rupturing the tumor capsule. If possible, early vascular control and ligation of adrenal vessels are helpful.
During the operation, excellent communication between the surgical and anesthesia teams is of paramount importance, especially during dissection and prior to resection of the mass. Furthermore, after removal of the mass, the anesthesia team should be prepared to resuscitate the patient with fluids and vasopressors as needed. In cases involving large tumors, such as this one, we believe an arterial line and central venous access should be used for better monitoring and resuscitation.
A literature search yielded a case report by Maharaj et al. which provided a summary of all giant pheochromocytomas (defined as tumors >10 cm) reported in the literature (9). The largest tumor reported by Maharaj et al. was 45×20 cm originally described by Grissom et al. in 1979 (10). Thirty-seven cases of giant pheochromocytomas were cited. Our case represents one of the largest reported pheochromocytomas in the United States.
References
- 1.Stein PP, Black HR. A simplified diagnostic approach to pheochromocytoma. A review of the literature and report of one institution’s experience. Medicine. 1991;70(1):46–66. doi: 10.1097/00005792-199101000-00004. [DOI] [PubMed] [Google Scholar]
- 2.Neumann HP, Berger DP, Sigmund G, Blum U, Schmidt D, Parmer RJ, Volk B, Kirste G. Pheochromocytomas, multiple endocrine neoplasia type 2, and von Hippel-Lindau disease. N Engl J Med. 1993;329(21):1531–1538. doi: 10.1056/NEJM199311183292103. [DOI] [PubMed] [Google Scholar]
- 3.Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. von Recklinghausen’s disease and pheochromocytomas. J Urol. 1999;162(5):1582–1586. [PubMed] [Google Scholar]
- 4.Goldstein RE, O’Neill JA Jr., Holcomb GW 3rd, Morgan WM 3rd, Neblett WW 3rd, Oates JA, Brown N, Nadeau J, Smith B, Page DL, Abumrad NN, Scott HW Jr. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229(6):755–766. doi: 10.1097/00000658-199906000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quan-Yang D, Yeh MW. The adrenal glands. Chapter 39: In: Textbook of Surgery: The Biological Basis of Modern Surgical Practice. Philadelphia: W.B: Saunders. 1997:pp. 1016–017. [Google Scholar]
- 6.Pacak K, Linehan WM, Eisenhofer G, Walther MM, Goldstein DS. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann Intern Med. 2001;134(4):315–329. doi: 10.7326/0003-4819-134-4-200102200-00016. [DOI] [PubMed] [Google Scholar]
- 7.Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, Naruse M, Pacak K, Young WF Jr, Endocrine Society. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915–1942. doi: 10.1210/jc.2014-1498. [DOI] [PubMed] [Google Scholar]
- 8.Young WF Jr., Kebebew E. Treatment of Pheochromocytoma in Adults. https://www.uptodate.com/contents/treatment-ofpheochromocytoma-in-adults [Google Scholar]
- 9.Maharaj R, Parbhu S, Ramcharan W, Baijoo S, Greaves W, Harnanan D, Warner WA. Giant cystic pheochromocytoma with low risk of malignancy: a case report and literature review. Case Rep Oncol Med. 2017 doi: 10.1155/2017/4638608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grissom JR, Yamase HT, Prosser PR. Giant pheo-chromocytoma with sarcoidosis. SouthMed J. 1979;72(12):1605–1607. doi: 10.1097/00007611-197912000-00035. [DOI] [PubMed] [Google Scholar]