

A novel method of studying chloroplast proteins, which combines error-prone PCR with mutant complementation, is demonstrated using petD in Chlamydomonas reinhardtii.
Abstract
Site-directed mutagenesis of chloroplast genes was developed three decades ago and has greatly advanced the field of photosynthesis research. Here, we describe a new approach for generating random chloroplast gene mutants that combines error-prone polymerase chain reaction of a gene of interest with chloroplast complementation of the knockout Chlamydomonas reinhardtii mutant. As a proof of concept, we targeted a 300-bp sequence of the petD gene that encodes subunit IV of the thylakoid membrane-bound cytochrome b6f complex. By sequencing chloroplast transformants, we revealed 149 mutations in the 300-bp target petD sequence that resulted in 92 amino acid substitutions in the 100-residue target subunit IV sequence. Our results show that this method is suited to the study of highly hydrophobic, multisubunit, and chloroplast-encoded proteins containing cofactors such as hemes, iron-sulfur clusters, and chlorophyll pigments. Moreover, we show that mutant screening and sequencing can be used to study photosynthetic mechanisms or to probe the mutational robustness of chloroplast-encoded proteins, and we propose that this method is a valuable tool for the directed evolution of enzymes in the chloroplast.
The mutagenesis of discrete nucleotide position(s) along a gene sequence is a powerful tool for the study of protein structure-function relationships through genetic screens. Many applications have been derived from this general concept. Organism-wide (in vivo) mutagenesis using UV light (Witkin, 1969), chemical mutagens (Hayatsu and Miura, 1970), or mutator plasmids (Selifonova et al., 2001; Badran and Liu, 2015) provide ways to study processes involving many interacting parts, such as metabolic pathways, or to perform directed evolution using a well-defined selective pressure. Other methods, such as error-prone PCR (epPCR; Zakour and Loeb, 1982; Leung et al., 1989; Cadwell and Joyce, 1992) and DNA shuffling (Crameri et al., 1998), are used in vitro to target the mutagenesis to specific DNA sequences. In addition to being noninvasive, these latter methods ensure better control over mutation rate and mutational spectrum while remaining compatible with large-scale approaches such as directed evolution. Two determinants in these kinds of genetic screens are the compatibility of the target organism with high-throughput methods and the selection or screening techniques used to isolate and identify a lost, affected, or evolved phenotype.
A plant cell contains up to 100 chloroplasts, each containing up to 100 copies of the chloroplast genome encompassing about 100 genes (Shinozaki et al., 1986). In contrast to plant cells, the unicellular alga Chlamydomonas reinhardtii contains only one chloroplast with approximately 80 copies of the chloroplast genome. This genome is sequenced, and chloroplast transformation techniques are widely available, making this model organism fully amenable to genetic analysis. Various chemicals have been found to increase the rate of random mutations in the chloroplast genome, such as the mutagen ICR-191 (Huang et al., 1981) or the thymidine analog 5-fluorodeoxyuridine, which reduces the number of chloroplast DNA molecules (Wurtz et al., 1979). 5-Fluorodeoxyuridine facilitates the dissemination of mutations throughout the plastid genomes that otherwise would have been counterselected against wild-type copies. Other agents such as metronidazole are toxic to the photosynthesizing cell and allow enrichment in mutants null for photosynthetic electron transport (Schmidt et al., 1977). These random mutagenesis techniques have proven very useful throughout the years, despite the random nature of their mutational effects. As almost all the loci on the chloroplast genome were characterized, new techniques were developed, such as chloroplast transformation by biolistics (Boynton et al., 1988) and homologous recombination of a transgene in the chloroplast genome. It thus became possible to target a specific plastid genome locus and introduce site-directed mutations. Selection on antibiotic-containing medium allowed markers (such as the spectinomycin resistance marker; Goldschmidt-Clermont, 1991) to be inserted into every copy of the chloroplast genome.
Thus, these latter techniques are specific to a genome locus and have been used to introduce selected deletions or point mutations in the chloroplast genome. A few examples can be found for C. reinhardtii, where the structure and function of PSI (Redding et al., 1998), PSII (Wang et al., 2002), or Rubisco (Chen et al., 1991) were studied using these methods. Going a step further, it would be highly valuable to have a technique for the random mutagenesis of specific chloroplast genes. This would be useful for structure-function studies of chloroplast proteins, testing protein-protein interactions or even the directed evolution of exogenous enzymes in the chloroplast, as is currently done in other biological hosts such as Escherichia coli or Saccharomyces cerevisiae. In contrast with those organisms, the chloroplast appears to be an excellent chassis for the production and analysis of highly hydrophobic compounds, including membrane proteins and liposoluble pigments and chemicals, because the thylakoids form abundant compartmentalized membrane systems with high packing densities.
A first attempt at randomly mutating a targeted chloroplast gene fragment was reported by Fischer et al. (1998), who succeeded in randomly mutagenizing a short region of PsaC (a peripheral subunit of PSI) to probe electron transfer reactions between PSI and Fd. The degenerate oligonucleotide-directed mutagenesis used in that study, and in the subsequent study by Naver et al. (2001), however, was very limiting because of the length restriction in the target sequence (42-mer oligonucleotides, with only five wobbles giving one substitution each). Here, we adapted epPCR for the random mutagenesis of the chloroplast petD gene that codes for a core, highly hydrophobic protein subunit of the cytochrome (cyt) b6f complex, which binds several cofactors (hemes, iron-sulfur cluster, chlorophyll, and carotenoid). Using this method, we analyze the robustness and plasticity of this subunit in the context of a multisubunit transmembrane complex. Our method has virtually no other limitation than the maximal length of amplification by PCR. This approach is potentially a game changer for photosynthesis and chloroplast biotechnology studies.
RESULTS
epPCR, Library Construction, and Amplification
petD, the chloroplast gene encoding subunit IV of the cyt b6f complex, was chosen for random mutagenesis. Random mutagenesis was done by epPCR using commercially available molecular biology kits and specific primers to target the desired petD sequence on the pWQH6 plasmid (Supplemental Fig. S1; Supplemental Table S1). After the epPCR, the amplicon library of petD variants was inserted back into vector pWQH6. The Agilent kit supplies reagents for a reconstruction PCR (rcPCR; EZClone Reaction), which uses the epPCR products as megaprimers and the host plasmid (here pWQH6) as a template to reconstruct each mutagenized gene fragment into a plasmid. The ease of this technique, allowing for an efficient and reliable construction of a plasmid library without any cloning steps, prompted us to develop our own rcPCR to be used for the reconstruction of any mutagenized fragment (see the optimal conditions detailed in the second line of the rcPCR column, Supplemental Table S2). The rcPCR product was then treated with the DpnI restriction enzyme, which specifically degrades methylated and hemimethylated DNA and, in this case, targeted the nonmutated pWQH6 plasmid DNA used as a template for the rcPCR.
E. coli cells were transformed with the product of this reaction in order to repair the staggered nicks left on the plasmids at the rcPCR step and amplify the plasmid library. Ten percent of the transformed cell suspension was plated on selective medium (containing 100 μg mL−1 ampicillin) to isolate clones and sequence petD variants. This allowed us to control the mutational frequency and assess library diversity (Table I). Initial trials using the Agilent kit (mutageneses 1–3) produced a high proportion of nonmutated sequences (greater than 50%) and up to three mutations per mutant. Reducing the amount of target DNA even 4-fold (between mutagenesis 1 and 3) did not have much effect on the mutational frequency. In mutagenesis 4, the amount of target DNA was reduced 20-fold compared with mutagenesis 1 and 30 epPCR cycles were performed. As expected, this decreased the proportion of nonmutated sequences (41%) and generated up to six mutations per mutant. To further reduce the percentage of nonmutated fragments, the epPCR was performed using condition 9 of the Clontech mutagenesis kit (mutagenesis 5). In this condition, no wild-type sequences were recovered among the 12 sequenced E. coli clones, and up to eight mutations per mutant were introduced into the petD gene.
Table I. Summary of the various random mutagenesis trials performed and sequencing results of E. coli and C. reinhardtii transformants.
| Mutagenesis No. | Kit Used | Error-Prone Conditions | E. coli Transformants | C. reinhardtii Transformants | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total DNA Template | Target DNA Template | Buffer Condition | No. of PCR Cycles | Total | Sequenced | Nonmutated | No. of Variant Sequences | Distribution Rangea | Total | Sequenced | Nonmutated | No. of Variant Sequences | Distribution Rangea | ||
| µg | ng | ||||||||||||||
| 1 | Agilent | 10 | 500 | n/a | 25 | 250 | 23 | 12 (52%) | 11 | 0–3 | nd | nd | nd | nd | nd |
| 2 | Agilent | 5 | 250 | n/a | 30 | nd | 17 | 10 (58%) | 7 | 0–2 | ∼400 | 18 | 14 (78%) | 3 | 0–1 |
| 3 | Agilent | 2.5 | 125 | n/a | 30 | nd | 18 | 10 (56%) | 8 | 0–2 | |||||
| 4 | Agilent | 0.5 | 25 | n/a | 30 | nd | 12 | 5 (41%) | 7 | 0–6 | ∼1,000 | 62 | 30 (48%) | 12 | 0–3 |
| 5 | Clontech | n/a | n/a | #9 | 25 | nd | 12 | 0% | 12 | 1–8 | ∼600 | 72 | 15 (21%) | 40 | 0–10 |
Minimal and maximal number of mutations per mutant. nd: not determined. n/a: not applicable.
Because chloroplast transformation requires a large quantity of DNA (0.5–2 µg per shot, i.e. 2–3 orders of magnitude more than bacterial transformation), the remaining 90% of the transformation volume was used for library amplification. E. coli transformants were grown in ampicillin-containing medium for this amplification, and random mutations were introduced in petD only and not in the ampicillin-resistant marker, so that no reduction in library diversity was expected. However, sequencing of C. reinhardtii transformants after chloroplast transformation with the amplified library showed that the amplification step led to a loss in petD variant diversity. In one particular batch of sequencing (data not shown and not included in the statistical analysis), some copies of petD were overly abundant: among 34 C. reinhardtii clones picked randomly on the transformation plates, 12 contained the same three mutations and eight contained another set of three identical mutations. Such a loss of diversity arose during library amplification when E. coli cells were grown in large batch cultures (200 mL) over extended periods of time, allowing many divisions (for the recovery of large amounts of DNA for chloroplast transformation). Indeed, random growth differences between E. coli clones are exacerbated as the number of generations increases, and this can lead to random overrepresentation of certain plasmid populations. We circumvented this problem by growing cells in smaller volumes (5-mL cultures) and, therefore, limiting the number of generations needed to reach the stationary phase. Alternatively, liquid gel amplification of the plasmid library can be performed (Elsaesser and Paysan, 2004). In our random mutagenesis trials (Table I), after switching to small batch cultures, an identical mutagenized petD sequence was sometimes retrieved from two or more different C. reinhardtii clones, a sign that sequence diversity was reduced during amplification but not to a level that would preclude further genetic screens or functional analysis of the mutants.
ΔpetD Complementation and Sequencing of C. reinhardtii Complemented Lines
The recovery of pWQH6 variants by miniprep from 5-mL batch cultures yielded rather low amounts of plasmid DNA (typically, 15 μg) compared with what is usually recommended for chloroplast transformation (2 μg per shot). Prior to transforming the C. reinhardtii ΔpetD strain with the library of petD variants, we optimized the protocol for complementation using pWQH6 carrying the wild-type petD gene in order to minimize the amount of plasmid DNA needed per shot. We found that doubling the number of host cells plated on the petri dish (20 × 106 instead of 10 × 106) compensated for a 4-fold decrease in the amount of plasmid DNA (500 ng per shot instead of 2 μg) without compromising complementation efficiency. In summary, after optimization, the product of the miniprep allowed for ∼30 chloroplast transformations and the recovery of a sufficiently high number of transformants for subsequent analysis.
C. reinhardtii transformants were selected under photoautrophic growth conditions, that is, based on their ability to recombine and replicate a viable version of the petD gene throughout the multicopy chloroplast genome, synthesize a stable subunit IV polypeptide, and assemble a functional cyt b6f complex. Overall, ∼2,000 transformants were obtained from mutageneses 2 to 5, with an average of 40 complemented clones per transformation. After 14 to 20 d of growth under phototrophic selection pressure, the petri dishes were placed under a chlorophyll fluorescence imaging system (Johnson et al., 2009) to assess photosynthetic electron flow and PSII quantum efficiency (Fig. 1). A photochemical quenching of 0.5 ± 0.1 under high light (600 µmol photons m−2 s−1) showed that all clones were efficient for photosynthesis. Clones ∼1 to 2 mm in colony size were chosen randomly from each transformation plate, restreaked on Tris-phosphate (TP)-agar medium, and resuspended in 15 µL of water for DNA extraction using the Chelex resin. PCR products of the amplification of the recombined petD gene were sequenced and analyzed using Geneious (Table I). In sequenced clones originating from mutageneses 1 to 3, the proportion of nonmutated petD genes was 78% (i.e. higher than the ∼57% found in E. coli), and the distribution peak was shifted toward fewer mutations. The increased mutation rate induced in mutagenesis 4 was confirmed in E. coli as a shift of the distribution toward larger numbers of mutations (up to six per 300 bp) and fewer nonmutated copies. It was also observed after library amplification and chloroplast transformation that 30 out of the 62 sequenced C. reinhardtii transformants contained the wild-type sequence (comprising 48%, compared with 78% in transformants originating from mutageneses 1–3), and mutants held up to three mutations per 300 bp (compared with a maximum of one per 300 bp in mutageneses 1–3). In the last mutagenesis trial using the Clontech kit, among the 72 C. reinhardtii transformants sequenced, only 15 clones (21%) were nonmutated, whereas the others contained up to 10 mutations.
Figure 1.

Screen capture of the interactive computer interface used for the screen for photosynthetic activity. A, Chlorophyll fluorescence imaging of random mutagenesis complementation clones selected on minimal medium. B, Chlorophyll fluorescence kinetics corresponding to clones in A renormalized between F0 shifted to 0 and Fm = 1. The illumination protocol was 3 s at 600 µmolphotons.m−2.s−1, followed by a saturating pulse and a 500-ms dark recovery. The red curve in B corresponds to the clone marked with a red cross in A.
Occurrence of Cotransformation at the Same Locus
Sequencing chromatograms revealed interesting features of chloroplast transformation using a nonhomogenous pool of plasmid DNA. When complementing a knockout mutant with a homogenous population of plasmids, any copy of the plasmid is expected to recombine with the chloroplast genome and be replicated with the same efficiency, so that homoplasmy is eventually reached. For an equimolar mix of two different plasmids, however, Kindle et al. (1991) reported the occurrence of cotransformation at two different chloroplast loci. In our experiments, petD variants can only recombine at one locus. In rare instances, the sequencing chromatograms of the petD gene recovered from our various mutants showed evidence of heteroplasmy. Figure 2 shows a comparison of the sequencing chromatograms of the wild-type petD gene and two mutants for the sequence surrounding bp 213. Mutant B has a clear double read (T and A) at that position, which indicates a single-nucleotide polymorphism (SNP). This case corresponds to a silent mutation, since ACT and ACA both code for a Thr, which may explain why this SNP was retained and not lost through counterselection. It nevertheless suggests that at least two plasmid molecules can penetrate the same chloroplast and recombine at the same loci on two different copies of the chloroplast DNA. It is probable that competition occurred between viable and nonviable petD variants inside a given chloroplast and that nonviable variants were eventually counterselected.
Figure 2.

Alignment of nucleotide sequences of C. reinhardtii wild type (WT) and two mutant petD genes surrounding bp 213. Chromatogram peaks are proportional to the relative concentration of each base at each position. Mutant A has a clear base attribution at position 213 showing a T-to-A substitution. Mutant B shows an ambiguous base attribution, with both T and A nucleotides having high readout values.
Evidence for the Counterselection of Nonviable petD Copies
The diversity of petD variants in E. coli cells is a direct representation of the diversity introduced by epPCR, since no selection is exerted on the petD sequence in this organism. From our sequencing data, it was clear that C. reinhardtii chloroplasts retained a higher proportion of nonmutated sequences and a lower number of mutations per mutant than those in E. coli cells. This is illustrated by comparing the Nonmutated and Distribution columns of Table I between the two organisms. This was also apparent after statistical analysis of the mutational frequency in the 114 C. reinhardtii transformants that were picked randomly and sequenced. Fifty-nine of these transformants did not show any mutation in the petD gene, whereas 55 transformants contained between one and 10 mutations. Figure 3 shows a bar graph of the distribution of the number of mutants containing m mutations. This graph was fitted to a Poisson distribution (Eq. 2; solid line in Fig. 3). The solid curve matches satisfactorily the data for m > 1, showing that the Poisson model was appropriate for fitting. The fit yielded an average number of mutations of λ = 3.1 for 300 bp (i.e. ∼10 per kb). The fit also predicts that the number of sequences retrieved with no mutations should have fallen in the range of two to three, which is in stark contrast with the 59 nonmutated sequences we obtained experimentally (gray bar in Fig. 3).
Figure 3.

Distribution of the number of mutations per mutant. The bar graph shows the distribution of the number of mutants against the number of mutations in the population of 114 petD mutants picked randomly and sequenced. The data were fitted to a Poisson distribution (black line).
Our conclusion is that some genetic diversity of the random petD DNA library was lost through the complementation of C. reinhardtii, a bias that is readily explained by the selection pressure exerted on the organism. Only a fraction of the sequence space of randomly mutagenized petD genes is expected to complement photoautotrophic growth after transformation of the ΔpetD strain. The rest of the petD sequence space is expected to produce nonviable subunit IV polypeptides, due, for example, to impaired folding of the apoprotein or insertion in the membrane, misassembly of the cofactors, altered redox potentials of the hemes (bL, bH, or ci), loss of proper interactions with the other subunits of the complex, or perturbed affinity for quinones at the Qo or Qi sites. Such mutations, which would have impeded cyt b6f assembly and accumulation or blocked electron and proton transfer, were not obtained after phototrophic selection.
Mutation Types, Gene Coverage, and Genetic Saturation
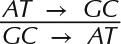
All petD variants, generated using both random mutagenesis kits, were aligned to the reference petD sequence, and SNP data were retrieved using Geneious software. We report 149 identified mutations in the 300-bp petD region targeted for random mutagenesis. The distributions of mutation types and effects are described in Table II, as well as two common measures of mutational bias. A-to-G and T-to-C transitions were the dominant substitutions, accounting for 25% and 37% of all mutations, respectively, and giving a high transition-to-transversion ratio (Ts/Tv = 2.1, theoretically equal to 0.5 for mutagenesis conditions completely lacking bias, since there are twice as many possible transversions as possible transitions). The measured AT→GC-to-GC→AT ratio of 8.3 also was a sign of mutational bias, as this ratio should equal 1 for completely unbiased epPCR conditions. We attribute these numbers to the statistical contribution of sequence variants generated with the Clontech kit. The method employed to reduce polymerase fidelity in this kit is such that high mutation rates (as are obtained by using epPCR condition 9) and low mutational bias are mutually exclusive.
Table II. Distribution of SNP types and effects.
| Mutation Types and Effects | Mutational Bias | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Transition (Ts) | Transversion (Tv) | Total | Percentage | 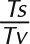 |
 |
|||||||||||
| A to G | G to A | T to C | C to T | A to T | A to C | T to A | T to G | G to C | G to T | C to G | C to A | |||||
| Nonsilent | 23 | 1 | 32 | 6 | 10 | 1 | 10 | 2 | 1 | 3 | 1 | 3 | 93 | 62.67 | 2.1 | 8.3 |
| Silent | 14 | 0 | 24 | 1 | 4 | 0 | 10 | 3 | 0 | 0 | 0 | 0 | 56 | 37.33 | ||
| Total | 37 | 1 | 56 | 7 | 14 | 1 | 20 | 5 | 1 | 3 | 1 | 3 | 149 | |||
| Percentage | 24.67 | 0.67 | 37.33 | 4.67 | 9.33 | 0.67 | 13.33 | 3.33 | 0.67 | 2.00 | 0.67 | 2.00 | ||||
The 149 identified mutations are represented as a bar graph in Figure 4A, which shows the number and types of SNPs at each mutated position along the petD sequence. The overall aspect of the graph suggests that the random mutagenesis conditions used in this study generated good sequence coverage. Mutations are equally spread along the petD gene, a sign that we were able to mutate equally any region of the 300-bp gene fragment. The protein sequences below show the substitutions in amino acids. Positions 87, 122, 124, 128, and 143 tolerated up to three different substitutions (asterisk in Fig. 4C). Figure 4C shows an alignment of subunit IV protein sequences from organisms possessing cyt bc1 or cyt b6f complexes, together with the important residues for b-type cyt turnover (Brasseur et al., 1996). Highly conserved catalytic (PEWY) or structural (PNKL) motifs in cyt b complexes were not affected by mutations, suggesting again that the phototrophic selection pressure had discarded these mutants.
Figure 4.

Distribution of mutations along petD and plasticity of subunit IV. A, Bar graph of the distribution and types of all nucleotide substitutions introduced in the petD gene by random mutagenesis. Bases are color coded, and substitutions that created nonsilent mutations are marked with yellow triangles. B, Index of protein plasticity calculated from the physicochemical distances between amino acids (Miyata et al., 1979). Each position along subunit IV was attributed a plasticity value by calculating the sum of the physicochemical distances between the original amino acid and the substituted amino acid(s). C, Alignment of the subunit IV sequence from C. reinhardtii and all substitutions obtained at each position with other cyt b sequences. Amino acids are color coded as follows: yellow, nonpolar/hydrophobic; green, polar uncharged; blue, positively charged; red, negatively charged. Symbols are used as follows: asterisks, tolerated up to three different substitutions; circles, unexpected substitutions; squares, electrostatic charges introduced in transmembrane helices F or G; diamonds, nonconservative substitutions in the subunit IV fg loop. Subunit IV is highly conserved throughout the green lineage, including in green algae (1), moss (2), higher plants (3), and cyanobacteria (4). It also aligns with subunit IV from the firmicutes Bacillus subtilis and Heliobacillus mobilis (5) and with the C-terminal part of cyt b from mitochondria (6), purple sulfur Allochromatium vinosum (7), or green sulfur Chlorobaculum tepidum (8) bacteria.
In different instances, certain residues were repeatedly substituted. For example, the Val-104Ala substitution was found in two different mutants from the same mutagenesis trial. This suggests that the mutation appeared in an early cycle of epPCR and was later combined with other random mutations eventually found in the triple mutant V104A-Q121R-F149S and the sextuple mutant V104A-L108H-F120L-T130S-L138F-L159S. In other cases, however, mutations were found repeatedly in mutants originating from strictly independent mutagenesis experiments. N118S, for example, was found on its own, but it also appeared in combination with T110A and I145T in a totally independent mutagenesis round. In another instance, which was even more peculiar, two sextuple mutants (L95P-V104E-F120S-T130A-L132S-I145V and V104A-L108H-F120L-T130S-L138F-L159S) carried different mutations at three common loci (Val-104, Phe-120, and Thr-130). These positions may correspond to flexible structural regions of the protein, easily tolerating substitutions, or represent tight hinges that cannot be moved independently from each other. We take these sequencing results in C. reinhardtii, showing a reduced sequence divergence, as a sign that genetic saturation was reached for the random mutagenesis of the petD gene under phototrophic selection pressure.
Robustness and Plasticity of a Macromolecular Protein Complex
Analyzing the spectrum of mutations that were introduced in petD and that restored phototrophy gives clues on the robustness and plasticity of subunit IV (Fig. 4B). Among the 149 mutations identified, 93 were nonsilent (Table II; Fig. 4A, yellow triangles in bar graph). The alignment of these nonsilent mutations in Figure 4, B and C, shows that transmembrane helices F and G tolerated few substitutions for bulky and/or charged amino acids and greatly favored substitutions that conserved amino acid properties (small and/or hydrophobic). In contrast, in the interhelix ef and fg loops and the C-terminal loop, containing more polar amino acids, substitutions modified residue polarity (Glu-74Gly, Val-84Glu, Gln-86Arg, Lys-119Glu, Gln-121Arg/His, Asn-122Lys, Tyr-124His, and Arg-125Ser) more often than they conserved polarity (Glu-74Asp, Arg-89His, and Lys-94Arg). This suggests that mutations in the regions exposed to the water interface are less likely to affect the function of the complex as a whole and that these regions are more robust and more plastic than the transmembrane regions. Expectedly, as mentioned above, catalytic and structural motifs that are generally conserved across species show lower plasticity.
When random substitutions in subunit IV, selected under photoautotrophic growth conditions, are considered regarding the 3D structure of the macromolecular complex (Fig. 5)((Stroebel et al., 2003), some structural constraints of the b-type cytochromes are clearly revealed. For example, the positively charged Lys-94 of subunit IV was only substituted to Arg (Fig. 4C). It is likely that Lys-94 forms an electrostatic interaction with Asp-156 of the cyt b6 cd loop, thus providing an attachment point between subunit IV helix F and the cyt b6 cd loop and contributing to the shaping of the Qo pocket. The Asp-156 residue is conserved or replaced by a Glu in b6f complexes of cyanobacteria and the green lineage. Interesting evidence of coevolution between these two interacting residues was found by looking at the unsplit cyt b subunit sequence of the green sulfur bacterium C. tepidum: this subunit bears a Lys and a Glu at the positions that coincide, respectively, with Asp-156 cyt b6 and Lys-94 subunit IV in b6f complexes.
Figure 5.

Mutational robustness heat map of subunit IV. A, Exterior view of the cyt b6f complex, with the peripheral region targeted for mutagenesis (helices F and G of subunit IV) colored according to the degree of variability (i.e. mutational robustness) at each position: from gray (no substitutions) to red (several and/or nonconservative substitutions), through green, yellow, and orange. B, Interior view of the targeted region (from the inside of the complex), showing some residues of interest and the symbols described in Figure 4. Atomic coordinates are from PDB 1Q90 (Stroebel et al., 2003).
Although they were rare, unexpected substitutions were found (circles in Figs. 4C and 5B). For example, hydrophobic residues pointing toward the core of the complex in the vicinity of the Qo site were replaced by charged residues (Glu, Arg, or Lys). Val-84 and Met-101 are situated, respectively, 3.9 and 3.7 Å from the hydrophobic tail of the stigmatellin inhibitor bound in place of the quinone at the Qo site. The tolerance of the Qo site to accommodate either positively charged or negatively charged residues in that pocket remains unclear and deserves further functional analysis.
Most of the other substitutions seemed fairly acceptable at the structural level. The other three electrostatic changes (squares in Figs. 4C and 5B) in helices F and G concerned residues situated at the interhelix interface (Val-104Glu and Leu-108His) or at the outer face of the complex (Trp-142Arg). For Val-104, we know from previous studies that replacement with Phe had no effect on the function of the complex (de Lacroix de Lavalette et al., 2008). In contrast, state transitions and electron transfer rates were affected in a mutant bearing a substitution of Gly-136, a residue that was unaffected in this study, likely because of mutant selection for phototrophy. Aromatic residue Phe-133 was substituted with a hydrophobic Leu, as reported previously by Yan et al. (2008), who showed that this aromatic residue was involved in maintaining the short lifetime of chlorophyll a*, and polar Thr-137 was changed to a hydrophobic Ala, whereas a hydrophobic Val is found at this position in Helibacillus mobilis.
Of outstanding interest were the substitutions obtained in the loop linking helices F and G. Not only does the fg loop vary between the cyt bc1 and b6f complexes, but it also easily tolerates nonconservative substitutions. From Lys-119 and extending to Arg-125, additions or removal of positive charges (Arg, Lys, or His) occurred, as well as changes to opposite charges (Lys-119Glu mutant; diamonds in Figs. 4C and 5B). Interestingly, although these substitutions did not affect the electron transfer rates of the complex, several of these impaired the mechanism of state transitions (Dumas et al., 2017).
DISCUSSION
In this work, we report the first attempt at combining random mutagenesis and chloroplast transformation. We relied on epPCR to introduce random mutations into the petD gene, then complemented the ΔpetD strain via biolistic transformation and selected transformants that had recovered phototrophic growth. The combination of these techniques allowed us to randomly mutate a specific sequence at the desired rate and to introduce these sequence variations at the desired genomic locus. Although we focused on a rather short sequence in this work (300 bp), the length of the target sequence is limited only by the maximal length of PCR amplification; thus, much longer DNA fragments can be targeted (up to ∼10 kb).
This protocol, validated and optimized for the C. reinhardtii chloroplast petD gene, is a new method for the study and engineering of chloroplast genes. Application to other chloroplast-containing organisms, including higher plants, represents the next step toward its widespread use in the fields of photosynthesis and chloroplast biology. One limitation for its application to higher plants might be the availability of chloroplast deletion mutants that can be retransformed with marker genes. However, as shown in tobacco (Nicotiana tabacum), such mutants can be isolated and retransformed with the efficient aadA marker gene (Monde et al., 2000; Kode et al., 2005). The method described here is compatible with genetic screens and opens up opportunities for the directed evolution of enzymes. When coupled to genetic screens for the study of the photosynthetic function, the epPCR of chloroplast genes can address functional questions in a straightforward manner. For example, a screen of chlorophyll fluorescence parameters was used to screen for mutants affected in state transitions (Dumas et al., 2017). Although the number of genetic variants created in this work (∼103 mutant clones) was sufficient to screen for a loss of function, we were far from having generated every possible permutation for three substitutions [19 × 3 × 100! / (100-3)! / 3! ≈ 9.106], which would have been required for a gain-of-function (Currin et al., 2015) screen. Owing to the low efficiency of chloroplast transformation, mutant generation and screening of this amplitude would represent a very work-intensive task. Nonetheless, if photosynthesis is to be considered as the new frontier for crop improvement (Ort et al., 2015), the directed evolution of chloroplast genes may very well represent a crucial step toward this grand challenge.
Cyt b from mitochondria, archaea, proteobacteria, and green sulfur bacteria is encoded by a single gene, whereas in chloroplasts, cyanobacteria, heliobacteria, bacilli, and haloarchaea, the corresponding sequence is split into two different genes, namely petB and petD, that encode cyt b6 and subunit IV, respectively. The complex core contains a number of transmembrane helices denoted from A to H. Cyt b6 encompasses helices A, B, C, and D, and subunit IV contains helices E, F, and G. Helix H is not found in the green clade of cyt b complexes (Nitschke et al., 2010). As noted on the list of mutants of cyt b complexes (Brasseur et al., 1996), the distribution of point mutations affecting the catalytic turnover of the enzyme is far from being random but, rather, pinpoints the regions of structural constraints for quinone binding or heme coordination. Interestingly, no mutations affecting electron or proton transfer were reported in helices F, G, and H (Brasseur et al., 1996), suggesting that these peripheral protein regions are more plastic than the core subunits B, C, D, and E. The mutants we report in this study are no exception to this rule, because they show normal turnover of the complex and allow for photoautotrophic growth. The robustness of subunit IV against mutations blocking electron transfer seemed to have allowed cyt b6f complexes to evolve a distinctive role in the regulation of state transitions. The adaptable interhelix F and G loop has been recruited, during the course of evolution, as a binding site for the Stt7 kinase involved in LHCII phosphorylation (Dumas et al., 2017).
MATERIALS AND METHODS
Strains and Media
A nonphototrophic strain of Chlamydomonas reinhardtii deleted for the chloroplast petD gene coding for subunit IV of the cyt b6f complex (ΔpetD) was used as a host strain for complementation (Kuras and Wollman, 1994). The strain was maintained on Tris-acetate-phosphate (TAP) medium in dark or very low light (∼10 µmolphotons.m−2.s−1), whereas complemented lines were selected on photoautotrophic growth in TP medium under 100 µmolphotons.m−2.s−1 illumination in air supplemented with 2% (v/v) CO2.
Plasmids and Primers
The previously described plasmid pWQH6 (Choquet et al., 2003; de Lacroix de Lavalette et al., 2008) was used as the epPCR template (see annotated map in Supplemental Fig. S1). Supplemental Table S1 lists the primers used for epPCR. Both primer pairs define the 5′ and 3′ boundaries of the sequence targeted for random mutagenesis, in this case going from bp 4,109 to 4,465 following pWQH6 map numbering or from bp 199 to 483 (3′ stop codon) following petD gene numbering.
Random Mutagenesis
Two molecular biology kits were used for random mutagenesis by epPCR: the GeneMorph II EZClone Domain Mutagenesis Kit (Agilent Technologies) and the Diversify PCR Random Mutagenesis Kit (Clontech). These rely on two different techniques to control the error rate of the polymerase. The first technique (Agilent kit) uses a high-error-rate DNA polymerase with a low mutational bias, and the mutation frequency is adjusted by varying the amount of template DNA and the number of epPCR cycles. The second technique (Clontech kit) acts directly on the fidelity of a Taq DNA polymerase. Manganese (in the form of MnSO4) is added to the epPCR and increases the mutation frequency in a concentration-dependent manner by binding to the DNA template and decreasing the fidelity of the enzyme. The mutation frequency can be increased further by increasing the dGTP concentration in order to skew the relative deoxyribonucleotide triphosphate concentrations. Supplemental Table S2 shows the epPCR conditions used for each kit. The epPCR products were loaded on a 2% (w/v) agarose gel, and the 750-bp fragment of interest was purified using the NucleoSpin Gel and PCR cleanup kit (Macherey-Nagel). Purified fragments were used as megaprimers in an rcPCR (conditions detailed in Supplemental Table S2). The reaction products were treated with 10 units of DpnI restriction enzyme and left to incubate at 37°C for 2 h. Two microliters of this reaction was used to transform 50 μL of XL10-Gold Ultracompetent Escherichia coli cells by heat shock. Ten percent of the transformation mixture was plated on Luria-Bertani agar medium containing 100 μg mL−1 ampicillin and incubated at 37°C overnight. Ampicillin-resistant clones were counted, and a subset was sequenced using the Pseq-f primer after colony growth in liquid culture and plasmid DNA recovery. The rest of the transformation mixture was used to amplify the plasmid library by inoculating either 5-mL or 200-mL Luria-Bertani batch cultures containing 100 μg mL−1 ampicillin and growing the cells overnight (37°C at 180 rpm). The amplified plasmid DNA library was recovered either by miniprep (for 5-mL cultures) using the NucleoSpin Plasmid kit (Macherey-Nagel) or by midiprep (for 200-mL cultures) using the NucleoBond Xtra Midi kit (Macherey-Nagel), and DNA concentration was measured on a NanoDrop spectrometer (GE). When the concentration was below 0.5 μg μL−1, the DNA preparation was concentrated on a SpeedVac (Savant ISS110; Thermo Scientific) to 0.5 μg μL−1 or greater.
Biolistic Transformation
The S550d Seashell Technology kit was used for the transformation of C. reinhardtii chloroplasts following a protocol adapted from the original technique described by Boynton et al. (1988). This kit contains 550-nm gold carrier particles at 50 mg mL−1 and binding and precipitation buffers. Additional materials to be supplied include autoclaved membrane holders and a bombardment chamber to which a partial vacuum and a high-pressure helium pulse can be applied. A freshly inoculated 200-mL TAP culture containing ΔpetD cells in exponential phase (between 1 × 106 and 4 × 106 cells mL−1) was used for chloroplast transformation. ΔpetD cells in TAP medium were centrifuged and resuspended in TP medium to a concentration of 200 × 106 cells mL−1. A total of 100 µL of this suspension was plated on TP-agar plates to give 20 × 106 cells per plate and per transformation. Five microliters of a 0.5 µg µL−1 pWQH6 plasmid DNA preparation containing the randomly mutated petD genes was added to 50 µL of binding buffer in a sterile Eppendorf tube. A total of 60 µL of S550d gold carrier particles at 50 mg mL−1 was added, and the tube was left to incubate on ice for 1 min. A total of 100 µL of precipitation buffer was added and incubated on ice for 1 min. The suspension was vortexed for 5 s and centrifuged at 13,000 rpm for 30 s. The pellet was washed with 500 µL of ice-cold 100% (v/v) ethanol and centrifuged at 13,000 rpm for 30 s. Fifty microliters of ice-cold 100% (v/v) ethanol was added to the pellet, which was then resuspended with a brief sonication (1–5 s, 25 V, and 25%–30% amplitude) to avoid particle aggregation. Ten microliters of gold particles coated with plasmid DNA was spread on a membrane holder, which was then placed inside a vacuum-sealed bombardment chamber along with a TP-agar plate placed ∼20 cm below the holder tip. The following parameters were used: 0.1 bar partial vacuum in the bombardment chamber, and 7 bars helium-shot pressure. After transformation, cells were left to recover in the dark for up to 1 d before being transferred to phototrophic conditions (40 μmol photons m−2 s−1 and air supplemented with 2% (v/v) CO2). Once the first transformants became visible (10–14 d), light was increased to 100 μmol photons m−2 s−1 to speed up growth and subsequent analysis.
Sequencing
petD sequences of E. coli transformants were obtained as described previously. The Chelex method (Walsh et al., 1991) was used to extract DNA from cells of C. reinhardtii transformants, and this DNA material was used to sequence the petD gene. Clones were resuspended in 15 µL of water so as to leave no aggregates. Ten microliters of 100% (v/v) ethanol was added, and the mix was incubated for 5 min at room temperature. A total of 80 µL of 5% (w/v) Chelex 100 solution (Bio-Rad) was added, and the sample was incubated for 8 min at 95°C. The sample tube was left open at room temperature to evaporate the ethanol. Three microliters of the suspension was then used as template for PCR (Supplemental Table S2), and the unpurified PCR product was sent for sequencing using the Pseq-f primer. All sequence analyses were performed with Geneious.
Statistical Analysis
epPCR data can be fitted to a binomial distribution or to a Poisson distribution when the mutational frequency stays low (Drummond et al., 2005). The probability p of observing k events in an interval is given by the equation:
where λ is the average number of mutations. The number n of mutants containing m mutations was fitted to equation:
where Γ is the generalization of the factorial function to noninteger values and coefficient a is used to adjust the amplitude.
Accession Numbers
The accession number for petD is X72919.1.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. pWQH6 plasmid map.
Supplemental Table S1. Primer list.
Supplemental Table S2. PCR conditions used to introduce mutations in petD, reconstruct the mutated fragments into plasmids, and sequence C. reinhardtii transformants.
Footnotes
L.D. was supported by a CEA IRTELIS PhD grant; F.Z.’s laboratory was supported by the Initiative d’Excellence program (DYNAMO, grant ANR-11-LABEX-0011-01); X.J. acknowledges a grant from the Agence Nationale de la Recherche ChloroPaths (ANR-14-CE05-0041-01); J.A. was supported by CNRS INSIS PhotoModes (72749).
References
- Badran AH, Liu DR (2015) Development of potent in vivo mutagenesis plasmids with broad mutational spectra. Nat Commun 6: 8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boynton JE, Gillham NW, Harris EH, Hosler JP, Johnson AM, Jones AR, Randolph-Anderson BL, Robertson D, Klein TM, Shark KB, et al. (1988) Chloroplast transformation in Chlamydomonas with high velocity microprojectiles. Science 240: 1534–1538 [DOI] [PubMed] [Google Scholar]
- Brasseur G, Saribaş AS, Daldal F (1996) A compilation of mutations located in the cytochrome b subunit of the bacterial and mitochondrial bc1 complex. Biochim Biophys Acta 1275: 61–69 [DOI] [PubMed] [Google Scholar]
- Cadwell RC, Joyce GF (1992) Randomization of genes by PCR mutagenesis. PCR Methods Appl 2: 28–33 [DOI] [PubMed] [Google Scholar]
- Chen ZX, Yu WZ, Lee JH, Diao R, Spreitzer RJ (1991) Complementing amino acid substitutions within loop 6 of the alpha/beta-barrel active site influence the CO2/O2 specificity of chloroplast ribulose-1,5-bisphosphate carboxylase/oxygenase. Biochemistry 30: 8846–8850 [DOI] [PubMed] [Google Scholar]
- Choquet Y, Zito F, Wostrikoff K, Wollman FA (2003) Cytochrome f translation in Chlamydomonas chloroplast is autoregulated by its carboxyl-terminal domain. Plant Cell 15: 1443–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crameri A, Raillard SA, Bermudez E, Stemmer WP (1998) DNA shuffling of a family of genes from diverse species accelerates directed evolution. Nature 391: 288–291 [DOI] [PubMed] [Google Scholar]
- Currin A, Swainston N, Day PJ, Kell DB (2015) Synthetic biology for the directed evolution of protein biocatalysts: navigating sequence space intelligently. Chem Soc Rev 44: 1172–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lacroix de Lavalette A, Finazzi G, Zito F (2008) b6f-associated chlorophyll: structural and dynamic contribution to the different cytochrome functions. Biochemistry 47: 5259–5265 [DOI] [PubMed] [Google Scholar]
- Drummond DA, Iverson BL, Georgiou G, Arnold FH (2005) Why high-error-rate random mutagenesis libraries are enriched in functional and improved proteins. J Mol Biol 350: 806–816 [DOI] [PubMed] [Google Scholar]
- Dumas L, Zito F, Blangy S, Auroy P, Johnson X, Peltier G, Alric J (2017) A stromal region of cytochrome b6f subunit IV is involved in the activation of the Stt7 kinase in Chlamydomonas. Proc Natl Acad Sci USA 114: 12063–12068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsaesser R, Paysan J (2004) Liquid gel amplification of complex plasmid libraries. Biotechniques 37: 200, 202 [DOI] [PubMed] [Google Scholar]
- Fischer N, Hippler M, Sétif P, Jacquot JP, Rochaix JD (1998) The PsaC subunit of photosystem I provides an essential lysine residue for fast electron transfer to ferredoxin. EMBO J 17: 849–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt-Clermont M. (1991) Transgenic expression of aminoglycoside adenine transferase in the chloroplast: a selectable marker of site-directed transformation of Chlamydomonas. Nucleic Acids Res 19: 4083–4089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayatsu H, Miura A (1970) The mutagenic action of sodium bisulfite. Biochem Biophys Res Commun 39: 156–160 [DOI] [PubMed] [Google Scholar]
- Huang B, Piperno G, Ramanis Z, Luck DJ (1981) Radial spokes of Chlamydomonas flagella: genetic analysis of assembly and function. J Cell Biol 88: 80–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson X, Vandystadt G, Bujaldon S, Wollman FA, Dubois R, Roussel P, Alric J, Béal D (2009) A new setup for in vivo fluorescence imaging of photosynthetic activity. Photosynth Res 102: 85–93 [DOI] [PubMed] [Google Scholar]
- Kindle KL, Richards KL, Stern DB (1991) Engineering the chloroplast genome: techniques and capabilities for chloroplast transformation in Chlamydomonas reinhardtii. Proc Natl Acad Sci USA 88: 1721–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kode V, Mudd EA, Iamtham S, Day A (2005) The tobacco plastid accD gene is essential and is required for leaf development. Plant J 44: 237–244 [DOI] [PubMed] [Google Scholar]
- Kuras R, Wollman FA (1994) The assembly of cytochrome b6/f complexes: an approach using genetic transformation of the green alga Chlamydomonas reinhardtii. EMBO J 13: 1019–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung DW, Chen E, Goeddel DV (1989) A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique 1: 11–15 [Google Scholar]
- Miyata T, Miyazawa S, Yasunaga T (1979) Two types of amino acid substitutions in protein evolution. J Mol Evol 12: 219–236 [DOI] [PubMed] [Google Scholar]
- Monde RA, Zito F, Olive J, Wollman FA, Stern DB (2000) Post-transcriptional defects in tobacco chloroplast mutants lacking the cytochrome b6/f complex. Plant J 21: 61–72 [DOI] [PubMed] [Google Scholar]
- Naver H, Boudreau E, Rochaix JD (2001) Functional studies of Ycf3: its role in assembly of photosystem I and interactions with some of its subunits. Plant Cell 13: 2731–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke W, van Lis R, Schoepp-Cothenet B, Baymann F (2010) The “green” phylogenetic clade of Rieske/Cytb complexes. Photosynth Res 104: 347–355 [DOI] [PubMed] [Google Scholar]
- Ort DR, Merchant SS, Alric J, Barkan A, Blankenship RE, Bock R, Croce R, Hanson MR, Hibberd JM, Long SP, et al. (2015) Redesigning photosynthesis to sustainably meet global food and bioenergy demand. Proc Natl Acad Sci USA 112: 8529–8536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redding K, MacMillan F, Leibl W, Brettel K, Hanley J, Rutherford AW, Breton J, Rochaix JD (1998) A systematic survey of conserved histidines in the core subunits of photosystem I by site-directed mutagenesis reveals the likely axial ligands of P700. EMBO J 17: 50–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt GW, Matlin KS, Chua NH (1977) A rapid procedure for selective enrichment of photosynthetic electron transport mutants. Proc Natl Acad Sci USA 74: 610–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selifonova O, Valle F, Schellenberger V (2001) Rapid evolution of novel traits in microorganisms. Appl Environ Microbiol 67: 3645–3649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T, Zaita N, Chunwongse J, Obokata J, Yamaguchi-Shinozaki K, et al. (1986) The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J 5: 2043–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroebel D, Choquet Y, Popot JL, Picot D (2003) An atypical haem in the cytochrome b(6)f complex. Nature 426: 413–418 [DOI] [PubMed] [Google Scholar]
- Walsh PS, Metzger DA, Higuchi R (1991) Chelex 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10: 506–513 [PubMed] [Google Scholar]
- Wang J, Gosztola D, Ruffle SV, Hemann C, Seibert M, Wasielewski MR, Hille R, Gustafson TL, Sayre RT (2002) Functional asymmetry of photosystem II D1 and D2 peripheral chlorophyll mutants of Chlamydomonas reinhardtii. Proc Natl Acad Sci USA 99: 4091–4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin EM. (1969) Ultraviolet-induced mutation and DNA repair. Annu Rev Microbiol 23: 487–514 [DOI] [PubMed] [Google Scholar]
- Wurtz EA, Sears BB, Rabert DK, Shepherd HS, Gillham NW, Boynton JE (1979) A specific increase in chloroplast gene mutations following growth of Chlamydomonas in 5-fluorodeoxyuridine. Mol Gen Genet 170: 235–242 [DOI] [PubMed] [Google Scholar]
- Yan J, Dashdorj N, Baniulis D, Yamashita E, Savikhin S, Cramer WA (2008) On the structural role of the aromatic residue environment of the chlorophyll a in the cytochrome b6f complex. Biochemistry 47: 3654–3661 [DOI] [PubMed] [Google Scholar]
- Zakour RA, Loeb LA (1982) Site-specific mutagenesis by error-directed DNA synthesis. Nature 295: 708–710 [DOI] [PubMed] [Google Scholar]