

Abstract
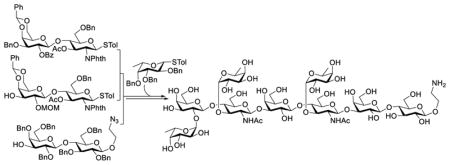
A robust, convergent, and efficient strategy was developed for the synthesis of the nonasaccharide cancer antigen, KH-1. This strategy featured a one-pot block assembly of the linear hexasaccharide backbone using three disaccharides followed by grafting of three fucose residues onto the backbone in one step.
Graphical Abstract
Cell surface glycans, which are usually present in forms of glycolipids, glycoproteins and other glycoconjugates, are key signaling molecules. Compared to normal cells, cancer cells display different glycosylation patterns, causing changes in communication, regulation, proliferation, survival and other cellular behaviors.1–3 The abnormal glycans, either unique or overexpressed, resulted from atypical glycosylations of cancer cells are known as tumor-associated carbohydrate antigens (TACAs)4–6 and have become useful targets for the development of novel cancer diagnostics and therapeutics, such as TACA-based therapeutic cancer vaccines or cancer immunotherapies.7–8 The working principle for therapeutic cancer vaccines is to elicit a specific immune response in cancer patients towards a certain cancer antigen to eventually allow for the patients’ immune system to kick in and eradicate cancer cells that expresses the exact TACA.
Numerous TACAs have been catalogued over the years,9 among which the KH-1 antigen, a nonasaccharide (Figure 1), is particularly intriguing. Although it was first isolated from adenocarcinoma cell,10 it has a wide presence. Importantly, the KH-1 antigen has not been detected on normal colonic cells, which makes it stand out from many other TACAs that are only overexpressed on cancer cells as compared to normal cells. This renders KH-1 an excellent candidate as a tumor-specific antigen for vaccine development.
Figure 1.

Structure of the KH-1 antigen
Following the first reports of chemical synthesis of the KH-1 antigen by Danishefsky11 and Schmidt12 independently in 1997, the Seeberger group described a successful solid-phase synthesis of KH-1 in 2004.13 Because of the dauntingly complex structure of KH-1, some groups took a shortcut to synthesize the Ley-Lex heptasaccharide14–15 (Figure 1) or Lex-Lex dimer as KH-1 mimics.16 These intense research efforts on KH-1 and its derivatives have been driven by the apparently strong immune responses induced by KH-1 as compared to monomeric Ley and Lex antigens.14 Thus, KH-1 is in high demand for biological studies due to its tumor specificity and its exceptional potency in eliciting immune responses.8, 14, 17 Chemical synthesis is still one of the primary means to access well-defined carbohydrates, but it falls short in providing sufficient quantities of the KH-1 antigen to satisfy an increasing demand, which has limited the full exploitation of KH-1 in anticancer vaccine development and other studies. Answering the call for improving the efficiency of KH-1 synthesis, this work aimed to develop a new synthetic strategy and to use it to prepare a sufficient quantity of KH-1 for cancer vaccine study.
As depicted in Figure 1, the KH-1 antigen can be seen as a heterodimer of the Ley and Lex antigens with an additional lactose unit present at the downstream end, to which a lipid ceramide is then linked. From a synthetic point of view, this nonasaccharide has a hexasaccharide backbone consisting of a lactose and two lactosamine units, onto which are grafted three fucose residues. Fucosylation is a common feature of blood type antigens and terminal fucose residues can be recognized by the mannose receptors on dendritic cells (DCs) thereby initiating immune responses.18–20 The stronger immunogenicity of the KH-1 antigen than Lewis antigens can be attributed to the higher degree of fucosylation. Fully aware of these structural properties, we planned a strategy featuring a linear block assembly of the hexasaccharide backbone followed by a onestep grafting of all three fucose units as outlined in Scheme 1. In addition, the synthetic target 1 was designed to carry an amino ethyl group at the downstream end, which could be used as a linker to facilitate chemoselective conjugation with other molecules for various applications and studies.
Scheme 1.

Retrosynthetic analysis of the synthetic target 1
Several issues were taken into consideration in the above design to minimize potential problems and secure synthetic efficiency. First, since the assembly of branched oligosaccharides was shown to be troublesome due to steric hindrance,21 which led to the failure in the coupling of a glycosyl acceptor to a Ley tetrasaccharide in the previous synthesis of KH-1,22 we planned to assemble the linear hexasaccharide backbone first, followed by branch grafting to minimize the potential influence of steric hindrance on the glycosylation reactions. Second, our synthetic design enabled linear elongation of the oligosaccharide chain through a preactivation-based one-pot glycosylation method developed by Huang and Ye,23 which has been successfully applied to many oligosaccharides, including our synthesis of complex β-glucan derivatives.24 The one-pot synthetic strategy employing thioglycosides as donors was projected to save several tedious glycosyl donor manipulation steps. Third, block assembly of the hexasaccharide backbone was expected to reduce the number of linear glycosylation steps and, in the meantime, to make use of the same or similar intermediates, such as 8 and 11, in the construction of the lactosamine building blocks. Finally, with careful design of protecting tactics, it would be possible to simultaneously unmask all of the fucose-linked positions in the oligosaccharide backbone to enable the introduction of three fucose residues in one step at a late stage.
Specifically, the to-be-fucosylated positions in the hexasaccharide backbone 3 were protected as acetyl esters to facilitate their one-step deacetylation. As mentioned earlier, we planned to assemble the hexasaccharide backbone 3 from disaccharides 4, 5, and 6 through one-pot glycosylation. Lactosamine derivatives 4 and 5, which shared the same core structure but were fucosylated at different positions, were orthogonally protected, e.g., using benzoyl (Bz) and allyl (All)/levulinoyl10 groups to protect the 2-O- and 3-O-positions of 7 and 9, respectively. Thus, 7 and 9 would be prepared from the same intermediate 11, whereas 6 could be derived from peracetylated lactose 10. In addition, 8 was designed to possess free 3,4-O-positions to offer improved reactivity as a glycosyl acceptor, but the 4-OH group was expected to be more reactive than the 3-OH group to enable regioselective glycosylation.25–26
Our synthesis commenced with the preparation of disaccharides 4 and 5 (Scheme 2). First, 1127 was converted into thioglycosides 7 and 9 through a series of similar transformations to orthogonally protect their 2-O- and 3-O-positions. Selective 3-O-benzylation and allylation directed by a tin complex were followed by 2-O-acylation to afford excellent yields of 7 and 9. Regioselective 3-O-benzylation or allylation has been well documented to prefer the equatorial hydroxyl group next to an axial hydroxyl group.28 Our confirmation of the regiochemistry of 7 and 9 was achieved by NMR spectrometry upon acylation of the 2-O-position, which significantly shifted their H-2 1H NMR signals to downfield. Glycosylation of 8 with either 7 or 9 went smoothly under the preactivation conditions,23 that is, activating 7 and 9 with p-toluenesulfenyl chloride (p-TolSCl) and silver triflate (AgOTf) followed by the addition of acceptor 8, produced 12 and 13. These glycosylation reactions were highly stereoselective (only β-linked disaccharides were isolated; JH1′2′ values for 12 and 13 were 10.5 and 7.5 Hz, respectively) likely due to the neighboring participation effect of the 2-O-benzoyl and levulinoyl groups in 7 and 9, respectively. These reactions also exhibited excellent regioselectivity, probably because of the presence of a 2-N,N-phalimido group in 8 that affected the reactivity of its 3-O-position, which was reported previously.25 Compounds 12 and 13 were finally acetylated to yield 4 and 14, respectively. The regiochemistry of these glycosylation reactions was again confirmed by the observed downfield shifts of the H-3 NMR signals of the glucosamine residue in 12 and 13 upon acetylation (from 3.70 and 3.73 ppm for 12 and 13 to 5.46 and 5.71 ppm for 4 and 14, respectively).
Scheme 2.

Synthesis of disaccharide building blocks
To differentiate the 2-O-position in 14 from all other acyl-protected positions deemed for fucosylation later on, the 2-O-levulinoyl group needed to be replaced with a base-stable protecting group, such as the benzyl group. Consequently, 14 was treated with hydrazine acetate to remove the levulinoyl group, and the product 15 was then subjected to benzylation. To our disappointment, the reaction under basic (BnBr/Bases), neutral (Ag2O/BnBr), and acidic (benzyl imidate/TMSOTf) conditions failed to provide the desired product, due to either instability of the 3′-O-ester functionality or the low reactivity of the 2-O-position as a result of steric hindrance in the highly substituted glucosamine residue. Therefore, the 2-hydroxyl group was protected with the less sterically demanding methoxymethyl (MOM) group to afford 16, which was a less appealing option as this required an additional step for the global deprotection as compared to that with benzyl group as the protecting group. Subsequently, the allyl group in 16 was removed on iridium-catalyzed olefin rearrangement and then HgO/HgCl2-promoted hydrolysis of the resultant vinyl ether to give 5 as a glycosyl acceptor. In the meantime, 6 was prepared according to a literature procedure.16
Starting from disaccharides 4, 5 and 6, hexasaccharide 3 was assembled via preactivation-based one-pot glycosylation (Scheme 3). Activation of thioglycoside donors was achieved with p-toluenesulfenyl triflate (TolSOTf) generated in situ from p-TolSCl and TMSOTf at −78 °C for 15 min, and glycosylation was carried out upon subsequent addition of the glycosyl acceptor together with 2,4,6-tri-tert-butylpyrimidine (TTBP) at −78 °C, and the reaction was monitored with TLC. The reaction of 4 with 5 was complete in an hour. Thereafter, the next cycle of activation/glycosylation using acceptor 6 was carried out. Eventually, hexasaccharide 3 was obtained in a 73% overall yield and as the only anomer (JH1,2 = 8.1 and 8.4 Hz for the two glucosamine residues). The structure of 3 was fully characterized with MS and NMR data.
Scheme 3.

Backbone assembly and fucosyl residue grafting for the synthesis of KH-1
To be readied for introducing the fucose residues, hexasaccharide 3 was heated with hydrazine in ethanol at reflux to simultaneously remove all benzoyl, acetyl and N,N-phthalyl groups, which deprotected the specific hydroxyl groups on the terminal galactose and two glucosamine residues. However, the amino groups were also exposed, which were selectively re-protected with acetyl groups upon acetic anhydride treatment in methanol to provide 17. At this point, all three fucose residues were introduced with thioglycoside 1816 as the glycosyl donor under the promotion of AgOTf and N-iodosuccinimide (NIS) to afford 2 in an excellent yield (88%) and stereoselectivity (JH1,2 = 3.8 Hz at 5.63 ppm; 3.6 Hz at 4.86 ppm, and 3.0 Hz at 4.85 ppm for the fucoses).
Global deprotection of 2 was carried out in two separate steps, starting with the optimization of reaction conditions to remove the MOM group. We found that commonly employed acidic conditions resulted in partial defucosylation because of the sensitivity of fucosidic linkages to acids, while a stoichiometric amount of the Lewis acid, B-bromocatecholborane, proved to be the best choice for removing the MOM group to provide the desired product 19 in a good yield (72%). Finally, all of the benzyl and benzylidene groups were removed smoothly via Pd/C-catalyzed hydrogenolysis to furnish 1, the KH-1 antigen with an ethylamine moiety at the downstream end.
In conclusion, KH-1, a complex TACA, was synthesized by a highly convergent and efficient strategy. This synthesis featured a block assembly of the hexasaccharide backbone via one-pot preactivation-based glycosylation of three disaccharide units, which was designed to minimize the number of steps of linear oligosaccharide assembly and circumvent tedious donor manipulations. Moreover, through thoughtful design of the protection tactics, all three fucose residues were grafted onto the hexasaccharide backbone in one step, which not only enabled a highly convergent synthesis but also helped avoid potential steric hindrance during other glycosylation reactions. These features of the synthesis made it not only efficient but also robust. Therefore, the target molecule 1 can be obtained in multi-milligram quantities to meet the demand for biological studies. For example, the synthetic KH-1 antigen is currently used to conjugate with carrier molecules and the resultant conjugates are evaluated as anticancer vaccines.
Supplementary Material
Acknowledgments
We appreciate the NIH/NCI support (grant # R01CA095142) for this research work.
Footnotes
Experimental details, spectra (1H, 13C, COSY, HSQC NMR and HRMS) of all new compounds. The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.Stowell SR, Ju T, Cummings RD. Annu Rev Pathol Mech Dis. 2015;10:473–510. doi: 10.1146/annurev-pathol-012414-040438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ohtsubo K, Marth JD. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 3.Fuster MM, Esko JD. Nat Rev Cancer. 2005;5:526–542. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- 4.Lloyd KO. Semin Cancer Biol. 1991;2:421–431. [PubMed] [Google Scholar]
- 5.Feizi T. Cancer Surv. 1985;4:245–269. [PubMed] [Google Scholar]
- 6.Hakomori S. Cancer Res. 1985;45:2405–2414. [PubMed] [Google Scholar]
- 7.Feng D, Shaikh AS, Wang F. ACS Chem Biol. 2016;11:850–863. doi: 10.1021/acschembio.6b00084. [DOI] [PubMed] [Google Scholar]
- 8.Ragupathi G, Deshpande PP, Coltart DM, Kim HM, Williams LJ, Danishefsky SJ, Livingston PO. Int J Cancer. 2002;99:207–212. doi: 10.1002/ijc.10305. [DOI] [PubMed] [Google Scholar]
- 9.Levery SB. In: Carbohydrate-Based Vaccines Immunotherapy. Guo Z, Boons G-J, editors. 2009. pp. 227–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nudelman E, Levery SB, Kaizu T, Hakomori S. J Biol Chem. 1986;261:11247–11253. [PubMed] [Google Scholar]
- 11.Deshpande PP, Danishefsky SJ. Nature. 1997;387:164–166. doi: 10.1038/387164a0. [DOI] [PubMed] [Google Scholar]
- 12.Hummel G, Schmidt RR. Tetrahedron Lett. 1997;38:1173–1176. [Google Scholar]
- 13.Love KR, Seeberger PH. Angew Chem Int Ed. 2004;43:602–605. doi: 10.1002/anie.200352539. [DOI] [PubMed] [Google Scholar]
- 14.Buskas T, Li Y, Boons GJ. Chem Eur J. 2005;11:5457–5467. doi: 10.1002/chem.200500412. [DOI] [PubMed] [Google Scholar]
- 15.Tsai BL, Han JL, Ren CT, Wu CY, Wong CH. Tetrahedron Lett. 2011;52:2132–2135. [Google Scholar]
- 16.Miermont A, Zeng Y, Jing Y, Ye XS, Huang X. J Org Chem. 2007;72:8958–8961. doi: 10.1021/jo701694k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deshpande PP, Kim HM, Zatorski A, Park TK, Ragupathi G, Livingston PO, Live D, Danishefsky SJ. J Am Chem Soc. 1998;120:1600–1614. [Google Scholar]
- 18.Schlesinger PH, Doebber TW, Mandell BF, White R, DeSchryver C, Rodman JS, Miller MJ, Stahl P. Biochem J. 1978;176:103–109. doi: 10.1042/bj1760103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Largent BL, Walton KM, Hoppe CA, Lee YC, Schnaar RL. J Biol Chem. 1984;259:1764–1769. [PubMed] [Google Scholar]
- 20.Apostolopoulos V, Barnes N, Pietersz GA, McKenzie IFC. Vaccine. 2000;18:3174–3184. doi: 10.1016/s0264-410x(00)00090-6. [DOI] [PubMed] [Google Scholar]
- 21.Mondal PK, Liao G, Mondal MA, Guo Z. Org Lett. 2015;17:1102–1105. doi: 10.1021/ol5036563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spassova MK, Bornmann WG, Ragupathi G, Sukenick G, Livingston PO, Danishefsky SJ. J Org Chem. 2005;70:3383–3395. doi: 10.1021/jo048016l. [DOI] [PubMed] [Google Scholar]
- 23.Huang X, Huang L, Wang H, Ye XS. Angew Chem Int Ed. 2004;43:5221–5224. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 24.Liao G, Zhou Z, Burgula S, Liao J, Yuan C, Wu Q, Guo Z. Bioconjugate Chem. 2015;26:466–476. doi: 10.1021/bc500575a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spijker NM, Van Boeckel CAA. Angew Chem Int Ed. 1991;103:179–182. [Google Scholar]
- 26.Gao J, Guo Z. Org Lett. 2016;18:5552–5555. doi: 10.1021/acs.orglett.6b02796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bartetzko MP, Schuhmacher F, Hahm HS, Seeberger PH, Pfrengle F. Org Lett. 2015;17:4344–4347. doi: 10.1021/acs.orglett.5b02185. [DOI] [PubMed] [Google Scholar]
- 28.Grindley TB. Adv Carbohydr Chem Biochem. 1998;53:17–142. doi: 10.1016/s0065-2318(08)60043-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.