

Abstract
We report the development of a quantum dot (QD)–peptide–fullerene (C60) electron transfer (ET)-based nanobioconjugate for the visualization of membrane potential in living cells. The bioconjugate is composed of (1) a central QD electron donor, (2) a membrane-inserting peptidyl linker, and (3) a C60 electron acceptor. The photoexcited QD donor engages in ET with the C60 acceptor, resulting in quenching of QD photoluminescence (PL) that tracks positively with the number of C60 moieties arrayed around the QD. The nature of the QD-capping ligand also modulates the quenching efficiency; a neutral ligand coating facilitates greater QD quenching than a negatively charged carboxylated ligand. Steady-state photophysical characterization confirms an ET-driven process between the donor–acceptor pair. When introduced to cells, the amphiphilic QD–peptide–C60 bioconjugate labels the plasma membrane by insertion of the peptide–C60 portion into the hydrophobic bilayer, while the hydrophilic QD sits on the exofacial side of the membrane. Depolarization of cellular membrane potential augments the ET process, which is manifested as further quenching of QD PL. We demonstrate in HeLa cells, PC12 cells, and primary cortical neurons significant QD PL quenching (ΔF/F0 of 2–20% depending on the QD–C60 separation distance) in response to membrane depolarization with KCl. Further, we show the ability to use the QD–peptide–C60 probe in combination with conventional voltage-sensitive dyes (VSDs) for simultaneous two-channel imaging of membrane potential. In in vivo imaging of cortical electrical stimulation, the optical response of the optimal QD–peptide–C60 configuration exhibits temporal responsivity to electrical stimulation similar to that of VSDs. Notably, however, the QD–peptide–C60 construct displays 20- to 40-fold greater ΔF/F0 than VSDs. The tractable nature of the QD–peptide–C60 system offers the advantages of ease of assembly, large ΔF/F0, enhanced photostability, and high throughput without the need for complicated organic synthesis or genetic engineering, respectively, that is required of traditional VSDs and fluorescent protein constructs.
Keywords: quantum dot, peptide, fullerene, membrane potential, voltage, neuron, electron transfer
Graphical abstract
The nanoparticle (NP)–biological interface continues to be an intensely studied research frontier with a primary goal being the realization of functional hybrid nano-scale materials that possess enhanced or augmented capabilities.1,2 Examples of such functionalities range from the use of the plasmonic thermal profile of gold nanoparticles (AuNPs) and nanoshells to specifically kill targeted cancer cells with incident light irradiation3,4 to the employment of semiconductor nanocrystals or quantum dots (QDs) as a scaffold to facilitate the enhanced activity of appended enzymes.5,6 Indeed, in both of these examples it is the inherent physicochemical properties of the NPs that facilitates the desired function. One arena in which the inherent properties of NPs have been predicted to make a significant impact is the real-time visualization of cellular membrane potential in electrically active cells (e.g., neurons and muscle cells). The primary goal of the United States’ Brain Research through Advancing Innovative Neurotechnologies (BRAIN) Initiative is to create a functional map of the human brain connectome—all of the interconnections of the tens of billions of neurons in the brain—as a means to better understand the brain’s function and dysfunction during disease and injury.7 The European Commission’s counterpart Human Brain Project seeks to replicate this brain activity map in silico.8 Success here could enable the development of the next generation of diagnostic and therapeutic reagents for the treatment of a range of brain pathologies. One of the key questions that has been raised, however, is whether or not the neuroscience community possesses the proper tools capable of visualizing the activity of thousands of cells simultaneously while maintaining single-cell resolution.9–12
Much of the driving force for this stems from the inherent limitations of currently available tools and techniques for the imaging/recording of membrane potential from large numbers of cells.13 For example, patch clamp, long the gold standard laboratory technique for recording and controlling membrane potential, is invasive, and it is limited to the interrogation of single cells.14 Microelectrode-based techniques are similarly invasive and record membrane potential extracellularly, which prohibits single-cell resolution.15 Fluorescence-based materials such as voltage-sensitive dyes (VSDs)16 and fluorescent protein constructs12 suffer from small changes in photoluminescence (PL) intensity (ΔF/F0), limited photostability, toxicity, or the requirement that cells be genetically manipulated to express the sensing construct. Further, Ca2+-sensitive fluorescent constructs measure intracellular Ca2+ release that occurs hundreds of milliseconds after the action potential and, thus, lack temporal resolution.17 One of the more promising genetically encoded voltage sensors are those based on archaerhodopsin, which display large PL changes (∼1% ΔF/F0 per mV) and can be used combinatorially with other spectrally resolvable opsin channel actuators for all-optical simultaneous stimulation and recording.18,19 Despite these advances, significant interest has arisen in the potential for NP materials to address some of the aforementioned critical issues.20
One particular class of NPs under consideration for the optical imaging of membrane potential are QDs, fluorescent materials synthesized on the nanoscale (<10 nm diameter) that exhibit quantum confinement. These nanocrystalline materials possess a number of photophysical attributes that make them ideal for imaging modulations in membrane potentials. These include QDs’ (1) bright, stable PL coupled with nanosecond fluorescence lifetimes, (2) quantum confined Stark effect in response to an electric field,21,23 (3) ability to be appended onto24,25 or localized in26 the plasma membrane bilayer, (4) large two-photon action cross sections that make them ideal for deep tissue imaging,27,28 (5) ability to engage in efficient Förster resonance energy transfer (FRET)29,30 and electron transfer (ET) processes,31,32 and (6) low toxicity when implemented with experimental brain platforms.33–36
Several reports, both theoretical37 and experimental,21–23 support the notion that QDs can function as voltage-sensitive probes. Indeed, recent work from our group showed the ability of photoexcited quasi-type II QDs (QDs with spatial segregation of electron–hole (e–h) pairs) to track, with excellent time-resolved fidelity, the millisecond voltage trace of a simulated firing cortical neuron.38 Yet, studies demonstrating the use of plasma membrane-inserted QDs for voltage sensing remain scant.39 This is largely due to the critical technical challenge of partitioning QDs coated with hydrophobic surface ligands into the ∼4 nm thick aliphatic region of the plasma membrane without perturbing membrane integrity or the function of membrane channel proteins responsible for maintaining membrane potential.2,26
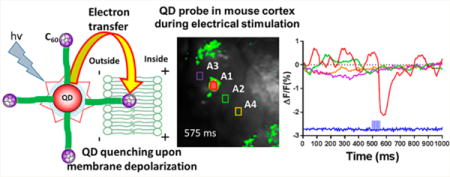
To overcome this technical hurdle, here we have designed QD–peptide–electron acceptor bioconjugates that obviate the need for direct insertion of the QD into the membrane bilayer. In the construct, a central hydrophilic QD (electron donor) serves as a scaffold for the self-assembly of multiple copies of a peptide–C60 fullerene conjugate (electron acceptor). This work builds on our previous observation that such QD–peptide–C60 conjugates engage in efficient ET-based quenching of QD PL that tracks with the number of peptide–C60 moieties displayed around the QD.31 The helix-forming peptide that appends the C60 to the QD is designed to facilitate membrane insertion such that the hydrophobic C60 is buried within the lipid bilayer of the membrane while the hydrophilic QD is localized on the extracellular face of the exofacial leaflet of the plasma membrane. Upon depolarization of the membrane potential, the basal rate of ET from the photoexcited QD electron donor to the fullerene acceptor is enhanced, resulting in the further quenching of QD PL, the magnitude of which tracks with the increasing potential of the cell’s plasma membrane (Figure 1C).
Figure 1.
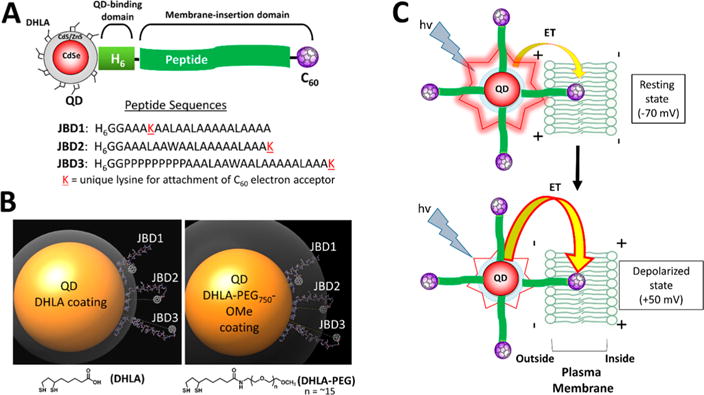
QD–peptide–C60 bioconjugates. (A) Modular design of QD–peptide–C60 bioconjugates. The core/shell QD serves as a central scaffold to which is appended peptides bearing a C60 fullerene at discrete positions. A unique lysine (K) allows for the controlled placement of the C60 moiety at different distances from the QD surface. An N-terminal polyhistidine tract (H6) mediates self-assembly of the peptide to the QD shell. Peptide sequences are written in N → C orientation. (B) Molecular models of QD–peptide–C60 bioconjugates. Shown is the 605 nm emitting core/shell QD (yellow sphere; 100 Å in diameter) capped with DHLA (left panel) and DHLA–PEG750–OMe (right panel) ligands for water solubility. The ligand layer is shown in gray and is 11 Å (DHLA) or 33 Å thick (DHLA–PEG750–OMe). Each QD is shown appended with peptides JBD1, JBD2, and JBD3 wherein the C60 electron acceptor is iteratively positioned at increasingly further distances from the QD surface. The predicted surface-to-surface distances from the QD to the C60 surface are as follows: JBD1, 10 Å; JBD2, 24 Å; JBD3, 42 Å. (C) Schematic of the response of QD–peptide–C60 bioconjugates to changes in membrane potential. Multiple peptide–C60 conjugates are arrayed around the central QD (electron donor), and they all engage in electron transfer (ET) with the QD; a percentage of the arrayed peptide–C60 are inserted into the membrane bilayer and contribute to depolarization-induced PL quenching. At resting potential, minimal ET from the photoexcited QD to those C60 embedded in the membrane bilayer results in bright QD emission (top). Depolarization of the membrane potential augments the rate of ET, causing a decrease in QD PL (bottom).
We present here the detailed photophysical analysis of this QD–peptide–C60 system both outside the cellular environment and when interfaced with the plasma membrane of cultured cell lines and primary neurons in vitro and in live mice in vivo. Our data clearly demonstrates that the efficiency of quenching of QD PL by the C60 electron acceptor is governed by the number of peptide–C60 conjugates arrayed around the central QD and the nature of the hydrophilic QD-capping ligand. In HeLa cells, the optimal QD–peptide–C60 construct exhibits a ∼20% ΔF/F0 upon membrane depolarization, which is comparable to a commercially available fluorescein–aniline photoinduced electron transfer voltage imaging probe under the same conditions. In primary mouse cortical neurons, the QD–peptide–C60 construct exhibits a ΔF/F0 of ∼7%. Importantly, cellular viabilities of >90% are observed in these studies, demonstrating minimal perturbation of cellular physiology. Finally, we employ the optimal QD–peptide–C60 probe for the real-time imaging of cortical electrical stimulation in live mice. Here, the fluorescence response of the QD–peptide–C60 probe exhibits temporal responsivity comparable to standard VSDs, yet with ΔF/F0 values that are 20- to 40-fold greater than those reported for VSDs.
RESULTS AND DISCUSSION
Rationale for QD–Peptide–C60-Based Membrane Potential Sensing Scheme
Our goal in this study was to design voltage-sensitive QD bioconjugates that engage in ET and can be interfaced with the plasma membrane of living cells such that membrane depolarization modulates ET, resulting in an optical readout. Specifically, depolarization of membrane potential results in a decrease in QD PL that tracks the transition from the resting state to the depolarized state (vide infra). Figure 1A shows schematically the design of the QD–peptide–C60 bioconjugate system and the three peptide variants used herein. The QD serves as the central scaffold onto which the peptidyl sequences (referred to as JBD1, JBD2, and JBD3) are self-assembled using metal affinity coordination of the N-terminal His6 motif to the QD surface.40 By virtue of their alanine/leucine-rich nature, the peptides are designed to be helix-forming to promote membrane insertion.41–43 Each peptidyl sequence expresses a unique lysine residue for the covalent attachment of one C60 electron acceptor per peptide via EDC chemistry. Peptide JBD1, with its lysine residue attachment point most proximal to the His6 QD binding domain, positions the C60 closest to the QD surface upon assembly (QD–C60 surface-to-surface distance (ssd), ∼10 Å). Peptide JBD3, which bears a nine-proline repeat that adopts a type II helical conformation,44–46 positions the C60 most distal from the QD surface (ssd, ∼42 Å). JBD2 positions the C60 at an intermediary distance from the QD (ssd, ∼24 Å). See SI Table S1 for additional physicochemical properties of the peptides used herein.
Two variations of hydrophilic QD-capping ligand provide flexibility in terms of mediating colloidal stability in aqueous media while allowing the peptide’s membrane inserting domain to extend from the QD surface. As evidenced by molecular modeling (Figure 1B), the shorter dihydrolipoic acid (DHLA) ligand (∼11 Å extended length) affords maximal extension of the attached C60 beyond the ligand layer while providing colloidal stability at slightly basic pH.47–49 The longer PEGylated (68 Å nm extended length) DHLA–PEG750–OMe ligand, which terminates in a methoxy group, avails stability over a wide pH range,50,51 but the longer extended length increases hydrodynamic volume while reducing the accessibility of the peptide–C60 to the surrounding aqueous environment. As shown in Figure 1C, the full QD–peptide–C60 ensemble is predicted to interface with the outer leaflet of the plasma membrane such that the peptide–C60 portion of the conjugate is embedded in the lipophilic bilayer. In this configuration, the photoexcited QD serves as the electron donor and is engaged in ET with the C60 moiety, a potent electron acceptor.52,53 At resting membrane potential (∼−70 mV) the differential positive charge of the outer membrane leaflet supports radiative recombination of photoexcited electrons and holes within the QD, resulting in bright QD luminescence. Upon membrane depolarization, however, photoexcited electrons from the QD are preferentially attracted to the C60 acceptor, resulting in quenching of QD PL.
For this study we prepared two samples of core/multishell QDs: (i) CdSe/CdS/ZnS and (ii) CdSe/CdS/CdZnS/ZnS QDs with emission maxima centered at 605 and 625 nm, respectively. Both QD samples are classified as quasi-type II QDs in which there is spatial separation between the e–h charge carriers such that photoexcited electrons should delocalize into the surrounding CdS layer. The outermost ZnS shell passivates the core to minimize surface defects and maintain QD quantum yield while simultaneously providing a surface for assembly of the His6-bearing peptides by the ratiometric metal-affinity coordination as described previously.54
Characterization of QD–Peptide–C60 Bioconjugates
The successful covalent attachment of the C60 to each of the peptides was confirmed by spectroscopy (SI Figure S1). The peptides alone displayed only modest absorption in the 250–300 nm spectral window. By contrast, the peptide–C60 conjugates showed significant absorbance across the 250–350 nm spectral range, consistent with the absorbance of the C60 fullerene.55 The His6-mediated assembly of the peptide C60 conjugates to the QD surface was characterized by dynamic light scattering (DLS) (SI Table S2). This analysis showed the concomitant increase in the hydrodynamic diameter of the QD coupled with a decrease in the diffusion coefficient when 30 copies of each peptide–C60 species were assembled onto the QD surface. Not surprisingly, the hydrodynamic diameter of the 605 nm QDs (13.8 nm) was larger than the hard diameter determined by transmission electron microscopy (TEM) (8.2 nm, SI Figure S2), as it includes contributions from the ligand capping layer and solvation sphere. Comparable results confirming successful assembly of the peptide–C60 to the surface of the 625 nm emitting QDs were also obtained. These results are consistent with our extensive previously published work showing the controlled, ratiometric assembly of His-tagged peptides and proteins to the QD surface and the long-term stability of the formed bioconjugates in cellular environments.25,27,48,49,56–58
Steady-State Photophysical Characterization of QD–Peptide–C60 Bioconjugates
Having confirmed our ability to controllably form QD–peptide–C60 bioconjugates, we next characterized the efficiency of the QD–peptide–C60 ET system using CdSe/CdS/ZnS QD (605 nm emission) as the electron donor. This emission window (along with the 625 nm QD) was chosen specifically to ensure that photoexcited ET from the QD donor to the C60 fullerene acceptor was more favorable than FRET. We have shown previously that while both ET and FRET processes between the QD donor and C60 acceptor are possible, the use of QDs whose emission is tuned to the 600–630 nm window significantly favors ET processes over FRET.31 Steady-state QD PL quenching data were collected as a function of increasing numbers of peptide–C60 (i.e., valence) arrayed around the central 605 nm QD capped with DHLA ligands. As evidenced in Figure 2A–C, across all three peptide–C60 species, the quenching efficiency tracked directly with the number of peptide–C60 assembled onto the QD scaffold. These results are consistent with our previous studies where the addition of increasing numbers of peptide–C60 allows for the iterative “tuning” of the quenching of QD PL.31 Notably, control experiments in which the 605 nm DHLA-capped QDs were assembled with non-C60 peptides showed no significant quenching of QD PL. Additional control experiments to assess the quenching of the QDs by free C60 in solution (at a ratio of 20 C60/QD) showed negligible quenching of QD PL, consistent with previous observations.31 These data confirm the necessity for attachment of the C60 to the QD surface via the peptide linker to mediate efficient quenching.
Figure 2.
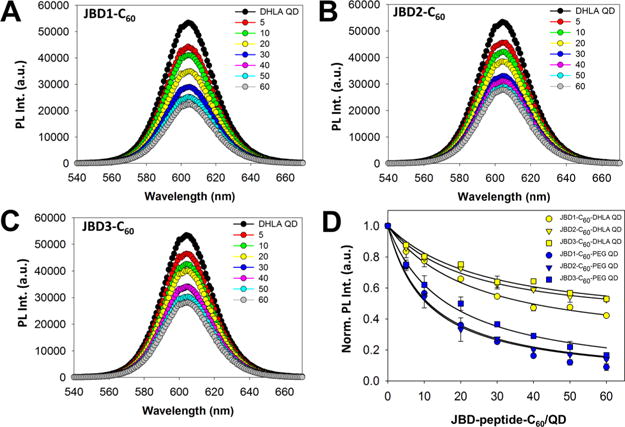
Steady-state PL analysis of QD–peptide–C60 conjugates. Representative PL spectra collected from 605 nm emitting DHLA-coated QDs assembled with increasing ratios of (A) JBD1-C60, (B) JBD2-C60, and (C) JBD3-C60 peptides. (D) Comparison of steady-state QD PL (normalized) from QD–JBD–C60 conjugates versus increasing ratio of peptide assembled per QD for DHLA–and DHLA–PEG750–OMe-coated QDs.
In the case of the three peptide–C60 species under investigation here, maximal quenching occurred as the peptide valence approached ∼60 peptides–C60/QD. While this peptide valence is slightly under the maximum number of peptides predicted to be able to assemble to the 605 nm QD surface (∼100 ± 20 peptides),59 clearly a peptide valence of ∼60 facilitated maximal quenching. Similar trends in QD PL quenching efficiency were observed for 605 QDs capped with DHLA–PEG750–OMe ligands (Figure 2D, SI Figure S3). The modulation in PL intensity can be described by the relationship
where IPL(p) and IPL(0) are the PL of the QD in the presence and absence of peptide–C60, respectively, p is the number of peptide–C60 assembled onto the QD surface, QY is the quantum yield of the QD in the absence of peptide–C60, τr is the radiative decay time of the exciton in the QD, and τe is the time of electron transfer from the QD donor to the C60 acceptor (see the SI for equation derivation). In these steady-state experiments we observed no significant dependence of QD PL quenching efficiency on the QD–C60 separation distance, suggesting that in solution the peptide–C60 interacts similarly with the QD surface across all three peptide–C60 conjugates. This strongly suggests that in the aqueous environment under which the steady-state PL data were collected, the peptides adopt a less structured conformation that allows the C60 moiety to approach very near the QD surface regardless of the peptide sequence. In the presence of biological membranes, however, the peptides adopt a more rigid helical structure that maintains the QD–C60 separation distance predicted by modeling. This is consistent with the behavior of the peptides in the presence of liposomal membranes as confirmed by circular dichroism analysis (vide infra).
Interestingly, in comparison to DHLA-capped QDs, the overall magnitude of quenching observed for the PEG-capped QDs was significantly greater. Consider again the data at the peptide–C60/QD valence of ∼40. At this ratio on PEG-capped QDs, QD PL quenching efficiency ranged from ∼83% (JBD1-C60) to ∼70% (JBD3-C60), corresponding to increases of 33% and 35%, respectively, in the quenching efficiency of these same peptides in the context of DHLA-capped QDs. It is likely that the carboxyl functions on the DHLA ligands played a key role in these observed differences. At the pH 8.5 conditions under which these experiments were performed, the carboxyl groups would be predominantly ionized and would likely abrogate the rate of ET between the QD and the C60 via electrostatic shielding. In contrast, the uncharged PEGylated ligand system would likely not elicit this effect. This trend continued for DHLA– and DHLA–PEG750–OMe-capped QDs with emis sion centered at 625 nm (SI Figure S4), demonstrating that the C60-induced PL quenching operated consistently across QD platforms. It is also worth noting the structural and “excluded-volume” roles played by the PEG ligands in this context. Previous studies have implicated the role of PEGylated ligands in mediating conformational changes in peptide structure when assembled onto the QD surface, which can alter peptide persistence length,44,60 resulting in shorter QD–C60 separation distances than those predicted by modeling. These effects likely contribute to the enhanced quenching of the QD by the C60 in the context of the PEG-capped QDs observed herein.
To confirm that the observed peptide–C60-induced QD PL quenching was dominated by non-FRET processes (i.e., ET), a wavelength-dependent quenching analysis was performed on the steady-state PL data. This analysis determines the magnitude of the wavelength dependence in QD quenching in QD donor–acceptor FRET systems, which is a hallmark of FRET systems that employ the QD as donor.61 We previously used this analysis to confirm that FRET was the predominant energy transfer process in QD–peptide–osmium bioconjugates.62 Herein, we performed this analysis on the QD–peptide–C60 ensembles (∼30 peptides assembled/QD) and found that the QD quenching was wavelength-independent for all three JBD peptide species when assembled on DHLA or DHLA–PEG750–OMe 605 nm QDs (SI Figure S5). Indeed, the flat slopes of the quenching efficiency plots coupled with the nearly symmetrical unquenched and quenched spectra confirmed this. This is not surprising given the fact that in the 605 nm spectral window of the QD, the absorbance of the C60 is negligible and does not afford any appreciable energy transfer from the QD donor to C60 acceptor.31 Further, the overlap integral between the 605 nm QD donor and C60 acceptor is minimal (J = 1.16 × 10−14 cm3 M−1) compared to lower wavelength emitting QDs.31 Cumulatively, these data strongly suggest that ET is the dominant mechanism in the observed QD PL quenching.
Characterization of Membrane Binding of Peptide–C60 and QD–Peptide–C60 Conjugates
Having established the ET-based nature of QD quenching by the C60, we next characterized the interaction of the peptide, peptide–C60, and QD–peptide–C60 conjugates with the plasma membrane of living cells. The peptides used in this study were designed to insert into biological membranes by virtue of their alanine/leucine-rich sequence, which promotes helix stabilization.41 To begin, we first characterized the unconjugated peptides’ (no C60) secondary structure alone in solution and in the presence of liposomal vesicles as analyzed by circular dichroism (CD). Peptides JBD2 and JBD3 were selected for this analysis, as the JBD2 sequence is nearly identical to JBD1 (except for the relocation of the internal lysine residue to the C-terminus and its replacement with leucine) and JBD3 contains a nine-proline repeat predicted to form a type II proline helix.45 The CD spectra for peptides JBD2 and JBD3 and the accompanying tabulated results are shown in SI Figures S6 and S7, respectively. Here, we looked for clear trends showing an increasing percentage of α-helical content of the peptide when in the presence of liposomes compared to when the peptide was in phosphate-buffered saline (PBS) buffer alone. Indeed, for both peptides an increase in α-helical content was observed. For peptide JBD3, the increase in helical structure was less pronounced but still notable. Nonetheless, for both peptides, helical character was the only value for which an increase was observed, consistent with the peptides’ adoption of a helical conformation in the presence of biological membranes. Similar observations have been made for other alanine-leucine-rich peptides.41,63
Next, we determined the membrane-inserting behavior of the peptide–C60 conjugates. To accomplish this, immunocyto-chemistry using a C60-specific primary monoclonal antibody was performed to probe the interaction of the His6-bearing JBD peptides and JBD–peptide–C60 conjugates with the membrane of A549 (adenocarcinomic human alveolar epithelial) cells. The primary antibody was probed with a secondary antibody–gold NP conjugate followed by silver staining to visualize the presence of the gold NP in the formed immunocomplex. As presented in the micrographs in Figure 3A and quantitatively in Figure 3B, the magnitude of the normalized silver staining intensity clearly tracked with the presence of the C60 moiety compared to the nonconjugated peptide. These data, together with the CD data showing helix formation in the presence of liposomes and the well-documented propensity of the C60 carbon allotrope to partition into biological membranes,64–66 gave us great confidence that the His6-tagged peptide–C60 conjugate would append hydrophilic coated QDs to the outer leaflet of the plasma membrane by virtue of its membrane-inserting properties.
Figure 3.
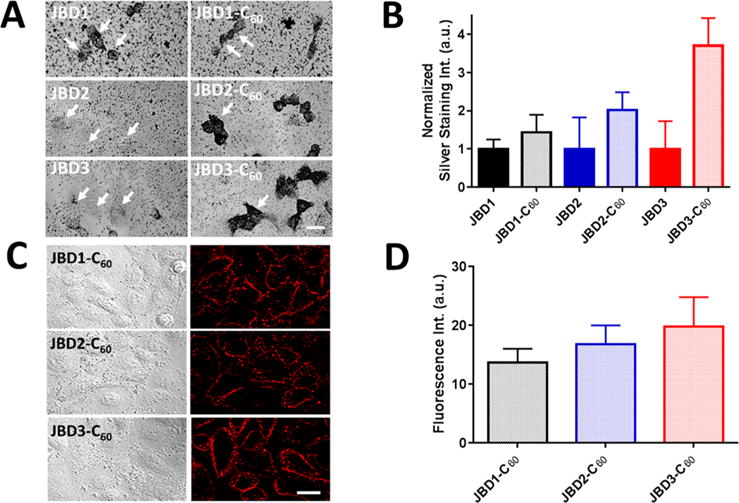
Membrane binding of JBD peptides, JBD–C60 peptide conjugates, and QD–DHLA–peptide–C60 conjugates. Immunocytochemistry combined with silver staining was used to visualize gold nanoparticle-bearing immunoplexes in A549 cells. (A) Cells were incubated with 10 μM JBD peptide minus C60 (left column) or 10 μM JBD peptide–C60 conjugate (right column), fixed/permeabilized, immunoprobed, and silver stained for the presence of the C60 moiety. White arrows point to individual cells. Scale bar, 20 μm. (B) Comparative quantitative analysis of silver stain intensity of the images shown in A. Silver stain intensity was quantified by treating individual cells in each image as separate regions of interest (ROIs). The bar graph shows the resulting average ± SEM of silver stain intensities corresponding to at least 40 cells for each sample (three independent experiments). (C) DIC (left) and confocal fluorescence (right) images of A549 cell monolayers labeled with 605 nm QD–DHLA–JBD peptide–C60 complexes as indicated. The micrographs show robust binding of the QD–peptide–C60 conjugates to the plasma membrane. Scale bar, 20 μm. (D) Quantification of membrane staining of the QD–peptide–C60 QD complexes in panel C. Fluorescence intensities were determined by drawing an ROI around the plasma membrane of individual cells. Bar graph shows the average ± SEM fluorescence intensities of 50 to 70 cells for each sample from four independent experiments. The results shown are background corrected for nonspecific binding of QD alone (no peptide).
To confirm the latter, we performed membrane labeling studies in A549 cells to determine the efficiency with which the self-assembled 605 nm QD–peptide–C60 bioconjugates decorated the plasma membrane. As evidenced by Figure 3C and D, we observed robust, bright membrane labeling immediately after 10 min of incubation of 20 nM QDs (20 JBD–C60 peptides/QD) with the cell monolayers. The pH of the DPBS incubation buffer was adjusted to 7.9 to facilitate enhanced colloidal stability of the DHLA-capped QDs. The labeling was highly efficient (confirmed by confocal microscopy) with 70% (QD–JBD2–C60) to 91% (QD–JBD1–C60) of cells positive for membrane labeling. Cells probed with QD–JBD3–C60 conjugates showed a labeling efficiency of 81%. This labeling morphology remained membranous for at least 30 min with minimal endocytosis of the QDs. Comparable membranous labeling was obtained with peptide/QD ratios of >20 (data not shown). DHLA-capped QDs bearing no peptide showed minimal nonspecific binding to the cell monolayers (<10% of the signal of that for QD with peptide) over the same incubation time course. The QDs capped with DHLA–PEG750–OMe ligands showed no significant binding whether or not they were decorated with peptide–C60 conjugates (data not shown). These observations are consistent with the important role of PEG ligands and coatings in minimizing nonspecific binding of a myriad of NPs both in vitro and in vivo.67–69 Cumulatively, these data confirmed the successful membrane labeling with the QD-DHLA–peptide–C60 conjugates, setting up their use as optical probes for monitoring modulations in membrane potential.
In Vitro Characterization of the Fluorescence Response of QD–Peptide–C60 Conjugates to Membrane Depolarization
Having confirmed the successful membrane labeling of cells with the QD–DHLA–peptide–C60 conjugates, we next sought to ascertain the optical response of the probes to changes in membrane potential. We employed HeLa cells for this purpose, as our initial depolarization experiments on A549 cells using a commercial voltage-sensitive probe (FluoVolt) did not show any significant membrane depolarization response upon addition of 140 mM KCl. The addition of extracellular isotonic 140 mM KCl induces cells into a sustained depolarized state, and it has been used in a wide array of studies for the membrane depolarization of multiple cell types.39,70–73 For these studies, 605 nm DHLA-capped QDs appended with 20 copies of JBD–C60 peptides were used. Not only did initial binding studies confirm excellent membrane labeling at this peptide valence, but this peptide/QD ratio also corresponded to ∼35% quenching of steady-state QD PL, allowing for further quenching upon membrane depolarization.
Figure 4A shows a representative example of the fluorescence intensities at resting potential (2.5 mM KCl) and after membrane depolarization (by addition of 140 mM KCl) in HeLa cells labeled with QD–DHLA–JBD1–C60 conjugates. Upon depolarization, a notable decrease in fluorescence intensity was observed for all three QD–peptide–C60 conjugates with distinct differences observed among the three probes. Figure 4B shows the quantified fluorescence intensities of all three QD–peptide–C60 conjugates pre- and post-depolarization. The average percent fluorescence decreases (ΔF/F0 ± standard error of the mean (SEM)) were determined to be QD–JBD1–C60, 20 ± 2% (41 cells queried); QD–JBD2–C60, 12 ± 2% (52 cells); and QD–JBD3–C60, 2 ± 1% (76 cells). Upon addition of 140 mM KCl, the membrane potential of HeLa cells is expected to rise from ∼−50 mV to ∼0 mV.74 Accordingly, the percent fluorescence decreases per mV we observed here correspond to estimated values of 0.40%, 0.24%, and 0.04% for JBD1, JBD2, and JBD3, respectively. The PL modulations in response to membrane depolarization observed here are consistent with those predicted by modeling the effects of changes in membrane potential on the electron tunneling efficiency in the QD–peptide–C60 conjugates (see the SI). Control experiments with HeLa cells labeled with QD–DHLA–JBD1 peptide (no C60; 20 peptides per QD) showed less than 3% decrease in QD PL upon depolarization (SI Figure S8).
Figure 4.
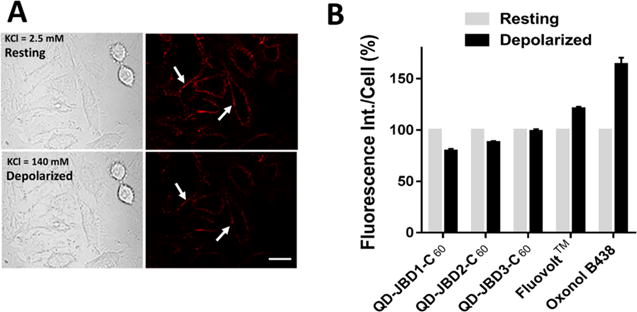
Imaging of membrane potential changes in cells labeled with QD–DHLA–JBD peptide–C60 bioconjugates. (A) Differential interference contrast (DIC) and confocal fluorescence images of HeLa cell monolayers labeled with 605 nm QD–DHLA–JBD1 peptide–C60 bioconjugates before (top) and after (bottom) depolarization with KCl solution. Labeled cell monolayers were cultured in live cell imaging solution (LCIS) containing 2.5 mM KCl (resting, top). Depolarization of membrane potential was performed by addition of LCIS containing 140 mM KCl (depolarized, bottom). Scale bar, 20 μm. Arrows highlight the same cell in each panel. (B) Quantification of the fluorescence intensity decrease from resting (2.5 mM KCl, gray bar) to depolarized (140 mM KCl, black bar) membrane potential for the QD–peptide–C60 conjugates. The fluorescence response increase of the commercial VSDs FluoVolt and oxonol B438 are shown for comparison.
For comparison, Figure 4B also shows the response of the fast-response VSD FluoVolt, which displayed a fluorescence increase as expected (ΔF/F0 of 21% ± 2% (39 cells; 0.41% ΔF/F0 per mV)). Reported values of ΔF/F0 per mV for this dye range from 0.1% to 0.48% in patch-clamped cortical neurons and HEK cells, respectively,75 values to which the QD–peptide–C60 conjugates reported here compare well. Relative to other fast-response VSDs (e.g., ANEPPS dyes (0.1% ΔF/F0 per mV76); RH-1691 (0.01% ΔF/F0 per mV77)) the fluorescence responses of the QD–peptide–C60 conjugates are significantly higher. The slow-response dibutylbarbituric-based VSD oxonol B438 exhibited a fluorescence increase of 64% (30 cells, 1.3% ΔF/F0 per mV). Despite its rather large ΔF/F0, oxonol B438 does not remain membrane-resident like other VSDs; it enters cells upon depolarization, where it binds to intracellular proteins and membranes, resulting in the influx of more dye and an increase in fluorescence.78 Thus, it was used here primarily as a positive control indicator of successful membrane depolarization.
The relative magnitude of the depolarization-induced fluorescence changes observed for the three QD–peptide–C60 conjugates generally tracked inversely with increasing QD–C60 separation distance predicted by modeling (Figure 1B). This points to the important role played by the separation distance between the QD and C60 in mediating differential responsivity to changes in membrane potential, particularly when the QD–peptide–C60 complex is interfaced with the plasma membrane. In this context, the lipid environment of the plasma membrane likely plays a critical structural role in maintaining peptide persistence length and optimal QD–C60 separation distance in the hydrophobic bilayer.79 It also strongly suggests that the loading conditions used consistently resulted in the proper orientation of the QD–DHLA–peptide–C60 complex in the plasma membrane wherein the hydrophilic QD is located on the cell surface and the hydrophobic peptide–C60 portion of the bioconjugate is buried within the aliphatic portion of the plasma membrane bilayer.
Importantly, the interfacing of the QD–DHLA–peptide–C60 conjugates with HeLa cells had minimal impact on their cellular viability. MTS proliferation assays were performed on HeLa cells incubated with QDs alone, JBD1–C60 peptide, or the full QD–DHLA–JBD1-C60 conjugate under the labeling conditions described in the Methods section. None of these materials induced significant toxicity, with viabilities of >85–90% noted in all cases (SI Figure S9). We also confirmed the successful labeling and biocompatibility of the QD–DHLA–JBD1–C60 conjugates using cultured networks of mouse cortical neurons (SI Figure S10). Following labeling of networks with the QD–DHLA–JBD1–C60 conjugates, cellular viabilities of >94% as compared to unlabeled control were determined by live/dead staining (five cultures and eight imaged regions of interest). These findings are consistent with previous studies detailing the largely innocuous nature of QD bioconjugates in biological systems48,49,56,80,81 and points to improvements in cellular viability relative to other organic voltage-sensitive dyes that are known to significantly impact cellular homeostasis.82,83
We further demonstrated the utility and flexibility of the QD–DHLA–JBD1–C60 conjugate for imaging membrane potential by performing a series of two-color depolarization experiments where the VSD oxonol B438 (green channel) and the QD conjugate (red channel) were used combinatorially to provide dual-channel visualization of membrane depolarization in HeLa cells (Figure 5). Here, upon KCl-induced depolarization, a ∼21% decrease in QD conjugate PL coupled with a concomitant increase in oxonol B438 emission was noted. Comparable results were obtained in PC12 (rat adrenal medulla pheochromocytoma) cells (SI Figure S11). Notably, the presence of the oxonol fluorophore did not abrogate the function of the QD–peptide–C60 probe. This dual-channel format was also replicated in cultured monolayers of mouse primary cortical neurons where the QD PL was quenched ∼7% upon depolarization, coupled with a corresponding ∼5-fold increase in the emission of the oxonol probe (Figure 6). In addition to its demonstrated utility across multiple cell lines and primary cells, the narrow, red-shifted emission profile of the QD–peptide–C60 probe (SI Figure S12) avails direct two-color membrane potential imaging that has heretofore necessitated complicated deconvolution or ratiometric imaging due largely to the broad emission profiles of voltage-sensitive dyes.84,85 Further, the simultaneous, differential PL responses of the oxonol (PL “on”) and QD (PL “off”) probes are easily discernible using standard filter sets.
Figure 5.
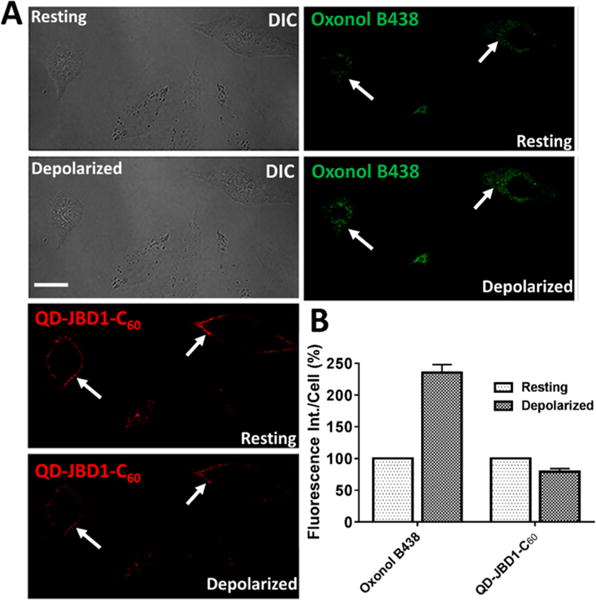
Dual-channel mode imaging of cellular membrane potential in HeLa cells using QD–JBD peptide–C60 bioconjugates and oxonol. (A) HeLa cells were colabeled with 605 nm QD–DHLA–JBD1–C60 conjugates (red) and oxonol B438 (green) and were depolarized by KCl perfusion. Shown are the DIC and confocal fluorescence images of HeLa cell monolayers before and after depolarization with KCl solution. Scale bar, 20 μm. White arrows highlight the same cell in each panel. (B) Quantification of the relative change in the fluorescence intensity of the QD–peptide–C60 complexes and oxonol upon addition of KCl solution. Fluorescence intensity of the cells was obtained by drawing the ROI around individual cell plasma membranes, and the results represent the mean ± SEM fluorescence intensity of 26 cells for each channel across three separate experiments.
Figure 6.
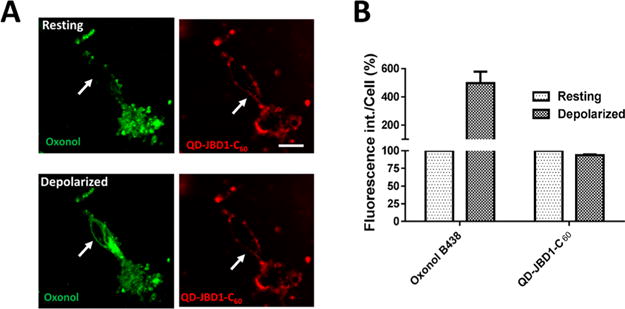
Dual-channel mode imaging of cellular membrane potential in primary mouse neuronal cells using QD–JBD peptide–C60 bioconjugates and oxonol. (A) Confocal fluorescence images of primary mouse cortical neurons colabeled with QD–DHLA–JBD1–C60 (red) and oxonol (green) before (top) and after (bottom) KCl perfusion. Scale bar, 20 μm. White arrows denote the same spot in cells pre- and post-depolarization. (B) Quantification of the relative changes in the fluorescence intensity of the QD–JBD1–C60 complexes and oxonol upon addition of KCl solution. Fluorescence intensity of the cells was obtained by drawing ROIs around individual neuron membranes. The graph shows the average fluorescence intensity (±SEM) obtained from 39 cells from five independent experiments.
In Vivo Characterization of the Fluorescence Response of QD–Peptide–C60 Conjugates to Cortical Brain Function
Given their significant photostability and large two-photon action cross sections, one of the very attractive applications of voltage-sensitive QD-based probes is the real-time imaging of in vivo brain function. To assess the sensitivity and temporal responsivity of our probes, we performed a series of cortical brain function imaging experiments in anesthetized B6 mice. Solutions of QD–JBD1–C60 (or control samples of QD–JBD1 (no C60) or QDs alone) were injected into multiple regions of the right or left parietal cortex (SI Figure S13) followed by the region selective introduction of electrical stimulation (100 μA) from a bipolar tungsten electrode. To reduce noise and to assess the ability of the QD–peptide–C60 probe to respond to repetitive rounds of stimulation, optical signals were averaged over 50 successive trials in each animal tested. In each trial, the stimulus consisted of seven pulses (each 3 ms duration at 200 Hz, 12 s between each trial). Figure 7A shows the relevant time-resolved optical images collected from mouse cortex injected with QD–DHLA–JBD1–C60 conjugates. The temporal fluorescence intensity profiles of four regions of interest (ROIs) were queried before, during, and after the stimulus (575 ms frame expanded in Figure 7B for clarity). Note that the responding ROI (A1) is located proximal to one of the stimulating electrodes, while the nonresponsive ROIs are distal from the inserted electrode. As evidenced by the plots in Figure 7C, ROI A1 responded nearly immediately upon application of the stimulus and displayed maximal fluorescence decrease (ΔF/F0 = ∼2.1%) at ∼100 ms after stimulation onset. The response decayed upon removal of the stimulation, returning to baseline within 80–100 ms after removal of the applied stimulation. The variability of the response of the conjugate was also assessed across multiple animals, where the average percent PL response across three animals (±SEM) was 2.1 ± 0.05 (SI Figure S14). Panels D and E in Figure 7 present the time-resolved fluorescence responses of mice injected with two control constructs: QD–JBD1 peptide (no C60, panel D) and QDs alone (panel E), respectively (full data in SI Figure S12). Clearly, these two materials were nonresponsive to the applied stimulation, demonstrating the necessity of the full ensemble QD–peptide–C60 donor–acceptor conjugate for function.
Figure 7.
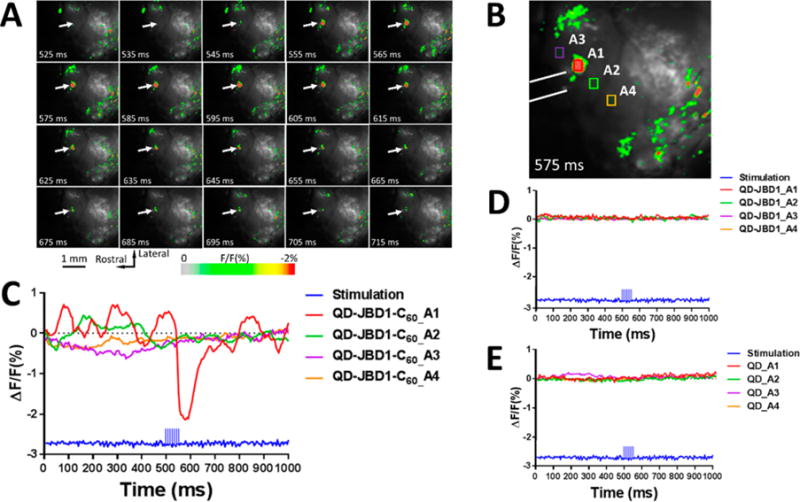
In vivo optical imaging of cortical electrical stimulation using QD–JBD1–C60 bioconjugates. (A) Images of mouse cortex injected with QD–JBD1–C60 conjugates during electrical stimulation. Images were captured at 200 Hz frame rate, and numbers indicate the acquisition time point. Arrow points to ROI A1 shown in panel B; image is false-colored to correspond to scale at bottom. (B) Frame at 575 ms shows fluorescence response of four ROIs. White lines show the position of the bipolar tungsten electrode. (C) Time-resolved fluorescence response of the four ROIs in B showing the averaged response of 50 trials. Panels D and E show the responses of four ROIs in mouse cortex injected with control samples QD–JBD1 peptide (no C60) (panel D) and QDs alone (panel E).
In comparison to popular in vivo VSDs under these same experimental conditions, the QD–JBD1–C60 probe compares well in terms of its responsivity dynamics. For example, the so-called “blue dyes” RH-795 (λem max. = 695 nm) and RH-1691 (λem max. = 675 nm) are specifically designed to have minimal light scattering and be less sensitive to contamination from hemodynamic components of the vascular system.82,86 These dyes respond optically within ∼10 ms to applied electrical stimulation and return to baseline within 50–100 ms,77 comparable to the QD–JBD1–C60 probe. However, the QD–JBD1–C60 conjugate system responds with a 20- to 40-fold larger ΔF/F0 than the RH dyes (which typically exhibit ΔF/F0 values of only ∼0.05–0.1%).77 Clearly the QD–peptide–C60 probe offers significant improvement in signal strength with no sacrifice in temporal responsivity.
CONCLUSIONS
The desire to map the functional connectome of the human brain to better diagnose and treat brain injury and disease has necessitated the development of new imaging modalities that afford capabilities beyond currently available techniques and materials. Currently employed approaches for the in vitro or in vivo monitoring/imaging of neuronal cell activity (e.g., action potentials) rely on patch clamp or fluorescent dyes/protein constructs and are limited by their lack of throughput, susceptibility to photobleaching, intracellular instability, or the need for the generation of transfected cells or transgenic animals.13 Ideally, next-generation optical probes to circumvent these issues would possess most, if not all, of the following criteria: (1) optical responsivity to biologically relevant electric field strengths, (2) bright, stable PL for long-term imaging, (3) the ability to be targeted specifically and noninvasively to neurons across multiple experimental platforms (monolayer, tissue slice, and in vivo whole animal), (4) large two-photon action cross sections that avail deep tissue imaging, and (5) the ability to engage in efficient energy or charge transfer with nanosecond lifetime. Clearly, the realization of the above attributes requires an understanding of new functional hybrid materials and how to integrate them, with fine control, with cells, tissues, and organisms.
Taking these desired criteria into account, here we set out to build on our previous body of work where we showed the utility of QD bioconjugates in a number of applications with implications for neuronal cell imaging. These include the use of QD–peptide assemblies for the specific labeling of the plasma membrane in monolayer cultured cells,24,25 the targeted labeling of neurons in tissue slices,34,35 the in vivo tracking of brain development,36 and the use of QD-coated patch pipets for multiphoton excitation-assisted deep-tissue visualization of live mouse electrophysiology.33 These demonstrations, coupled with our previous work that showed the ability of QD–fullerene donor–acceptor systems to engage in efficient ET,31 motivated us to ascertain whether this ET system could be used for the real-time visualization of changes in membrane potential in live cells.
In this work, we designed a QD–peptide–C60 bioconjugate system wherein each of the three components plays a pivotal role. The core/shell QD is specifically designed to be of the type II configuration to promote spatial segregation of electron–hole charge carriers. This configuration was selected to promote enhanced sensitivity to applied electric fields, as we have reported previously.38 The C60 is an efficient electron acceptor that quenches the PL of the photoexcited QD quantitatively as a function of the number of C60 acceptors assembled on the QD surface.31 Finally, the alanine/leucinerich peptide bridge serves to append the C60 acceptor at discrete fixed distances while also mediating membrane insertion as confirmed by CD analysis and immunostaining. Steady-state PL quenching analysis confirmed that the efficiency of QD quenching was intimately dependent on the number of peptide–C60 acceptors arrayed around the central QD donor. This is consistent with ET theory and has been observed in donor–acceptor systems where the peptide plays the role of functional spacer and facilitates ET via tunneling.87,88 In contrast to the work of Virkki et al.,32 who studied the QD quenching efficiency of C60 attached directly to the capping ligand, our approach here provides finer control over the number of C60s in the ensemble bioconjugate.
The nature of the ligand played a critical role in the quenching efficiency in the QD–C60 donor–acceptor system, as has been reported elsewhere in the literature for other QD-based ET systems.89–91 The shorter, negatively charged DHLA ligand mediated a moderate degree of QD quenching, while for the same peptide/ratio combination the quenching induced by the uncharged DHLA–PEG750–OMe ligand was significantly higher. In this context, it is likely that the negative charges on the DHLA ligands abrogate the efficient quenching of the QD PL. The Weiss group made similar observations in their studies of electron exchange between PbSe QDs and a negatively charged anthraquinone where the introduction of increasing numbers of charged 6-mercaptohexanoate ligands resulted in a concomitant decrease in the rate of ET.92
Just as the ligand coating influenced the QD quenching efficiency, it was also a critical determinant of the interaction of the assembled QD–peptide–C60 conjugates with cells. DHLA–PEG-capped QDs assembled with any of the three JBD–C60 conjugates showed no detectable QD binding to the plasma membrane. Molecular modeling strongly suggested the steric shielding of the peptide–C60 conjugates in the context of this ligand coating. DHLA-capped QDs, however, presented the peptide–C60 conjugate beyond the ligand shell and extending from the QD surface. These observations are consistent with previous reports showing the effect of the capping ligand layer on the display and packing of peptides and proteins on the QD surface when assembled via the Hisn-based metal affinity strategy used herein.59,93 It is fortuitous in our case that the DHLA ligand coating that affords more efficient binding of the QD–peptide–C60 bioconjugates to the plasma membrane also mediates only a moderate degree of QD quenching. The intermediary degree of quenching in this ligand system (∼35% quenching at 20 JBD1 peptides/QD) avails a sufficient dynamic range for further QD quenching upon depolarization of membrane potential.
We functionally demonstrated the utility of the QD–peptide–C60 system for the imaging of membrane potential changes in cultured HeLa and PC12 cells and in primary cortical neurons. In these systems, we showed the compatibility of the QD–peptide–C60 probe with standard voltage-sensitive dyes for two-color combinatorial membrane potential imaging. We further showed their ability to be used as photostable labeling probes in networks of cortical neurons cultured on microelectrode arrays (MEAs). Finally, in vivo imaging of cortical electrical stimulation functionally demonstrated the superior imaging capability of the QD–peptide–C60 probe compared to traditional VSDs, where a 20- to 40-fold improvement in ΔF/F0 was realized with no sacrifice in responsivity.
Previous reports have demonstrated the ability to interface QD-based ET constructs with live cells for imaging applications. Medintz et al. used QDs engaged in ET with a peptide–dopamine conjugate self-assembled to the QD surface to monitor intracellular pH changes in real time.58 In those studies, the QDs were physically microinjected into the cellular cytosol to visualize drug-induced alkalosis. In the present report, the QD–peptide–C60 bioconjugate spans the outer leaflet of the plasma membrane, and the starting degree of quenching of the photoexcited QD by the C60 moiety at resting potential can be modulated by the number of C60 acceptors arrayed around the central QD donor.
The QD–peptide–C60 system we describe here offers several distinct advantages over VSDs and genetically encoded voltage-sensitive fluorescent protein constructs. First, the ΔF/F0 values observed are larger than commonly employed VSDs (vide supra). Second, its modular nature affords facile assembly and tractability that cannot be readily achieved with organic dyes that require complicated synthesis and purification schemes.75,85,94,95 Third, the QD quenching efficiency is tunable and easily controlled through rational peptide design as demonstrated here. Fourth, as the system employs QDs in the >600 nm emission window as electron donors, cellular autofluorescence is minimized. Of particular note is the large two-photon action cross sections of QDs (often 103–104 times greater than organic dyes),28,96 which could allow deeper tissue imaging combined with voltage sensing.33 Finally, in contrast to previous reports that have used QDs as optical voltage-sensing probes,39 our approach described here is not dependent on the insertion of the QDs into the membrane bilayer. This is a considerable challenge given the hard diameter of QDs (typically 5–10 nm) relative to the 4 nm expanse of the aliphatic portion of the bilayer. Membrane-partitioning QDs require hydrophobic coatings, and their delivery depends on the use of fusogenic delivery vesicles for membrane insertion.26 We expect that the QD–peptide–C60 system described here will find great utility in an array of voltage-sensing schemes. Given the significantly higher photostability and two-photon action cross sections of QDs compared to VSDs, we anticipate the expanded use of the QD–peptide–C60 constructs described herein for deeper tissue imaging of in vivo brain function.
METHODS/EXPERIMENTAL
Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used as received unless otherwise mentioned. Dulbecco’s phosphate buffered saline (DPBS), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, 1 M), Dulbecco’s modified Eagle medium (DMEM), modified Eagle medium (MEM), fetal bovine serum (FBS), DMEM containing 25 mM HEPES (DMEM–HEPES), antibiotic–antimycotic, DiBAC4(3) (bis(1,3-dibutylbarbituric acid)trimethine oxonol (B438)), and FluoVolt membrane potential kit were purchased from Life Technologies (Grand Island, NY, USA). Primary fullerene mouse monoclonal IgG and goat anti-mouse polyclonal–gold nanoparticle conjugate were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). 1,2-Dipalmitoyl-snglycero-3-phosphocholine was from Avanti Polar Lipids (Alabaster, AL, USA).
Quantum Dots
CdSe/CdS/ZnS and CdSe/CdS/CdZnS/ZnS QDs with emission maxima at 605 and 625 nm, respectively, were synthesized according to previously published procedures.97–100 DHLA and DHLA–PEG750–OMe were prepared according to established protocols.49,51,99,101 A two-step ligand exchange procedure was used to coat these QDs with DHLA or DHLA–PEG750–OMe ligands, adopted from our previous work for the transfer of QDs to water with pyridine-appended polymers.102 Briefly, the QDs (5 nmol) were precipitated with acetone and centrifuged at 3500 rpm for 5 min, and the supernatant was discarded. The QD pellet was mixed with ethanol (0.5 mL), chloroform (0.3 mL), and 2-(2-aminoethoxy)-ethanol (0.15 mL) and then heated at 60 °C for 1 h under nitrogen with stirring to form a clear solution. After cooling to room temperature, the QDs were precipitated with a mixture of ethyl acetate, 2-propanol, and hexanes. The QDs were centrifuged at 3500 rpm for 5 min, and the supernatant discarded. The QD pellets were then mixed with ethanol (0.5 to 1 mL) and the desired ligand at approximately 30 000 equiv of ligand per QD. The mixture was sealed under nitrogen and stirred at 60 °C overnight. The QDs were then purified according to published protocols.103 DHLA and DHLA–PEG750–OMe coated QDs were stored in sodium tetraborate (4–5 mM) and DI water solutions, respectively, in the dark at 4 °C.
Transmission Electron Microscopy of QDs
Structural characterization of asprepared QDs was carried out using a JEOL 2200-FX analytical high-resolution transmission electron microscope with a 200 kV accelerating voltage. Samples for TEM were prepared by spreading a drop (5–10 μL at a concentration of ∼1 μM) of the filtered QD dispersion onto an ultrathin carbon support film on a 300 mesh Au grid (Ted Pella, Inc.) and letting it dry. Individual particle sizes were measured using a Gatan Digital Micrograph (Pleasanton, CA, USA). Average sizes along with standard deviations were extracted from analysis of ∼100 QDs.
Peptide–C60 Conjugates
Peptides were labeled with C60 and purified according to the procedure detailed in ref 31 with slight modifications. Briefly, peptide (1 mg, 1 equiv), C60-pyrrolidine tris-acid (Sigma-Aldrich) (6 equiv), and N-hydroxysuccinimide (84 equiv) were placed in a 20 mL scintillation vial with 2 mL of 95% DMSO/H2O. Triethylamine (8 equiv) was added to the mixture with stirring. After a few minutes N,N′-diisopropylcarbodiimide (66 equiv) was added, and the mixture was stirred overnight (∼16 to 20 h) at room temperature in the absence of light. To isolate the C60-labeled peptide, 0.5 mL of DI water and 2.5 mL of Ni2+-nitrilotriacetic acid agarose resin (Qiagen, Valencia, CA, USA) was added to the reaction mixture and stirred for 1–2 h. The resin was then filtered on a glass frit and washed with DMSO (∼6 × 7 mL aliquots, or until the filtrate was colorless) and then DI water (∼4 × 7 mL aliquots). The C60-labeled peptide was then eluted from the resin using an imidazole (600–900 mM) solution in 90% DMSO/H2O (∼4 × 1 mL aliquots). The combined filtrate was desalted on a preprimed oligonucleotide purification cartridge (OPC) and dried to a pellet.40 After desalting, the bound peptide was eluted with neat acetonitrile (∼4 × 0.1 mL aliquots) and then dried. The peptide pellet was dissolved in DMSO, quantified by UV–vis spectrophotometry (C60 absorbance at 350 nm, 34 300 M−1 cm−1), and then stored at −20 °C. The conjugation of the C60 to the peptide was confirmed by differential absorbance spectroscopy using unconjugated peptide as a reference.
Molecular Modeling of QD–Peptide–C60 Conjugates
All models were created using tools in UCSF Chimera, version 1.4.1. Energy minimization was carried out in Chimera using built-in features including ANTECHAMBER (version 1.27) and the AM1-BCC method of calculating charges.104 The model of the C60 fullerene was constructed from the structure of (η5-pentaphenyl-C60fullerene)-(η5-cyclopentadienyl)iron carbon disulfide.105 The covalently attached phenyl groups were removed from the fullerene using the tools in Chimera, and the resulting model was energy minimized as described above. The C60 fullerene was further modified to contain a bridging group on the region formerly occupied by the phenyl groups. A model for peptide JBD2 (see Figure 1 for peptide sequences) was first created in Chimera. On the basis of prior work the His6 region was modeled with an extended conformation, which is consistent with the four to six coordination bonds the His6 tail forms with the QD surface. The remaining portion of the peptide was modeled as a helix based on both modeling and similarity to other short peptides of know conformation. Following initial peptide folding the peptide model was energy minimized using tools in Chimera. The helical conformation of the AALAAWAALAAAAALAAAK region was verified using Mobyle106 and PSIPRED.107 The models for the C60 fullerene and peptide linker were joined, and the full model was energy minimized. In a similar manner the JBD3 peptide containing a Pro9 segment was built in Chimera. The helical region of peptide JBD2 was extended by inserting an Ala-Pro9 segment just before the His6 QD binding region to yield the full JBD3 sequence. The Pro9 was constrained to be helical based on prior work.44,108 Lastly a model for the JBD1 sequence was constructed in Chimera with residues 1–20 constrained to be α-helical and the His6 region extended. In this model the C60 was attached to the ε-amino group in the internal lysine residue. All models were docked to the surface of the QD with a diameter of 100 Å with an 11 Å DHLA layer on the QD surface. Comparable models were generated where the QD was coated with a DHLA–PEG750–OMe ligand layer (∼33 Å thick). Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).
Self-Assembly of Quantum Dot–Peptide Conjugates for Optical Characterization
The QD–peptide–C60 conjugates were prepared via self-assembly using the conditions described previously.31 Briefly, the frozen, concentrated C60-labeled peptide solutions (in DMSO) were thawed, sonicated, and then used to form diluted peptide stock solutions in DMSO (∼10 μM). The following is an example for preparing the conjugates with DHLA–and PEG750–OMe-coated QDs. First, QDs (∼10 pmol, 0.26 μM) in 14 mM sodium tetraborate buffer (pH ∼8.5) were mixed with DMSO, and then appropriate volumes of C60-labeled peptide stock solutions (10 μM) were added to achieve the desired peptide/QD ratio with a final volume of 110 μL. The initial volume of DMSO added was adjusted for each ratio of peptide/QD to achieve a final DMSO content of 65% (v/v). The final QD concentration was 90 nM. These solutions were mixed and assembled overnight before analysis. Successful self-assembly of the peptide–C60 conjugates to the QD surface was confirmed by measuring particle size and distribution via DLS using a ZetaSizer NanoSeries equipped with a HeNe laser source (λ = 633 nm) (Malvern Instruments Ltd., Worcestershire, UK) and analyzed using Dispersion Technology Software (DTS, Malvern Instruments Ltd.).
Optical Characterization of QD–Peptide–C60 Conjugates
Steady-state photoluminescence measurements were performed on a Tecan Infinite M1000 fluorescence multifunction plate reader. Electronic absorption spectra were collected on an HP 8453 diode array spectrophotometer (Agilent Technologies, Santa Clara, CA, USA).
Liposome Synthesis
Liposomes composed of 1,2-dipalmitoyl-snglycerol-3-phosphocholine phospholipids were prepared through thin film hydration. The phospholipids were dissolved in chloroform to a final concentration of ∼10–20 mM. The lipids were then placed in a round-bottom flask, and the solvent was removed overnight by Rotovap under vacuum to produce a thin lipid film. The lipids were rehydrated in ∼5 mL of warmed PBS (preheated to ∼45 °C) for 1 h. The lipid suspension was agitated by shaking for 30 min and then placed in a bath sonicator for a minute to produce liposomal vesicles.
Analysis of Peptide Structure by Circular Dichroism
CD experiments were carried out using a Jasco J-815 CD spectrometer (Jasco, Easton, MD, USA). A peptide concentration of 5 μM was used for CD measurements. CD spectra were recorded in phosphate buffer, pH 7.0, in the presence or absence of liposomes (∼250 μM lipids). Wavelength scans were performed from 190 to 260 nm in a thermally controlled (20 °C) quartz cell having a 0.1 cm path length. Each CD spectrum was the average of three scans done at a scan rate of 50 nm/ min using a data pitch of 1 nm, data integration time of 8 s, bandwidth of 1 nm, and a scan speed of 50 nm per min. The background spectrum (buffer only in the cuvette) was measured first, followed by that of the peptide or peptide/liposome solution. Collected CD data were normalized, and the ellipticity value (θ, in mdeg) was converted to standard units of deg·cm2/dmol (designated as [θ]) using [θ] = θ/(path length in mm × molar concentration of peptide × number of residues). Data on the molar ellipticity as a function of wavelength that were obtained from the CD scans were deconvoluted using the SP-22X algorithm and analyzed using the CONTIN/LL software packages to yield the predicted percentages of secondary structural components. These programs analyze the ellipticity values at each wavelength and compare them with a library of proteins with known secondary structure to estimate the percentages of the various components.
Cell Culture
A549 and HeLa cells (ATCC, Manassas, VA, USA) were cultured in DMEM and MEM, respectively. Each basal medium was amended to contain 10% FBS, 1× antibiotic–antimycotic, and 1 mM sodium pyruvate to yield the complete growth medium formulation. All cellular delivery/staining experiments were performed using cells between passages 5 and 15. In general, cells were seeded to 35 mm culture dishes fitted with 14 mm #1.0 cover glass inserts (MatTek Corp., Ashland, MA, USA) at a density of ∼7 × 104 cells/mL (total 3 mL/dish). The dishes were precoated with fibronectin (10–20 μg/mL) in DPBS before adding the cell suspension, and the cells were cultured for 24 h prior to use in labeling experiments.
For primary neuronal cultures, glass dishes were pretreated with 50 μg/mL poly-D-lysine (Sigma-Aldrich) by placing 50 μL drops in the center of the dishes overnight, rinsing twice with DI water, and allowing to air-dry. Next, 50 μL drops of 20 μg/mL laminin (Sigma-Aldrich) in DPBS (without Ca2+ or Mg2+; Thermo Fisher Scientific Inc., Waltham, MA, USA) were placed at the center of the dishes for 1 h, then removed, and the dishes air-dried.109 E18 murine cortices were obtained commercially (BrainBits LLC, Springfield, IL, USA) or surgically isolated and cultured as previously described.107 Briefly, embryonic cortices were surgically dissected, minced, and placed in serum-free DMEM containing 20 U/mL papain (Sigma) in papain activation buffer (1.1 mM EDTA, 67 μM mercaptoethanol, and 5 mM L-cysteine) and 200 U/mL DNase (Thermo Scientific) for 20 min at 37 °C. Enzymatically dissociated tissue was then triturated using a fire-polished Pasteur pipet until the suspension appeared uniform (20–30 times). Following trituration, the cells and dissociation solution were centrifuged at 250g for 5 min, and the resulting pellet was resuspended in 1 mL of complete cell medium (high-glucose DMEM + Glutamax, 2% B27, 8 μg/mL L-ascorbic acid, 5% horse serum, 5% fetal bovine serum) for cell counting. Additional medium was then added to obtain a cell concentration of 1 × 106/mL. An aliquot (50 μL) of cell solution was added to the center of each well and kept at 37 °C, 10% CO2, and 90% humidity for approximately 2 h to facilitate cell adhesion, then flooded with an additional 1 mL of complete medium. Cortical cultures underwent 50% medium exchanges every 48 h prior to QD labeling and live/dead staining experiments. Following 7 days in culture, complete cell medium was replaced with serum-free medium to cease the proliferation of non-neuronal cells.
Determination of Membrane-Binding Efficiency of Peptides and Peptide–C60 Conjugates by Silver Staining
Growth media was removed, and A549 cell monolayers and the cells were washed with DMEM–HEPES. The cells were then incubated with 5–10 μM peptide or peptide–C60 conjugates for 10–15 min at 37 °C. Following washing with DMEM–HEPES, the cells were fixed with paraformaldehyde (3.7% in DPBS) for 15 min followed by permeabilization using 0.01% Triton/DPBS for 15 min at room temperature. Then the cells were washed again with DMEM–HEPES and incubated with mouse anti-C60 monoclonal Ab (3.3 μg/mL in 0.1% gelatin/DPBS) for 45 min at 37 °C followed by incubation with secondary goat anti-mouse (2 μg/mL in 0.1% gelatin/DPBS) for 45 min at 37 °C. The cells were washed with DMEM–HEPES and water and stained for 5 min using a silver staining/enhancer kit (Sigma-Aldrich). A control sample without peptides was also prepared following the same procedure. The samples were imaged by differential interference contrast (DIC), and the degree of staining was quantified by selecting ROIs for individual cells using ImageJ software ver. 1.50i (National Institutes of Health, USA).
Preparation of QD–DHLA–Peptide–C60 Complexes for Membrane Labeling and Depolarization Studies
The QD–DHLA–peptide–C60 complexes were prepared by adding 2 μL of a 3 μM QD stock solution (final QD concentration 20 nM) to 297 μL of peptide solution (400 nM final concentration) in PBS (pH 7.9–8.2). The final QD:peptide–C60 ratio was 1:20. The mixtures were incubated for 15 min at room temperature to form QD–peptide assemblies. The complexes were briefly centrifuged and used immediately to label cellular membranes.
Cellular Labeling with QD–DHLA–Peptide–C60 Complexes
Complete growth medium was removed from cell monolayers, and the cells were washed with DMEM–HEPES. Solutions (typically ∼150 μL) of QD–DHLA (20 nM QD) or QD–DHLA–peptide–C60 complexes (20 nM QD appended with 20 peptides/QD; 400 nM peptide concentration) were incubated on the cells for 10–15 min at 37 °C followed by washing with DPBS. The cells were fixed with paraformaldehyde (3.7% in DPBS) for 15 min at room temperature, stored in 0.05% NaN3/PBS at 4 °C, and imaged within 48 h. Live cell imaging during depolarization studies was performed as described below.
KCl Depolarization Studies
Depolarization experiments were performed on live HeLa, PC-12, and primary murine neuronal cells. The cells were labeled with QD–DHLA–peptide–C60 complexes as described above. For comparison, two commercial voltage-sensitive probes were also studied: the slow response dye, oxonol (B438), or the fast response probe, FluoVolt. Cells were labeled with oxonol by incubation with 125 μL of 1 μM oxonol in DMEM–HEPES for 5 min at 4 °C. Labeling of cells with FluoVolt was done according to the manufacturer’s supplied protocol. After staining cells were washed with DPBS. Live cell imaging solution (200 μL, Life Technologies) was added to cover the cell monolayers. Cells were imaged (vide infra) for florescence response to membrane depolarization induced by the addition of isotonic 140 mM KCl solution. In these experiments, 140 mM KCl was perfused into the culture dish over 3 min, and the KCl concentration was allowed to stabilize for another 2 min. Fluorescence was quantified at the beginning and end of this regimen. Fluorescence quantification of images was done using NIS-Elements AR 4.3 software (Nikon Co. Ltd., Tokyo, Japan) by sampling multiple RIOs in individual cells.
Microscopy
Imaging of fixed cell samples and live cell depolarization experiments was performed by DIC and confocal laser scanning microscopy using a Nikon A1RSi confocal microscope equipped with a 405 nm diode laser with fluorescence detection channels set to 500–550 nm (green) and 570–620 nm (red) with dichroic mirrors at 405/488/561/640 nm. All images were collected using a Plan Apo 60× objective. Laser power, PMT gain, and threshold for each channel were held constant across different samples to allow for quantitative analysis. The confocal images were acquired as a z-stack comprising sequential/series optical x–y sections taken at 0.5–1.5 μm z-intervals using NIS-Elements AR 4.3 software (Nikon Co. Ltd., Tokyo, Japan). The images were processed for publication with Adobe Photoshop CS4 (ver. 11.0).
Cellular Proliferation Assays
The cytotoxicity of QDs and QD–peptide–C60 complexes was determined on HeLa cells using the CellTiter 96 Aqueous One Solution MTS cell proliferation assay (Promega, Madison, WI, USA). This assay is based upon the conversion of a tetrazolium substrate to a soluble formazan product by viable cells. HeLa cells were seeded to the wells of 96-well tissue culture plates (∼5 × 103 cells/well). QDs and QD–peptide–C60 complexes were incubated on the cells for 15 min to emulate the labeling conditions used herein. After incubation, the materials were replaced with complete growth medium and the cells were cultured for 72 h. After this proliferation period, 20 μL of the tetrazolium substrate was added to each well, and color formation proceeded at 37 °C for 4h. The absorbance of the formazan product was read at 590 and 620 nm (for subtraction of nonspecific background absorbance) using a Tecan Infinite M1000 (Tecan, US) microtiter plate reader. Absorbance values with the background subtracted were reported as percent of control cell proliferation (degree of proliferation of cells in cell culture media only).
Cortical Neuron Live/Dead Assays
Live cell staining was carried out using Calcein AM (Thermo Fisher). Briefly, cells were washed three times in PBS and incubated in serum-free complete cell medium containing 2 μM Calcein AM at 37 °C for 10 min.
In Vivo Optical Recording of Cortical Brain Function
Experimental procedures were all in accordance with the National Institutes of Health guidelines for the care of experimental animals and an experimental protocol approved by the Animal Use and Care Committee of the University of Maryland School of Medicine. Imaging was performed on a total of five adult mice (B6 male and female, 25–30 g body weight, age 5–10 weeks): QD–JBD1–C60 (2 mice), QD–JBD1 (1 mouse), QD alone (2 mice). All animals were anesthetized with urethane (1.2 g/kg) and fixed on a stereotactic frame. A 3 × 3 mm section of the cranial bone above the left or right parietal cortex was removed using a dental drill. The exposed dural surface was cleaned with a hemostatic sponge and washed with artificial cerebral spinal fluid (ACSF). After craniotomy, 3–6 intracortical injections (0.5 μL each) of QD (1 μM QD) or QD–peptide solution (QDs assembled with 20 peptide/QD) were performed using a Nanojet II injector device fitted with a glass micropipet (20–50 μm diameter) mounted on a micromanipulator that allowed precise injections 0.3 mm below the dural surface. After injection the cortex was washed with ACSF, covered with high-density silicone oil, and sealed with a 0.1 mm thick cover glass to suppress brain pulsations caused by cardiovascular and respiratory movements. For imaging, the CCD camera was positioned above the recording area and directed such that its optical axis was perpendicular to the cortical surface. The focusing plane was set 100 μm below the dural surface. During recording, the imaging area of the cortex was illuminated with a tungsten filament lamp (12 V, 100 WA, Zeiss) passed through an excitation filter (490 ± 30 nm) and a dichroic mirror at 575 nm. Imaging was performed through a tandem configuration of two 50 mm lenses, at a frame rate of 200 Hz, and recorded using a MiCAM-02 system (SciMedia, Costa Mesa, CA, USA).
For electrical stimulation, a bipolar tungsten electrode was introduced into the cortex at a depth of 0.1–0.2 mm. An electrical stimulus (100 μA) was computer-generated using an ISO-Flex stimulator (A.M.P.I., Israel) and consisted of seven pulses (each 3 ms duration, 100 Hz frequency). At the start of each optical recording, a grayscale image of the opened cortical region was captured and saved as a graphic file. Each experimental session consisted of 50 trials (500 frames/trial) with the electrical stimulus (one stimulus per trial) presented at the 100th frame (500 ms). The interval between trials was 12 s. Fluorescence changes were calculated as ΔF/F0 in ROIs using Brain Vision Analyzer (Brain Products GmbH, Germany). Images of areas activated by electrical stimulation were pseudocolored for publication.
Data analysis
The data were statistically analyzed by the univariate analysis of variance (ANOVA) using GraphPad Prism 6.01 software for Windows (La Jolla, CA, USA). For multiple comparisons, Bonferroni’s post hoc test was applied. All average values were given ± standard error of mean unless otherwise mentioned. The acceptable probability for significance was p < 0.05.
Supplementary Material
Acknowledgments
The authors acknowledge the NRL NSI and Base Funding Program (Work Units MA041-06-41-T008-15 and MA041-06-41-4943). O.K.N. and Y.C. were supported by postdoctoral fellowships from the National Research Council and the American Society for Engineering Education, respectively. R.V. acknowledges the Office of Naval Research (Grant No. 0001416WX01849). Yu Chen acknowledges the support from NSF CBET-1254743 (CAREER Award). V.T.’s and R.S.E.’s research was supported by NIH/NINDS (R01 NS084818). L.D.F. is a Ph.D. candidate in the Fischell Department of Bioengineering, University of Maryland, College Park, MD, USA. The Maryland NanoCenter and its NispLab supported TEM measurements.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.7b00954.
Additional experimental details and results including QD and peptide analysis, bioconjugate photophysical analysis, and in vitro and in vivo imaging controls (PDF)
ORCID
Igor L. Medintz: 0000-0002-8902-4687
James B. Delehanty: 0000-0001-9245-3936
Notes
The authors declare no competing financial interest.
References
- 1.Nel AE, Madler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M. Understanding Biophysicochemical Interactions at the Nano-Bio Interface. Nat Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 2.Dennis AM, Delehanty JB, Medintz IL. Emerging Physicochemical Phenomena along with New Opportunities at the Biomolecular–Nanoparticle Interface. J Phys Chem Lett. 2016;7:2139–2150. doi: 10.1021/acs.jpclett.6b00570. [DOI] [PubMed] [Google Scholar]
- 3.Lal S, Clare SE, Halas NJ. Nanoshell-Enabled Photothermal Cancer Therapy: Impending Clinical Impact. Acc Chem Res. 2008;41:1842–1851. doi: 10.1021/ar800150g. [DOI] [PubMed] [Google Scholar]
- 4.Bardhan R, Lal S, Joshi A, Halas NJ. Theranostic Nanoshells: From Probe Design to Imaging and Treatment of Cancer. Acc Chem Res. 2011;44:936–946. doi: 10.1021/ar200023x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breger JC, Ancona MG, Walper SA, Oh E, Susumu K, Stewart MH, Deschamps JR, Medintz IL. Understanding How Nanoparticle Attachment Enhances Phosphotriesterase Kinetic Efficiency. ACS Nano. 2015;9:8491–8503. doi: 10.1021/acsnano.5b03459. [DOI] [PubMed] [Google Scholar]
- 6.Ding S, Cargill AA, Medintz IL, Claussen JC. Increasing the Activity of Immobilized Enzymes with Nanoparticle Conjugation. Curr Opin Biotechnol. 2015;34:242–250. doi: 10.1016/j.copbio.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 7.Weiss PS. President Obama Announces the BRAIN Initiative. ACS Nano. 2013;7:2873–2874. doi: 10.1021/nn401796f. [DOI] [PubMed] [Google Scholar]
- 8.The Human Brain Project. https://www.humanbrainproject.eu (accessed January 12, 2017).
- 9.Alivisatos AP, Chun M, Church, George M, Greenspan, Ralph J, Roukes, Michael L, Yuste R. The Brain Activity Map Project and the Challenge of Functional Connectomics. Neuron. 2012;74:970–974. doi: 10.1016/j.neuron.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrews AM, Weiss PS. Nano in the Brain: Nano-Neuroscience. ACS Nano. 2012;6:8463–8464. doi: 10.1021/nn304724q. [DOI] [PubMed] [Google Scholar]
- 11.Antic SD, Empson RM, Knöpfel T. Voltage Imaging to Understand Connections and Functions of Neuronal Circuits. J Neurophysiol. 2016;116:135–152. doi: 10.1152/jn.00226.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Storace D, Rad MS, Kang B, Cohen LB, Hughes T, Baker BJ. Toward Better Genetically Encoded Sensors of Membrane Potential. Trends Neurosci. 2016;39:277–289. doi: 10.1016/j.tins.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterka DS, Takahashi H, Yuste R. Imaging Voltage in Neurons. Neuron. 2011;69:9–21. doi: 10.1016/j.neuron.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakmann Ba, Neher E. Patch Clamp Techniques for Studying Ionic Channels in Excitable Membranes. Annu Rev Physiol. 1984;46:455–472. doi: 10.1146/annurev.ph.46.030184.002323. [DOI] [PubMed] [Google Scholar]
- 15.Rios G, Lubenov EV, Chi D, Roukes ML, Siapas AG. Nanofabricated Neural Probes for Dense 3-D Recordings of Brain Activity. Nano Lett. 2016;16:6857–6862. doi: 10.1021/acs.nanolett.6b02673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsytsarev V, Liao LD, Kong KV, Liu YH, Erzurumlu RS, Olivo M, Thakor NV. Recent Progress in Voltage-Sensitive Dye Imaging for Neuroscience. J Nanosci Nanotechnol. 2014;14:4733–44. doi: 10.1166/jnn.2014.9531. [DOI] [PubMed] [Google Scholar]
- 17.Broussard GJ, Liang R, Tian L. Monitoring Activity in Neural Circuits with Genetically Encoded Indicators. Front Mol Neurosci. 2014;7:1–17. doi: 10.3389/fnmol.2014.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maclaurin D, Venkatachalam V, Lee H, Cohen AE. Mechanism of Voltage-Sensitive Fluorescence in a Microbial Rhodopsin. Proc Natl Acad Sci U S A. 2013;110:5939–5944. doi: 10.1073/pnas.1215595110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hochbaum DR, Zhao Y, Farhi SL, Klapoetke N, Werley CA, Kapoor V, Zou P, Kralj JM, Maclaurin D, Smedemark-Margulies N, Saulnier JL, Boulting GL, Straub C, Cho YK, Melkonian M, Wong GK-S, Harrison DJ, Murthy VN, Sabatini BL, Boyden ES, et al. All-Optical Electrophysiology in Mammalian Neurons Using Engineered Microbial Rhodopsins. Nat Methods. 2014;11:825–833. doi: 10.1038/nmeth.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alivisatos AP, Andrews AM, Boyden ES, Chun M, Church GM, Deisseroth K, Donoghue JP, Fraser SE, Lippincott-Schwartz J, Looger LL, Masmanidis S, McEuen PL, Nurmikko AV, Park H, Peterka DS, Reid C, Roukes ML, Scherer A, Schnitzer M, Sejnowski TJ, Shepard KL, et al. Nanotools for Neuroscience and Brain Activity Mapping. ACS Nano. 2013;7:1850–1866. doi: 10.1021/nn4012847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park K, Deutsch Z, Li JJ, Oron D, Weiss S. Single Molecule Quantum-Confined Stark Effect Measurements of Semiconductor Nanoparticles at Room Temperature. ACS Nano. 2012;6:10013–10023. doi: 10.1021/nn303719m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Empedocles SA, Bawendi MG. Quantum-Confined Stark Effect in Single CdSe Nanocrystallite Quantum Dots. Science. 1997;278:2114–2117. doi: 10.1126/science.278.5346.2114. [DOI] [PubMed] [Google Scholar]
- 23.Krauss TD, Brus LE. Electronic Properties of Single Semiconductor Nanocrystals: Optical and Electrostatic Force Microscopy Measurements. Mater Sci Eng, B. 2000;69:289–294. [Google Scholar]
- 24.Delehanty JB, Blanco-Canosa JB, Bradburne CE, Susumu K, Stewart MH, Prasuhn DE, Dawson PE, Medintz IL. Site-Specific Cellular Delivery of Quantum Dots with Chemoselectively-Assembled Modular Peptides. Chem Commun. 2013;49:7878–7880. doi: 10.1039/c3cc42781a. [DOI] [PubMed] [Google Scholar]
- 25.Boeneman K, Delehanty JB, Blanco-Canosa JB, Susumu K, Stewart MH, Oh E, Huston AL, Dawson G, Ingale S, Walters R, Domowicz M, Deschamps JR, Algar WR, DiMaggio S, Manono J, Spillmann CM, Thompson D, Jennings TL, Dawson PE, Medintz IL. Selecting Improved Peptidyl Motifs for Cytosolic Delivery of Disparate Protein and Nanoparticle Materials. ACS Nano. 2013;7:3778–3796. doi: 10.1021/nn400702r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gopalakrishnan G, Danelon C, Izewska P, Prummer M, Bolinger PY, Geissbuhler I, Demurtas D, Dubochet J, Vogel H. Multifunctional Lipid/Quantum Dot Hybrid Nanocontainers for Controlled Targeting of Live Cells. Angew Chem, Int Ed. 2006;45:5478–5483. doi: 10.1002/anie.200600545. [DOI] [PubMed] [Google Scholar]
- 27.Clapp AR, Pons T, Medintz IL, Delehanty JB, Melinger JS, Tiefenbrunn T, Dawson PE, Fisher BR, O’Rourke B, Mattoussi H. Two-Photon Excitation of Quantum Dot-Based Fluorescence Resonance Energy Transfer and Its Applications. Adv Mater. 2007;19:1921–1926. [Google Scholar]
- 28.Resch-Genger U, Grabolle M, Cavaliere-Jaricot S, Nitschke R, Nann T. Quantum Dots Versus Organic Dyes as Fluorescent Labels. Nat Methods. 2008;5:763–775. doi: 10.1038/nmeth.1248. [DOI] [PubMed] [Google Scholar]
- 29.Clapp AR, Medintz IL, Mauro JM, Fisher BR, Bawendi MG, Mattoussi H. Fluorescence Resonance Energy Transfer Between Quantum Dot Donors and Dye-Labeled Protein Acceptors. J Am Chem Soc. 2004;126:301–310. doi: 10.1021/ja037088b. [DOI] [PubMed] [Google Scholar]
- 30.Medintz IL, Pons T, Susumu K, Boeneman K, Dennis A, Farrell D, Deschamps JR, Melinger JS, Bao G, Mattoussi H. Resonance Energy Transfer Between Luminescent Quantum Dots and Diverse Fluorescent Protein Acceptors. J Phys Chem C. 2009;113:18552–18561. doi: 10.1021/jp9060329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stewart MH, Huston AL, Scott AM, Oh E, Algar WR, Deschamps JR, Susumu K, Jain V, Prasuhn DE, Blanco-Canosa J, Dawson PE, Medintz IL. Competition Between Förster Resonance Energy Transfer and Electron Transfer in Stoichiometrically Assembled Semiconductor Quantum Dot–Fullerene Conjugates. ACS Nano. 2013;7:9489–9505. doi: 10.1021/nn403872x. [DOI] [PubMed] [Google Scholar]
- 32.Virkki K, Demir S, Lemmetyinen H, Tkachenko NV. Photoinduced Electron Transfer in CdSe/ZnS Quantum Dot–Fullerene Hybrids. J Phys Chem C. 2015;119:17561–17572. [Google Scholar]
- 33.Andrasfalvy BK, Galinanes GL, Huber D, Barbic M, Macklin JJ, Susumu K, Delehanty JB, Huston AL, Makara JK, Medintz IL. Quantum Dot-Based Multiphoton Fluorescent Pipettes for Targeted Neuronal Electrophysiology. Nat Methods. 2014;11:1237–1241. doi: 10.1038/nmeth.3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walters R, Kraig RP, Medintz I, Delehanty JB, Stewart MH, Susumu K, Huston AL, Dawson PE, Dawson G. Nanoparticle Targeting to Neurons in a Rat Hippocampal Slice Culture Model. ASN Neuro. 2012;4:383–392. doi: 10.1042/AN20120042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walters R, Medintz IL, Delehanty JB, Stewart MH, Susumu K, Huston AL, Dawson PE, Dawson G. The Role of Negative Charge in the Delivery of Quantum Dots to Neurons. ASN Neuro. 2015;7:1–12. doi: 10.1177/1759091415592389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agarwal R, Domowicz MS, Schwartz NB, Henry J, Medintz I, Delehanty JB, Stewart MH, Susumu K, Huston AL, Deschamps JR, Dawson PE, Palomo V, Dawson G. Delivery and Tracking of Quantum Dot Peptide Bioconjugates in an Intact Developing Avian Brain. ACS Chem Neurosci. 2015;6:494–504. doi: 10.1021/acschemneuro.5b00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marshall JD, Schnitzer MJ. Optical Strategies for Sensing Neuronal Voltage Using Quantum Dots and Other Semiconductor Nanocrystals. ACS Nano. 2013;7:4601–4609. doi: 10.1021/nn401410k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rowland CE, Susumu K, Stewart MH, Oh E, Makinen AJ, O’Shaughnessy TJ, Kushto G, Wolak MA, Erickson JS, Efros AL, Huston AL, Delehanty JB. Electric Field Modulation of Semiconductor Quantum Dot Photoluminescence: Insights into the Design of Robust Voltage-Sensitive Cellular Imaging Probes. Nano Lett. 2015;15:6848–6854. doi: 10.1021/acs.nanolett.5b02725. [DOI] [PubMed] [Google Scholar]
- 39.Molokanova E, B JA, Zhao W, Naasani I, Ignatius MJ, Treadway JA, Savtchenko A. Quantum Dots Move Beyond Fluorescence Imaging. Biophotonics Int. 2008:26–31. [Google Scholar]
- 40.Sapsford KE, Farrell D, Sun S, Rasooly A, Mattoussi H, Medintz IL. Monitoring of Enzymatic Proteolysis on a Electroluminescent-CCD Microchip Platform Using Quantum Dot-Peptide Substrates. Sens Actuators, B. 2009;139:13–21. [Google Scholar]
- 41.Lyu PC, Sherman JC, Chen A, Kallenbach NR. Alpha-Helix Stabilization by Natural and Unnatural Amino Acids with Alkyl Side Chains. Proc Natl Acad Sci U S A. 1991;88:5317–5320. doi: 10.1073/pnas.88.12.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karle IL, Flippen-Anderson JL, Sukumar M, Balaram P. Helix Packing of Leucine-Rich Peptides: A Parallel Leucine Ladder in the Structure of Boc-Aib-Leu-Aib-Aib-Leu-Leu-Leu-Aib-Leu-Aib-OMe. Proteins: Struct, Funct, Genet. 1992;12:324–330. doi: 10.1002/prot.340120404. [DOI] [PubMed] [Google Scholar]
- 43.Marqusee S, Robbins VH, Baldwin RL. Unusually Stable Helix Formation in Short Alanine-Based Peptides. Proc Natl Acad Sci U S A. 1989;86:5286–5290. doi: 10.1073/pnas.86.14.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gemmill KB, Díaz SA, Blanco-Canosa JB, Deschamps JR, Pons T, Liu HW, Deniz A, Melinger J, Oh E, Susumu K, Stewart MH, Hastman DA, North SH, Delehanty JB, Dawson PE, Medintz IL. Examining the Polyproline Nanoscopic Ruler in the Context of Quantum Dots. Chem Mater. 2015;27:6222–6237. [Google Scholar]
- 45.Rabanal F, Ludevid MD, Pons M, Giralt E. CD of Proline-Rich Polypeptides: Application to the Study of the Repetitive Domain of Maize Glutelin-2. Biopolymers. 1993;33:1019–1028. doi: 10.1002/bip.360330704. [DOI] [PubMed] [Google Scholar]
- 46.Pujals S, Giralt E. Proline-Rich, Amphipathic Cell-Penetrating Peptides. Adv Drug Delivery Rev. 2008;60:473–484. doi: 10.1016/j.addr.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 47.Mattoussi H, Mauro JM, Goldman ER, Anderson GP, Sundar VC, Mikulec FV, Bawendi MG. Self-Assembly of CdSe-ZnS Quantum Dot Bioconjugates Using an Engineered Recombinant Protein. J Am Chem Soc. 2000;122:12142–12150. [Google Scholar]
- 48.Bradburne CE, Delehanty JB, Boeneman Gemmill K, Mei BC, Mattoussi H, Susumu K, Blanco-Canosa JB, Dawson PE, Medintz IL. Cytotoxicity of Quantum Dots Used for In Vitro Cellular Labeling: Role of QD Surface Ligand, Delivery Modality, Cell Type, and Direct Comparison to Organic Fluorophores. Bioconjugate Chem. 2013;24:1570–1583. doi: 10.1021/bc4001917. [DOI] [PubMed] [Google Scholar]
- 49.Delehanty JB, Medintz IL, Pons T, Brunel FM, Dawson PE, Mattoussi H. Self-Assembled Quantum Dot-Peptide Bio-conjugates for Selective Intracellular Delivery. Bioconjugate Chem. 2006;17:920–927. doi: 10.1021/bc060044i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Susumu K, Uyeda HT, Medintz IL, Pons T, Delehanty JB, Mattoussi H. Enhancing the Stability and Biological Functionalities of Quantum Dots via Compact Multifunctional Ligands. J Am Chem Soc. 2007;129:13987–96. doi: 10.1021/ja0749744. [DOI] [PubMed] [Google Scholar]
- 51.Mei BC, Susumu K, Medintz IL, Delehanty JB, Mountziaris TJ, Mattoussi H. Modular Poly(Ethylene Glycol) Ligands for Biocompatible Semiconductor and Gold Nanocrystals with Extended pH and Ionic Stability. J Mater Chem. 2008;18:4949–4958. [Google Scholar]
- 52.Haddon RC, Palmer RE, Kroto HW, Sermon PA. The Fullerenes: Powerful Carbon-Based Electron Acceptors [and Discussion] Philos Trans R Soc, A. 1993;343:53–62. [Google Scholar]
- 53.Guldi DM. Fullerenes: Three Dimensional Electron Acceptor Materials. Chem Commun. 2000:321–327. [Google Scholar]
- 54.Blanco-Canosa JB, Wu M, Susumu K, Petryayeva E, Jennings TL, Dawson PE, Algar WR, Medintz IL. Recent Progress in the Bioconjugation of Quantum Dots. Coord Chem Rev. 2014;263–264:101–137. [Google Scholar]
- 55.Dong H, Zhu H, Meng Q, Gong X, Hu W. Organic Photoresponse Materials and Devices. Chem Soc Rev. 2012;41:1754–1808. doi: 10.1039/c1cs15205j. [DOI] [PubMed] [Google Scholar]
- 56.Delehanty JB, Bradburne CE, Boeneman K, Susumu K, Farrell D, Blanco-Canosa JB, Dawson G, Dawson PE, Medintz IL. Delivering Quantum Dot-Peptide Bioconjugates to the Cellular Cytosol: Escaping from the Endolysosomal System. Integr Biol. 2010;2:265–277. doi: 10.1039/c0ib00002g. [DOI] [PubMed] [Google Scholar]
- 57.Medintz IL, Pons T, Delehanty JB, Susumu K, Brunel FM, Dawson PE, Mattoussi H. Intracellular Delivery of Quantum Dot-Protein Cargos Mediated by Cell Penetrating Peptides. Bioconjugate Chem. 2008;19:1785–1795. doi: 10.1021/bc800089r. [DOI] [PubMed] [Google Scholar]
- 58.Medintz IL, Stewart MH, Trammell SA, Susumu K, Delehanty JB, Mei BC, Melinger JS, Blanco-Canosa JB, Dawson PE, Mattoussi H. Quantum-Dot/Dopamine Bioconjugates Function as Redox Coupled Assemblies for In Vitro and Intracellular pH Sensing. Nat Mater. 2010;9:676–684. doi: 10.1038/nmat2811. [DOI] [PubMed] [Google Scholar]
- 59.Prasuhn DE, Deschamps JR, Susumu K, Stewart MH, Boeneman K, Blanco-Canosa JB, Dawson PE, Medintz IL. Polyvalent Display and Packing of Peptides and Proteins on Semiconductor Quantum Dots: Predicted Versus Experimental Results. Small. 2010;6:555–564. doi: 10.1002/smll.200901845. [DOI] [PubMed] [Google Scholar]
- 60.Kuznetsova I, Zaslavsky B, Breydo L, Turoverov K, Uversky V. Beyond the Excluded Volume Effects: Mechanistic Complexity of the Crowded Milieu. Molecules. 2015;20:1377. doi: 10.3390/molecules20011377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pons T, Medintz IL, Sykora M, Mattoussi H. Spectrally Resolved Energy Transfer Using Quantum Dot Donors: Ensemble and Single-Molecule Photoluminescence Studies. Phys Rev B: Condens Matter Mater Phys. 2006;73:245302–245307. [Google Scholar]
- 62.Stewart MH, Huston AL, Scott AM, Efros AL, Melinger JS, Gemmill KB, Trammell SA, Blanco-Canosa JB, Dawson PE, Medintz IL. Complex Förster Energy Transfer Interactions Between Semiconductor Quantum Dots and a Redox-Active Osmium Assembly. ACS Nano. 2012;6:5330–5347. doi: 10.1021/nn301177h. [DOI] [PubMed] [Google Scholar]
- 63.Ahmad A, Azmi S, Srivastava S, Kumar A, Tripathi JK, Mishra NN, Shukla PK, Ghosh JK. Design and Characterization of Short Antimicrobial Peptides Using Leucine Zipper Templates with Selectivity Towards Microorganisms. Amino Acids. 2014;46:2531–2543. doi: 10.1007/s00726-014-1802-3. [DOI] [PubMed] [Google Scholar]
- 64.Ikeda A, Kiguchi K, Shigematsu T, Nobusawa K, Kikuchi J-i, Akiyama M. Location of [60]Fullerene Incorporation in Lipid Membranes. Chem Commun. 2011;47:12095–12097. doi: 10.1039/c1cc14650e. [DOI] [PubMed] [Google Scholar]
- 65.Russ KA, Elvati P, Parsonage TL, Dews A, Jarvis JA, Ray M, Schneider B, Smith PJS, Williamson PTF, Violi A, Philbert MA. C60 Fullerene Localization and Membrane Interactions in RAW 2647 Immortalized Mouse Macrophages. Nanoscale. 2016;8:4134–4144. doi: 10.1039/c5nr07003a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang S, Mu Y, Zhang JZH, Xu W. Effect of Self-Assembly of Fullerene Nano-Particles on Lipid Membrane. PLoS One. 2013;8:e77436. doi: 10.1371/journal.pone.0077436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tong R, Kohane DS. New Strategies in Cancer Nano-medicine. Annu Rev Pharmacol Toxicol. 2016;56:41–57. doi: 10.1146/annurev-pharmtox-010715-103456. [DOI] [PubMed] [Google Scholar]
- 68.Kobayashi K, Wei J, Iida R, Ijiro K, Niikura K. Surface Engineering of Nanoparticles for Therapeutic Applications. Polym J. 2014;46:460–468. [Google Scholar]
- 69.Jokerst JV, Lobovkina T, Zare RN, Gambhir SS. Nanoparticle PEGylation for Imaging and Therapy. Nanomedicine. 2011;6:715–728. doi: 10.2217/nnm.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Macías W, Carlson R, Rajadhyaksha A, Barczak A, Konradi C. Potassium Chloride Depolarization Mediates CREB Phosphorylation in Striatal Neurons in an NMDA Receptor-Dependent Manner. Brain Res. 2001;890:222–232. doi: 10.1016/s0006-8993(00)03163-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levitan ES, Gealy R, Trimmer JS, Takimoto K. Membrane Depolarization Inhibits Kv1.5 Voltage-Gated K Channel Gene Transcription and Protein Expression in Pituitary Cells. J Biol Chem. 1995;270:6036–6041. doi: 10.1074/jbc.270.11.6036. [DOI] [PubMed] [Google Scholar]
- 72.Obata T, Aomine M, Yamanaka Y. Potassium Chloride Depolarization Enhances MPP+-Induced Hydroxyl Radical Generation in the Rat Striatum. Brain Res. 2000;852:488–491. doi: 10.1016/s0006-8993(99)02238-6. [DOI] [PubMed] [Google Scholar]
- 73.Lindgren S. Effects of KCl-Induced Depolarization on the GABA Concentration in the Corpus Striatum and in the Substantia Nigra. J Neural Transm. 1986;66:161–170. doi: 10.1007/BF01260911. [DOI] [PubMed] [Google Scholar]
- 74.Zhang X, Jin Y, Plummer MR, Pooyan S, Gunaseelan S, Sinko PJ. Endocytosis and Membrane Potential Are Required for HeLa Cell Uptake of R.I.-CKTat9, a Retro-Inverso Tat Cell Penetrating Peptide. Mol Pharmaceutics. 2009;6:836–48. doi: 10.1021/mp800121f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Woodford CR, Frady EP, Smith RS, Morey B, Canzi G, Palida SF, Araneda RC, Kristan WB, Kubiak CP, Miller EW, Tsien RY. Improved PeT Molecules for Optically Sensing Voltage in Neurons. J Am Chem Soc. 2015;137:1817–1824. doi: 10.1021/ja510602z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loew LM, Cohen LB, Dix J, Fluhler EN, Montana V, Salama G, Wu JY. A Naphthyl Analog of the Aminostyryl Pyridinium Class of Potentiometric Membrane Dyes Shows Consistent Sensitivity in a Variety of Tissue, Cell, and Model Membrane Preparations. J Membr Biol. 1992;130:1–10. doi: 10.1007/BF00233734. [DOI] [PubMed] [Google Scholar]
- 77.Tsytsarev V, Premachandra K, Takeshita D, Bahar S. Imaging Cortical Electrical Stimulation In Vivo: Fast Intrinsic Optical Signal Versus Voltage-Sensitive Dyes. Opt Lett. 2008;33:1032–1034. doi: 10.1364/ol.33.001032. [DOI] [PubMed] [Google Scholar]
- 78.Epps DE, Wolfe ML, Groppi V. Characterization of the Steady-State and Dynamic Fluorescence Properties of the Potential-Sensitive Dye Bis-(1,3-Dibutylbarbituric Acid) Trimethine Oxonol (Dibac4(3)) in Model Systems and Cells. Chem Phys Lipids. 1994;69:137–150. doi: 10.1016/0009-3084(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 79.Liu LP, Li SC, Goto NK, Deber CM. Threshold Hydrophobicity Dictates Helical Conformations of Peptides in Membrane Environments. Biopolymers. 1996;39:465–470. doi: 10.1002/(sici)1097-0282(199609)39:3<465::aid-bip17>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 80.Wegner KD, Hildebrandt N. Quantum Dots: Bright and Versatile In Vitro and In Vivo Fluorescence Imaging Biosensors. Chem Soc Rev. 2015;44:4792–4834. doi: 10.1039/c4cs00532e. [DOI] [PubMed] [Google Scholar]
- 81.Nazarenus M, Zhang Q, Soliman MG, del Pino P, Pelaz B, Carregal-Romero S, Rejman J, Rothen-Rutishauser B, Clift MJD, Zellner R, Nienhaus GU, Delehanty JB, Medintz IL, Parak WJ. In Vitro Interaction of Colloidal Nanoparticles with Mammalian Cells: What Have We Learned Thus Far? Beilstein J Nanotechnol. 2014;5:1477–1490. doi: 10.3762/bjnano.5.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grandy TH, Greenfield SA, Devonshire IM. An Evaluation of In Vivo Voltage-Sensitive Dyes: Pharmacological Side Effects and Signal-to-Noise Ratios after Effective Removal of Brain-Pulsation Artifacts. J Neurophysiol. 2012;108:2931–2945. doi: 10.1152/jn.00512.2011. [DOI] [PubMed] [Google Scholar]
- 83.Mennerick S, Chisari M, Shu HJ, Taylor A, Vasek M, Eisenman LN, Zorumski CF. Diverse Voltage-Sensitive Dyes Modulate GABA(A) Receptor Function. J Neurosci. 2010;30:2871–2879. doi: 10.1523/JNEUROSCI.5607-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shynkar VV, Klymchenko AS, Duportail G, Demchenko AP, Mely Y. Two-Color Fluorescent Probes for Imaging the Dipole Potential of Cell Plasma Membranes. Biochim Biophys Acta, Biomembr. 2005;1712:128–136. doi: 10.1016/j.bbamem.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 85.Loew LM. Design and Use of Organic Voltage Sensitive Dyes. In: Canepari M, Zecevic D, editors. Membrane Potential Imaging in the Nervous System: Methods and Applications. Springer New York; New York, NY: 2011. pp. 13–23. [Google Scholar]
- 86.Grinvald A, Lieke EE, Frostig RD, Hildesheim R. Cortical Point-Spread Function And Long-Range Lateral Interactions Revealed by Real-Time Optical Imaging of Macaque Monkey Primary Visual Cortex. J Neurosci. 1994;14:2545. doi: 10.1523/JNEUROSCI.14-05-02545.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Onuchic JN, Kobayashi C, Baldridge KK. Quantum Tunneling in Biological Reactions: The Interplay Between Theory and Experiments. J Braz Chem Soc. 2008;19:206–210. [Google Scholar]
- 88.Marcus RA, Sutin N. Electron Transfers in Chemistry and Biology. Biochim Biophys Acta, Rev Bioenerg. 1985;811:265–322. [Google Scholar]
- 89.Morris-Cohen AJ, Aruda KO, Rasmussen AM, Canzi G, Seideman T, Kubiak CP, Weiss EA. Controlling the Rate of Electron Transfer Between a Quantum Dot and a Tri-Ruthenium Molecular Cluster by Tuning the Chemistry of the Interface. Phys Chem Chem Phys. 2012;14:13794–13801. doi: 10.1039/c2cp40827a. [DOI] [PubMed] [Google Scholar]
- 90.Hines DA, Kamat PV. Quantum Dot Surface Chemistry: Ligand Effects and Electron Transfer Reactions. J Phys Chem C. 2013;117:14418–14426. [Google Scholar]
- 91.Bley S, Diez M, Albrecht F, Resch S, Waldvogel SR, Menzel A, Zacharias M, Gutowski J, Voss T. Electron Tunneling from Colloidal CdSe Quantum Dots to ZnO Nanowires Studied by Time-Resolved Luminescence and Photoconductivity Experiments. J Phys Chem C. 2015;119:15627–15635. [Google Scholar]
- 92.He C, Weinberg DJ, Nepomnyashchii AB, Lian S, Weiss EA. Control of the Redox Activity of PbS Quantum Dots by Tuning Electrostatic Interactions at the Quantum Dot/Solvent Interface. J Am Chem Soc. 2016;138:8847–8854. doi: 10.1021/jacs.6b03970. [DOI] [PubMed] [Google Scholar]
- 93.Dennis AM, Sotto DC, Mei BC, Medintz IL, Mattoussi H, Bao G. Surface Ligand Effects on Metal-Affinity Coordination to Quantum Dots: Implications for Nanoprobe Self-Assembly. Bioconjugate Chem. 2010;21:1160–1170. doi: 10.1021/bc900500m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Miller EW, Lin JY, Frady EP, Steinbach PA, Kristan WB, Tsien RY. Optically Monitoring Voltage in Neurons by Photo-Induced Electron Transfer Through molecular wires. Proc Natl Acad Sci U S A. 2012;109:2114–2119. doi: 10.1073/pnas.1120694109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huang YL, Walker AS, Miller EW. A Photostable Silicon Rhodamine Platform for Optical Voltage Sensing. J Am Chem Soc. 2015;137:10767–10776. doi: 10.1021/jacs.5b06644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fisher JAN, Salzberg BM, Yodh AG. Near Infrared Two-Photon Excitation Cross-Sections of Voltage-Sensitive Dyes. J Neurosci Methods. 2005;148:94–102. doi: 10.1016/j.jneumeth.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 97.Clapp AR, Goldman ER, Mattoussi H. Capping of Cdse-Zns Quantum Dots with DHLA and Subsequent Conjugation with Proteins. Nat Protoc. 2006;1:1258–66. doi: 10.1038/nprot.2006.184. [DOI] [PubMed] [Google Scholar]
- 98.Snee PT, Chan YH, Nocera DG, Bawendi MG. Whispering-Gallery-Mode Lasing from a Semiconductor Nanocrystal/ Microsphere Resonator Composite. Adv Mater. 2005;17:1131–1136. [Google Scholar]
- 99.Li JJ, Wang YA, Guo WZ, Keay JC, Mishima TD, Johnson MB, Peng XG. Large-Scale Synthesis of Nearly Monodisperse CdSe/CdS Core/Shell Nanocrystals Using Air-Stable Reagents via Successive Ion Layer Adsorption and Reaction. J Am Chem Soc. 2003;125:12567–12575. doi: 10.1021/ja0363563. [DOI] [PubMed] [Google Scholar]
- 100.Blackman B, Battaglia D, Peng XG. Bright and Water-Soluble Near Ir-Emitting Cdse/Cdte/ZnSe Type-II/Type-I Nano-crystals, Tuning the Efficiency and Stability by Growth. Chem Mater. 2008;20:4847–4853. [Google Scholar]
- 101.Uyeda HT, Medintz IL, Jaiswal JK, Simon SM, Mattoussi H. Synthesis of Compact Multidentate Ligands to Prepare Stable Hydrophilic Quantum Dot Fluorophores. J Am Chem Soc. 2005;127:3870–8. doi: 10.1021/ja044031w. [DOI] [PubMed] [Google Scholar]
- 102.Susumu K, Oh E, Delehanty JB, Pinaud F, Gemmill KB, Walper S, Breger J, Schroeder MJ, Stewart MH, Jain V, Whitaker CM, Huston AL, Medintz IL. A New Family of Pyridine-Appended Multidentate Polymers As Hydrophilic Surface Ligands for Preparing Stable Biocompatible Quantum Dots. Chem Mater. 2014;26:5327–5344. [Google Scholar]
- 103.Mei BC, Susumu K, Medintz IL, Mattoussi H. Polyethylene Glycol-Based Bidentate Ligands to Enhance Quantum Dot and Gold Nanoparticle Stability in Biological Media. Nat Protoc. 2009;4:412–23. doi: 10.1038/nprot.2008.243. [DOI] [PubMed] [Google Scholar]
- 104.Wang J, Wang W, Kollman PA, Case DA. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J Mol Graphics Modell. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 105.Herber RH, Nowik I, Matsuo Y, Toganoh M, Kuninobu Y, Nakamura E. Mössbauer Spectroscopy of Bucky Ferrocenes: Lattice Dynamics and Motional Anisotropy of the Metal Atom. Inorg Chem. 2005;44:5629–5635. doi: 10.1021/ic050251e. [DOI] [PubMed] [Google Scholar]
- 106.Néron B, Ménager H, Maufrais C, Joly N, Maupetit J, Letort S, Carrere S, Tuffery P, Letondal C. Mobyle: A New Full Web Bioinformatics Framework. Bioinformatics. 2009;25:3005–3011. doi: 10.1093/bioinformatics/btp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Buchan DWA, Minneci F, Nugent TCO, Bryson K, Jones DT. Scalable Web Services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 2013;41:349–357. doi: 10.1093/nar/gkt381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Adzhubei AA, Sternberg MJE. Left-Handed Polyproline-Ii Helices Commonly Occur in Globular-Proteins. J Mol Biol. 1993;229:472–493. doi: 10.1006/jmbi.1993.1047. [DOI] [PubMed] [Google Scholar]
- 109.Knaack GL, Charkhkar H, Hamilton FW, Peixoto N, O’Shaughnessy TJ, Pancrazio JJ. Differential Responses to ω-agatoxin IVA in Murine Frontal Cortex and Spinal Cord Derived Neuronal Networks. NeuroToxicology. 2013;37:19–25. doi: 10.1016/j.neuro.2013.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.