

Abstract
Aging increases the risk of abrupt declines in cognitive function after an event that triggers immune system activation (e.g. surgery, infection, or injury). This phenomenon is poorly understood, but rodent models may provide clues. We have previously shown that aging (24-mo-old) F344xBN rats generally do not show significant physical or cognitive impairments. However, their brains mount an exaggerated inflammatory response to signals triggered by a peripheral immune challenge (an intraperitoneal injection of Escherichia coli or laparotomy). Their hippocampal levels of the proinflammatory cytokine IL-1β are significantly elevated for at least 8 d, but generally less than 14 d, after infection or surgery. This IL-1β elevation is mirrored by prolonged deficits in a hippocampus-dependent long-term memory task. In contrast, young (3-mo-old) counterparts exhibit only transient elevations in IL-1β that drop to near baseline levels within 24 h. We previously demonstrated that theta burst–evoked late-phase long-term potentiation (L-LTP)—a BDNF-dependent form of synaptic plasticity—is impaired in hippocampal area CA1 of aged animals 4 d after infection. Also, levels of mature brain-derived neurotrophic factor (mBDNF)—the protein isoform required for stabilization of L-LTP—are reduced in hippocampal synaptoneurosomes of aged animals at the same time point. In this study, we investigated whether the deficits in L-LTP and mBDNF persist in parallel with the elevation in IL-1β and impairment in memory. This was the case, consistent with the idea that an exaggerated brain inflammatory response may compromise memory consolidation in part by altering availability of mBDNF to stabilize memory-related synaptic plasticity.
Keywords: BDNF, Delirium, Hippocampus, Interleukin-1beta, LTP, Microglia
Significance Statement
Not all cognitive decline is gradual. Older individuals—even those previously healthy and high functioning—are more likely to experience an abrupt decline in mental function (termed delirium) after immune challenge. Even if this is temporary, it is associated with increased risk of ultimately developing dementia. Although clinically important, this phenomenon is much less studied than gradual senescence and aging-associated neurodegenerative disorders. Here we use a naturalistic rodent model to further test the hypothesis that the combination of age and an immune challenge may trigger an exaggerated inflammatory state in the brain, which in turn, disrupts molecular systems critical for memory. These studies may provide mechanistic insights into the earliest stages of inflammation-driven failures of memory-related synaptic plasticity.
Introduction
When we think about age-related cognitive decline, we tend to think of gradual decrepitude or overt neurodegenerative disease, as in Alzheimer’s disease. However, cognitive decline is not always gradual. Rapid decline can be triggered by activation of the peripheral immune system. Proinflammatory cytokines (e.g., IL-1β and TNF-α) produced by peripheral immune activation can communicate with the brain via both humoral and neural routes, triggering a cascade of effects in the CNS including microglial activation and de novo production of proinflammatory cytokines (Maier et al., 2001; Konsman et al., 2002; Dantzer et al., 2008). Interestingly, aging has been shown to sensitize the brain inflammatory response to a variety of experimental immune challenges [e.g., Escherichia coli, surgery, lipopolysaccharide (LPS)], increasing the size and duration of the resulting spike in proinflammatory cytokines in the hippocampus (Godbout et al., 2005; Chen et al., 2008; Barrientos et al., 2009).
At 24 mo, Fisher 344/Brown Norway (F344xBN) rats are generally healthy, aging but not senescent. We have previously shown that a single i.p. injection of E. coli produces prolonged elevations in IL-1β in the hippocampi of aging 24-mo-old F344xBN rats, but not in 3-mo-old rats (Barrientos et al., 2009). The exaggerated elevation in IL-1β does not impair the initial learning of the test tasks or formation of short-term memories. Instead, it is associated with profound and specific deficits in tasks requiring consolidation of hippocampus-dependent long-term memory (Barrientos et al., 2009). As the levels of IL-1β drop, these deficits fade. Similarly, blocking IL-1β signaling in the brain with an intra-cisterna magna infusion of an IL-1 receptor antagonist (Frank et al., 2010) blocks the memory deficits.
Previously, we examined the effects of age and infection on memory-related synaptic plasticity and levels of hippocampal BDNF (and related proteins) at a single time point, 4 d after the E. coli injection. This time point was chosen for several reasons: (1) both the young and aged animals have recovered from the overt symptoms of illness (e.g., fever, loss of appetite, etc.; Barrientos et al., 2006); (2) levels of hippocampal IL-1β are still significantly elevated in the aged rats but have returned to near pre-infection levels in the young rats (Barrientos et al., 2009); and (3) the aged rats show significant deficits in hippocampus-dependent long-term memory, but the young rats do not (Barrientos et al., 2006). We measured a BDNF-dependent, memory-related, long-term form of synaptic plasticity, theta burst–evoked L-LTP in hippocampal area CA1. Deficits in theta-frequency LTP in area CA1 have been shown to distinguish cognitively impaired from unimpaired aged Fischer 344 rats (e.g., Tombaugh et al., 2002). We found that a recent history of infection was associated with reduced theta burst L-LTP in the young rats and that aging greatly exacerbated this effect (Chapman et al., 2010). We also found that levels of mature BDNF (mBDNF, the cleaved protein isoform required for long-lasting forms of memory and LTP; Pang et al., 2004; Barnes and Thomas, 2008) were significantly reduced in the hippocampal synaptoneurosomes prepared from aged rats 4 d after E. coli injection (Cortese et al., 2011). Like the deficit in long-term memory (Frank et al., 2010), the deficits in L-LTP and mBDNF could be prevented by interfering with IL-1β signaling in the brain (Chapman et al., 2010; Cortese et al., 2011).
In this study, we extended our examination of theta burst L-LTP and mBDNF to longer time periods (8, 14 and 21 d after infection). The goal was to determine if the deficits in synaptic plasticity and mBDNF would resolve, and if they did, to compare the time courses of their recovery with those of the alterations in IL-1β and hippocampus-dependent long-term memory. The results show that the changes in L-LTP and mBDNF paralleled the changes in IL-1β and memory over time. This suggests that prolonged inflammatory responses in the brain might affect memory-related plasticity of hippocampal synapses, in part by modulating levels of mBDNF and downstream effectors required to stabilize synaptic plasticity.
Materials and Methods
Experimental animals
The animals in this study were 3- and 24-mo-old male Fisher 344/Brown Norway F1 hybrid rats from National Institute on Aging Aged Rodent Colony. They were housed in pairs with ad libitum access to water and food and were maintained on 12-h light-dark cycle. The animals were allowed to acclimate to the animal facility for a minimum of 10 d before the experiments were begun. All experiments complied with protocols approved by the University of Colorado and Temple University Animal Care & Use Committees.
The infection model
Stock E. coli cultures (15746; ATCC) were thawed and cultured in 40 ml brain–heart infusion (BHI; Difco Laboratories) at 37°C overnight. The number of bacteria in individual cultures was extrapolated from previously determined growth curves. The cultures were centrifuged at 3000 rpm for 15 min, the supernatants were discarded, and the bacterial pellets were suspended in sterile PBS to achieve a final dose of 1.0 × 1010 colony-forming units (CFU)/mL in 250 μl. All animals were given an intraperitoneal injection of 250 μl of either E. coli or vehicle (sterile PBS).
Slice preparation
Rats were decapitated, and hippocampi were collected 8 (± 1), 14 (± 1), or 21 (± 1) d after injection of E. coli or saline. Experiments on tissue from E. coli– or saline-injected animals collected at the different time points were interleaved. Transverse hippocampal slices (400 μm) were prepared employing conventional techniques (Patterson et al., 1992, 1996). Slices were maintained in an interface chamber at 28°C with perfusion of oxygenated artificial cerebral spinal fluid (ACSF; in mm: 124.0 NaCl, 4.4 KCl, 26.0 NaHCO3, 1.0 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4, and 10 glucose). Slices were left in the chamber to recover for at least 3 h before recording.
Electrophysiology
Bipolar stimulating (FHC: CBBRC75) and ACSF-filled glass recording (A-M Systems: 603000) electrodes were placed in stratum radiatum to record field excitatory postsynaptic potentials (fEPSPs) from Schaffer collateral–CA1 synapses. All stimuli were set to evoke fEPSP slopes equal to one-third of the maximum in each slice. Test stimuli were delivered every minute, and test responses were recorded for 30 min before starting the experiment to ensure stability. Slices were then tetanized using a theta-burst protocol: 12 bursts of four pulses at 100 Hz, delivered 200 ms apart. The same stimulus intensity was used for tetanization and evoking test responses. Field EPSP recordings were normalized to pre-tetanus baselines and averaged for each group. Error bars indicate SEM. These data were further analyzed by factorial ANOVA, followed by Turkey’s HSD post hoc tests (GraphPad Prism 7).
Synaptoneurosome preparation
Rats were decapitated, and hippocampi were collected 8 or 14 d after E. coli or saline injection. Tissue was minced in 500 μl homogenization buffer (HB) with protease and phosphatase inhibitors [in m: 1 Tris, 1 sucrose, 0.5 EDTA, 0.25 EGTA, 0.5 NaF, 1 benzamidine, and 0.1 AEBSF (4-(2-aminoethyl)benzenesulfonyl fluoride)] and homogenized with a glass tissue grinder and a Teflon pestle. The homogenate was centrifuged at 960 × g for 15 min to pellet nuclear material and unbroken cells. The remaining supernatant was further centrifuged at 10,000 × g for 15 min, yielding an S2 cytosolic supernatant and a P2 crude synaptoneurosomal pellet (pre- and postsynaptic components). The synaptoneurosomal pellet was washed gently in 100 μl HB, homogenized with a 0.5-ml plastic pestle in 100 μl HB and 10 μl of 10× sodium chloride-TRIS-EDTA (1× final concentration), and sonicated. The final P2 fraction obtained using this procedure is enriched for presynaptic and postsynaptic proteins, terminal mitochondria, and cytoplasm and synaptic vesicles (Booth and Clark, 1978; Whittaker, 1993). Synaptic enrichment was confirmed using synaptic markers synaptophysin and postsynaptic density 95 (PSD95). Protein content was determined by the BCA protein assay (Bio-Rad).
Western blots
All procedures used here have been previously described (Cortese et al., 2011). Synaptoneurosomal samples were denatured in 4× Laemmli buffer and heated at 70°C for 5 min. The resulting protein samples (40 μg each) were loaded onto 4–12% NuPage Bis-Tris SDS-polyacrylamide gels (Invitrogen) and transferred onto polyvinylidene fluoride membranes (Millipore). Membranes were blocked in 5% milk/PBST (PBS with Tween 20) at room temperature for 30 min.
All primary antibodies were incubated overnight at 4°C and then washed 3 × 10 min with PBST. The primary antibodies (and dilutions) used were proBDNF (1:500; ab72440; Abcam), mature BDNF (1:1000; sc-546; Santa Cruz Biotechnology), phospho-TrkB (1:700; pTrkBY816, antisera gift from Moses Chao, New York University School of Medicine, New York, NY) and total (phosphorylated and unphosphorylated) TrkB (1:1000; sc-8316; Santa Cruz Biotechnology), phospho-PLCγ1 (phospholipase C γ1; 1:1000; 07-2134) and total PLCγ1 (1:500; 05–366; Millipore), and phospho-ERK (extracellular response kinase; 1:1000; 9101) and total ERK (1:1000; 9102; Cell Signaling Technology).
To confirm enrichment for synaptic components, blots were probed with synaptic markers, synaptophysin (1:1000; sc-12737; Santa Cruz Biotechnology) and PSD95 (1:1000; United Biomedical). β-Tubulin (1:100,000; MAB1637; Millipore Bioscience Research Reagents) and β-actin (1:5000; sc-47778; Santa Cruz Biotechnology) were used as loading controls. The identity of the BDNF isoform bands in synaptoneurosomes was confirmed by comparison with bands from HeLa cells transfected with a plasmid overexpressing BDNF, producing both the pro- and mature form.
Secondary antibodies were purchased from GE Health care and Bio-Rad and were diluted in the range of 1:5000–1:10,000. Incubations were at room temperature for 1 h, followed by 3 × 10 min washes. Super Signal West Pico Chemiluminescent (Pierce) was applied, and blots were exposed to autoradiography film (Denville Scientific). Blots were then stripped in Restore Western Blot Stripping Buffer (Pierce) for 15 min, washed 3 × 10 min in PBST, and subjected to standard Western blotting conditions.
ImageJ was used to quantify the protein bands, and all bands were normalized to their actin controls. For TrkB, PLCγ, and ERK, the ratio of the phosphorylated form to total expression of each protein was determined. We have previously found that the combined effects of age and infection uniquely disrupt BDNF-dependent memory and synaptic plasticity (Barrientos et al., 2006; Chapman et al., 2010) and also reduce mBDNF (and related proteins) 4–5 d after infection (Cortese et al., 2011). For the protein level time course, we therefore used an unpaired t test (GraphPad QuickCals) to determine if the level of the protein of interest in the aged + E. coli group differed from the level of the protein in the other groups. The p value listed for each protein (or phosphorylation state ratio) is for an unpaired t test comparing the mean of the aging + E. coli group to the mean of the combined values of the other test groups.
Results
Aging and a peripheral immune challenge interact to reduce theta burst–evoked L-LTP
In an earlier report (Chapman et al., 2010), we examined the impact of age and infection on synaptic function at Schaffer collateral-CA1 synapses in hippocampal slices from aged and young rats, 4–5 d after an i.p. injection of E. coli or saline. We found no significant differences between groups in basal synaptic transmission or early-phase long-term potentiation (E-LTP). We used two different stimulus protocols to induce late-phase long-term potentiation (L-LTP): either four trains of high-frequency stimulation, which induces robust activation of numerous plasticity-related signaling cascades (Bliss et al., 2007), or a more naturalistic theta burst stimulation, which mimics theta frequency burst firing of CA1 neurons during spatial exploration (O’Keefe, 2007). We found that four-train L-LTP was not significantly affected by age or infection. However, full expression of theta burst L-LTP was suppressed by a recent history of infection, and aging greatly exacerbated this effect.
The immune challenge–evoked elevations in IL-1β and the deficits in long-term memory both last >8 d, but typically <14 d, in aged animals (Barrientos et al., 2009). It seemed plausible that this might also be true of the deficits in theta burst L-LTP.
Theta burst–evoked L-LTP was still impaired in aged animals 8 d after infection
We examined the impact of age and infection on synaptic function at Schaffer collateral–CA1 synapses in hippocampal slices from aged and young rats, 8 d after an i.p. injection of E. coli or saline. As before (Chapman et al., 2010), input-output curves showed no significant difference across the four groups at any stimulating input (Page, infection = 0.95 at 5 V, 0.87 at 7 V, 0.85 at 10 V, 0.87 at 12 V, and 0.86 at 15 V; Fig. 1A). There was no significant difference in post-tetanic potentiation (immediately after theta burst stimulation) between the groups (Page, infection = 0.4130; young/vehicle = 216.9 ± 38.2%, young/E. coli = 230.3 ± 24.1%, aged/vehicle = 223.2 ± 32.5%, and aged/E. coli = 179.9 ± 15.4%; Fig. 2A). However, theta burst L-LTP was still severely impaired in aged rats 8 d after infection (Page, infection = 0.0070; percentage baseline 3 h after tetanus: young/vehicle = 145.3 ± 9.5%, young/E. coli = 145.2 ± 9.1%, aged/vehicle = 157.5 ± 11.5%, and aged/E. coli = 101.0 ± 9.0%; Fig. 2A). Turkey’s post hoc tests supported this statistical analysis, showing significance when the aged E. coli group was compared to young saline (p = 0.0101), young E. coli (p = 0.0180), or aged Saline (p = 0.0021).
Figure 1.
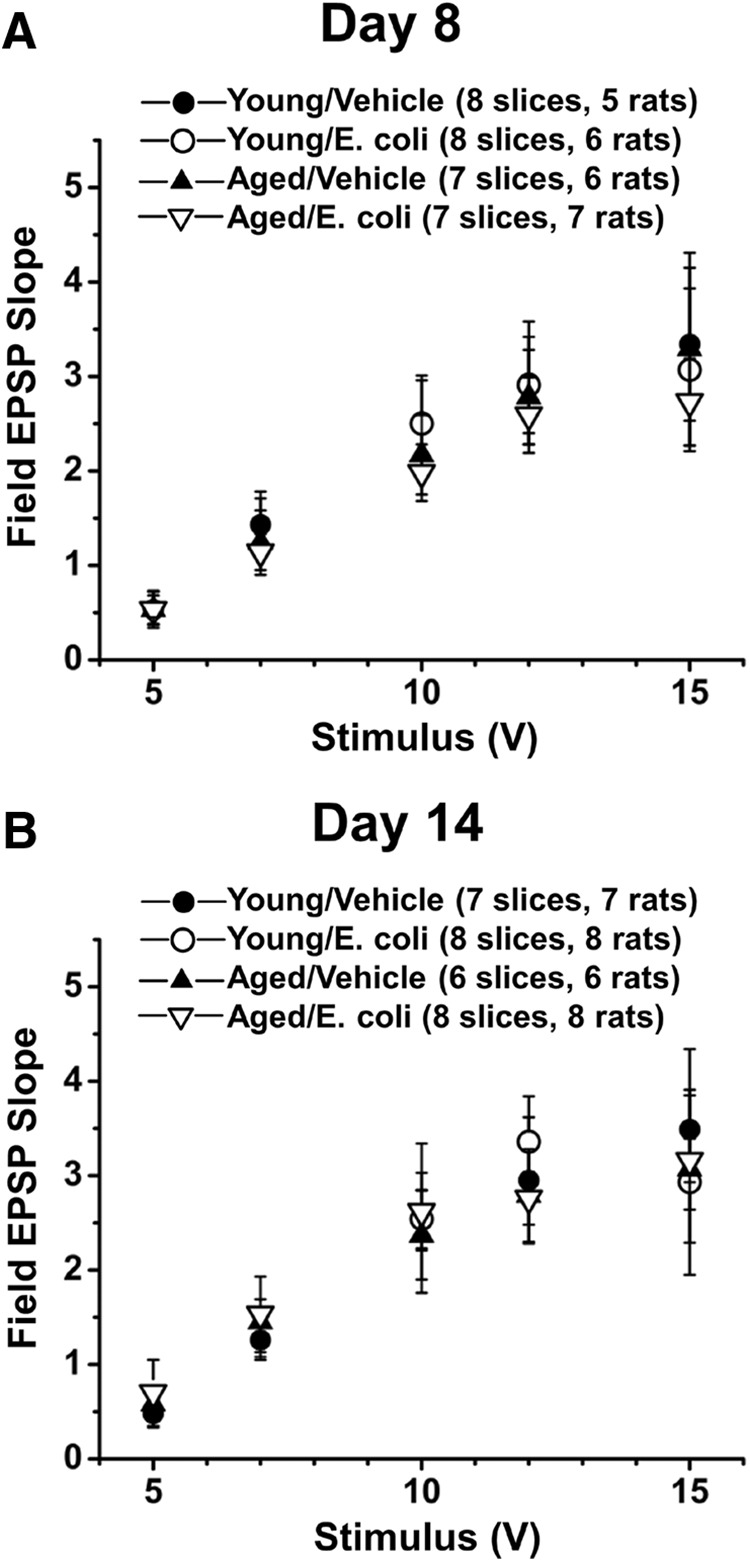
Stimulus–response curves are not altered by age or a history of infection. Plots of fEPSP slopes (in millivolts per millisecond) at various stimulation intensities for hippocampal slices from young and aged rats with and without a recent history of infection show no significant differences in basal synaptic transmission in area CA1. Input-output curves are shown for slice collected 8 d (A) and 14 d (B) after injection of E. coli or saline.
Figure 2.
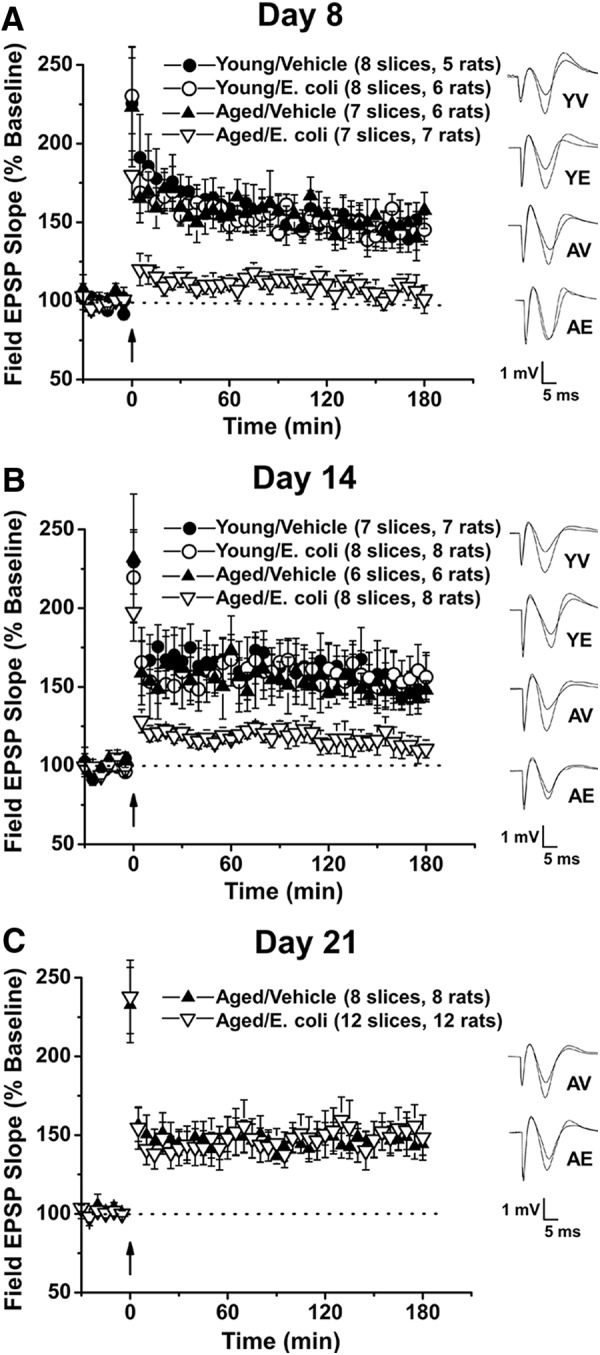
Aging and a peripheral immune challenge interact to produce prolonged, but temporary, reductions in L-LTP. Hippocampal slices were collected from young (3 mo) and aged (24 mo) rats 8, 14, or 21 d after injection of E. coli or saline (vehicle). L-LTP was induced in Schaffer collateral-CA1 synapses using theta-burst stimulation (12 bursts of 4 pulses at 100 Hz, delivered 200 ms apart). fEPSP slopes were normalized to pretetanus baselines, averaged, and plotted for each group. Error bars indicate SEM. Insets show representative traces before and 3 h after tetanus. A, L-LTP is severely impaired in slices from aged, but not young, rats 8 d after E. coli injection. B, L-LTP is still reduced but recovering in aged rats at 14 d. C, The L-LTP is back to baselines at 21 d.
Theta burst-evoked L-LTP was recovering in aged animals 14 d after infection
The effects of age and infection were subtler at 14 d. Input-output curves indicated no significant difference across the four groups (Page, infection = 0.77 at 5 V, 0.78 at 7 V, 0.73 at 10 V, 0.60 at 12 V, and 0.66 at 15 V; Fig. 1B). There was no significant effect on posttetanic potentiation (Page, infection = 0.8091; young/vehicle = 229.4 ± 20.4%, Young/E. coli = 209.2 ± 32.9%, aged/vehicle = 231.7 ± 40.6%, and aged/E. coli = 197.4 ± 18.3%; Fig. 2B). The initial statistical analysis of L-LTP revealed no significance in the combined effects of age and infection (Page, infection = 0.0757; percentage baseline 3 h after tetanus: young/vehicle = 155.4 ± 14.6%, young/E. coli = 156.2 ± 14.4%, aged/vehicle = 148.0 ± 5.9%, and aged/E. coli = 110.6 ± 5.7%; Fig. 2B). However, the group means and graphs suggest some remaining reduction in the L-LTP of aged E. coli animals; Tukey’s post hoc tests indicated that the aged E. coli group differed from young saline (p = 0.0169) and young E. coli (p = 0.0220). This was not the case when the aged E. coli group was compared to the aged saline group (p = 0.0542). Together, these results suggest significant, but incomplete, recovery in the capacity for L-LTP at 14 d.
Theta burst–evoked L-LTP had returned to control levels in aged animals 21 d after infection
We extended our investigation to 21 d to determine if the E. coli evoked deficits in L-LTP in the aged rats would fully resolve (Fig. 2C). There was no significant difference in post-tetanic potentiation at 21 d (aged/vehicle vs. aged/E. coli: p = 0.9081; aged/vehicle = 232.7 ± 23.9%, and aged/E. coli = 237.8 ± 23.3%). The results show normal levels of L-LTP in aged E. coli animals at this time point (aged/vehicle vs. aged/E. coli: p = 0.8232; percentage baseline 3 h after tetanus: aged/vehicle = 144.2 ± 6.9%, and aged/E. coli = 148.4 ± 14.3%).
Levels of the mature BDNF protein isoform were significantly reduced in hippocampal synaptoneurosomes prepared from aged rats 8 d, but not 14 d after infection
The forms of long-lasting memory and synaptic plasticity compromised by age and infection are highly dependent on BDNF (Tyler et al., 2002; Chao, 2003; Lu, 2003; Bramham and Messaoudi, 2005). We have therefore hypothesized that aging and infection might compromise production or processing of BDNF protein, reducing the availability of BDNF for memory-related plasticity processes at synaptic sites (Cortese et al., 2011). BDNF is synthesized as a precursor, proBDNF, which undergoes post-translational cleavage to produce mature BDNF (mBDNF), the protein isoform required for long-lasting forms of memory and LTP (Pang et al., 2004; Barnes and Thomas, 2008).
We previously demonstrated that levels of the mBDNF protein were significantly reduced in synaptoneurosomes prepared from the hippocampi of aged rats 4–5 d after injection of E. coli (Cortese et al., 2011). In this study, we examined later time points—8 and 14 d after injection—to determine if levels of mBDNF in aged animals would recover as levels of IL-1β dropped toward pre-infection baselines (Barrientos et al., 2009). Western blot analysis with an antibody against the mature domain of BDNF (Lee et al., 2001) supported this hypothesis. At 8 d, levels of mBDNF (Fig. 3A) were still significantly reduced in hippocampal synaptoneurosomes from the aged E. coli group compared to the other groups (t(18) = 2.3427, p = 0.0308). In contrast, levels of mBDNF were back to normal at 14 d (Fig. 4A), showing no statistical significance (t(14) = 0.0936, p = 0.9267). Meanwhile, Western blot analysis with an antibody against a specific proBDNF signal revealed no significant difference at 8 d (Fig. 3A; t(14) = 0.1983, p = 0.8457) or 14 d (Fig. 4A; t(10) = 0.4676, p = 0.6501).
Figure 3.
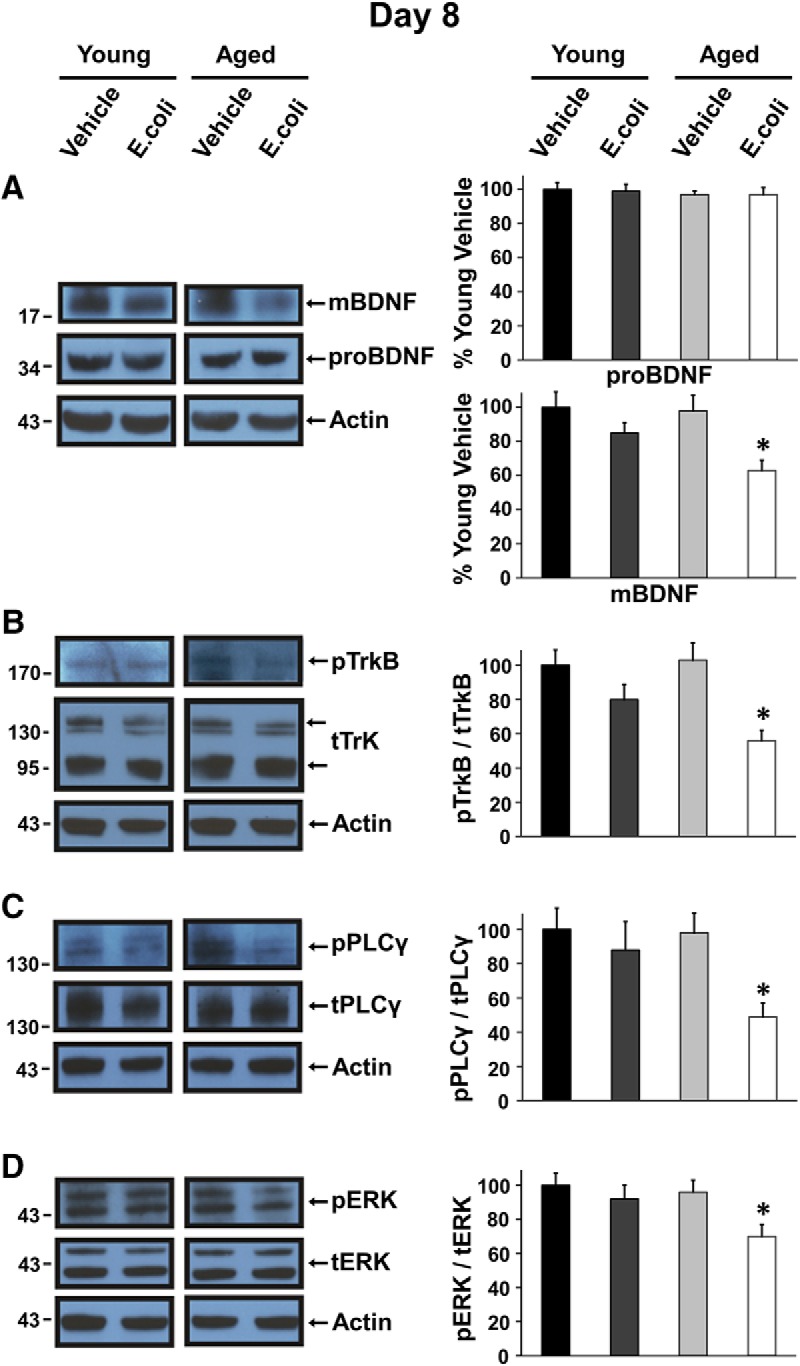
Levels of the mature BDNF protein isoform and activation of TrkB and downstream signaling are reduced in aged rats 8 d after infection. Western blot analysis was conducted using hippocampal synaptoneurosomes prepared from young and aged rats 8 d after injection of E. coli or vehicle; representative examples are shown. A, levels of mBDNF, but not proBDNF, are diminished in the aged animals 8 d after infection. B–D, Graphs present the ratio of phosphorylated TrkB (pTrkB), PLCγ1 (pPLCγ), or ERK (pERK) to total expression of TrkB (tTrkB), PLCγ1 (tPLCγ), or ERK (tERK). Levels of phosphorylated TrkB (B), PLC γ1 (C), and ERK (D) are significantly lower in the aged animals 8 d after infection. Protein bands were quantified using ImageJ, normalized to their actin controls, and expressed as percentages of mean protein levels from young vehicle-injected animals. Error bars represent SEM. All graphs here and below represent at least three independent experiments, with 1–2 animals per group in each experiment.
Figure 4.
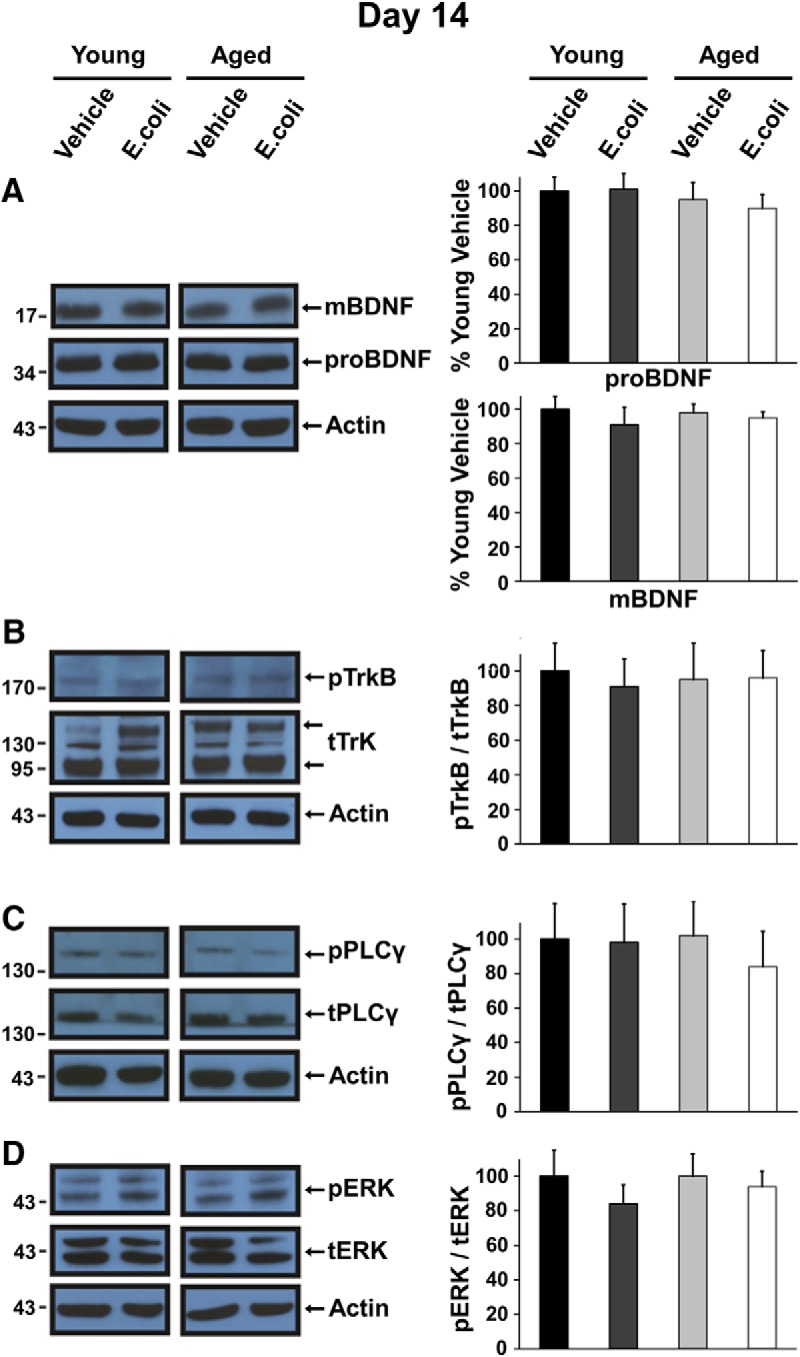
Levels of mBDNF and activation of TrkB, PLCγ1, and ERK in aged rats have returned to normal 14 d after E. coli injection. Western blot analysis of hippocampal synaptoneurosomes prepared from young and aged rats 14 d after injection of E. coli or vehicle; representative examples are shown. A–D, Aged E. coli animals show no significant difference in levels of mBDNF (A) or activation of TrkB (B), PLC γ1 (C), or ERK (D). Quantification was as described in the legend for Fig. 3.
Age and infection interact to reduce activation of TrkB and downstream signaling systems, but this effect is not permanent
Mature BDNF binds to the tropomyosin-related kinase B receptor (TrkB), triggering a cascade of phosphorylation events, starting with the receptor, which can activate downstream signaling pathways including the phospholipase C-γ1 (PLCγ1) and the Ras/extracellular signal-regulated kinase (ERK) pathways (Patapoutian and Reichardt, 2001; Segal, 2003). These pathways ultimately lead to the transcription and translation events required for L-LTP (Finkbeiner et al., 1997; Minichiello, 2009).
We previously reported significantly reduced levels of mBDNF in synaptoneurosomes prepared from the hippocampi of aged rats 4–5 d after injection of E. coli (Cortese et al., 2011). Consistent with the decreased availability of mBDNF, we also found significantly reduced activation of TrkB and the PLCγ1 and ERK downstream signaling pathways (Cortese et al., 2011). Here, we examined the impact of age and infection on activation of TrkB and downstream signaling 8 and 14 d after injection.
Analysis of Western blots using an antibody against phosphorylated TrkB (pTrkB) and an antibody against total TrkB (tTrkB; sc-8316 antibody; Santa Cruz Biotechnology) showed that the ratio of pTrkB/tTrkB was significantly reduced in synaptoneurosomes from aged E. coli rats at 8 d compared to the other groups (Fig. 3B; t(10) = 2.2692, p = 0.0466). At 14 d (Fig. 4B), levels of phospho-TrkB were back to baselines in the aged E. coli (t(10) = 0.0470, p = 0.9635).
Activation of PLCγ1 and ERK was also examined, and these results were consistent with the changes in levels of mBDNF and activation of TrkB. The ratio of phosphorylated PLCγ1 (pPLCγ1; 07-2134 antibody; Millipore) to total PLCγ1 (tPLCγ1; 05-366 antibody; Millipore) was significantly reduced in synaptoneurosomes of aged E. coli injected rats at 8 d (Fig. 3C; t(14) = 2.1706, p = 0.0477), but was back to baselines at 14 d (Fig. 4C; t(10) = 0.5000, p = 0.6279). The ratio of phosphorylated ERK (pERK; 910 antibody1; Cell Signaling) to total ERK (tERK; 9102 antibody; Cell Signaling) was profoundly reduced in synaptoneurosomes of aged E. coli injected rats at 8 d (Fig. 3D; t(18) = 2.5149, p = 0.0216) but back to normal at 14 d (Fig. 4D; t(10) = 0.0047, p = 0.9964).
Discussion
We have previously demonstrated that in 24-mo-old F344xBN rats, a single i.p. injection of E. coli triggers an exaggerated hippocampal production of IL-1β (Barrientos et al., 2009) that is associated with profound deficits in contextual fear conditioning, a hippocampus-dependent memory task (Barrientos et al., 2006), in theta burst–evoked L-LTP (Chapman et al., 2010), and in mBDNF/TrkB signaling (Cortese et al., 2011). Blunting the effects of IL-1β in the brains of aged animals using the IL-1 receptor antagonist IL-1Ra blocks all of these deficits (Chapman et al., 2010; Frank et al., 2010; Cortese et al., 2011). We have also determined that the elevation in IL-1β and the associated memory deficits do subside, but slowly—they last more than a week, but typically <2 weeks (Barrientos et al., 2009). Here we extend these observations, further exploring the strength of these correlations over time and asking if the infection-induced deficits in theta burst L-LTP and mBDNF will also subside and follow the same time course of recovery as the alterations in IL-1β and memory.
Our principle new findings are that (1) theta burst L-LTP remained profoundly compromised in aged animals 8 d after E. coli injection, but the much milder suppression observed in young animals 4–5 d after injection (Chapman et al., 2010) had resolved; there were also still significant deficits in mBDNF levels and signaling in the aged animals at this time point; (2) theta burst LTP in the aged animals showed significant, but incomplete, recovery 14 d after the E. coli injection, and mBDNF levels and signaling in aged animals were no longer significantly impaired; and (3) 21 d after the E. coli injection, theta burst LTP in the aged animals had completely recovered. Thus, the exaggerated elevation of IL-1β is precisely mirrored by the deficits in memory and in mBDNF/TrkB signaling: as levels of hippocampal IL-1β decline, memory and mBDNF/TrkB signaling recover. There was a slight lag in the full recovery of theta burst L-LTP, consistent with reports of a critical threshold level of BDNF being required to set the conditions necessary for full expression of BDNF-dependent forms of long-lasting synaptic plasticity such as theta burst L-LTP (Korte et al., 1995, 1996; Patterson et al., 1996; Pang et al., 2004).
Taken together, these results (summarized in Fig. 5) are consistent with the hypothesis that the exaggerated hippocampal inflammatory response produced by age and an infection might decrease availability of BDNF at hippocampal synapses, and thus contribute to deficits in forms of long-lasting memory and synaptic plasticity that require BDNF for their complete expression.
Figure 5.
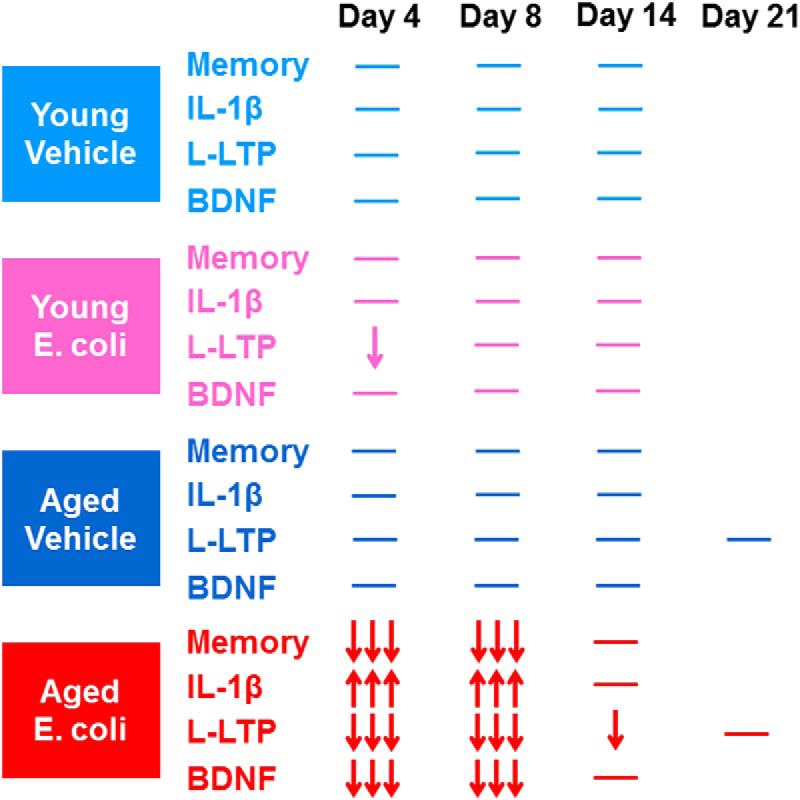
A summary of the effects of age and infection at multiple time points after injection of E. coli or saline. Memory = hippocampus-dependent long-term memory; IL-1β = levels of proinflammatory cytokine interleukin-1β in hippocampal synaptoneurosomes; L-LTP = theta burst–evoked L-LTP in the hippocampal CA1 area; and BDNF = levels of mature BDNF and activity of related proteins in hippocampal synaptoneurosomes. Upward arrows indicate an increase, and downward arrows show a reduction. Three arrows represent severe deficits, and one arrow means impairments to a lesser degree. Horizontal lines indicate baseline values. Data summarized for day 4 are drawn from earlier publications: Memory and IL-1β (Barrientos et al., 2006, 2009), L-LTP (Chapman et al., 2010), and BDNF (Cortese et al., 2011).
Many studies have examined the effects of normal aging on memory, synaptic plasticity, and BDNF signaling, with varied results. It is now recognized that variability in cognitive and synaptic functioning, and in BDNF signaling, increases with increasing age in individuals and populations. Aging is frequently, though not always, associated with some degree of cognitive decline and with disruptions in related forms of synaptic plasticity; there appears to be considerable variability depending on the experimental protocols used and the species, strains, and ages of the subjects (Landfield and Lynch, 1977; Gage et al., 1984; Barnes and McNaughton, 1985; Deupree et al., 1991; Diana et al., 1995; Gallagher et al., 2003; Tombaugh et al., 2005; Lynch et al., 2006; Sharma et al., 2015). Data from human autopsy studies and animal models examining variability in cognitive functioning with aging suggest that when deficits in hippocampus-dependent memory occur, they do not arise from a loss of hippocampal neurons, or initially even from a loss of synapses, but rather from more subtle alterations in synaptic efficacy (Lister and Barnes, 2009). For example, in hippocampal area CA1, the basic mechanisms for synaptic modification persist in old age, but the threshold for producing long-lasting, memory-related plasticity increases. High-frequency stimulation protocols can still induce L-LTP, but milder, more naturalistic types of stimulation are less likely to do so (Lynch et al., 2006). Because BDNF is a key mediator of synaptic efficacy in circuits critical for cognition (Bramham and Messaoudi, 2005; Leal et al., 2015), it has long been suspected that disruption of BDNF signaling systems might play a significant role in aging-associated cognitive decline. Somewhat surprisingly, it now appears that basal levels of BDNF and its receptor TrkB in the brain do not change very much as a result of aging alone (Pang and Lu, 2004), although significant changes are seen in some neurodegenerative diseases (Zuccato and Cattaneo, 2009).
Clearly, age is not the only important variable in age-related cognitive decline. The present results add to a growing body of evidence suggesting that much of this variability arises from complex interactions of age with genetics, lifestyle, and life history. Aging sensitizes the brain immune response (Godbout et al., 2005; Chen et al., 2008; Barrientos et al., 2009), and increases the vulnerability of systems for memory-related plasticity to immune challenges. This may represent an important source of variability in cognitive function in the aging brain.
Aberrantly elevated levels of proinflammatory molecules such as IL-1β can compromise memory and synaptic plasticity. Interleukin-1β, its receptor, and the natural IL-1 receptor antagonist are all present at relatively high levels in the hippocampus (Lechan et al., 1990; Takao et al., 1990; Ban et al., 1991). This expression pattern suggests that IL-1 signaling may play a significant role in modulating hippocampal functions, and that memory-related plasticity processes in the hippocampus may be particularly vulnerable to dysregulated IL-1 signaling. This may be particularly true in aging, since sensitivity to IL-1β is augmented in aged hippocampal synapses (Prieto et al., 2015). Low basal levels of IL-1β appear to be required for long-term memory and synaptic plasticity in the healthy hippocampus (Yirmiya and Goshen, 2011). However, performance on hippocampus-dependent memory tasks can be seriously compromised by manipulations that result in too much IL-1β. These include intraventricular (Oitzl et al., 1993) and hippocampal (Barrientos et al., 2002) IL-1 administration, multiweek elevation of IL-1β in the hippocampi of transgenic mice (Hein et al., 2010), and elevation of endogenous IL-1β evoked by infections (Gibertini et al., 1995; Barrientos et al., 2006; Chen et al., 2008) or psychological stressors (Pugh et al., 1999). Similarly, in young rodents, experimental elevation of IL-1β can block full expression of LTP in several areas of the hippocampus (Lynch, 2010). Application of high levels of IL-1β to rodent hippocampal slices reduced LTP in areas CA1 (Bellinger et al., 1993; Ross et al., 2003) and CA3 (Katsuki et al., 1990) and in the dentate (Cunningham et al., 1996; Coogan and O’Connor, 1997). In vivo LTP in the dentate was inhibited by intraventricular injection of IL-1β (Murray and Lynch, 1998; Kelly et al., 2003) or an i.p. injection of LPS, a potent endotoxin that triggers strong immune responses (Lynch, 2004; Barry et al., 2005).
The memory and plasticity processes compromised by excess IL-1β are highly dependent on BDNF, and there is increasing evidence that experimental elevation of proinflammatory cytokines in the brain can diminish the availability of BDNF—potentially from both neuronal and microglial (Parkhurst et al., 2013) sources—for memory-related processes (Patterson, 2015). Infusing IL-1β into the hippocampus decreases its capacity for transcription of BDNF after fear learning (Barrientos et al., 2004), while infusion of IL-1Ra protects it during social isolation stress (Barrientos et al., 2003). Perhaps not surprisingly, intraperitoneal injection of high levels of IL-1β or LPS produced an acute (within 4 h) reduction in hippocampal BDNF mRNA (Lapchak et al., 1993). However, expression of specific activity– and plasticity-associated BDNF mRNA transcripts, and the capacity to recruit these transcripts after fear learning, was still reduced in the hippocampi of aged rats 4 d after i.p. E. coli (Chapman et al., 2012). Aberrantly elevated levels of cytokines also appear to compromise production of the BDNF protein and downstream signaling. Levels of BDNF protein in the hippocampus showed a dose-dependent reduction 7 h after i.p. LPS (Guan and Fang, 2006). A high dose of LPS injected i.p. into young mice is reported to produce a small (15%) reduction in both proBDNF and mature BDNF in brain synaptoneurosomes 3 d later (Schnydrig et al., 2007). Intraperitoneal injection of E. coli produces a large reduction in mBDNF and TrkB signaling in hippocampal synaptoneurosomes from aged rats 4–5 d later (Cortese et al., 2011). There are also indications that excessive IL-1β may sometimes interfere with the neuroprotective effect of BDNF-induced signal transduction, in addition to compromising its plasticity-related functions (Tong et al., 2008, 2012; Cortese et al., 2011; Chapman et al., 2012).
The hypothesis that exaggerated brain inflammatory responses might disrupt BNDF-dependent synaptic plasticity and neuroprotective processes has broad implications for understanding, preventing, or treating cognitive dysfunction in a variety of disorders associated with neuroinflammation or dysregulated brain immune responses, but may be particularly informative in the context considered here. Very few studies have focused on the mechanisms of acute cognitive decline (termed delirium) following an inflammatory event, despite its clinical prevalence and association with markedly increased risk of progression to and acceleration of dementia (Fong et al., 2009; Cunningham, 2011). The immune challenge–triggered cognitive decline we model here in rodents resembles that observed in human delirium. There is a common aging-associated vulnerability, and the pathology shares a similar trajectory and time course; there may well be elements of a common etiology.
Synthesis
Reviewing Editor: Alfonso Araque, University of Minnesota
Decisions are customarily a result of the Reviewing Editor and the peer reviewers coming together and discussing their recommendations until a consensus is reached. When revisions are invited, a fact-based synthesis statement explaining their decision and outlining what is needed to prepare a revision will be listed below. The following reviewer(s) agreed to reveal their identity: Jonathan Godbout, Sreedharan Sajikumar
The reviewers have found merit and interest on the findings reported. I concur with the reviewers' comments expressing the interest of the study. However, they also expressed some concerns about several issues that need to be addressed and clarified. I also consider pertinent the concerns expressed. We believe that addressing those concerns, which may require less than 2 months' work and will definitely improve the manuscript, is necessary to firmly validate the results.
Specific comments of the reviewers:
Reviewer 1.
The combination of age and an immune challenge trigger an exaggerated inflammatory state in the brain, which in turn disrupts memory-related synaptic plasticity. This mechanism is important for understanding inflammation-driven failures in memory-related synaptic plasticity. The authors, Tanaka et al., used electrophysiology and protein assays to determine the effects of age and E. coli infection on long-term potentiation (LTP). This manuscript, titled "Aging and an immune challenge interact to produce prolonged, but not permanent reductions in hippocampal L-LTP and mBDNF in a rodent model with features of delirium", follows up with a previous paper examining these effects at 4-5 days post-infection, continuing their studies to 8 and 14 days post-infection. These data suggest the deficits in late-phase LTP (L-LTP) and mBDNF/TrkB signaling persist at 8 days post-infection in aging mice, but they are (for the most part) resolved by 14 days post-infection.
1. Along with the main effects and interactions for the electrophysiology data, please provide the results of the post-hoc analysis. Additionally, the insignificance in Figure 1B seems to come from increased error in the Aged/Vehicle group, as all other means do not seem to change from Day 8 to 14. Treating time as a variable may increase the power of this analysis (show L-LTP remains depressed until Day 21).
2. More statistics are required for Western blot analysis. The methods state, "The p value listed for each protein (or phosphorylation state ratio) is for an unpaired t test comparing the mean of the aging + E. coli group to the mean of the summed values of the other test groups." First, the word "summed" should be substituted with "combined". Second, ANOVA and post hoc comparisons should be performed to ensure the combined groups are not significantly different from one another if this combination is indeed to be performed. This approach also calls into question why a standard ANOVA comparison is not being performed to ensure the difference is not due to a main effect of age that is lost when combining two young groups with one aging group. Moreover, the statistics reported for Figure 3D do not seem correct. It is claimed that the levels of pERK are "back to normal" with p = 0.0047, which indicates a highly significant difference.
3. Why are Adult animals omitted from the Day 21 electrophysiology? This changes the statistical analyses between time-points.
4. The Results subsections for electrophysiology could be combined into a single subsection to make result presentation more fluid.
5. The Western blot bar graph legends do not fit the graphs if the bar colors are all different. The pro- vs. m-BDNF bars should also be separated, as the comparison being highlighted in this figure is pro- vs. m-BDNF, not age or infection as it should be.
6. The manuscript contains several typographical errors and inconsistencies that should be addressed (e.g. "tetanous", Molar = "M" not "m", "synaptic makers", "interact to reductions", "infecxtion", "significance effect", "key mediatory").
Reviewer 2.
The study by Tanaka et al., show that brain inflammatory response elicited by intraperitoneal injection of E. coli has an effect on long-term plasticity in aging and is dependent on the availability of mBDNF.
The study is interesting and well designed. The authors should address the following points to improve the impact of their studies:
1) It has been reported earlier that microglial BDNF increases neuronal TrkB phosphorylation (PMID: 2436028), thus it would helpful if the authors should specify whether the BNDF changes observed in their studies after intraperitoneal injection of E. coli is due to microglia activation or due to neuronal changes.
2) The authors show that the magnitude of TBS- LTP is identical in young and aged vehicle (Figure 1 A & B). There are many studies reported earlier the depleted level of BDNF with aging thus the authors should observe a decrease in the magnitude of TBS-LTP. Indeed , using strong tetanization protocol it has been demonstrated earlier that the magnitude of late- LTP is less in aged rats compared to young , in addition synaptic tagging and capture processes were also impaired (PMID: 26577291 , PMID: 28065806). It should be helpful for the readers to include this point in the discussion.
3) The authors stated that they did not observe any changes in the basal synaptic responses in young and aged animals. Interestingly the analog traces show drastic changes in the presynaptic (fibre) volley. For instance in Figure. 1 A and B group YV and AL groups. These differences provide an indication that stimulation intensities were not within a comparable range. It would be helpful to provide a graph showing input -output relations of different groups to further confirm that the basal synaptic transmission were identical in all groups.
References
- Ban E, Milon G, Prudhomme N, Fillion G, Haour F (1991) Receptors for interleukin-1 (alpha and beta) in mouse brain: mapping and neuronal localization in hippocampus. Neuroscience 43:21–30. [DOI] [PubMed] [Google Scholar]
- Barnes CA, McNaughton BL (1985) An age comparison of the rates of acquisition and forgetting of spatial information in relation to long-term enhancement of hippocampal synapses. Behav Neurosci 99:1040–1048. [DOI] [PubMed] [Google Scholar]
- Barnes P, Thomas KL (2008) Proteolysis of proBDNF is a key regulator in the formation of memory. PLoS One 3:e3248. 10.1371/journal.pone.0003248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Frank MG, Hein AM, Higgins EA, Watkins LR, Rudy JW, Maier SF (2009) Time course of hippocampal IL-1 beta and memory consolidation impairments in aging rats following peripheral infection. Brain Behav Immun 23:46–54. 10.1016/j.bbi.2008.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos RM, Higgins EA, Biedenkapp JC, Sprunger DB, Wright-Hardesty KJ, Watkins LR, Rudy JW, Maier SF (2006) Peripheral infection and aging interact to impair hippocampal memory consolidation. Neurobiol Aging 27:723–732. 10.1016/j.neurobiolaging.2005.03.010 [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Higgins EA, Sprunger DB, Watkins LR, Rudy JW, Maier SF (2002) Memory for context is impaired by a post context exposure injection of interleukin-1 beta into dorsal hippocampus. Behav Brain Res 134:291–298. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Sprunger DB, Campeau S, Higgins EA, Watkins LR, Rudy JW, Maier SF (2003) Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist. Neuroscience 121:847–853. [DOI] [PubMed] [Google Scholar]
- Barrientos RM, Sprunger DB, Campeau S, Watkins LR, Rudy JW, Maier SF (2004) BDNF mRNA expression in rat hippocampus following contextual learning is blocked by intrahippocampal IL-1beta administration. J Neuroimmunol 155:119–126. 10.1016/j.jneuroim.2004.06.009 [DOI] [PubMed] [Google Scholar]
- Barry CE, Nolan Y, Clarke RM, Lynch A, Lynch MA (2005) Activation of c-Jun-N-terminal kinase is critical in mediating lipopolysaccharide-induced changes in the rat hippocampus. J Neurochem 93:221–231. 10.1111/j.1471-4159.2004.03011.x [DOI] [PubMed] [Google Scholar]
- Bellinger FP, Madamba S, Siggins GR (1993) Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res 628:227–234. [DOI] [PubMed] [Google Scholar]
- Bliss T, Collingridge G, Morris RG (2007) Synaptic Plasticity in the Hippocampus In: The Hippocampus Book (Andersen P, Morris RG, Amaral D, O’Keefe J, eds), pp 343–475. Oxford, NY: Oxford University Press. [Google Scholar]
- Booth RF, Clark JB (1978) A rapid method for the preparation of relatively pure metabolically competent synaptosomes from rat brain. Biochem J 176:365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E (2005) BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol 76:99–125. 10.1016/j.pneurobio.2005.06.003 [DOI] [PubMed] [Google Scholar]
- Chao MV (2003) Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci 4:299–309. 10.1038/nrn1078 [DOI] [PubMed] [Google Scholar]
- Chapman TR, Barrientos RM, Ahrendsen JT, Hoover JM, Maier SF, Patterson SL (2012) Aging and infection reduce expression of specific brain-derived neurotrophic factor mRNAs in hippocampus. Neurobiol Aging 33:832.e1–814. 10.1016/j.neurobiolaging.2011.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman TR, Barrientos RM, Ahrendsen JT, Maier SF, Patterson SL (2010) Synaptic correlates of increased cognitive vulnerability with aging: peripheral immune challenge and aging interact to disrupt theta-burst late-phase long-term potentiation in hippocampal area CA1. J Neurosci 30:7598–7603. 10.1523/JNEUROSCI.5172-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW (2008) Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun 22:301–311. 10.1016/j.bbi.2007.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coogan A, O’Connor JJ (1997) Inhibition of NMDA receptor-mediated synaptic transmission in the rat dentate gyrus in vitro by IL-1 beta. Neuroreport 8:2107–2110. [DOI] [PubMed] [Google Scholar]
- Cortese GP, Barrientos RM, Maier SF, Patterson SL (2011) Aging and a peripheral immune challenge interact to reduce mature brain-derived neurotrophic factor and activation of TrkB, PLCgamma1, and ERK in hippocampal synaptoneurosomes. J Neurosci 31:4274–4279. 10.1523/JNEUROSCI.5818-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C (2011) Systemic inflammation and delirium: important co-factors in the progression of dementia. Biochem Soc Trans 39:945–953. 10.1042/BST0390945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham AJ, Murray CA, O’Neill LA, Lynch MA, O’Connor JJ (1996) Interleukin-1 beta (IL-1 beta) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci Lett 203:17–20. 10.1016/0304-3940(95)12252-4 [DOI] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008) From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deupree DL, Turner DA, Watters CL (1991) Spatial performance correlates with in vitro potentiation in young and aged Fischer 344 rats. Brain Res 554:1–9. [DOI] [PubMed] [Google Scholar]
- Diana G, Domenici MR, Scotti de Carolis A, Loizzo A, Sagratella S (1995) Reduced hippocampal CA1 Ca(2+)-induced long-term potentiation is associated with age-dependent impairment of spatial learning. Brain Res 686:107–110. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19:1031–1047. 10.1016/S0896-6273(00)80395-5 [DOI] [PubMed] [Google Scholar]
- Fong TG, Tulebaev SR, Inouye SK (2009) Delirium in elderly adults: diagnosis, prevention and treatment. Nat Rev Neurol 5:210–220. 10.1038/nrneurol.2009.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MG, Barrientos RM, Hein AM, Biedenkapp JC, Watkins LR, Maier SF (2010) IL-1RA blocks E. coli-induced suppression of Arc and long-term memory in aged F344xBN F1 rats. Brain Behav Immun 24:254–262. 10.1016/j.bbi.2009.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH, Dunnett SB, Björklund A (1984) Spatial learning and motor deficits in aged rats. Neurobiol Aging 5:43–48. [DOI] [PubMed] [Google Scholar]
- Gallagher M, Bizon JL, Hoyt EC, Helm KA, Lund PK (2003) Effects of aging on the hippocampal formation in a naturally occurring animal model of mild cognitive impairment. Exp Gerontol 38:71–77. 10.1016/S0531-5565(02)00159-6 [DOI] [PubMed] [Google Scholar]
- Gibertini M, Newton C, Friedman H, Klein TW (1995) Spatial learning impairment in mice infected with Legionella pneumophila or administered exogenous interleukin-1-beta. Brain Behav Immun 9:113–128. 10.1006/brbi.1995.1012 [DOI] [PubMed] [Google Scholar]
- Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, Johnson RW (2005) Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J 19:1329–1331. [DOI] [PubMed] [Google Scholar]
- Guan Z, Fang J (2006) Peripheral immune activation by lipopolysaccharide decreases neurotrophins in the cortex and hippocampus in rats. Brain Behav Immun 20:64–71. 10.1016/j.bbi.2005.04.005 [DOI] [PubMed] [Google Scholar]
- Hein AM, Stasko MR, Matousek SB, Scott-McKean JJ, Maier SF, Olschowka JA, Costa AC, O’Banion MK (2010) Sustained hippocampal IL-1beta overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav Immun 24:243–253. 10.1016/j.bbi.2009.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuki H, Nakai S, Hirai Y, Akaji K, Kiso Y, Satoh M (1990) Interleukin-1 beta inhibits long-term potentiation in the CA3 region of mouse hippocampal slices. Eur J Pharmacol 181:323–326. [DOI] [PubMed] [Google Scholar]
- Kelly A, Vereker E, Nolan Y, Brady M, Barry C, Loscher CE, Mills KH, Lynch MA (2003) Activation of p38 plays a pivotal role in the inhibitory effect of lipopolysaccharide and interleukin-1 beta on long term potentiation in rat dentate gyrus. J Biol Chem 278:19453–19462. 10.1074/jbc.M301938200 [DOI] [PubMed] [Google Scholar]
- Konsman JP, Parnet P, Dantzer R (2002) Cytokine-induced sickness behaviour: mechanisms and implications. Trends Neurosci 25:154–159. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T (1995) Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A 92:8856–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T (1996) Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci U S A 93:12547–12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landfield PW, Lynch G (1977) Impaired monosynaptic potentiation in in vitro hippocampal slices from aged, memory-deficient rats. J Gerontol 32:523–533. [DOI] [PubMed] [Google Scholar]
- Lapchak PA, Araujo DM, Hefti F (1993) Systemic interleukin-1 beta decreases brain-derived neurotrophic factor messenger RNA expression in the rat hippocampal formation. Neuroscience 53:297–301. [DOI] [PubMed] [Google Scholar]
- Leal G, Afonso PM, Salazar IL, Duarte CB (2015) Regulation of hippocampal synaptic plasticity by BDNF. Brain Res 1621:82–101. 10.1016/j.BrainRes.2014.10.019 [DOI] [PubMed] [Google Scholar]
- Lechan RM, Toni R, Clark BD, Cannon JG, Shaw AR, Dinarello CA, Reichlin S (1990) Immunoreactive interleukin-1 beta localization in the rat forebrain. Brain Res 514:135–140. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL (2001) Regulation of cell survival by secreted proneurotrophins. Science 294:1945–1948. 10.1126/science.1065057 [DOI] [PubMed] [Google Scholar]
- Lister JP, Barnes CA (2009) Neurobiological changes in the hippocampus during normative aging. Arch Neurol 66:829–833. 10.1001/archneurol.2009.125 [DOI] [PubMed] [Google Scholar]
- Lu B (2003) BDNF and activity-dependent synaptic modulation. Learn Mem 10:86–98. 10.1101/lm.54603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA (2004) Long-term potentiation and memory. Physiol Rev 84:87–136. 10.1152/physrev.00014.2003 [DOI] [PubMed] [Google Scholar]
- Lynch MA (2010) Age-related neuroinflammatory changes negatively impact on neuronal function. Front Aging Neurosci 1:6. 10.3389/neuro.24.006.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Gall CM (2006) Synaptic plasticity in early aging. Ageing Res Rev 5:255–280. 10.1016/j.arr.2006.03.008 [DOI] [PubMed] [Google Scholar]
- Maier SF, Watkins LD, Nance DM (2001) Multiple routes of action of interleukin-1 on the nervous system. In: Psychoneuroimmunology, 3rd Edition (Ader R, Felton DL, Cohen N, eds), pp 563–583. New York: Academic Press. [Google Scholar]
- Minichiello L (2009) TrkB signalling pathways in LTP and learning. Nat Rev Neurosci 10:850–860. 10.1038/nrn2738 [DOI] [PubMed] [Google Scholar]
- Murray CA, Lynch MA (1998) Evidence that increased hippocampal expression of the cytokine interleukin-1 beta is a common trigger for age- and stress-induced impairments in long-term potentiation. J Neurosci 18:2974–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oitzl MS, van Oers H, Schöbitz B, de Kloet ER (1993) Interleukin-1 beta, but not interleukin-6, impairs spatial navigation learning. Brain Res 613:160–163. [DOI] [PubMed] [Google Scholar]
- O’Keefe J (2007) Hippocampal Neurophysiology In the Behaving Animal In: The Hippocampus Book (Andersen P, Morris RG, Amaral D, O’Keefe J, eds), pp 475–549. Oxford, NY: Oxford University Press. [Google Scholar]
- Pang PT, Lu B (2004) Regulation of late-phase LTP and long-term memory in normal and aging hippocampus: role of secreted proteins tPA and BDNF. Ageing Res Rev 3:407–430. 10.1016/j.arr.2004.07.002 [DOI] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B (2004) Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 306:487–491. 10.1126/science.1100135 [DOI] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155:1596–1609. 10.1016/j.cell.2013.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF (2001) Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol 11:272–280. [DOI] [PubMed] [Google Scholar]
- Patterson SL (2015) Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1beta, BDNF and synaptic plasticity. Neuropharmacology 96:11–18. 10.1016/j.neuropharm.2014.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER (1996) Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16:1137–1145. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M (1992) Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron 9:1081–1088. [DOI] [PubMed] [Google Scholar]
- Prieto GA, Snigdha S, Baglietto-Vargas D, Smith ED, Berchtold NC, Tong L, Ajami D, LaFerla FM, Rebek J, Jr, Cotman CW (2015) Synapse-specific IL-1 receptor subunit reconfiguration augments vulnerability to IL-1beta in the aged hippocampus. Proc Natl Acad Sci U S A 112:E5078–E5087. 10.1073/pnas.1514486112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh CR, Nguyen KT, Gonyea JL, Fleshner M, Wakins LR, Maier SF, Rudy JW (1999) Role of interleukin-1 beta in impairment of contextual fear conditioning caused by social isolation. Behav Brain Res 106:109–118. [DOI] [PubMed] [Google Scholar]
- Ross FM, Allan SM, Rothwell NJ, Verkhratsky A (2003) A dual role for interleukin-1 in LTP in mouse hippocampal slices. J Neuroimmunol 144:61–67. [DOI] [PubMed] [Google Scholar]
- Schnydrig S, Korner L, Landweer S, Ernst B, Walker G, Otten U, Kunz D (2007) Peripheral lipopolysaccharide administration transiently affects expression of brain-derived neurotrophic factor, corticotropin and proopiomelanocortin in mouse brain. Neurosci Lett 429:69–73. 10.1016/j.neulet.2007.09.067 [DOI] [PubMed] [Google Scholar]
- Segal RA (2003) Selectivity in neurotrophin signaling: theme and variations. Annu Rev Neurosci 26:299–330. 10.1146/annurev.neuro.26.041002.131421 [DOI] [PubMed] [Google Scholar]
- Sharma M, Shetty MS, Arumugam TV, Sajikumar S (2015) Histone deacetylase 3 inhibition re-establishes synaptic tagging and capture in aging through the activation of nuclear factor kappa B. Sci Rep 5:16616. 10.1038/srep16616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao T, Tracey DE, Mitchell WM, De Souza EB (1990) Interleukin-1 receptors in mouse brain: characterization and neuronal localization. Endocrinology 127:3070–3078. 10.1210/endo-127-6-3070 [DOI] [PubMed] [Google Scholar]
- Tombaugh GC, Rowe WB, Chow AR, Michael TH, Rose GM (2002) Theta-frequency synaptic potentiation in CA1 in vitro distinguishes cognitively impaired from unimpaired aged Fischer 344 rats. J Neurosci 22:9932–9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombaugh GC, Rowe WB, Rose GM (2005) The slow afterhyperpolarization in hippocampal CA1 neurons covaries with spatial learning ability in aged Fisher 344 rats. J Neurosci 25:2609–2616. 10.1523/JNEUROSCI.5023-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L, Balazs R, Soiampornkul R, Thangnipon W, Cotman CW (2008) Interleukin-1 beta impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol Aging 29:1380–1393. 10.1016/j.neurobiolaging.2007.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L, Prieto GA, Kramár EA, Smith ED, Cribbs DH, Lynch G, Cotman CW (2012) Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1beta via p38 mitogen-activated protein kinase. J Neurosci 32:17714–17724. 10.1523/JNEUROSCI.1253-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler WJ, Alonso M, Bramham CR, Pozzo-Miller LD (2002) From acquisition to consolidation: on the role of brain-derived neurotrophic factor signaling in hippocampal-dependent learning. Learn Mem 9:224–237. 10.1101/lm.51202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker VP (1993) Thirty years of synaptosome research. J Neurocytol 22:735–742. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I (2011) Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25:181–213. 10.1016/j.bbi.2010.10.015 [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E (2009) Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol 5:311–322. 10.1038/nrneurol.2009.54 [DOI] [PubMed] [Google Scholar]