

ABSTRACT
Nephron progenitor cells (NPCs) are Six2-positive metanephric mesenchyme cells, which undergo self-renewal and differentiation to give rise to nephrons until the end of nephrogenesis. Histone deacetylases (HDACs) are a group of epigenetic regulators that control cell fate, but their role in balancing NPC renewal and differentiation is unknown. Here, we report that NPC-specific deletion of Hdac1 and Hdac2 genes in mice results in early postnatal lethality owing to renal hypodysplasia and loss of NPCs. HDAC1/2 interact with the NPC renewal regulators Six2, Osr1 and Sall1, and are co-bound along with Six2 on the Six2 enhancer. Although the mutant NPCs differentiate into renal vesicles (RVs), Hdac1/2 mutant kidneys lack nascent nephrons or mature glomeruli, a phenocopy of Lhx1 mutants. Transcriptional profiling and network analysis identified disrupted expression of Lhx1 and its downstream genes, Dll1 and Hnf1a/4a, as key mediators of the renal phenotype. Finally, although HDAC1/2-deficient NPCs and RVs overexpress hyperacetylated p53, Trp53 deletion failed to rescue the renal dysgenesis. We conclude that the epigenetic regulators HDAC1 and HDAC2 control nephrogenesis via interactions with the transcriptional programs of nephron progenitors and renal vesicles.
KEY WORDS: Epigenetics, Histone deacetylase, Kidney development, Nephron progenitors
Summary: Histone deacetylases 1 and 2 perform redundant, sequential and essential roles in the balance of nephron progenitor cell self-renewal and differentiation, as well as in progression of nephrogenesis.
INTRODUCTION
Kidney development requires precise integration of various progenitor cell populations. In ∼1/500 births, some abnormality occurs in kidney development, leading to congenital anomalies of the kidney and urinary tract (CAKUT) (Schedl, 2007). CAKUT accounts for up to 30% of end-stage renal disease in children less than 4 years of age (North American Pediatric Renal Trials and Collaborative Studies 2008 Annual Report; https://web.emmes.com/study/ped/annlrept/Annual%20Report%20-2008.pdf). Moreover, CAKUT increases the risk of development of hypertension and other cardiovascular diseases in adulthood (Wuhl et al., 2013). The formation of a sufficient number of nephrons is crucial for final kidney function in the adult and requires a delicate balance between nephron progenitor cell (NPC) self-renewal and differentiation. Conversely, unrestrained NPC expansion and arrested differentiation lead to Wilms' tumor, an embryonic tumor of the kidney (Kreidberg and Hartwig, 2008).
Six2, a homeodomain transcription factor, is a key factor within the kidney metanephric mesenchyme that maintains the NPC pool (Kobayashi et al., 2008; Self et al., 2006). In Six2 null mice, ectopic renal vesicles (the earliest epithelialized forms of nascent nephrons) develop at the onset of nephrogenesis and the progenitor pool is rapidly lost (Self et al., 2006). Many transcriptional regulators – such as Osr1, WT1 and Sall1/Mi-2b (Chd4)/nucleosome remodeling and deacetylase (NuRD), which function to maintain the NPC pool – display genetic interactions with Six2 (Basta et al., 2014; Denner and Rauchman, 2013; Hartwig et al., 2010; Kanda et al., 2014; Xu et al., 2014). In addition, it has been shown that Six2 regulates self-renewal and commitment of NPCs through sharing gene regulatory networks with Wnt proteins (Park et al., 2012). However, the details of how these networks operate to maintain the multipotency of nephron progenitors are not well understood. This knowledge is necessary to understand mechanisms of nephrogenesis and for therapeutic intervention of kidney diseases.
Recent years have witnessed an expanded awareness of the crucial role of epigenetic mechanisms in health and disease (Egger et al., 2004). During development, epigenetic mechanisms – such as DNA methylation, histone modifications and miRNA biogenesis – program the genome in a particular cell by alteration of chromatin structure and DNA accessibility to the transcriptional machinery. Disruptions of these epigenetic mechanisms can lead to dysregulation of gene function, without altering the DNA sequence itself (Egger et al., 2004). As epigenetic abnormalities depend on the interplay between genes and the environment, they are often phenotypically variable, which fits well with the broad phenotypic spectrum of CAKUT. Therefore, understanding the epigenetic basis of kidney development might provide new insights into the pathological mechanisms of CAKUT and, hopefully, open new avenues to treatment or prevention of CAKUT, through pharmaceutical agents that target epigenetic modifiers. Histone deacetylases (HDACs) are an evolutionarily conserved group of enzymes that remove acetyl groups from histones as well as nonhistone proteins [e.g. p53 (tumor protein p53)]. HDACs regulate gene expression in a highly selective way, and exhibit both repressive and activating effects (Haberland et al., 2009). To date, 18 mammalian HDACs have been identified. HDAC1 and HDAC2 share high sequence identity of ∼83% (de Ruijter et al., 2003) and regulate gene expression as the catalytic core of three major multiprotein co-repressor complexes: Sin3 (Sin3a), NuRD and co-repressor for element-1-silencing transcription factor (CoREST; Rcor2) (Kelly and Cowley, 2013). During embryogenesis, HDAC1 and HDAC2 play both redundant and distinct functions in a tissue-specific manner (Brunmeir et al., 2009; Jacob et al., 2011, 2014; LeBoeuf et al., 2010; Turgeon et al., 2013; Winter et al., 2013; Ye et al., 2009). Our previous studies showed that HDAC activity is required for key developmental pathways regulating overall renal growth and differentiation and ureteric bud (UB) branching (Chen et al., 2011, 2015). In the present work, using conditional targeting in Six2+ NPCs, we unraveled novel roles of HDAC1/2 in the control of NPC maintenance. Moreover, our findings implicate HDAC1/2 in the regulation of the differentiation program of renal vesicles (RVs) into nascent nephrons. Thus, HDAC1/2 regulate nephron endowment through actions on multiple steps of nephrogenesis.
RESULTS
NPC-specific deletion of Hdac1 and Hdac2
To gain insights into the role of HDAC1/2 in NPC maintenance and differentiation, we crossed Six2eGFPCre (Six2GC) mice (Kobayashi et al., 2008) with Hdac1flox/flox;Hdac2flox/flox mice (Montgomery et al., 2007). To test the efficacy of Six2-driven Cre-mediated excision, we examined the expression of HDAC1 and HDAC2 proteins in wild-type (WT) and mutant kidneys. As previously reported by our group (Chen et al., 2011), HDAC1/2 are nuclear proteins abundantly expressed in the nephrogenic zone (Fig. 1A,C,E). In Six2GC;Hdac1flox/flox;Hdac2flox/flox (herein referred to as HDAC1/2 mutant) mice, HDAC1/2 are not detected in the cap mesenchyme (CM) but are maintained in the surrounding UB branches and stromal cells (Fig. 1B,D,F). In accordance with the key functions of HDAC1/2 in deacetylation of histones and p53, the acetylation levels of H3K9, H4 (K5, K8, K12 and K16), and p53 (K386) are substantially increased in the NPC and derivatives [pretubular aggregates (PTAs) and RVs] of HDAC1/2 mutant kidneys (Fig. 2A-F). Collectively, these results demonstrate the efficient deletion of Hdac1 and Hdac2 from the Six2+ nephron progenitor pool.
Fig. 1.
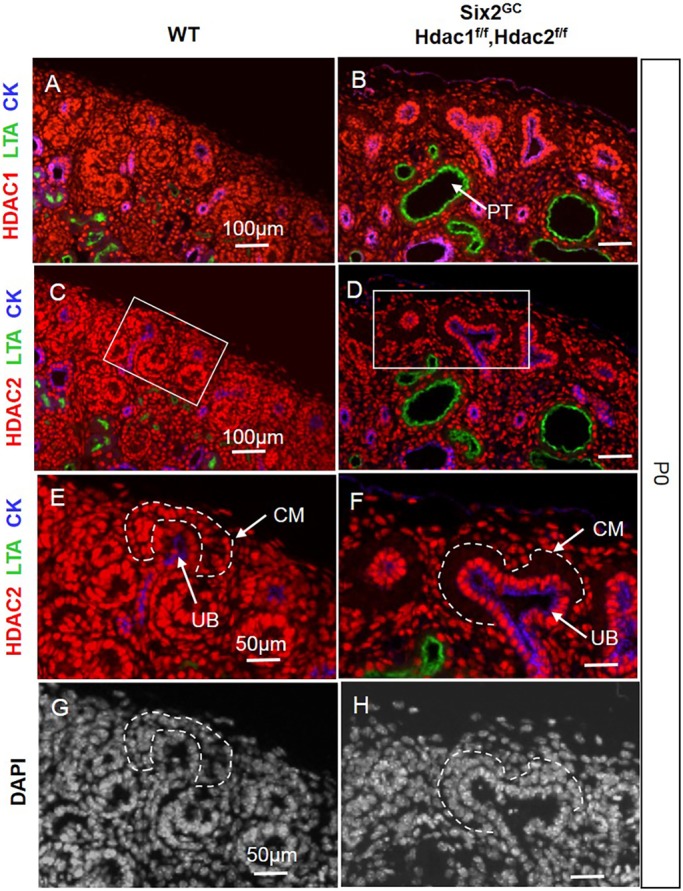
Deletion of Hdac1 and Hdac2 genes in the CM. (A,C,E,G) Consecutive section immunofluorescence (IF) at P0 showing the relatively abundant nuclear expression of HDAC1/2 proteins in the nephrogenic zone within the CM, UB and stroma. (B,D,F,H) Conditional Six2-Cre-mediated deletion of Hdac1/2 genes results in efficient loss of HDAC1/2 proteins from the CM. Boxes in C and D are shown enlarged in E and F, respectively. The scale bar information is the same in the left-hand and right-hand panels. CK, pancytokeratin; CM, cap mesenchyme; LTA, Lotus tetragonolobus lectin; PT, proximal tubule; UB, ureteric bud branches.
Fig. 2.
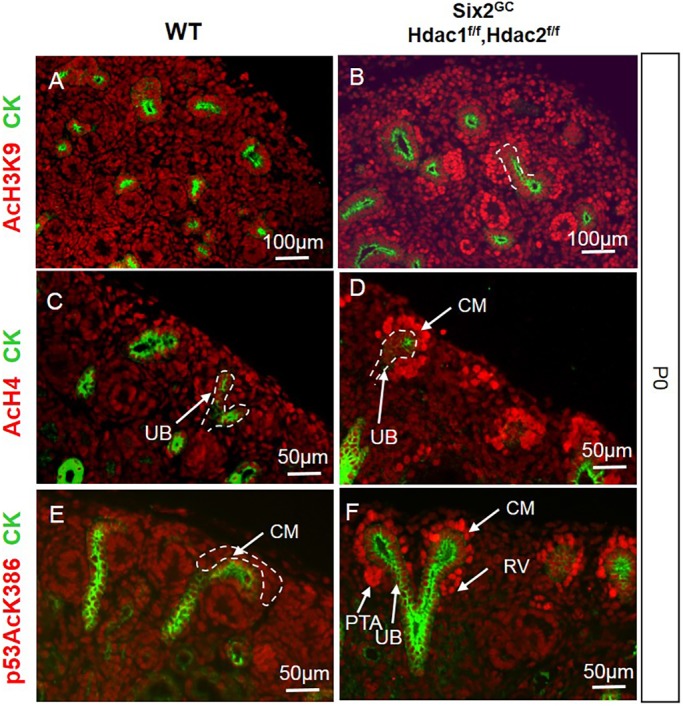
NPC-specific deletion of Hdac1 and Hdac2 causes hyperacetylation of histones H3 and H4 and p53. (A,C,E) AcH3K9, AcH4 and p53AcK386 are expressed at relatively low levels in all cell types of the developing kidney. (B,D,F) In HDAC1/2 mutant kidneys, there is upregulation of acetylated H3K9, H4 and p53 in NPCs and derived nascent tubules. AcH3K9, histone H3 (acetyl K9), AcH4, acetyl-histone H4; CK, pancytokeratin; CM, cap mesenchyme; p53AcK386, acetyl-Lys386 p53; PTA, pretubular aggregate; RV, renal vesicle; UB, ureteric bud branch.
NPC-specific deletion of Hdac1 and Hdac2 causes renal hypodysplasia
Mice with NPC-specific double deletion of Hdac1 and Hdac2 (all four alleles) were born in normal Mendelian ratios; however, they died soon after birth. At birth, Hdac1/2 mutant mice exhibited bilaterally small kidneys with full penetrance and there were some obvious petechial hemorrhagic spots on the surface of the mutant kidneys (Fig. 3A-C). Histological examination of mutant kidneys at postnatal day (P) 0 showed small kidney size, absence of the nephrogenic zone, lack of nascent nephrons and glomeruli, and formation of multiple cysts (Fig. 3D-I). Lotus tetragonolobus lectin (LTA) staining determined that the majority of cortical renal cysts originate from proximal tubules (LTA-positive tubules, Fig. 1B,D). In contrast, one allele of either Hdac1 or Hdac2 is sufficient to ensure nephrogenesis (Fig. 3B,E,H) and survival until adulthood, although these mice show subtle phenotypes including fewer nephrons with variable penetrance (data not shown).
Fig. 3.
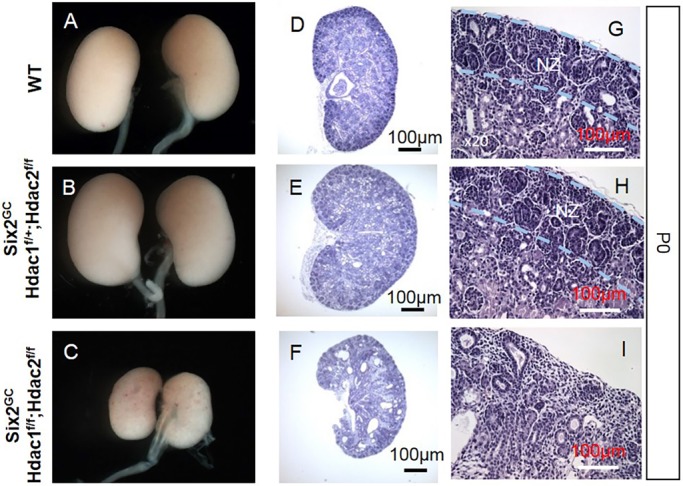
NPC-specific deletion of Hdac1 and Hdac2 causes severe renal hypodysplasia. (A-I) Gross and histological morphology in wild-type (WT) (A,D,G), and conditional compound heterozygous (B,E,H) and homozygous null (C,F,I) HDAC1/2 mutant mice. Six2-Cre-mediated deletion of HDAC1/2 impairs renal growth and patterning owing to loss of the nephrogenic zone (NZ) and cystic tubular degeneration.
NPC-specific deletion of Hdac1/2 inhibits cell proliferation but not survival
The defects in nephrogenesis in HDAC1/2 mutant kidneys could have partly resulted from decreased cell proliferation and/or increased apoptosis. Proliferating cell nuclear antigen (PCNA) is a sliding clamp that serves as a loading platform for many proteins involved in DNA replication and DNA repair (Strzalka and Ziemienowicz, 2011). PCNA protein expression and synthesis is linked with cell proliferation, and PCNA associates with nuclear histone deacetylase activity (Milutinovic et al., 2002). Moreover, cell proliferation defects are commonly found in most HDAC1/2 knockout or knockdown models (Haberland et al., 2009; Kelly and Cowley, 2013). Immunostaining for PCNA at embryonic day (E) 16.5 and P0 revealed that HDAC1 and HDAC2 are essential for proliferation of NPCs and their derivatives in the nephrogenic zone (Fig. 4A-D). Quantitatively, Hdac1/2 deletion resulted in a ∼75% reduction in the number of proliferating cells per CM (P<0.05, n=3 per group) (Fig. 4E). We also observed that the remnant Six2 cells seem to form a single cell layer surrounding the UB tips in the mutant CM niches (Fig. 4B, Fig. S1). We do not have an explanation for this unusual observation and are not aware of other mutants exhibiting this abnormality in patterning of the CM around the UB tip. Whether this represents an intrinsic defect in the organization of HDAC1/2 mutant Six2+ cells, or results from abnormal organization of the UB tips, remains to be elucidated. We next investigated whether HDAC1/2-deficient NPCs exhibit increased levels of apoptosis. Co-immunostaining of active caspase 3 and Six2, as well as terminal deoxynucleotidyl transferase (rTdT) mediated biotin-dUTP nick end-labeling (TUNEL) assay, at E14.5 (not shown) and P0 showed that deletion of Hdac1/2 had no obvious effect on cell survival in the NPCs (Fig. S1). Taken together, these results indicate that HDAC1 and HDAC2 are essential for NPC growth, but not survival.
Fig. 4.
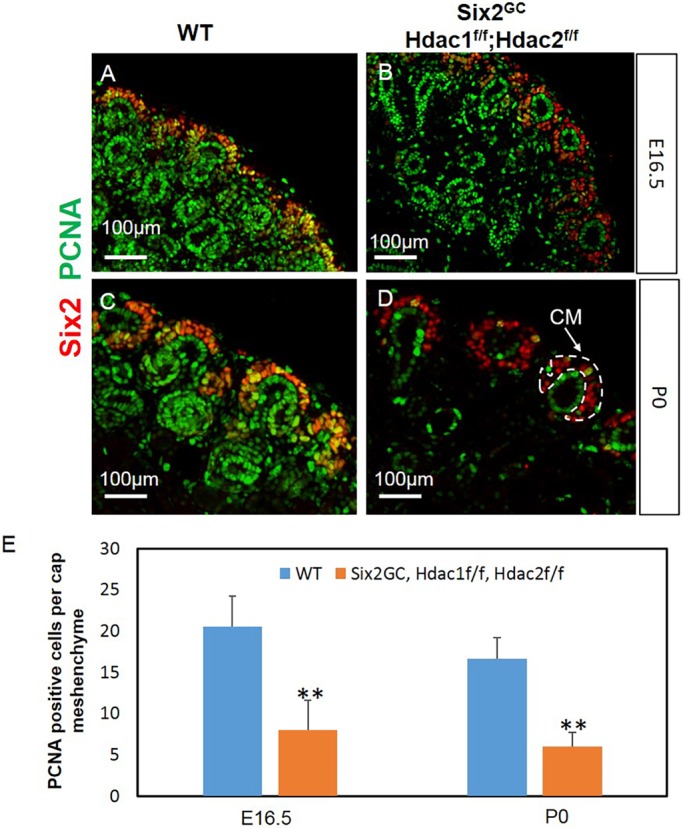
NPC-specific deletion of Hdac1 and Hdac2 inhibits cellular proliferation in the CM and nephrogenic zone. Section IF for PCNA and the NPC-specific marker Six2 at E16.5 (A,B) or P0 (C,D). There is reduction/loss of proliferating cells in the CM and nephrogenic zone in HDAC1/2 mutants compared with controls. Original magnification ×20. (E) Quantification of PCNA staining showed significantly decreased cell proliferation in E16.5 and P0 mutant CM. Data are mean±s.e.m. **P<0.05; n=3 animals per group.
p53 hyperacetylation is not a mediator of renal dysgenesis in HDAC1/2 mutants
In addition to their chromatin modifying activities, HDAC1/2 de-acetylate the transcription factor p53. p53 is induced by cell stress via post-translational modifications, including acetylation of its C-terminus by CBP (Crebbp)/p300 (Ep300). Hyperacetylated p53 has been linked to transcriptional activation, which in turn induces cell cycle arrest and/or apoptosis. We therefore tested whether the observed p53 hyperacetylation in NPCs and derivatives resulting from HDAC1/2 deletion contributes to the renal dysgenesis. We generated triple mutant Six2GC;Hdac1flox/flox;Hdac2flox/flox;p53+/− and Six2GC;Hdac1flox/flox;Hdac2flox/flox;p53−/− mouse strains and examined the pups at P0. The results showed that triple mutant kidneys continue to exhibit depletion of NPCs and arrest of tubular differentiation (Fig. S2). In fact, loss of p53 exaggerated renal cystogenesis in this model (Fig. S2A-D). Thus, genetic p53 deletion fails to rescue the HDAC/12 mutant renal phenotype.
NPC-specific deletion of Hdac1/2 represses the NPC self-renewal genes
We next assessed the molecular phenotype resulting from deletion of Hdac1/2 in the NPCs. Immunostaining of Six2, Pax2, Sall1 and WT1 and in situ hybridization (ISH) of Osr1, markers and key regulators of the CM, demonstrated that progenitor gene expression and the NPC pool are dramatically reduced or absent in E14.5, E16.5 and P0 HDAC1/2 mutant compared with WT kidneys (Fig. 5A-N, Fig. S3A,B).
Fig. 5.
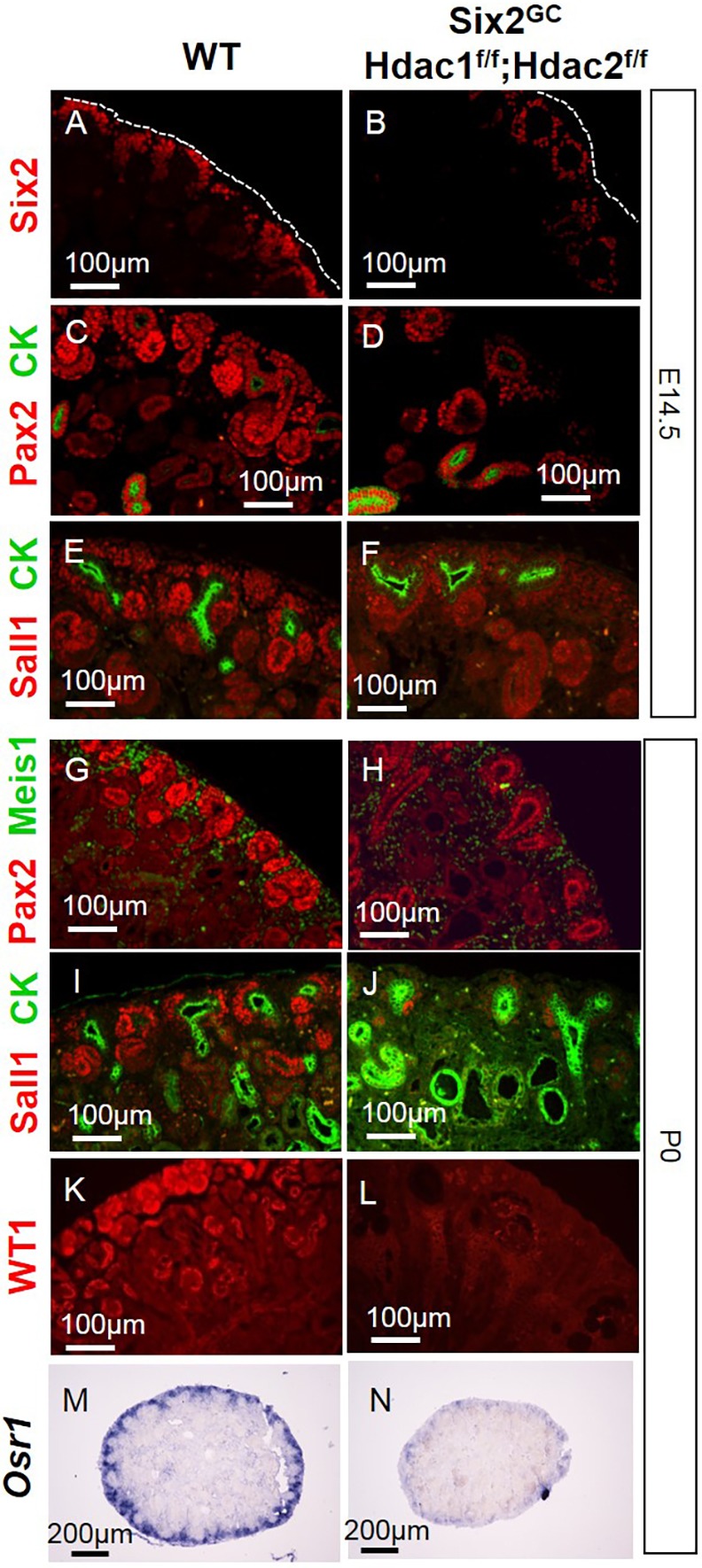
NPC-specific deletion of Hdac1 and Hdac2 depletes nephron progenitors and results in loss of NPC markers. Section IF at E14.5 (A-F) or P0 (G-N) reveals decreased expression of Six2, Osr1, Pax2, Sall1 and WT1. Section ISH shows decreases expression of Osr1 at P0 (M,N) CK, cytokeratin.
HDAC1/2 interact with NPC regulators and are bound to the Six2 enhancer
We next investigated whether HDAC1/2 interact biochemically with the NPC regulators Six2, Osr1 and Sall1 in transfected human embryonic kidney (HEK) 293T cells. Immunoprecipitation of either Flag-HDAC1 or Flag-HDAC2 pulled down Myc-tagged Six2 (Fig. 6A). Conversely, immunoprecipitation of Myc-tagged Six2 pulled down Flag-HDAC1 and Flag-HDAC2 (Fig. 6A). Osr1 acts downstream of and interacts synergistically with Six2 to maintain NPCs (Xu et al., 2014). We tested whether co-expressed Myc-Osr1 is able to interact with transfected Flag-HDAC1 and Flag-HDAC2. Co-immunoprecipitation showed robust interactions of Osr1 with both HDAC1 and HDAC2 (Fig. 6B). Furthermore, immunoprecipitation of Flag-Sall1 pulled down endogenous HDAC1, HDAC2 and transfected Myc-Six2 (Fig. 6C). In addition, we also detected endogenous interaction between Six2 and HDAC1 and HDAC2 in mouse E16.5 kidneys (Fig. 6D), P0 kidneys and E16.5 NPCs (Fig. S4). Of note, immunoprecipitation of HDAC1 pulled down a small amount of endogenous HDAC1 and Six2, whereas HDAC2 immunoprecipitation pulled down HDAC1 and an obviously higher amount of Six2. Collectively, these data suggest that HDAC1 and HDAC2 interact with Six2, Sall1 and Osr1, which are essential players in the balance of NPC self-renewal and differentiation.
Fig. 6.

HDAC1/2 interact with NPC transcriptional regulators and bind the Six2 enhancer. (A-C) The designed plasmids (+) were transfected into HEK 293T cells and the cell lysates were subject to Flag-tag immunoprecipitation. Flag-HDAC1/2 co-immunoprecipitate with Myc-Six2 (A); Flag-HDAC1/2 co-immunoprecipitate with Myc-Osr1 (B); Flag-Sall1 and Myc-Six2 co-immunoprecipitate with endogenous HDAC1/2 (C). (D) Endogenous interaction between Six2 and HDAC1/2 was detected in E16.5 kidneys. (E) Chromatin immunoprecipitation-PCR showing HDAC1/2 and Six2 co-occupancy of the Six2 enhancer.
Chromatin immunoprecipitation (ChIP) followed by NextGen sequencing in embryonic kidneys revealed that Sall1 and Six2 co-occupy many loci containing genes essential for kidney development, as well as Sall1 and Six2 themselves (Kanda et al., 2014). Interestingly, in the Six2 gene, Sall1 binding sites lie within 500 bp of the Six2 binding site (Kanda et al., 2014). The Six2/Sall1-bound region corresponds to that reported by Park et al., and is located 60 kb upstream of the Six2 transcription start site (Park et al., 2012). This region directs faithful spatial and temporal expression of a reporter in transgenic mice (Park et al., 2012). We tested whether HDAC1/2 and Six2 are bound to the Six2 enhancer in NPCs. We isolated NPCs from E16.5 kidneys by magnetic-activated cell sorting (Brown et al., 2015) and performed ChIP-PCR using anti-HDAC1, anti-HDAC2 and anti-Six2 antibodies. The isolated NPCs are highly enriched with Six2 (90%), with minor contamination with stromal cells (Meis1) (Fig. S5). The results showed that HDAC1/2 and Six2 co-occupy the Six2 enhancer (Fig. 6E). The specificity of the HDAC1 antibody was further validated by ChIP-PCR using positive and negative control primer sets (Fig. S6). Because HDAC1/2 and Six2 proteins interact (Fig. 6A), these findings suggest a model in which Six2 recruits HDAC1/2 to the Six2 enhancer, and this interaction might be necessary for regulation of Six2 expression.
HDAC1/2 are required for Lhx1 gene expression and renal vesicle differentiation
In E14.5-E16.5 HDAC1/2 mutant kidneys, early nephron precursors such as pre-tubular aggregates and RVs form. However, only rare comma- and S-shaped nascent nephrons or proximal tubules were detected (Fig. 7A-F), suggesting that HDAC1/2 are required for the RVs to progress to comma- and S-shaped nephrons and eventually into segmented epithelial nephrons.
Fig. 7.

NPC-specific deletion of Hdac1 and Hdac2 arrests nephrogenesis at the RV stage. (A-F) Section IF at E14.5 (A,B) and E16.5 (C-F). Laminin staining outlines the basement membrane of nascent nephrons showing that nephrogenesis is arrested at the RV stage in double HDAC1/2 mutant kidneys as early as E14.5. Few and scattered proximal tubules are formed in mutant kidneys. CK, pancytokeratin; LTA, Lotus tetragonolobus lectin; RV, renal vesicle; SB, S-shaped body.
Genome-wide transcriptome analysis of whole kidney RNA (see below) revealed reduced expression of Lhx1 in HDAC1/2 mutant kidneys. Lhx1, also known as LIM-class homeodomain transcription factor 1 (Lim-1), is expressed in the intermediate mesoderm, nephric duct, mesonephric tubules, ureteric bud, pretubular aggregates and RVs (Kobayashi et al., 2005; Pedersen et al., 2005). Lhx1 function is required for patterning and RV maturation into comma- and S-shaped bodies because tubulogenesis is blocked at the RV stage in Lhx1 null mice (Kobayashi et al., 2005; Pedersen et al., 2005). Accordingly, we examined Lhx1 expression in HDAC1/2 mutant kidneys. In E16.5 WT kidneys, Lhx1 protein is expressed in the RVs and nascent nephrons (Fig. 8A,C). In contrast, Lhx1 expression is abrogated in the HDAC1/2-deficient RVs and nascent nephrons (Fig. 8B,D-F). Interestingly, the few remnant RVs and nascent nephrons observed in the HDAC1/2 mutant kidneys appear to have escaped Cre-mediated recombination (Fig. 8G,H).
Fig. 8.

RVs lacking HDAC1/2 fail to express the transcription factor Lhx1. (A-F) Section IF for Six2/Lhx1 (A,B), HDAC2/Lhx1 (C,D), and laminin/HDAC2 (E,F) showing abrogated Lhx1 protein expression in nascent nephrons of HDAC1/2-mutant kidneys, which correlates spatially with loss of HDAC1/2. (G,H) The few instances in which Lhx1 expression was preserved correlate with nascent nephrons that escaped Cre-mediated excision of HDAC1/2. Arrows indicate the different stages of nascent nephrons: renal vesicle (RV), comma-shaped body (CB) and S-shaped body (SB).
NPC-specific deletion of Hdac1/2 downregulates Pax8 and Fgf8 in RVs
During nephrogenesis, Lhx1 is downstream of Fgf8, Pax8 and Wnt4, whereas Wnt4 lies downstream of Pax8/Fgf8 and upstream of Lhx1 (Grieshammer et al., 2005; Kispert et al., 1998; Kobayashi et al., 2005; Narlis et al., 2007; Park et al., 2007; Pedersen et al., 2005; Perantoni et al., 2005; Stark et al., 1994). Section ISH in E16.5 and P0 mice demonstrated that both Fgf8 and Pax8 are markedly downregulated in the HDAC1/2 mutant versus control RVs (Fig. 9A-D,G,H). Also, we confirmed abrogated Lhx1 expression in the mutant RVs (Fig. 9E,F,I,J). In contrast, Wnt4 mRNA expression [ISH and quantitative PCR (qPCR)] is maintained in the mutant RVs but not in NPCs (Fig. 10A-C). Thus, loss of Lhx1 expression in HDAC1/2 mutant RVs is Wnt4 independent, but at least partly caused by decreased expression of Pax8/Fgf8.
Fig. 9.
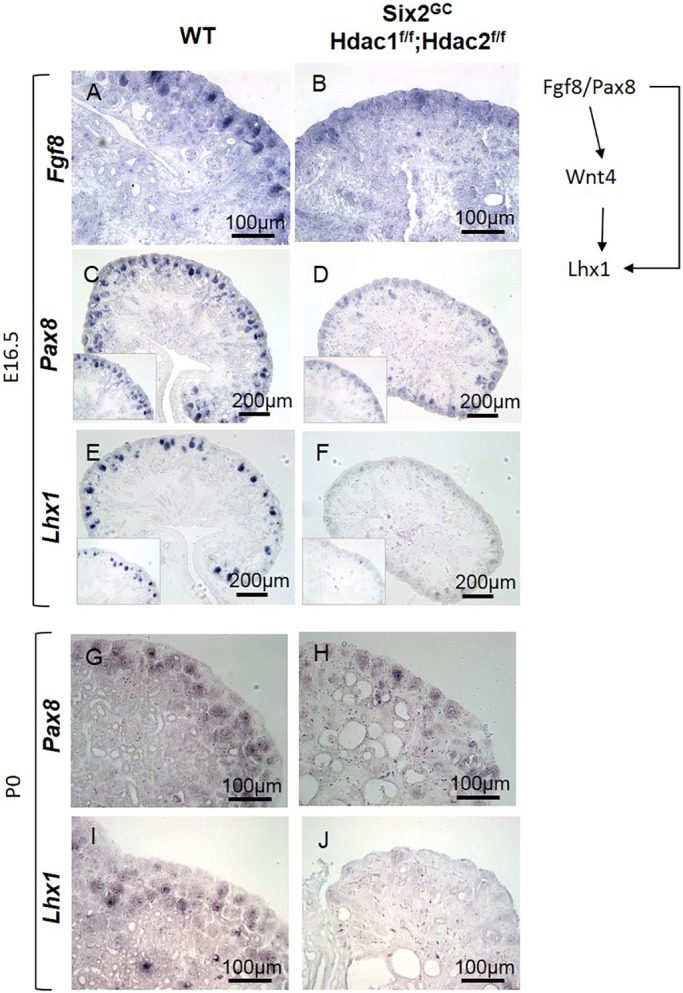
HDAC1/2 deletion disrupts the RV regulatory network. (A-J) Section ISH at E16.5 (A-F) and P0 (G-J), showing early and persistent repression of Pax8, Fgf8 and Lhx1, three major factors required for differentiation of RVs to nascent nephrons, in HDAC1/2 mutant kidneys.
Fig. 10.

Arrested RV differentiation in HDAC1/2 mutant kidneys is Wnt4 independent. (A-C) Section ISH shows maintained expression of Wnt4 in the mutant RVs at E16.5 (A,B) and enhanced total kidney Wnt4 by RT-qPCR (C). *P<0.05; n=4. (D,E) Section IF at P0 using antibodies against Lef1. In WT kidneys, Lef1 is expressed in RVs (arrowheads) and in a thin layer of stromal cells surrounding the UB branches. In HDAC1/2 mutant kidneys, Lef1 is ectopically expressed in the stroma (small arrows) and is upregulated in the peri-UB stroma. (F) FACS-isolated NPCs from WT (n+3) and HDAC1/2 mutant (n=2) were cultured in differentiation medium (with 3.5 µM CHIR) for 48 h. ddPCR showed that the Lef1 RNA copy number of mutant NPCs is not significantly different from that of WT cells, whereas the Six2 RNA copy number of mutant NPC is significantly lower than that of WT cells. Data are mean±s.e.m.
Interestingly, we found enhanced expression of the canonical Wnt protein target, Lef1, in the stromal mesenchyme surrounding the UB branches as well as in the outer cortical stroma of HDAC1/2 mutant kidneys (Fig. 10D,E). This increase does not appear to be mediated by excess Wnt9b expression in the UB (Fig. S7). However, because it is difficult to clearly compare the Lef1 levels between WT and mutant NPCs from Lef1 immunostaining alone, we examined the effect of HDAC1 and HDAC2 deletion on Lef1 gene expression in fluorescence-activated cell sorting (FACS)-isolated Six2-GFP+ NPCs from E16.5 WT and HDAC1/2 mutants. NPCs were cultured in differentiation (KO) medium (Brown et al., 2011, 2013) in the presence of 3.5 µM CHIR for 48 h. Lef1 RNA copy number, quantitated by droplet digital PCR (ddPCR), was not different in CHIR-treated HDAC1/2 mutant and WT NPCs (Fig. 10F). In contrast, Six2 RNA copy number was 50% lower in mutant CHIR-treated than in WT CHIR-treated NPCs. Collectively, these findings indicate that loss of HDAC1/2 in NPCs and derived cells activates stromal Wnt signaling, suggesting the presence of intercompartmental crosstalk. Of note, our transcriptome profiling identified multiple stromal genes (Pbx1, Meis1, Foxd1, Fat4) that are upregulated in the HDAC1/2 mutant kidneys. Our data also suggest that Six2 expression is dependent on HDAC1/2 in the setting of activated Wnt signaling.
Transcriptome analysis of whole kidney RNA
To further understand the molecular pathogenesis of the renal phenotype and to elucidate the developmental pathways regulated by HDAC1 and HDAC2, we carried out a genome-wide microarray analysis of RNA samples extracted from E15.5 WT and Hdac1/2 kidneys. The raw and analyzed data have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE84305. The results revealed that 649 transcripts (1.17%) are significantly altered in double-mutant kidneys (≥1.5-fold, P<0.05, n=4 independent experiments), of which 349 (69.24%) were upregulated (range +1.50- to +8.56-fold) and 155 transcripts (30.76%) were downregulated (range −1.50- to −12.40-fold) (Fig. S8A).
To analyze the pathways and biological processes that are sensitive to the loss of HDAC1/2, Ingenuity Pathway Analysis (IPA) was performed on the differentially expressed transcripts. This analysis indicated that the most significantly enriched pathways are concerned with cancer, embryonic development and organ development (Fig. S8B-D). A complete list of genes for each category and pathway is shown in Table S1. Further analysis using the Biological Networks Gene Ontology (BiNGO) tool revealed that many genes involved in kidney development processes are altered in HDAC1/2 mutant kidneys; for example, key factors involved in differentiation of the proximal and distal nephrons, such as Lhx1 (−1.6-fold), Hnf1a (−1.90-fold), Hnf4a (−2.85-fold), Irx1 (−2.346-fold), and the Notch signaling pathway [Dll1 (−2.18-fold), Hes5 (−2.24-fold), Hey1 (−1.67-fold) and Osr2 (−1.70-fold)] (Table S2). Network analysis placed Lhx1 upstream of Hnf1a and Hnf4a as well as many components of the Notch pathway, such as Hes5 and Dll1 (Fig. 11A,B). Section ISH at E16.5 and P0 confirmed significant downregulation of Hnf1a and Hnf4a in HDAC1/2 mutant nascent tubules but preservation of Hnf1b (Fig. 11C). Similarly, the Notch ligands Dll1 and Jag1, and several components of the Notch signaling pathway, including Lfng, Osr2, Hes1 and Hes5 were repressed (Fig. 11D). Consistent with downregulation of the Lhx1/Hnf1a/Hnf4a network, there was a significant decrease in expression of the epithelial differentiation genes, such as Nphs2, Slc34a1, Slc22a6, Slc37a4, CA4 (Car4) and Cdh6. In addition to the genes validated by section immunofluorescence (IF), ISH and reverse transcription (RT)-qPCR, NanoString gene expression analysis confirmed nine out of 11 randomly selected genes (Table S2). Unlike the differences in differentiation gene expression, which were readily identified using microarray analysis of whole kidney RNA, only a few progenitor genes were detected [e.g. decreased expression of Cited1 (−6.35-fold) and c-Myc (Myc) (−1.59-fold)]. Section ISH and section IF readily detected changes in expression of these genes (e.g. Pax2, Six2, Sall1 and Osr1) (Figs 4 and 5).
Fig. 11.

Downregulation of the Notch/HNF network in Six2GC;Hdacflox/flox,Hdac2flox/flox kidneys. (A,B) Network analysis of differentially expressed genes shows altered (mostly decreased) genes that are downstream of Lhx1 and Hnf1a/4a. Green, downregulated; red, upregulated. Numbers indicate fold change versus WT. (C,D) Section ISH showing downregulation of Hhf1a/4a (but not Hnf1b) and Notch genes in HDAC1/2 mutant kidneys. HNF1A/HNF4A control expression of a network of genes involved in metabolic and transport functions in the proximal tubules. Decreased HNF1A-mediated gene expression is likely secondary to downregulation of Notch signaling, which controls segmental tubular differentiation.
DISCUSSION
The present study provides comprehensive insights into the role of HDAC1 and HDAC2 in nephrogenesis. NPC-specific deletion of Hdac1/2 genes caused downregulation of key progenitor genes, including Six2, Pax2, Sall1, Wt1, Osr1, c-Myc and Cited1, and premature depletion of NPCs. Our biochemical and ChIP analyses revealed that HDAC1/2 interact with Six2, Osr1 and Sall1, a network of transcriptional regulators that maintain the balance of NPC proliferation and differentiation, and that Six2 is a potential in vivo target of HDAC1/2. Previous studies demonstrated that Sall1 is upstream of Six2, and Sall1 and Six2 are required for gene expression and cell renewal in the CM (Basta et al., 2014; Kanda et al., 2014). Six2 or Sall1 deletion results in depletion of nephron progenitors (Basta et al., 2014; Kanda et al., 2014; Kobayashi et al., 2008). Also, conditional deletion of the NuRD-specific component Mi2b (Chd4) in NPCs led to depletion of the CM (Denner and Rauchman, 2013). Sall1 and Mi2b exhibit a strong genetic interaction in the developing kidney, supporting a cooperative role for Sall1 and NuRD in maintaining NPCs (Denner and Rauchman, 2013). Because HDAC1 and HDAC2 are key components of the NuRD complex, our data support a model in which the Sall1/Six2/HDAC complex is recruited to the Six2 enhancer to maintain high Six2 expression in the NPCs.
Osr1 acts downstream of and interacts synergistically with Six2 and Groucho family transcriptional co-repressors to maintain the NPC pool via repression of Wnt4-directed nephrogenic differentiation (Xu et al., 2014). In Osr1 mutant kidneys, Wnt4 is ectopically activated throughout the CM, which undergoes premature mesenchyme-to-epithelium transition (Xu et al., 2014). Although our results showed protein-protein interactions between Osr1 and HDAC1/2, and Osr1 and HDAC1/2 target the Wnt4 enhancer region, we did not observe ectopic Wnt4 activation or RV formation in the CM. We surmise that Osr1/Groucho interactions compensate for the loss of HDAC1/2 in NPC, thus preventing ectopic Wnt4 activation in the CM.
In line with NPC depletion of HDAC1/2 mutant kidneys and the known pro-proliferative functions of HDAC1/2, we found that HDAC1/2 are crucial for NPC growth. Surprisingly, HDAC1/2 are not required to protect against p53-mediated apoptosis in NPCs. These findings are in sharp contrast to the effects of Hdac1/2 deletion in the UB epithelium, where p53 hyperacetylation was accompanied by increased UB cell apoptosis, and concomitant deletion of p53 partially rescued the defect in UB branching morphogenesis (Chen et al., 2015). Here, we show that accumulation of acetylated p53 in NPCs does not induce aberrant apoptosis and concomitant deletion of p53 fails to rescue the renal dysgenesis in HDAC1/2 mutants. Collectively, these findings point to the differential and context-dependent functions of HDAC1/2 in NPCs versus UB epithelium, which warrants further studies.
In addition to premature NPC depletion, deletion of Hdac1/2 blocks nephrogenesis at RV stage. This effect appears to be mediated via downregulation of the Lhx1/HNF/Notch network. Wnt4 and Lhx1 are both expressed in the RV, and Lhx1 is genetically downstream of Wnt4 (Kispert et al., 1998; Kobayashi et al., 2005; Stark et al., 1994). It is believed that the Wnt4-dependent transcriptional program leading to Lhx1 activation serves to differentiate the pre-tubular aggregate into the epithelialized RVs (Halt and Vainio, 2014). Here, we show that deletion of Hdac1/2 in NPCs abrogates Lhx1 expression in RVs. Although downregulation of Fgf8 and Pax8 in HDAC1/2 mutant RVs can mediate Lhx1 repression, it is conceivable that Lhx1 is directly regulated by HDAC1/2 and is worthy of future study.
Aberrant Wnt4 expression in RVs in the face of downregulated expression of Pax8/Fgf8/Pax2/WT1 (upstream activators of Wnt4) was a surprising finding in this study. This suggests the presence of noncanonical upstream regulators of Wnt4 gene expression within the RV that are unmasked by the loss of HDAC1/2. The nature of these factors remains to be determined. Our data also indicate that Wnt9b expression in the UB branches is not affected by the loss of HDAC1/2 in NPCs. Thus, the cause for maintained/enhanced Wnt4 expression in mutant HDAC1/2 RVs remains to be determined. Because our findings suggested that HDAC1/2 perform RV-specific functions, we attempted to delete Hdac1/2 genes specifically from the RVs using Wnt4-Cre/GFP mice. Although GFP was expressed appropriately in the RVs, we were unable to eliminate HDAC1/2 proteins, presumably as a result of the long half of the proteins, as each generation of nascent RVs express abundant HDAC1/2 prior to the deletion occurring. Genome-wide analysis of HDAC1/2-bound genes in NPCs versus RVs will be necessary to illuminate the direct versus indirect pathways regulated by HDACs during nephrogenesis.
In addition to Lhx1, many components of the Notch signaling pathway are downregulated in HDAC1/2 mutant kidneys. Notch signaling plays a key role in segmentation of the nephron and in the progression of pretubular aggregates/RVs towards comma- and S-shaped bodies during nephrogenesis (Kopan et al., 2007). γ-Secretase releases the Notch intramembrane domain (NICD), which, along with RBPJ, mediates the transcriptional effects of Notch proteins. Knockout of PSEN proteins (essential component of γ-secretase) or treatment with γ-secretase inhibitors allows formation of pretubular aggregates/RVs but not comma- and S-shaped bodies (Cheng et al., 2003; Wang et al., 2003). In addition, Notch2-deficient RVs initiate the segmentation process but fail to establish the proximal fate (Cheng et al., 2007), and more recently, the distal fate as well (Chung et al., 2016). Inactivation of Lhx1 causes loss of Dll1, and Dll1 hypomorphic mice have a severe reduction in nephron numbers and the loss of proximal segments (Kobayashi et al., 2005). Following loss of Lhx1, at least three other Notch signaling components, Jag1, Hes5 and musashi homolg2 (Msi2), are repressed (Kobayashi et al., 2005). Our immunostaining and ISH results revealed that the expression level of Notch ligands (Dll1 and Jag1) and many Notch protein targets (Hes1, Hes5 and Osr2) that are important for nephrogenesis are also dramatically downregulated. Based on these results, we conclude that impaired Notch signaling contributes significantly to the developmental arrest at the RV stage and tubulogenesis failure of HDAC1/2 mutant kidneys.
In summary, the present study demonstrates that HDAC1/2 perform redundant, sequential and essential roles in the balance of NPC self-renewal and differentiation as well as in progression of nephrogenesis (Fig. 12). In wild-type NPCs, HDAC1/2, working in concert with Six2, Sall1 and Osr1, maintain expression of progenitor genes cell autonomously favoring the expansion of the multipotent nephron progenitor population. Additionally, HDAC1/2, presumably working together with other transcriptional regulators, are required to maintain the Pax8/Fgf8/Lhx1/HNF/Notch transcriptional regulatory network required for RV differentiation to nascent nephrons. It remains to be determined whether the effects of HDAC1/2 on NPC and RV transcriptional networks are mediated via locus-specific control/binding or more generalized effects on the epigenome. Future studies uncovering HDAC1/2 target genes in NPCs and RVs are clearly warranted to further understand the epigenetic regulation of nephrogenesis.
Fig. 12.
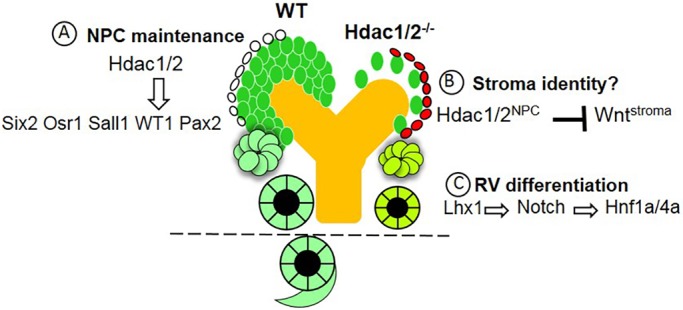
Working model for the actions of HDAC1/2 in the regulation of nephrogenesis. HDAC1/2 perform sequential functions during NPC and epithelial differentiation. (A) In the NPCs, HDAC1/2 are recruited (possibly by Six2/Sall1) to Six2 enhancer where they serve a positive regulatory role. HDAC1/2 also interact with Osr1 in the maintenance of NPCs. (B) HDAC1/2 in the NPCs and/or nascent nephrons restrain Wnt protein activity in adjacent stroma via unknown mechanisms. (C) HDAC1/2 directly or indirectly regulate the RV transcriptional network (Pax8/Fgf8/Lhx1) upstream of the Notch/HNF1A/HNF4A pathway. The dotted line indicates that NPC-specific deletion of HDAC1/2 arrests nephrogenesis at the RV stage.
MATERIALS AND METHODS
Mice
Mice bearing conditional null alleles of Hdac1flox/flox and Hdac2flox/flox (Montgomery et al., 2007) were crossed to Six2-CreEGFP transgenic mice (Six2-Cretg/+) (Kobayashi et al., 2008) to delete the Hdac1 and Hdac2 genes, singly or in combination, specifically in the NPCs. Wnt4GFPCre BAC transgenic mice (Shan et al., 2010) were used to cross with Hdac1flox/flox;Hdac2flox/flox mice to inactivate Hdac1/2 in the RVs. Trp53−/− mice were obtained from the Jackson (JAX) Laboratory.
Histology and immunohistochemistry
Kidneys were fixed in 10% buffered formalin, embedded in paraffin and sectioned at 4 μm. Histological analyses were performed by standard Hematoxylin and Eosin (H&E) staining. Section IF was performed as previously described (Chen et al., 2011, 2015). Antigen retrieval was accomplished by placing slides in 10 mM of boiling sodium citrate, pH 6.0, for 20 min. The following primary antibodies were used: anti-HDAC1 (1:100, 3601, BioVision), anti-HDAC2 (1:100, 3602, BioVision), anti-cytokeratin (1:200, C2562, Sigma-Aldrich), anti-Six2 (1:200, 11562-1-AP, Proteintechgroup), anti-Pax2 (1:200, 616000, Invitrogen), anti-PCNA (clone PC10, 1:200; DAKO Corp.), anti-cleaved caspase 3 (1:100, 9661s, Cell Signaling Technology), anti-acH4 (1:100, 06-866, Millipore), anti-acH3K9 (1:100, ab4441, Abcam), anti-p53AcK386 (1:100, ab52172, Abcam), anti-Jag1 (H-114; 1:100, sc-8303, Santa Cruz Biotechnology), Lhx1 (1:100, 4F2-C, Developmental Studies Hybridoma Bank), anti-Lef1 (1:100, 2230s, Cell Signaling Technology), anti-GFP (1:100, ab13970, Abcam), anti-E-cadherin (1:100, 610181, BD Biosciences), anti-Sall1 (1:100, ab31526, Abcam), anti-WT1 (1:100, ab15247, Abcam) and anti-laminin (1:100, L9393, Sigma-Aldrich). In negative controls, the primary antibody was omitted or replaced by nonimmune serum. For IF, the secondary antibodies were Alexa Fluor 488-conjugated anti-rabbit and Alexa Fluor 594-conjugated anti-rabbit (1:2000, Invitrogen) and anti-mouse fluorescein isothiocyanate (FITC) (1:200, Sigma-Aldrich). In addition, FITC-conjugated Lotus tetragonolobus lectin agglutinin (1:100, Vector Laboratories) was used to label the apical brush border of proximal tubules. Nuclei were counterstained by 4′,6-diamidino-2-phenylindole (DAPI) (1:500, D1306, Invitrogen). The immunofluorescent images were captured using a 3D or deconvolution microscope (Leica DMRXA2).
Section ISH
ISH was performed using digoxigenin-labeled antisense probes on kidney tissue fixed with 4% paraformaldehyde (PFA) as previously described (Chen et al., 2011). For section ISH, the kidney tissues were collected in DEPC-treated PBS, fixed in 4% PFA in diethyl pyrocarbonate (DEPC)-treated PBS overnight at 4°C, dehydrated in a series of alcohol, cleared in xylene and embedded in paraffin wax. Sections were cut to 10 μm thickness. After rehydration in 0.1% Tween in PBS, the samples were digested with proteinase K, and then refixed in 4% PFA, followed by 0.2% glutaraldehyde, followed by three washes in PBS. After a 3-h incubation in hybridization solution, the explants were hybridized with the digoxigenin-labeled antisense probes (∼1 μg of probe/vial) overnight at 65°C. The next day, the samples were sequentially washed with hybridization solution, 2× saline sodium citrate (SSC), pH 4.5, 2× SSC, pH 7.0, 0.1% CHAPS, maleic acid buffer and PBS at room temperature. The slides were incubated with preblocked antibody (1:10,000, anti-Dig alkaline phosphatase, Roche Applied Science) at 4°C overnight. The following day, after sequential washes of 0.1% bovine serum albumin in PBS, PBS and AP1 buffer at room temperature, the samples were stained by BM Purple (Roche Applied Science) at 4°C. When the desired level of staining was reached, the reaction was stopped by two washes of Stop Solution for 15 min each. The plasmids for Dll1, Hes1, Hes5 probe preparation were gifts from Dr Ryoichiro Kageyama (Kita et al., 2007). The experimental and control samples were put in the same reaction vessel to allow for proper comparison. All the experiments, including ISH, immunostaining and TUNEL were repeated at least three times.
TUNEL assay
Apoptosis was assessed using TUNEL and was carried out using an in situ apoptosis detection kit (Trevigen) according to the manufacturer's guidelines. Four-micrometer paraffin sections were fixed in methanol-free PFA before and after proteinase K treatment at 20 μg/ml for 8-10 min at room temperature. The sections were incubated with the nucleotide mixture (which included fluorescein-tagged dUTP) and rTdT enzyme for 1 h at 37°C. The slides were mounted using Vectashield with DAPI (Vector Laboratories). The images were captured using a deconvolution fluorescent microscope.
Cell culture and transient transfection
HEK 293T cells were obtained from the laboratory of Dr Hua Lu (Tulane University, New Orleans, LA, USA, and Johns Hopkins University Cell Center, Baltimore, MD, USA). Cells were grown in high-glucose Dulbecco's modified Eagle medium (DMEM) with stable glutamine supplemented with 10% fetal bovine serum (FBS) and 10 mg/ml antibiotics (penicillin and streptomycin). All cells were maintained at 37°C in a 5% CO2 humidified atmosphere. Cells seeded on the plate overnight were transfected with plasmids as indicated in figure legends using Lipofectamine LTX with Plus Reagent following the manufacturer's protocol (Thermo Fisher Scientific). Cells were harvested at 48-72 h post-transfection. The plasmid Flag-Sall1 was a gift from Dr Ryuichi Nishinakamura (Kanda et al., 2014), Flag-Six2 and Myc-Six2 were gifts from Dr Joo-Seop Park (Park et al., 2012), and Flag-Osr1 was a gift from Dr Rulan Jian (Xu et al., 2014).
Immunoprecipitation and western blotting
Immunoprecipitation (IP) was conducted using antibodies as indicated in the figure legends. Briefly, ∼500-1000 μg of protein was incubated with the indicated antibody at 4°C for 4 h or overnight. Protein A or G beads (Santa Cruz Biotechnology) were then added, and the mixture was incubated at 4°C for an additional 1-2 h. Beads were washed at least three times with lysis buffer. Bound proteins, as well as the whole-cell extracted proteins, were detected by western blotting. The following primary antibodies were used: anti-HDAC1 (rabbit polyclonal, 1:1000, BioVision) and anti-HDAC2 (rabbit polyclonal, 1:1000, BioVision), anti-Flag (1:1000, Sigma-Aldrich), anti-Myc (1:1000, Sigma-Aldrich) and mouse anti-β-actin (1:5000, Sigma-Aldrich).
Isolation of NPCs
Six2+/Cited1+ NPCs were isolated from E16.5 kidneys by magnetic-activated cell sorting (Brown et al., 2015). Briefly, after dissecting kidneys and removing the capsule, the kidneys were digested in collagenase A/pancreatin enzyme digest solution at 37°C for 15 min. After digestion, FBS was added to the tube containing kidneys to stop the enzyme reaction. The cell suspension was transferred to new microfuge tubes and cells were collected by centrifugation at 300 g for 5 min. Subsequently, the cells were re-suspended and filtered through a 30-µm pre-separation filter. Magnetic depletion was carried out through the addition of anti-CD105-PE, anti-CD140-PE, anti-Ter119-PE and anti-CD326-PE. Finally, the NPCs (Cited1+ NPCs) were collected as the negative fractions.
RNA isolation and ddPCR
E16.5 NPCs were isolated, plated and expanded on Matrigel-coated plates in a monolayer in keratinocyte serum-free medium (Gibco) supplemented with 50 ng/ml FGF2 (R&D Systems) as described (Brown et al., 2011, 2013) for 48 h. Total RNA was isolated using an RNA isolation kit (Qiagen). RNA concentration was quantified using Nanodrop 2000 (Thermo Fisher Scientific). ddPCR was performed on a Bio-Rad ddPCR system to determine Lef1 and Six2 gene expression. All reagents for the One-Step RT-ddPCR system were purchased from Bio-Rad to generate complementary DNA (cDNA) and quantify gene expression. Droplets were analyzed on the QX200 droplet reader and target cDNA concentration was determined using QuantaSoft analysis software (Bio-Rad).
ChIP
ChIP experiments were performed using an EZ ChIP chromatin immunoprecipitation kit (17-371, Millipore) according to the manufacturer's protocol. Immunoprecipitation was performed with ChIP-grade antibodies to HDAC1 (ab7028, Abcam), HDAC2 (ab7029, Abcam), Six2 (11562-1-AP, ProteinTech). Rabbit IgG (ab46540, Abcam) was used as a control antibody. The chromatin-antibody complexes were captured on protein G-coupled Dynabeads (Invitrogen). After washing and elution of the complexes from the beads, the DNA-protein cross-links were reversed at 65°C overnight. Next, the precipitated DNA was treated with RNase A and proteinase K and purified using spin columns. The purified DNA along with input genomic DNA (1:100) were analyzed by PCR. The primer sequences used for PCR were:
Six2Enh Forward: 5′-ACCGGATGGAAAGGCTTTAT-3′
Six2Enh Reverse: 5′-GGGCTGTTCCAGCTACAGAG-3′
Genome-wide microarray analysis
Microarray analysis was performed according to established protocols (Schanstra et al., 2007). Briefly, fluorescently labeled cRNA was generated from 0.5 μg total RNA in each reaction using a Fluorescent Direct Label Kit (Agilent) and 1.0 mM cyanine 3′- or 5′-labeled dCTP (PerkinElmer). Hybridization was performed using an Oligonucleotide Microarray Hybridization and In Situ Hybridization Plus Kit (Agilent). The labeled cRNA was hybridized to Agilent 44K whole mouse genome oligonucleotide microarray (containing ∼41,000 probes) as previously described (Schanstra et al., 2007). The arrays were scanned using a dual-laser DNA microarray scanner (Agilent). The data were then extracted from images using Feature Extraction software 6.1 (Agilent). Microarray data are available at GEO under accession number GSE84305.
Data analysis
MultiExperiment Viewer v4.9 software was used to generate lists of genes differentially expressed between WT and HDAC1/2 mutant kidneys, using P<0.05 and a minimum 1.5-fold change in gene expression. Genes were classified according to their function using IPA software and BiNGO classification systems as previously described (Chen et al., 2011, 2015). Additional analysis of the microarray data was accomplished using IPA software.
Supplementary Material
Acknowledgements
We thank Dr Eric Olson (UT Southwestern) for Hdac1flox/flox;Hdac2flox/flox mice and Andrew McMahon for Six2-Cre mice. We also thank the Tulane Renal and Hypertension Center of Excellence Core facilities.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: H.L., S.S.E.-D.; Methodology: S.C., Y.L., C.-H.C., J.L., Z.S.; Validation: Y.L.; Formal analysis: H.L.; Investigation: H.L., S.C., X.Y., C.-H.C., J.L., Z.S.; Resources: S.S.E.-D.; Data curation: H.L., S.C., X.Y.; Writing - original draft: H.L., S.S.E.-D.; Writing - review & editing: S.C., Z.S., S.S.E.-D.; Supervision: S.S.E.-D.; Project administration: H.L., S.S.E.-D.; Funding acquisition: H.L., S.S.E.-D.
Funding
This work was supported by the Foundation for the National Institutes of Health [1P50 DK096373], the National Institutes of Health [P30GM103337; 1RO1 DK114500 to S.S.E.-D.], and the American Heart Association [17SDG33660072 to H.L.]. Deposited in PMC for release after 12 months.
Data availability
Microarray analysis data are available at GEO under accession number GSE84305.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.153619.supplemental
References
- Basta J. M., Robbins L., Kiefer S. M., Dorsett D. and Rauchman M. (2014). Sall1 balances self-renewal and differentiation of renal progenitor cells. Development 141, 1047-1058. 10.1242/dev.095851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. C., Adams D., de Caestecker M., Yang X., Friesel R. and Oxburgh L. (2011). FGF/EGF signaling regulates the renewal of early nephron progenitors during embryonic development. Development 138, 5099-5112. 10.1242/dev.065995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. C., Muthukrishnan S. D., Guay J. A., Adams D. C., Schafer D. A., Fetting J. L. and Oxburgh L. (2013). Role for compartmentalization in nephron progenitor differentiation. Proc. Natl. Acad. Sci. USA 110, 4640-4645. 10.1073/pnas.1213971110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. C., Muthukrishnan S. D. and Oxburgh L. (2015). A synthetic niche for nephron progenitor cells. Dev. Cell 34, 229-241. 10.1016/j.devcel.2015.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunmeir R., Lagger S. and Seiser C. (2009). Histone deacetylase HDAC1/HDAC2-controlled embryonic development and cell differentiation. Int. J. Dev. Biol. 53, 275-289. 10.1387/ijdb.082649rb [DOI] [PubMed] [Google Scholar]
- Chen S., Bellew C., Yao X., Stefkova J., Dipp S., Saifudeen Z., Bachvarov D. and El-Dahr S. S. (2011). Histone deacetylase (HDAC) activity is critical for embryonic kidney gene expression, growth, and differentiation. J. Biol. Chem. 286, 32775-32789. 10.1074/jbc.M111.248278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Yao X., Li Y., Saifudeen Z., Bachvarov D. and El-Dahr S. S. (2015). Histone deacetylase 1 and 2 regulate Wnt and p53 pathways in the ureteric bud epithelium. Development 142, 1180-1192. 10.1242/dev.113506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H.-T., Miner J. H., Lin M., Tansey M. G., Roth K. and Kopan R. (2003). Gamma-secretase activity is dispensable for mesenchyme-to-epithelium transition but required for podocyte and proximal tubule formation in developing mouse kidney. Development 130, 5031-5042. 10.1242/dev.00697 [DOI] [PubMed] [Google Scholar]
- Cheng H.-T., Kim M., Valerius M. T., Surendran K., Schuster-Gossler K., Gossler A., McMahon A. P. and Kopan R. (2007). Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development 134, 801-811. 10.1242/dev.02773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E., Deacon P., Marable S., Shin J. and Park J.-S. (2016). Notch signaling promotes nephrogenesis by downregulating Six2. Development 143, 3907-3913. 10.1242/dev.143503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ruijter A. J., van Gennip A. H., Caron H. N., Kemp S. and van Kuilenburg A. B. (2003). Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370, 737-749. 10.1042/bj20021321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denner D. R. and Rauchman M. (2013). Mi-2/NuRD is required in renal progenitor cells during embryonic kidney development. Dev. Biol. 375, 105-116. 10.1016/j.ydbio.2012.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger G., Liang G., Aparicio A. and Jones P. A. (2004). Epigenetics in human disease and prospects for epigenetic therapy. Nature 429, 457-463. 10.1038/nature02625 [DOI] [PubMed] [Google Scholar]
- Grieshammer U., Cebrian C., Ilagan R., Meyers E., Herzlinger D. and Martin G. R. (2005). FGF8 is required for cell survival at distinct stages of nephrogenesis and for regulation of gene expression in nascent nephrons. Development 132, 3847-3857. 10.1242/dev.01944 [DOI] [PubMed] [Google Scholar]
- Haberland M., Montgomery R. L. and Olson E. N. (2009). The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32-42. 10.1038/nrg2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halt K. and Vainio S. (2014). Coordination of kidney organogenesis by Wnt signaling. Pediatr. Nephrol. 29, 737-744. 10.1007/s00467-013-2733-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig S., Ho J., Pandey P., Macisaac K., Taglienti M., Xiang M., Alterovitz G., Ramoni M., Fraenkel E. and Kreidberg J. A. (2010). Genomic characterization of Wilms’ tumor suppressor 1 targets in nephron progenitor cells during kidney development. Development 137, 1189-1203. 10.1242/dev.045732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob C., Christen C. N., Pereira J. A., Somandin C., Baggiolini A., Lötscher P., Özçelik M., Tricaud N., Meijer D., Yamaguchi T. et al. (2011). HDAC1 and HDAC2 control the transcriptional program of myelination and the survival of Schwann cells. Nat. Neurosci. 14, 429-436. 10.1038/nn.2762 [DOI] [PubMed] [Google Scholar]
- Jacob C., Lotscher P., Engler S., Baggiolini A., Varum Tavares S., Brugger V., John N., Buchmann-Moller S., Snider P. L., Conway S. J. et al. (2014). HDAC1 and HDAC2 control the specification of neural crest cells into peripheral glia. The J. Neurosci. 34, 6112-6122. 10.1523/JNEUROSCI.5212-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda S., Tanigawa S., Ohmori T., Taguchi A., Kudo K., Suzuki Y., Sato Y., Hino S., Sander M., Perantoni A. O. et al. (2014). Sall1 maintains nephron progenitors and nascent nephrons by acting as both an activator and a repressor. J. Am. Soc. Nephrol. 25, 2584-2595. 10.1681/ASN.2013080896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly R. D. W. and Cowley S. M. (2013). The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem. Soc. Trans. 41, 741-749. 10.1042/BST20130010 [DOI] [PubMed] [Google Scholar]
- Kispert A., Vainio S. and McMahon A. P. (1998). Wnt-4 is a mesenchymal signal for epithelial transformation of metanephric mesenchyme in the developing kidney. Development 125, 4225-4234. [DOI] [PubMed] [Google Scholar]
- Kita A., Imayoshi I., Hojo M., Kitagawa M., Kokubu H., Ohsawa R., Ohtsuka T., Kageyama R. and Hashimoto N. (2007). Hes1 and Hes5 control the progenitor pool, intermediate lobe specification, and posterior lobe formation in the pituitary development. Mol. Endocrinol. 21, 1458-1466. 10.1210/me.2007-0039 [DOI] [PubMed] [Google Scholar]
- Kobayashi A., Kwan K.-M., Carroll T. J., McMahon A. P., Mendelsohn C. L. and Behringer R. R. (2005). Distinct and sequential tissue-specific activities of the LIM-class homeobox gene Lim1 for tubular morphogenesis during kidney development. Development 132, 2809-2823. 10.1242/dev.01858 [DOI] [PubMed] [Google Scholar]
- Kobayashi A., Valerius M. T., Mugford J. W., Carroll T. J., Self M., Oliver G. and McMahon A. P. (2008). Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3, 169-181. 10.1016/j.stem.2008.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R., Cheng H.-T. and Surendran K. (2007). Molecular insights into segmentation along the proximal distal axis of the nephron. J. Am. Soc. Nephrol. 18, 2014-2020. 10.1681/ASN.2007040453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreidberg J. A. and Hartwig S. (2008). Wilms’ tumor-1: a riddle wrapped in a mystery, inside a kidney. Kidney Int. 74, 411-412. 10.1038/ki.2008.307 [DOI] [PubMed] [Google Scholar]
- LeBoeuf M., Terrell A., Trivedi S., Sinha S., Epstein J. A., Olson E. N., Morrisey E. E. and Millar S. E. (2010). Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev. Cell 19, 807-818. 10.1016/j.devcel.2010.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milutinovic S., Zhuang Q. and Szyf M. (2002). Proliferating cell nuclear antigen associates with histone deacetylase activity, integrating DNA replication and chromatin modification. J. Biol. Chem. 277, 20974-20978. 10.1074/jbc.M202504200 [DOI] [PubMed] [Google Scholar]
- Montgomery R. L., Davis C. A., Potthoff M. J., Haberland M., Fielitz J., Qi X., Hill J. A., Richardson J. A. and Olson E. N. (2007). Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 21, 1790-1802. 10.1101/gad.1563807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlis M., Grote D., Gaitan Y., Boualia S. K. and Bouchard M. (2007). Pax2 and pax8 regulate branching morphogenesis and nephron differentiation in the developing kidney. J. Am. Soc. Nephrol. 18, 1121-1129. 10.1681/ASN.2006070739 [DOI] [PubMed] [Google Scholar]
- Park J.-S., Valerius M. T. and McMahon A. P. (2007). Wnt/beta-catenin signaling regulates nephron induction during mouse kidney development. Development 134, 2533-2539. 10.1242/dev.006155 [DOI] [PubMed] [Google Scholar]
- Park J.-S., Ma W., O'Brien L. L., Chung E., Guo J.-J., Cheng J.-G., Valerius M. T., McMahon J. A., Wong W. H. and McMahon A. P. (2012). Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev. Cell 23, 637-651. 10.1016/j.devcel.2012.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen A., Skjong C. and Shawlot W. (2005). Lim1 is required for nephric duct extension and ureteric bud morphogenesis. Dev. Biol. 288, 571-581. 10.1016/j.ydbio.2005.09.027 [DOI] [PubMed] [Google Scholar]
- Perantoni A. O., Timofeeva O., Naillat F., Richman C., Pajni-Underwood S., Wilson C., Vainio S., Dove L. F. and Lewandoski M. (2005). Inactivation of FGF8 in early mesoderm reveals an essential role in kidney development. Development 132, 3859-3871. 10.1242/dev.01945 [DOI] [PubMed] [Google Scholar]
- Schanstra J. P., Bachvarova M., Neau E., Bascands J. L. and Bachvarov D. (2007). Gene expression profiling in the remnant kidney model of wild type and kinin B1 and B2 receptor knockout mice. Kidney Int. 72, 442-454. 10.1038/sj.ki.5002172 [DOI] [PubMed] [Google Scholar]
- Schedl A. (2007). Renal abnormalities and their developmental origin. Nat. Rev. Genet. 8, 791-802. 10.1038/nrg2205 [DOI] [PubMed] [Google Scholar]
- Self M., Lagutin O. V., Bowling B., Hendrix J., Cai Y., Dressler G. R. and Oliver G. (2006). Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 25, 5214-5228. 10.1038/sj.emboj.7601381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J., Jokela T., Skovorodkin I. and Vainio S. (2010). Mapping of the fate of cell lineages generated from cells that express the Wnt4 gene by time-lapse during kidney development. Differentiation 79, 57-64. 10.1016/j.diff.2009.08.006 [DOI] [PubMed] [Google Scholar]
- Stark K., Vainio S., Vassileva G. and McMahon A. P. (1994). Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 372, 679-683. 10.1038/372679a0 [DOI] [PubMed] [Google Scholar]
- Strzalka W. and Ziemienowicz A. (2011). Proliferating cell nuclear antigen (PCNA): a key factor in DNA replication and cell cycle regulation. Ann. Bot. 107, 1127-1140. 10.1093/aob/mcq243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turgeon N., Blais M., Gagné J.-M., Tardif V., Boudreau F., Perreault N. and Asselin C. (2013). HDAC1 and HDAC2 restrain the intestinal inflammatory response by regulating intestinal epithelial cell differentiation. PLoS ONE 8, e73785 10.1371/journal.pone.0073785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Pereira F. A., Beasley D. and Zheng H. (2003). Presenilins are required for the formation of comma- and S-shaped bodies during nephrogenesis. Development 130, 5019-5029. 10.1242/dev.00682 [DOI] [PubMed] [Google Scholar]
- Winter M., Moser M. A., Meunier D., Fischer C., Machat G., Mattes K., Lichtenberger B. M., Brunmeir R., Weissmann S., Murko C. et al. (2013). Divergent roles of HDAC1 and HDAC2 in the regulation of epidermal development and tumorigenesis. EMBO J. 32, 3176-3191. 10.1038/emboj.2013.243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuhl E., van Stralen K. J., Verrina E., Bjerre A., Wanner C., Heaf J. G., Zurriaga O., Hoitsma A., Niaudet P., Palsson R. et al. (2013). Timing and outcome of renal replacement therapy in patients with congenital malformations of the kidney and urinary tract. Clin. J. Am. Soc. Nephrol. 8, 67-74. 10.2215/CJN.03310412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Liu H., Park J.-S., Lan Y. and Jiang R. (2014). Osr1 acts downstream of and interacts synergistically with Six2 to maintain nephron progenitor cells during kidney organogenesis. Development 141, 1442-1452. 10.1242/dev.103283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F., Chen Y., Hoang T. N., Montgomery R. L., Zhao X.-H., Bu H., Hu T., Taketo M. M., van Es J. H., Clevers H. et al. (2009). HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat. Neurosci. 12, 829-838. 10.1038/nn.2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.