

Abstract
In this Scientific Perspectives we first review the recent advances in our understanding of the functional architecture of basal ganglia circuits. Then we argue that these data can best be explained by a model in which basal ganglia act to control the gain of movement kinematics to shape performance based on prior experience, which we refer to as a history‐dependent gain computation. Finally, we discuss how insights from the history‐dependent gain model might translate from the bench to the bedside, primarily the implications for the design of adaptive deep brain stimulation. Thus, we explicate the key empirical and conceptual support for a normative, computational model with substantial explanatory power for the broad role of basal ganglia circuits in health and disease. © 2018 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
Keywords: basal ganglia, bradykinesia, dopamine, mouse models, neural circuits
A voluntary, purposive action requires one to extract information from experience and act on that information by executing the requisite movements to obtain desired outcomes. The subcortical circuits of the basal ganglia (BG) are a critical nexus in which inference, planning, selection, and execution all appear to be integrated to control volition. However, despite it being clear for centuries that disruptions to BG function produce profound disruptions to purposive movements,1 a detailed account of the circuit mechanisms by which these deficits follow from perturbation remains elusive.
Loss of midbrain dopamine neurons results in parkinsonism in vertebrates. Parkinsonism is typified by its cardinal symptoms of bradykinesia/akinesia, rigidity, and tremor.2 The primary efferent target of dopamine neurons in the substantia nigra pars compacta that preferentially degenerate in Parkinson's disease (PD) 3 is the dorsal striatum.4 As a result, much interest has been focused on attempting to account for the mechanisms by which dopamine‐depletion produces the profound deficits in voluntary movement typical of parkinsonism.5 Bradykinesia, the slowing of movement speed, is directly attributed to the loss of dopaminergic innervation6 and dopamine replacement therapy using l‐dopa can ameliorate bradykinesia in PD patients7 (although movement velocity does not fully recover with dopamine replacement therapy or deep brain stimulation)8, 9. Further, the presence of augmented dopamine levels increased the speed and amplitude of voluntary movements.10, 11
Studies of healthy BG function support its role in the control of movement kinematics. In a number of experiments in primates12, 13, 14 and rodents,15, 16, 17 the most consistent and profound effects of perturbation of BG activity are changes in movement speed.18, 19 Perturbations in movement kinematics can even occur in the absence of any alterations in selection or sequencing of actions,14 and acute suppression of activity only after movement initiation (presumably after action selection) is also sufficient to produce kinematic changes.13, 15 These data thus argue for an essential role of BG and dopamine in the control of movement vigor. We use “movement vigor” to refer to a general property of movement kinematics reflected in various, often correlated measurements (eg, speed and amplitude). This is by analogy to descriptions of “response vigor”20 that refer to a general property of task engagement indexed by a specific measurement (eg, rate of lever pressing).19
Although initial models proposed that dopamine signals per se were necessary for initiating movement,10 it has subsequently become clear that parkinsonism is the consequence of persistent disrupted circuit function mediated in large part by plastic changes following dopamine depletion.21, 22 Indeed, PD is now widely considered the paradigmatic “circuit disorder.”23 We note that dopamine clearly plays important roles outside of BG circuits, which will not be the focus of this discussion. Here, we propose that understanding the specific circuit functions (what we refer to as computations) implemented by BG and modulated by dopamine is critical to understanding both normal functioning and the pathological dysfunction characteristic of PD. As interventions increasingly attempt to restore circuit function, we believe that the refinement of intervention will need to be informed by mechanistic models of the computations implemented in BG.
Reentrant and Convergent Pathways of Basal Ganglia
BG are an interconnected set of subcortical nuclei found essentially unchanged in their core circuitry and cell types across all studied vertebrates.24 The primary input nuclei, striatum (STR) and subthalamic nucleus (STN), receive the majority of their afferent excitatory input from the neocortex, anterior and intralaminar thalamic nuclei, and amygdala.4 The STR receives corticostriatal input from layer II/III pyramidal neurons and both major classes (intratelencephalic and corticofugal) of layer V excitatory projection neurons.25 STN receives input from corticofugal projection neurons.26 The primary output nuclei, substantia nigra pars reticulata (SNr) and globus pallidus internal segment (GPi), send reentrant projections to the thalamus.27 Although it receives less emphasis, BG output nuclei also send descending projections to multiple targets in the diencephalon and brain stem.4, 27, 28, 29 Indeed, although the reentrant thalamic projections from primate SNr have received intense interest, a sizable majority of SNr output neurons project to the superior colliculus.27
Modern viral strategies,29, 30, 31, 32, 33, 34 empowered by molecular genetic tools and new imaging methods, now allow for detailed anatomical tracing of complete neurons and axon pathways throughout the rodent brain.35, 36, 37 These techniques have begun to reveal the convergent nature of descending BG output pathways. Deep layer cortical neurons give rise to the corticofugal projection that leaves the cortex and descends via the internal capsule. En route many of these axons elaborate collaterals36, 38, 39, 40 that synapse within dorsal STR (Fig. 1a‐c). The same axons project densely to deep layers of superior colliculus and pontine nuclei.26, 29, 36 These target regions coincide with regions that receive descending SNr output27, 28 (Fig. 1a‐c). We recognize that the widely accepted functional architecture of the BG is characterized by reentrant loops from the cortex through the BG and thalamus.41 Here we highlight the highly conserved42 feed‐forward convergent pathways to premotor structures in the midbrain and brain stem19, 43 (Fig. 1a,d). We further note that the superior colliculus is an important premotor structure for upper body movements generally,44 including the limbs.45, 46, 47 This latter feature suggests that descending (polysynaptic) cortical commands to the spinal cord reflect both descending extra‐BG commands and BG output integrated in premotor structures of the midbrain and hindbrain19 (Fig. 1d,e).
Figure 1.
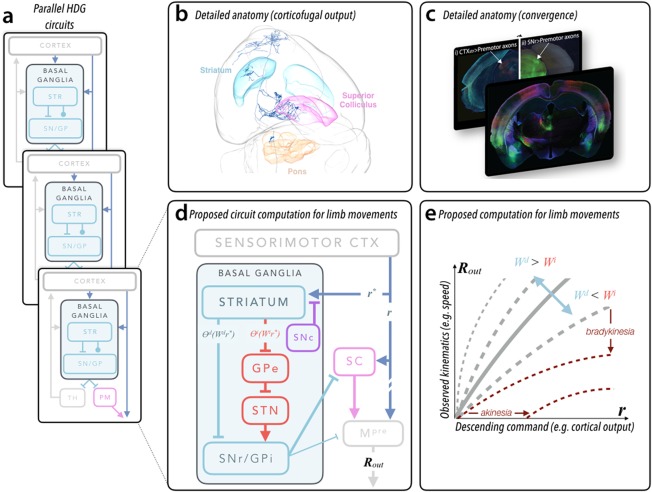
Proposed functional architecture of basal ganglia. (a) Schematic of basal ganglia organization highlighting the parallel organization of both reentrant loop architecture (as in eference101 and the feed‐forward, convergent pathways discussed in this article. Whereas the reentrant loop circuits flow through the ascending basal ganglia output to thalamus (TH), the feed‐forward, convergent pathways flow through specific premotor (PM) structures in the midbrain and brain stem (more akin to the feed‐forward pathways in lower vertebrates102. (b) Detailed anatomical evidence for convergence of feed‐forward pathways. A rendering of a single corticofugal neuron in the anterior sensorimotor cortex and its axonal arborization through the entire mouse brain reconstructed using the method in reference36 and rendered with software developed by the MouseLight project (https://www.janelia.org/project-team/mouselight). Important brain regions discussed in the text are indicated. Striatum is cyan, deep layers of superior colliculus are pink, and the basal pontine nuclei are orange. (c) Fluorescent images in (i) and the front panel obtained from injection of a retrograde virus expressing fluorescent proteins in the dorsal striatum reveal cortical inputs and clearly illuminate the corticofugal pathway (intermingled axon bundles below thalamus). Modified from reference 29. Corticofugal axons can be followed in to the superior colliculus in a more posterior section (i). Comparison with tissue in which the substantia nigra pars reticulata was infected with an anterograde virus (ii) reveals convergent termination zones (indicated by arrows; image modified from data in reference 63). (d) Schematic diagram of cortical‐BG circuitry described in the text and focusing on the convergent pathways relevant to movements of the forelimb in mice (similar to recent proposals19. Labels on individual pathways represent terms in computation depicted in (e) and in the main text. r and r* describe the cortical output (eg, vector of firing rates) providing excitatory input to subcortical premotor structures (broadly defined) and striatum, respectively. As mentioned in the text, r and r* are correlated but not identical. The output of the direct and indirect pathway striatal projection neurons are expressed as an input‐output function (θi/d) of the cortical input (r*) multiplied by the synaptic strengths of corticostriatal inputs (Wi/d). The net output (Rout) — a motor command — reflects integration of basal ganglia output and direct cortical output (see main text for details). (e) Here we plot the predicted change in observed movement kinematics — a consequence of R out — as a function of the cortical output, r. The shape of the curve is drawn for different ratios of average strength of the direct (Wd) and indirect (Wi) corticostriatal synaptic strengths. When this becomes dramatically altered (ie, following dopamine depletion), the slope of the curves is dramatically reduced (dark red, dashed lines). We also annotate the specific changes in this computation that map to the clinically observed symptoms of bradykinesia (reduction in average speed) and akinesia (a paucity of movement; assuming Rout ≤ 0 results in an absence of movement). We note that the shape of the curves depends on nonlinearities in input‐output functions θ; for simplicity, here we assume the same nonlinearity modifies r* and r. Example simulations of models implementing this function can be found in references 56 and 63.
BG output is a function of descending cortical activity carried via corticostriatal projections transformed by the intrinsic opponent circuitry of the BG.4, 48 The direct and indirect projection pathways of STR are the primary targets of corticostriatal input and recipients of dense innervation from midbrain dopaminergic neurons. The direct and indirect pathways arise from spiny projection neurons (dSPNs and iSPNs, respectively). dSPNs project primarily to BG output nuclei (eg. SNr), and iSPNs project primarily to the internal nuclei of the BG (for a review of the intrinsic anatomy of the BG, see reference 4). dSPNs and iSPNs are characterized by differential expression of a number of key receptors and signaling pathways in addition to their distinct anatomical projections.48 Most notably, dSPNs are characterized by expression of D1‐type dopamine receptors and iSPNs by D2‐type receptors.49
In its canonical form, dSPNs and iSPNs were proposed to be antagonistic, with either one or the other being dominantly active and enhancing the initiation of action or the suppression of unwanted actions, respectively.50 Consistent with this model, direct stimulation of dSPNs enhances locomotor behavior, and stimulation of iSPNs suppresses movement.51 Plastic changes in the excitability of dSPNs and iSPNs following pathological disruption of dopamine signaling was proposed to explain hypokinetic (bradykinesia; excess afferent drive of iSPNs) and hyperkinetic (chorea; excess afferent drive of dSPNs) behaviors.48, 52 Indeed, it is the case that blockade of excitatory input to iSPNs can ameliorate the effects of dopamine‐depletion.53 However, recent work has shown that dSPN and iSPN activity is not antagonistic during movement, but rather coactive.54, 55 This has shifted emphasis toward understanding how opponent dSPN and iSPN output may be integrated to shape behavior.19, 56 For example, the ratio of dSPN/iSPN modulation during movement is likely critical.57
Activity in Basal Ganglia Circuits Represents Movement Kinematics
The study of PD patients has been critical to our understanding of the function of BG. For example, Mazzoni and colleagues asked l‐dopa/agonist‐treated off‐drug patients to make movements of the hand from a central starting point to an eccentric target within a prescribed range of speeds.58 This revealed that patients are biased toward slow movements, but also demonstrated that patients can move with the same speed and accuracy as control subjects (recently confirmed in a similar design by reference 59). A subsequent study by Baraduc and colleagues allowed subjects to move a 1D joystick to a cued eccentric position without a prescribed range of movement speed.9 In this version of a free choice, variable‐amplitude operant task (hereafter, a VAO task), they found that untreated PD patients moved at systematically slower speeds, yet their movements were described by control laws that were indistinguishable from non‐PD subjects. Note that again in these studies as in perturbation experiments,14 patients did not demonstrate impairment in other aspects of task performance, such as selecting an appropriate action.
In rodents, it has been established that dopamine depletion leads to impaired performance in tasks that require increasing effort to obtain reward.60 However, such tasks typically used designs that made it difficult to quantify movement kinematics in detail. Inspired by the human studies above, we developed an approach to train thirsty mice to move a spring‐loaded joystick to eccentric positions in exchange for a water reward. This task provides a quantifiable behavior with high spatiotemporal resolution in a model organism in which we can use modern molecular genetic methods to dissect BG circuit function.61 It has also allowed us to analyze the kinematics and trajectories of discrete forelimb movements in a VAO task suited to mice.15, 56, 62, 63 Dopamine depletion produces systematic bias in performance toward slower, smaller amplitude movements in mice,15 as it does in PD patients9 (Fig. 2a,b). Indeed, closely paralleling the results from Baraduc and colleagues, this slowness of movement manifests as systematic slowing across all examined movement amplitudes (Fig. 2b).
Figure 2.
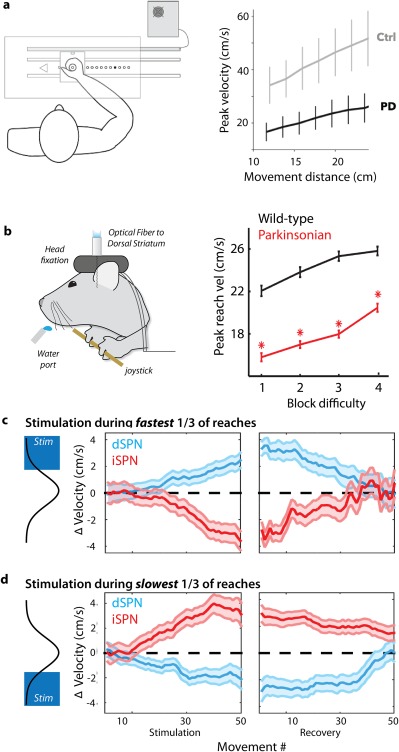
Regulation of movement vigor by opponent BG pathways. (a) Dopamine depletion from Parkinson's disease in patients (unmedicated patients with chronic DBS stimulators inactivate during the experiment) produces a robust slowing of movement across a broad range of movement vigor relative to control subjects (Ctrl). Modified from reference 9. Parkinsonian patients and controls were instructed to move a joystick to several distances indicated by an LED illuminated at the target eccentricity (example of a human VAO task). (b) Mice were required to perform a similar VAO task, moving a joystick to increasing amplitudes. Dopamine depletion in the MitoPark103 murine model of PD (“parkinsonian”) spares the ability to select and initiate the proper action, but reduces the speed of movement across a range of movement vigor. (c, d) Selective, closed‐loop optogenetic stimulation (pulsed for 450 milliseconds, < 10 milliseconds after movement onset) of direct pathway dSPNs (blue) or indirect pathway iSPNs (red) during the fastest‐reaching movements induces a cumulative shift in peak reach speed compared with control sessions (zero). Changes were a form of learning that persisted for tens of trials of the no‐stimulation recovery period. This effect extends to all reaches, not only those reaches during which stimulation occurred (as evidenced by a lack of change in in the width of the SEM bars throughout the session). The converse was observed when stimulation occurred during the slowest movements — direct pathway stimulation induced a slowing of movement, whereas indirect pathway stimulation induced a speeding of movement. Modified from reference 56.
What circuit computations might account for systematic slowing of movement following dopamine depletion? Although it is well established that the chronic absence of dopamine signaling produces dramatic structural and biophysical changes in SPNs,64 relatively little is known about changes in the activity of SPNs during movement. Theories premised on the antagonistic role of direct and indirect pathways suggest that excessive activation of iSPNs prior to and during movement initiation should be primarily responsible.50 In animals with normal dopamine levels, perturbation of SPN activity during movement is sufficient to slow movement — putatively equivalent to bradykinesia13, 15 — and suppression of BG output is also sufficient to alter movement speed and amplitude.14, 63 These observations suggest that dopamine depletion could alter movement vigor by chronically reducing the gain of SPN activity during movement execution. Recent work from our group has provided direct evidence that this is indeed the case and has argued against excessive antagonistic activity being the cause of bradykinesia.15 To understand this evidence, it is necessary to first consider in some detail the predicted SPN activity during movement in these two alternatives.
If it is the case that STR activity can specifically regulate movement vigor, then activity should be a function of the phase of movement.65, 66 To build an intuition for why this is so, imagine trying to push a child on a swing. If one wants to increase the amplitude of one's swing, it is critical not only to push, but to push at the appropriate phase of the swing to have the desired effect. First, we note that although there has been a tendency to focus on the modulation of STR activity prior to movement initiation, the most robust modulation of SPNs15, 67 and downstream target regions68, 69 occurs during movement execution. Although the onset of movement is convenient for alignment of perimovement neural activity, it can be misleading because movement durations are generally variable. Two recent studies overcame this obstacle by aligning activity to phase of either a discrete limb movement15 or a locomotor sequence.16 As would be required in a brain area controlling the kinematics of ongoing movement, it was observed in both studies that a substantial fraction of STR neurons are active during the acceleration and deceleration phases of movement that determine vigor. Importantly, in each case the magnitude of the activity modulation in STR neurons active in an early phase of movement correlated with movement speed and amplitude. Finally, pharmacological restoration of dopaminergic tone with l‐dopa reduced bradykinesia and partially restored robust modulation of SPN activity in early phases of movement execution15 (in addition to other, likely maladaptive,70, 71 changes). In contrast, there was not clear evidence for an aberrant large average increase above baseline of SPN activity throughout movement, arguing against the proposal that bradykinesia results from multiple conflicting actions being executed.50 Nonetheless, premovement, baseline activity of SPNs was increased in dopamine‐depleted animals15 (recently shown to be specifically in iSPNs71) suggesting that excessive BG output could be associated with akinesia — a point we will return to below.
Learned Changes in Basal Ganglia Circuits Underlying Changes in Movement Vigor
To this point, we have discussed the regulation of movement kinematics by a fixed BG modulation of descending motor commands. However, the profound effects of dopamine depletion indicate that plastic changes in BG responses to afferent input underlie at least part of the aberrant movement vigor. We note that acute pharmacological restoration of dopaminergic tone via systemic l‐dopa administration restored the functional response properties of STR neurons in the VAO task, but only after tens of minutes.15 Thus, these data suggested that the responses of STR neurons and their tuning to movement kinematics are both capable of persistently changing over a course of tens of minutes and that such changes are dopamine dependent.
Previous work had suggested that direct extracellular stimulation of STR in primates could be used to drive learning.72 In mice it was shown that such effects were likely cell‐type dependent (direct or indirect pathway).73 However, the specificity of the learned changes in behavior that could be produced and the formal rules governing stimulation‐induced learning were unclear. Thus, we asked whether direct modulation of STR activity could be sufficient to induce history‐dependent changes in movement vigor. To tease apart this mechanism, we designed an experiment in which we selectively stimulated either pathway (dSPN or iSPN) in a closed loop with a specific kinematic readout of vigor: movement speed. This selective closed‐loop stimulation provided the opportunity to address whether and how striatal SPNs might be sufficient to produce persistent changes in movement vigor.
We took several steps to isolate the putative changes to the stimulated neuron population. First, relatively weak stimulation intensities that did not alter the concurrent movement were chosen. Second, stimulation was delivered while SPN neurons would normally be active, beginning a few milliseconds after movement initiation and lasting only for the average movement duration. Third, taking advantage of classic microstimulation experimental design,74 animals collected rewards on all trials, allowing stimulation to bias behavior independent of extrinsic reinforcement. However, it is important to note that we did not stimulate only SPNs that were correlated with forelimb movements, as might have been expected to be necessary. Recordings performed from the same region of striatum suggest that only ∼30%‐40% of neurons are robustly activated during forelimb movements.15, 56, 75 We reasoned that an interaction between afferent input during movement (conferring response specificity) and exogenous stimulation (conferring cell‐type specificity) could be sufficient to produce specific behavioral changes.
We found that closed‐loop optogenetic stimulation of SPNs could reliably induce a learned shift in movement speed that became more pronounced with each successive stimulation (Fig. 2c). This shift was present in unstimulated reaches and persisted for many trials (>10 minutes) after stimulation had been terminated.56 We found that stimulation of dSPN and iSPN populations produced opponent changes in speed — increases and decreases, respectively. Importantly, these changes were largely restricted to the parameter used to trigger closed‐loop stimulation and speed, with no effect on uncorrelated kinematic features or general aspects of task performance such as rate of reach initiation. These results strongly suggest that dSTR is a locus of plasticity that is sufficient to control a specific movement parameter,56 in addition to more general associative learning.74, 75
Previous work has focused on the role of BG (often STR) in the selection of actions. Selection models argue that actions that yield robust dSPN activity are increased in their probability, and actions that yield robust iSPN activity are reduced in probability.52, 73 A general form of a selection model has been invoked to explain changes in movement kinematics by assuming that BG play a role in selecting movements with specific kinematics from a palette of possible movement speeds.58 However, with specific regard to movement kinematics, several features of the selection model appear inconsistent with our stimulation data56 and behavior in the VAO task.15
Here we focus on one intuitive issue with a selection model: how should one explain the systematic slowing of movement following iSPN stimulation? Canonically, iSPN activity is supposed to reduce the probability of selecting actions that lead to iSPN activation. In the context of closed‐loop stimulation of iSPN during fast movements, this should reduce the probability of engaging in the task and making a forelimb movement. Alternatively, iSPN stimulation could produce a selective reduction in fast movements, altering the distribution of movement velocities (eg, reducing variance). Neither effect was observed.56 A follow‐up experiment presented a robust counter to selection models: what happens if stimulation is delivered on every movement? If iSPN stimulation selects against an action or is inherently action‐suppressing, as some have suggested, this would seem to demand a reduction in the number of movements. However, no change in any aspect of movement kinematics or number of movements was observed when this test was performed.56
We have observed that selective closed‐loop stimulation on fast movements produced a cumulative, persistent change in speed, whereas the same stimulation delivered on all movements produced no change in speed or in the probability of movement. These results identified dSTR as a locus of plasticity, but suggested, in concert with other studies, that the control of kinematics may work through a mechanism distinct from the selection mechanisms previously proposed. Here we articulate in detail a hypothesis about the functional architecture of BG: the BG circuit implements a gain of cortical output. In the context of movement, BG apply a gain to the cortical motor commands proportional to the relative strength of the afferent inputs to dSPN and iSPN from premotor and motor cortical areas. The strength of these inputs is set by simple reinforcement‐driven plasticity rules that allow movement vigor to be adapted to the recent history of reward obtained (see HDG Model: A Proposed Computational Logic of Basal Ganglia Circuits section).
To evaluate this model, we next tested a provocative prediction. Closed‐loop dSPN stimulation on the slowest, rather than fastest, reaches should cause a cumulative slowing of movement, whereas iSPN stimulation should cause a counterintuitive invigoration of movement. Indeed, we observed a symmetric “flip” in the effect of stimulation when the contingency was changed from fastest to slowest56 (Fig. 2d), consistent with a model in which changes in the relative intensity of dSPN and iSPN activity can govern the net movement vigor. We further used a computational simulation of a cortical‐BG circuit to show that a model in which the pathways act as a push‐pull gain controller was sufficient to quantitatively account for these data.56, 63
HDG Model: A Proposed Computational Logic of Basal Ganglia Circuits
So far, we have explored anatomical and functional data in the BG that we argue can be best explained by a model in which the gain of descending motor commands can be modified by the feed‐forward convergent BG output onto subcortical premotor areas. The gain applied by the BG, we propose, is determined by the relative strength of excitatory (putatively corticostriatal) inputs to dSPN and iSPN, and the strength of the input is determined by dopamine‐ and activity‐ dependent plasticity. The dopamine dependence of this plasticity ensures that the gain applied by the BG is a function of the recent reward history. Thus, we refer to this as the history‐dependent gain (HDG) model.
We pause to note that this is a general, computational account of BG function rather than a specific claim that BG exclusively control movement vigor. In the context of purposive action, we elaborate an argument that a history‐dependent gain manifests as a central role for the BG in the control of movement vigor. Motor output provides us the ability to use overt, quantitative behavioral measurements to reveal computational principles of BG. Our focus on movement vigor is also motivated by the systematic decreases in movement vigor, bradykinesia, being a cardinal symptom of PD,10 suggesting that it is at least a very important contribution of the BG to behavior generally. However, a computation need not map onto a circumscribed behavioral role. A history‐dependent gain computation will manifest as different behavioral correlates depending on the specific task under study (see Implications of the HDG Model section).
Classic data suggest that the relationship between the activity of midbrain premotor neurons (like the superior colliculus) and movement, like neurons in motor cortices,76 is consistent with population vector models.77, 78, 79 In such models the net firing rate, properly, the length of the population vector, is proportional to the speed of movement.80, 81 Thus, if we neglect for the moment the specific timing of activity within the phase of movement discussed in the Activity in Basal Ganglia Circuits Represents Movement Kinematics section, the speed of movement can be described by monotonic functions with relationships of the form (Fig. 1c):
| (1) |
where,
descending motor command ∝ β r command
These schematized equations describe a computation in which changes in the weights of corticostriatal synapses (W) onto the direct (W d) and indirect (W i) SPNs produce changes in the integrated output (transformed by the input‐output function θ) to premotor structures. This computation is akin to a history‐dependent gain applied to cortical output because the magnitude of the BG output is itself a function of the descending cortical output (ie, ∝ r command). We provide a graphical explanation of how this computation can be mapped onto a cortico‐BG circuit and manifests as an observed change in movement kinematics (Fig. 1d,e).
The HDG model predicts that selective suppression of direct pathway SPN activity (ie, an increase in BG output) during movement should be sufficient to reduce movement speed. A classic study from Horak and Anderson demonstrated that microsimulation of a BG output nucleus, GPi, could alter movement kinematics after the onset of muscle contraction.13 This observation was consistent with the effect of acute pharmacological suppression or lesion12, 68 of GPi activity; however, questions about the effect of microstimulation persisted (for example, what sort of change in activity was induced by stimulation?). Thus, we sought to perform an analogous experiment using acute optogenetic suppression of dSPN activity selectively during movement execution. We found that suppression of dSPN activity was indeed sufficient to induce bradykinetic movements.15
Direct pathway STR neurons inhibit the tonically active, inhibitory output nuclei, SNr/GPi, of BG. The decreased speed of movements during acute suppression of direct pathway STR output strongly suggested that BG output during execution of movements is inversely correlated with vigor. We have recently confirmed this observation by using chemogenetics to make cell‐type‐specific pharmacological suppression of SNr output projection neurons during performance of the VAO task. Consistent with the effects of direct pathway suppression and the predictions of the HDG model, tonic suppression of SNr output increases movement speed and amplitude.63 However, we note that the effects of suppressing SNr output are complicated by the SNr and GPi projection neurons also elaborating recurrent collaterals that potently inhibit other projection neurons.82, 83 These recurrent collaterals had long been postulated to mediate lateral inhibition, putatively subserving an action selection mechanism.28 In contrast, we recently found that this recurrent inhibition appears to mediate a form of divisive gain control more consistent with feedback inhibition than lateral inhibition.82 Implementation of the HDG model incorporating feedback gain control could quantitatively account for changes in movement vigor following chemogenetic perturbation that were inconsistent with lateral inhibition models.63 At the moment, it is less clear whether the HDG model can similarly account for altered movement vigor following focal inactivation of GPi subregions in primates.14
The HDG model makes another quantitative prediction. Because STR activity (and thus BG output) is a function of corticostriatal inputs carrying motor command correlates ( ), it should be possible to decode movement kinematics from recordings of population activity in striatum. Although we and others have shown that either the mean or maximal firing rate of individual SPN neurons varied trial to trial as a function of movement vigor,15, 16 single neuron activity is nonetheless quite variable. It was thus unclear whether continuous modulation of the firing rate reflected continuous modulation of movement kinematics. Recently, we took advantage of simultaneous recordings of striatal populations (>20 neurons). We found that a low‐dimensional representation of population dynamics could be used as the input to a decoder that faithfully recovered the continuous kinematics of forelimb movements (unpublished; presented in reference 75). Importantly, we found that the decoder was predictive (activity predicted movement about 150 milliseconds prior to observed kinematics), consistent with the notion that striatal activity is (partially) causal for observed movement vigor. Decoding of locomotor speed17, 84 as well as more detailed kinematics85 has also been described.
Finally, the HDG model predicts that dopamine‐dependent changes in the weight of corticostriatal inputs (W D1+ ‐ W D2+) can alter movement kinematics. This is consistent with the observation that learning produced by closed‐loop stimulation of SPNs (see Activity in Basal Ganglia Circuits Represents Movement Kinematics section) requires functional dopamine receptors.56 We have discovered both an algorithmic learning rule and implementation of that rule in a model neural network that can account for how changes in movement vigor follow from changes in reward in the case of the VAO task and optogenetic stimulation.56, 63 Although a full discussion is beyond the scope of this article, the key insight of the learning rule is quite simple: the average BG output should be changed in proportion to how different the current movement vigor was from the expected movement vigor and modulated by reward. We referred to this algorithmic learning rule as “MeSH” (Mean Shift with Homeostasis). The key idea for implementation is that potentiation and depression in corticostriatal synapses are balanced in both dSPNs and iSPNs. This balance produces apparently stable movement vigor during constant reward rate, but allows for bidirectional changes in movement vigor in response to changes in reward rate.56 This account diverges from classic accounts that posit that the sign of plasticity is determined by the identity of the dopamine receptor (eg, dopamine would exclusively enhance vigor via potentiation in D1 + neurons). We believe our model of balanced plasticity more accurately reflects the important discovery that dopamine mediates long‐term potentiation and depression at corticostriatal synapses48 in both dSPNs and iSPNs.86, 87 However, our provisional implementation of MeSH does require some specific features of plasticity that are yet to be experimentally evaluated.
Implications of the HDG Model
In this Scientific Perspectives, we have presented a range of recent data, primarily from rodents, that can be accounted for by a parsimonious functional model of BG: history‐dependent gain (HDG). Perhaps one of the most surprising insights to follow from our data and the HDG model is that BG are constantly adapting even when behavior appears stationary. We tend to equate learning with a gated process that is present or absent depending on outcomes. The HDG model derived from experimental data suggests that we should view the BG as regulating a dynamic equilibrium in which the equilibrium point is controlled by the recent history of outcomes (often rewards) signaled by midbrain dopamine activity. From this perspective, bradykinesia reflects a new (aberrant) equilibrium point, and it is frustratingly robust because it is maintained by an equilibrium process.
In the context of a thorough evaluation of BG models by Schroll and Hamker, the HDG model provides computational implementation addressing a broad range of properties desired in a model of the BG (Table 1). As described here, our model remains incomplete in its interpretation of some features of BG anatomy (eg, thalamostriatal input, “hyperdirect” pathway) that have been important components of other models22 while highlighting an oft‐neglected aspect of BG anatomy — the feed‐forward, convergent outputs). This reflects both our desire to explain one computation in detail and a critical requirement for future work. We recognize that some PD‐related behavioral changes may not be a direct consequence of BG dysfunction. Nonetheless, here we explore the additional explanatory power of an HDG computation to PD symptoms beyond our specific explication with respect to bradykinesia. Then we conclude by discussing how closed‐loop deep brain stimulation in patients, inspired by our experience with mice, might improve outcomes while reducing stimulation requirements.
Table 1.
Qualitative model comparisons
| Authors address/implement | Authors provide a functional interpretation for | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Authors provide either partial or complete computational implementation | BG motor movement frequency | BG motor movement vigor | BG cognitive functions | Plasticity | Closed loops | Open loops | Direct pathway | Short indirect pathway | Long indirect pathway | Hyperdirect pathway | Output convergent pathways | Output feedback pathways | |
| Albin et al, 1989 | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✓ | ✗ | ✗ | ✗ |
| DeLong, 1990 | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✓ | ✗ | ✗ | ✗ |
| Mink, 1996 | ✗ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ | (✓) | ✗ | ✓ |
| Bar‐Gad et al, 2000 | ✓ | ✗ | ✗ | ✗ | ✓ | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Suri et al, 2001 | ✓ | ✓ | ✗ | ✗ | ✓ | ✗ | ✓ | ✓ | ✗ | ✓ | ✗ | ✗ | ✗ |
| Gurney et al, 2001a; Humphries et al, 2006 | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✓ | n/a | n/a | n/a | n/a | ✗ | ✗ |
| Nambu, 2004 | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✓ | ✗ | ✓ | ✓ | ✗ | ✗ | |
| Brown et al., 2004 | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ |
| Frank, 2006; Wiecki and Frank, 2013 | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ |
| Ashby et al, 2007 | ✓ | ✓ | ✗ | ✓ | ✓ | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Stocco et al, 2010 | ✓ | ✓ | ✗ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✗ | ✗ |
| Chersi et al, 2013 | ✓ | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ | ✓ | ✗ | ✗ |
| Schroll et al, 2013 | ✓ | ✓ | ✗ | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✗ | ✗ |
| History‐dependent gain | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ | ✗ | ✓ | ✗ | ✓ | ✓ |
Modified from Schroll and Hamker, 2013.
Akinesia
In Equation (1), nothing prohibits “BG output” from being a larger magnitude than “descending motor command.” As such, the HDG model would predict that a negative movement speed could be possible despite it being physically meaningless. We propose that negative speed might be equivalent to the absence of movement (Fig. 1e). In the healthy state, BG output and descending motor commands are presumably calibrated such that mismatches in magnitude do not frequently occur. However, in the diseased state, the strong bias in striatal plasticity and/or maladaptive changes may be sufficient to disrupt such calibration and produce excessive BG output. As one can readily appreciate, this is not dissimilar to the explanation of akinesia in canonical models; however, in our account, akinesia is an extreme of the process that produces bradykinesia. This may be contrasted with a common converse conceptualization: bradykinesia is a consequence of a failure to robustly select among competing actions.50 Our account would also suggest that akinesia may be dissociable from bradykinesia. Preventing a large negative BG output could ameliorate akinesia but would also prevent BG activity from applying a gain to movement commands and thus would not ameliorate bradykinesia. We have recently observed that silencing excitatory input to all striatal neurons ameliorated akinesia without any reversal of bradykinesia in a hemiparkinsonian mouse model,53 consistent with this interpretation.
Rigidity
In the Learned Changes in Basal Ganglia Circuits Underlying Changes in Movement Vigor section, we noted that for BG to be involved in controlling movement vigor, it would need to have activity modulated according movement phase or an estimate of the state of the periphery (eg, limb).65 One key sensory feedback component for state estimation, in the context of the limbs, is the transcortical long‐latency stretch reflex.88 Changes in the long‐latency stretch reflex are the primary correlate of rigidity in PD patients.89, 90 This reflex pathway is thought to run from the spinal cord back through motor cortical structures, returning to the spinal cord presumably through corticospinal projections. We have proposed that BG modulate the gain of descending signals carried at least in part by corticospinal neurons. Thus, BG could also modulate the gain of long‐latency stretch reflex signals. We propose that aberrant control of the gain of this reflex pathway is a cause of the comorbidity of rigidity and bradykinesia often observed in PD patients.
Bradyphrenia
Bradyphrenia (slowness of thought) can be dissociated from bradykinesia in tasks with distinct cognitive and motor aspects.91, 92, 93 Like bradykinesia, bradyphrenia has been shown to be mediated by dopamine.94 We conjecture that bradyphrenia might be similarly explained by reduced gain on a cognitive process. For example, decisions are thought to be consistent with an integration process in which the integration of evidence constitutes a decision variable that governs observed behavioral choices.95 The greater the evidence, the faster the decision variable approaches a decision threshold. The decision to act can also be influenced by other processes (eg, urgency).96 Without taking a particular stance on the distinctions between these accounts, there is empirical support for both evidence accumulation and urgency being represented in BG.97, 98 One could envision the BG applying a gain to either or both of these cognitive variables.99 Reduced gain, akin to the reduced gain on movement vigor following dopamine depletion, would be evidenced by an increased time to render a decision, one feature of bradyphrenia.
Adaptive DBS
Perhaps the best evaluation of the HDG model, and one that may stand to impart the greatest clinical benefit, is application of these principles to design adaptive (ie, closed‐loop) deep brain stimulation (aDBS) for patients. DBS is a surgical intervention that employs selective stimulation of BG nuclei rather than the current standard using a constant stimulus. Despite the demonstration of the feasibility of aDBS, the specifics of how, what, and when to selectively stimulate are only now being identified.100 Although there have been several successful innovations in stimulation protocols, which focus primarily on responding to maladaptive neuronal events as they occur, many fail to provide additional benefit to the patient. Our discovery that bidirectional simulation‐induced changes in learned movement vigor implies that aDBS may be able to harness learning effects to improve the duration or magnitude of amelioration. Akin to how we used closed‐loop stimulation in the mouse, we envision stimulation triggered by physiological (eg, electromyography), behavioral (eg, peripheral accelerometers), or intracranial (eg, ECog) signals to improve treatment efficacy by harnessing the power to produce persistent changes in activity. For instance,99 based on our observations in the mouse, it is proposed that selective stimulation of the motor cortex during slow movements could induce greater movement velocity. This paradigm would effectively exploit the pathway‐specific alterations in PD to produce indirect pathway specificity and possibly overcome previous unsuccessful attempts at aDBS in motor cortex.100
Conclusion
We have described a unifying computational model of BG function with substantial explanatory power, specific predictions, and potential insights into the treatment of BG disease manifestations. Implicitly, we are also highlighting the potential impact of research involving model organisms to rapidly advance our understanding of the computational principles of BG function. Some types of basic discoveries are more readily possible in the mouse because of the exquisite control over cell types and circuit components. We propose that translation inspired by computational principles of circuits may circumvent some of the well‐known challenges of translating molecular or pharmacological interventions from rodent to human.
Author disclosures
J.T.D. is a Janelia Group Leader of the Howard Hughes Medical Institute. J.T.D. and E.A.Y. jointly edited the article and produced the figures; J.T.D. wrote the first draft.
Relevant conflicts of interest/financial disclosures: none.
Funding agencies: Howard Hughes Medical Institute (to J.T.D.).
References
- 1. Meyer A, Hierons R. A Note on Thomas Willis' Views on the Corpus Striatum and the Internal Capsule. J Neurol Sci 1964;1(6):547‐554. [DOI] [PubMed] [Google Scholar]
- 2. Jankovic J. Parkinson's disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry 2008;79(4):368‐376. [DOI] [PubMed] [Google Scholar]
- 3. Kish SJ, Shannak K, Hornykiewicz O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson's disease. Pathophysiologic and clinical implications. N Engl J Med 1988;318(14):876‐880. [DOI] [PubMed] [Google Scholar]
- 4. Dudman JT, Gerfen CR, The Basal Ganglia, in the Rat Nervous System. Paxinos G, ed. Amsterdam: Elsevier; 2015. [Google Scholar]
- 5. Blesa J, Phani S, Jackson‐Lewis V, Przedborski S. Classic and new animal models of Parkinson's disease. J Biomed Biotechnol 2012;2012:845618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weinrich M, Koch K, Garcia F, Angel RW. Axial versus distal motor impairment in Parkinson's disease. Neurology 1988;38(4):540‐545. [DOI] [PubMed] [Google Scholar]
- 7. Chong TT, Bonnelle V, Manohar S, Veromann KR, et al. Dopamine enhances willingness to exert effort for reward in Parkinson's disease. Cortex 2015;69:40‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vaillancourt DE, Prodoehl J, Verhagen Metman L, Bakay RA, Corcos DM. Effects of deep brain stimulation and medication on bradykinesia and muscle activation in Parkinson's disease. Brain 2004;127(Pt 3):491‐504. [DOI] [PubMed] [Google Scholar]
- 9. Baraduc P, Thobois S, Gan J, Brousolle E, Desmurget M. A common optimization principle for motor execution in healthy subjects and parkinsonian patients. J Neurosci 2013;33(2):665‐677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berardelli A, et al. Pathophysiology of bradykinesia in Parkinson's disease. Brain 2001;124(Pt 11):2131‐2146. [DOI] [PubMed] [Google Scholar]
- 11. Cagniard B, Peeler JA, Britt JP, McGehee DS, Marinelli M, Zhuang X. Dopamine scales performance in the absence of new learning. Neuron 2006;51(5):541‐547. [DOI] [PubMed] [Google Scholar]
- 12. Horak FB, Anderson ME. Influence of globus pallidus on arm movements in monkeys. I. Effects of kainic acid‐induced lesions. J Neurophysiol 1984;52(2):290‐304. [DOI] [PubMed] [Google Scholar]
- 13. Horak FB, Anderson ME. Influence of globus pallidus on arm movements in monkeys. II. Effects of stimulation. J Neurophysiol 1984;52(2):305‐322. [DOI] [PubMed] [Google Scholar]
- 14. Desmurget M, Turner RS. Motor sequences and the basal ganglia: kinematics, not habits. J Neurosci 2010;30(22):7685‐7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Panigrahi B, Martin KA, Li Y, et al. Dopamine Is Required for the Neural Representation and Control of Movement Vigor. Cell 2015;162(6):1418‐1430. [DOI] [PubMed] [Google Scholar]
- 16. Rueda‐Orozco PE, Robbe D. The striatum multiplexes contextual and kinematic information to constrain motor habits execution. Nat Neurosci 2015;18(3):453‐460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Costa RM, Cohen D, Nicolelis MAL Differential corticostriatal plasticity during fast and slow motor skill learning in mice. Curr Biol 2004:1124‐1134. [DOI] [PubMed] [Google Scholar]
- 18. Turner RS, Desmurget M. Basal ganglia contributions to motor control: a vigorous tutor. Curr Opin Neurobiol 2010;20(6):704‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dudman JT, Krakauer JW. The basal ganglia: from motor commands to the control of vigor. Curr Opin Neurobiol 2016;37:158‐166. [DOI] [PubMed] [Google Scholar]
- 20. Niv Y, Daw ND, Joel D, Dayan P. Tonic dopamine: opportunity costs and the control of response vigor. Psychopharmacology (Berl) 2007;191(3):507‐520. [DOI] [PubMed] [Google Scholar]
- 21. DeLong MR, Wichmann T. Circuits and circuit disorders of the basal ganglia. Arch Neurol 2007;64(1):20‐24. [DOI] [PubMed] [Google Scholar]
- 22. Schroll H, Hamker FH. Computational models of basal‐ganglia pathway functions: focus on functional neuroanatomy. Front Syst Neurosci 2013;7:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bargmann CI. How the new neuroscience will advance medicine. JAMA 2015;314(3):221‐222. [DOI] [PubMed] [Google Scholar]
- 24. Stephenson‐Jones M, Samuelsson L, Robertson B, Grillner S. Evolutionary conservation of the basal ganglia as a common vertebrate mechanism for action selection. Curr Biol 2011;21(13):1081‐1091. [DOI] [PubMed] [Google Scholar]
- 25. Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci 1992;15:285‐320. [DOI] [PubMed] [Google Scholar]
- 26. Kita T, Kita H. The subthalamic nucleus is one of multiple innervation sites for long‐range corticofugal axons: a single‐axon tracing study in the rat. J Neurosci 2012;32(17):5990‐5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Parent A, Mackey A, Smith Y, Boucher R. The output organization of the substantia nigra in primate as revealed by a retrograde double labeling method. Brain Res Bull 1983;10(4):529‐537. [DOI] [PubMed] [Google Scholar]
- 28. Deniau JM, Mailly P, Maurice N, Charpier S. The pars reticulata of the substantia nigra: a window to basal ganglia output. Prog Brain Res 2007;160:151‐172. [DOI] [PubMed] [Google Scholar]
- 29. Tervo DG, Hwang BY, Viswanathan S, et al. A Designer AAV Variant Permits Efficient Retrograde Access to Projection Neurons. Neuron 2016;92(2):372‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chan KY, Jang MJ, Yoo BB, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci 2017;20(8):1172‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deverman BE, Pravdo PL, Simpson BP, et al. Cre‐dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol 2016;34(2):204‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wall NR, Wickersham JR, Celin A, De La Parra, Callway EM. Monosynaptic circuit tracing in vivo through Cre‐dependent targeting and complementation of modified rabies virus. Proc Natl Acad Sci U S A 2010;107(50):21848‐21853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wickersham I.R, Lyon DC, Barnard DJ, et al. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron 2007;53(5):639‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zingg B, Chou XL, Zhang ZG, Liang F, Tao HW, Zhang LI. AAV‐Mediated Anterograde Transsynaptic Tagging: Mapping Corticocollicular Input‐Defined Neural Pathways for Defense Behaviors. Neuron 2017;93(1):33‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oh SW, Harris JA, Ng L, et al. A mesoscale connectome of the mouse brain. Nature 2014;508(7495):207‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Economo MN, Clack NG, Lavis LD, et al. A platform for brain‐wide imaging and reconstruction of individual neurons. Elife 2016;5:e10566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ragan T, Kadiri LR, Venkataraju KU, et al. Serial two‐photon tomography for automated ex vivo mouse brain imaging. Nat Methods 2012;9(3):255‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kita H, Kitai S. Intracellular study of rat globus pallidus neurons: membrane properties and responses to neostriatal, subthalamic and nigral stimulation, in Brain Res 1991;564(2)296‐305. [DOI] [PubMed] [Google Scholar]
- 39. Parent M, Parent A. Single‐axon tracing study of corticostriatal projections arising from primary motor cortex in primates. J Comp Neurol 2006;496(2):202‐213. [DOI] [PubMed] [Google Scholar]
- 40. Zheng T, Wilson CJ. Corticostriatal combinatorics: the implications of corticostriatal axonal arborizations. J Neurophysiol 2002;87(2):1007‐1017. [DOI] [PubMed] [Google Scholar]
- 41. Haber SN. Calzavara R. The cortico‐basal ganglia integrative network: the role of the thalamus, in Brain Res Bull 2009;69‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hopkins DA, Niessen LW. Substantia nigra projections to the reticular formation, superior colliculus and central gray in the rat, cat and monkey. Neurosci Lett 1976;2(5):253‐259. [DOI] [PubMed] [Google Scholar]
- 43. Hikosaka O. Basal ganglia mechanisms of reward‐oriented eye movement. Ann N Y Acad Sci 2007;1104:229‐249. [DOI] [PubMed] [Google Scholar]
- 44. Loeb GE, Tsianos GA. Major remaining gaps in models of sensorimotor systems. Front Comput Neurosci 2015;9:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Werner W, Dannenberg S, Hoffmann KP. Arm‐movement‐related neurons in the primate superior colliculus and underlying reticular formation: comparison of neuronal activity with EMGs of muscles of the shoulder, arm and trunk during reaching. Exp Brain Res, 1997;115(2):191‐205. [DOI] [PubMed] [Google Scholar]
- 46. Rubelowski JM, Menge M, Distler C, Rothermel M, Hoffmann KP. Connections of the superior colliculus to shoulder muscles of the rat: a dual tracing study. Front Neuroanat 2013;7:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Philipp R, Hoffmann KP. Arm movements induced by electrical microstimulation in the superior colliculus of the macaque monkey. J Neurosci 2014;34(9):3350‐3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gerfen C, Surmeier D. Dichotomous modulation of striatal direct and indirect pathway neurons by dopamine. Annu Rev Neurosci 2010;34(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gerfen CR, Engber TM, Mahan LC, et al. D1 and D2 dopamine receptor‐regulated gene expression of striatonigral and striatopallidal neurons. Science 1990;250(4986):1429‐1432. [DOI] [PubMed] [Google Scholar]
- 50. Mink JW. The basal ganglia: focused selection and inhibition of competing motor programs. Progress in Neurobiology 1996;50(4):381‐425. [DOI] [PubMed] [Google Scholar]
- 51. Kravitz AV, Freeze BS, Parker PR, et al. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 2010;466(7306):622‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci 1989;12(10):366‐375. [DOI] [PubMed] [Google Scholar]
- 53. Shields, B.C. , et al., Deconstructing behavioral neuropharmacology with cellular specificity. Science, 2017. 356(6333). [DOI] [PubMed] [Google Scholar]
- 54. Cui G, Jun SB, Pham MD, Vogel SS, Lovinger DM, Costa RM. Concurrent activation of striatal direct and indirect pathways during action initiation. Nature 2013;494(7436):238‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Isomura Y, Takekawa T, Harukuni R, et al. Reward‐modulated motor information in identified striatum neurons. J Neurosci 2013;33(25):10209‐10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yttri EA, Dudman JT. Opponent and bidirectional selection of movement parameters in the basal ganglia. Nature 2016;533(7603):402‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Meng C, et al. Parallel striatal pathways collaboratively control the dynamics and fate of actions. Washington, DC: Society for Neuroscience; 2017. [Google Scholar]
- 58. Mazzoni P, Hristova A, Krakauer JW. Why don't we move faster? Parkinson's disease, movement vigor, and implicit motivation. J Neurosci 2007;27(27):7105‐7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Manohar SG, Chong TT, Apps MA, Batla M, et al. Reward Pays the Cost of Noise Reduction in Motor and Cognitive Control. Curr Biol 2015;25(13):1707‐1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Salamone JD, Correa M, Nunes EJ, Randall PA, Pardo M. The behavioral pharmacology of effort‐related choice behavior: dopamine, adenosine and beyond. J Exp Anal Behav 2012;97(1):125‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gerfen CR, Paletzki R, Heintz N. GENSAT BAC cre‐recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron, 2013;80(6):1368‐1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Osborne JE, Dudman JT. RIVETS: a mechanical system for in vivo and in vitro electrophysiology and imaging. PLoS One 2014;9(2):e89007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brown J, Martin KA, Dudman JT. Behavioral evidence for feedback gain control by the inhibitory microcircuit of the substantia nigra. bioRxiv 2016. [Google Scholar]
- 64. Zhai S, Tanimura A, Graves SM, Shen W, Sumeier DJ. Striatal synapses, circuits, and Parkinson's disease. Curr Opin Neurobiol 2017;48:9‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Novak KE, Miller LE, Houk JC. The use of overlapping submovements in the control of rapid hand movements. Exp Brain Res 2002;144(3):351‐364. [DOI] [PubMed] [Google Scholar]
- 66. Salimi‐Badr A, Ebadzadeh MM, Darlot C. A possible correlation between the basal ganglia motor function and the inverse kinematics calculation. J Comput Neurosci 2017;43(3):295‐318. [DOI] [PubMed] [Google Scholar]
- 67. Bar‐Gad I, Morris G, Bergman H. Information processing, dimensionality reduction and reinforcement learning in the basal ganglia. Prog Neurobiol 2003;71(6):439‐473. [DOI] [PubMed] [Google Scholar]
- 68. Anderson ME, and Horak FB. Influence of the globus pallidus on arm movements in monkeys. III. Timing of movement‐related information. J Neurophysiol 1985;54(2):433‐448. [DOI] [PubMed] [Google Scholar]
- 69. Barter JW, Li S, Sukharnikova T, Rossi MA, Bartholomew RA, Yin HH. Basal ganglia outputs map instantaneous position coordinates during behavior. J Neurosci 2015;35(6):2703‐2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fieblinger T, Graves SM, Sebel LE, et al. Cell type‐specific plasticity of striatal projection neurons in parkinsonism and L‐DOPA‐induced dyskinesia. Nat Commun 2014;5:5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Parker JG, et al. Diametric neural ensemble deficits in parkinsonian and dyskinetic states. Nature, 2017. [in press]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Williams ZM, Eskandar EN. Selective enhancement of associative learning by microstimulation of the anterior caudate. Nat Neurosci 2006;9(4):562‐568. [DOI] [PubMed] [Google Scholar]
- 73. Kravitz AV, Tye LD, Kreitzer AC. Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci 2012;15(6):816‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Salzman CD, Britten KH, Newsome WT. Cortical microstimulation influences perceptual judgements of motion direction. Nature 1990;346(6280):174‐177. [DOI] [PubMed] [Google Scholar]
- 75. Yttri EA, Dudman JT. Neural correlates of bidirectional kinetic control and reinforcement in the basal ganglia. In Society for Neuroscience. 2015.
- 76. Georgopoulos AP, Schwartz AB, Kettner RE. Neuronal population coding of movement direction. Science 1986;233(4771):1416‐1419. [DOI] [PubMed] [Google Scholar]
- 77. Kutz DF, Dannenberg S, Werner W, Hoffmann KP. Population coding of arm‐movement‐related neurons in and below the superior colliculus of Macaca mulatta. Biol Cybern 1997;76(5):331‐337. [DOI] [PubMed] [Google Scholar]
- 78. Lee C, Rohrer WH, and Sparks DL. Population coding of saccadic eye movements by neurons in the superior colliculus. Nature 1988;332(6162):357‐360. [DOI] [PubMed] [Google Scholar]
- 79. du Lac S, Knudsen EI. Neural maps of head movement vector and speed in the optic tectum of the barn owl. J Neurophysiol 1990;63(1):131‐146. [DOI] [PubMed] [Google Scholar]
- 80. Sparks DL, Lee C, Rohrer WH. Population coding of the direction, amplitude, and velocity of saccadic eye movements by neurons in the superior colliculus. Cold Spring Harb Symp Quant Biol 1990;55:805‐811. [DOI] [PubMed] [Google Scholar]
- 81. Georgopoulos AP, Carpenter AF. Coding of movements in the motor cortex. Curr Opin Neurobiol 2015;33:34‐39. [DOI] [PubMed] [Google Scholar]
- 82. Brown J, Pan WX, Dudman JT. The inhibitory microcircuit of the substantia nigra provides feedback gain control of the basal ganglia output. Elife 2014;3:e02397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tepper JM, Abercrombie ED, Bolam JP. GABA and the basal ganglia: from molecules to systems?, in Progress in Brain Research. Amsterdam, Netherlands: Elsevier; 2007:339. [Google Scholar]
- 84. Barbera G, Liang B, Zhang L, et al. Spatially Compact Neural Clusters in the Dorsal Striatum Encode Locomotion Relevant Information. Neuron 2016;92(1):202‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Taouali W, Rueda‐Orozco PE, Robbe D. A minority‐ruled population coding of kinematics in the striatum. bioRxiv 2017. [Google Scholar]
- 86. Pawlak V, Kerr JN. Dopamine receptor activation is required for corticostriatal spike‐timing‐dependent plasticity. J Neurosci, 2008;28(10):2435‐2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science 2008;321(5890):848‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pruszynski JA, Scott SH. Optimal feedback control and the long‐latency stretch response. Exp Brain Res 2012;218(3):341‐359. [DOI] [PubMed] [Google Scholar]
- 89. Rothwell JC, Obeso JA, Traub MM, Marsden CD. The behaviour of the long‐latency stretch reflex in patients with Parkinson's disease. J Neurol Neurosurg Psychiatry 1983;46(1):35‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Berardelli A, Sabra AF, Hallett M. Physiological mechanisms of rigidity in Parkinson's disease. J Neurol Neurosurg Psychiatry 1983;46(1):45‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Vlagsma TT, Koerts J, Tucha O, et al. Mental slowness in patients with Parkinson's disease: associations with cognitive functions? J Clin Exp Neuropsychol 2016;38(8):844‐852. [DOI] [PubMed] [Google Scholar]
- 92. Brown RG, Marsden CD. Dual task performance and processing resources in normal subjects and patients with Parkinson's disease. Brain 1991;114(Pt 1A):215‐231. [PubMed] [Google Scholar]
- 93. Oliveira RM, Gurd JM, Nixon P, Marshall JC, Passingham RE. Hypometria in Parkinson's disease: automatic versus controlled processing. Mov Disord 1998;13(3):422‐427. [DOI] [PubMed] [Google Scholar]
- 94. Rogers D, Lees AJ, Smith F, Trimble M, Stern GM. Bradyphrenia in Parkinson's disease and psychomotor retardation in depressive illness. An experimental study. Brain 1987;110(Pt 3):761‐776. [DOI] [PubMed] [Google Scholar]
- 95. Ding L, Gold JI. The basal ganglia's contributions to perceptual decision making. Neuron 2013;79(4):640‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Thura D, Beauregrd‐Racine J, Fradet CW, Cisek P. Decision making by urgency gating: theory and experimental support. J Neurophysiol 2012;108(11):2912‐2930. [DOI] [PubMed] [Google Scholar]
- 97. Ding L, Gold JI. Caudate encodes multiple computations for perceptual decisions. J Neurosci 2010;30(47):15747‐15759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Thura D, Cisek P. The basal ganglia do not select reach targets but control the urgency of commitment. Neuron 2017;95(5):1160‐1170.e5. [DOI] [PubMed] [Google Scholar]
- 99. Dunovan K, Verstynen T. Believer‐skeptic meets actor‐critic: rethinking the role of basal ganglia pathways during decision‐making and reinforcement learning. Front Neurosci 2016;10:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Malekmohammadi M, Herron J, Velisar A, et al. Kinematic adaptive deep brain stimulation for resting tremor in Parkinson's disease. Mov Disord 2016;31(3):426‐428. [DOI] [PubMed] [Google Scholar]
- 101. Alexander GE, DeLong MR, Strick PL. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Annu Rev Neurosci 1986;9:357‐381. [DOI] [PubMed] [Google Scholar]
- 102. Robertson B, Suryarayana SM, Saitoh K, et al. The lamprey blueprint of the mammalian nervous system. Prog Brain Res 2014;212:337‐349. [DOI] [PubMed] [Google Scholar]
- 103. Ekstrand MI, Terzioglu M, Gelter D, et al. Progressive parkinsonism in mice with respiratory‐chain‐deficient dopamine neurons. Proc Natl Acad Sci U S A 2007;104(4):1325‐1330. [DOI] [PMC free article] [PubMed] [Google Scholar]