

Abstract
By introducing a range of 2H labels into pyridine and the para‐substituted agents, methyl isonicotinate and isonicotinamide, we significantly improve their NMR detectability in conjunction with the signal amplification by reversible exchange process. We describe how the rates of T 1 relaxation for the remaining 1H nuclei are increased and show how this leads to a concomitant increase in the level of 1H and 13C hyperpolarization that can ultimately be detected.
Keywords: hyperpolarisation, NMR, parahydrogen, SABRE
1. INTRODUCTION
Hyperpolarization techniques overcome the inherent insensitivity of nuclear magnetic resonance (NMR) and magnetic resonance imaging by manipulating the spin distribution across Zeeman‐split energy levels away from equilibrium where a typical population difference used to produce a magnetic resonance (MR) signal can be less than 1 in 100,000. The employment of hyperpolarized states can therefore result in very substantial MR signal gains. This process can be achieved through a number of methods, the most common of which are dynamic nuclear polarization1, 2, 3 and parahydrogen induced polarization.4, 5, 6 Signal amplification by reversible exchange (SABRE)7 has recently emerged as a powerful alternative that can rapidly and repeatedly hyperpolarize a substrate through the interaction of parahydrogen with an iridium catalyst. In SABRE, parahydrogen (p‐H2) adds to a precatalyst, such as [IrCl(COD)(IMes)] (1, where IMes = 1,3‐bis(2,4,6‐trimethylphenyl)imidazol‐2‐ylidene and COD = cis,cis‐cycloocta‐1,5‐diene), in the presence of a coordinating substrate (sub) to form an iridium hydride complex such as [Ir(H)2(IMes)(sub)3]Cl (2)8, 9 (see Scheme 1). Polarization from the parahydrogen derived hydride ligands is then transferred into the bound substrate through the J‐coupling network that exists in this complex.10 Throughout the process, parahydrogen and the substrate, which are located in the bulk solution, are in reversible exchange with the corresponding ligands that are bound to the iridium complex. This results in a build‐up of hyperpolarized substrate in bulk solution and hence the signal gain grows with parahydrogen exposure time, although a limit is reached because relaxation acts to destroy the hyperpolarization that has been created through SABRE.11 As the rates of ligand exchange are relatively fast, substantial hyperpolarization levels can be produced in just a few seconds. SABRE has been demonstrated for a wide range of substrates which include pyridine and its derivatives,9, 12, 13, 14, 15, 16 pyrazines,16, 17 imidazoles,18, 19 pyrazoles,20, 21 pyridazines,22, 23, 24 pyrimidines,25 amino acids,26 diazirines,27 and acetonitrile.28 It hyperpolarizes not only their 1H nuclei but the wide array of heteronuclei that they contain.19, 24, 25, 29, 30
Scheme 1.
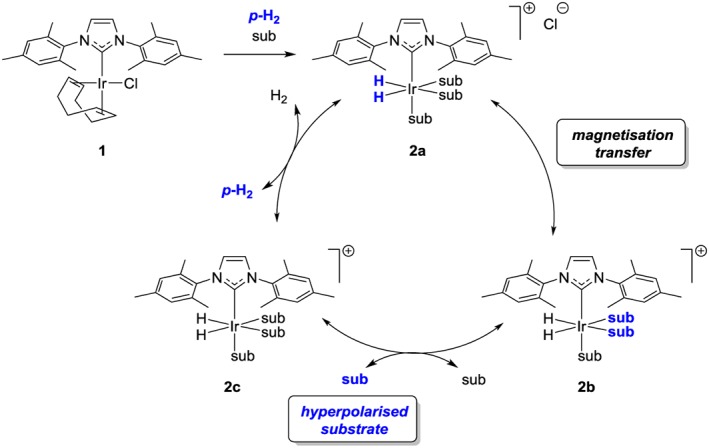
Schematic representation of the signal amplification by reversible exchange (SABRE) process wherein the substrate (sub) achieves hyperpolarization through interactions within the metal complex 2a
It has recently been shown that the selective, partial 2H labelling of substrates such as pyridine,13, 31 nicotinamide and methyl nicotinate greatly extends the T 1 relaxation times of the remaining 1H nuclei through inhibition of scalar relaxation processes.14, 32, 33 In addition, through 2H labelling of the IMes ligand of the iridium catalyst, the relaxation times of the bound substrate increase which further results in a dramatic improvement in the 1H hyperpolarization levels that can be achieved through SABRE. In fact, they exceed 50% polarization when 5 bar of parahydrogen is used in conjunction with a co‐ligand.14 In this current work, we seek to report further developments that employ this 2H labelling strategy by applying it to the related compounds pyridine, methyl isonicotinate, and isonicotinamide. We seek to ascertain its effect on the efficacy of the resulting 1H and 13C hyperpolarization levels that can be achieved through SABRE. For synthetic procedures and characterisation data please see Supporting information.
2. RESULTS AND DISCUSSION
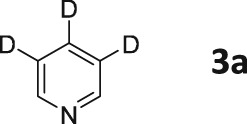
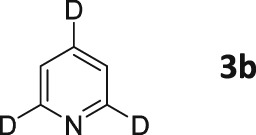
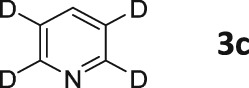
Pyridine (3) was one of the first compounds to be hyperpolarized by the SABRE method.7, 12, 15 Because of its relatively simple structure, we recognized that there was an opportunity to investigate the effect of selective 2H labelling on the level of hyperpolarization. A selection of partially deuterated pyridines shown in Figure 1 were therefore synthesized.34, 35 This approach gave us access to three isotopologues of pyridine, 3a, 3b, and 3c, in which 1H nuclei are retained at the ortho, meta, and para positions of the ring, respectively.
Figure 1.
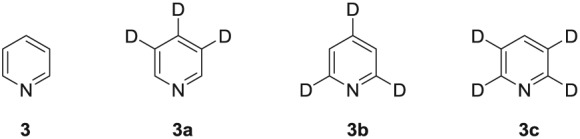
2H‐labelled pyridine isotopologues used in this study
When protio‐pyridine 3 (20 mM, 4 eq.) is mixed with [IrCl(COD)IMes] (5 mM) and parahydrogen in ethanol‐d 6 and shaken at 65 gauss for 10 s followed by immediate transport into the spectrometer for detection, the signal for Ha in Figure 2 appears with an intensity that is approximately 2,400 times larger than that observed under Boltzmann conditions in the corresponding thermally equilibrated control measurement. In contrast, the corresponding signals for Hb and Hc are 1,120 and 1,258 times larger than those of the corresponding reference signals (at 9.4 T). Figure 2 (left) illustrates a typical measurement. The new signal intensity reflects the detection of 3.9% Ha polarization, created after just 10 s of exposure to 3 bar of parahydrogen. Furthermore, we note that if 20 s were needed to repeat the control measurement which uses thermally equilibrated polarization levels, it would take 1,333 days of data averaging to reproduce the hyperpolarized signal intensity. It is for this reason that hyperpolarization reflects a method that could transform clinical diagnosis as it may facilitate the facile detection of markers that probe underlying physiology.36
Figure 2.
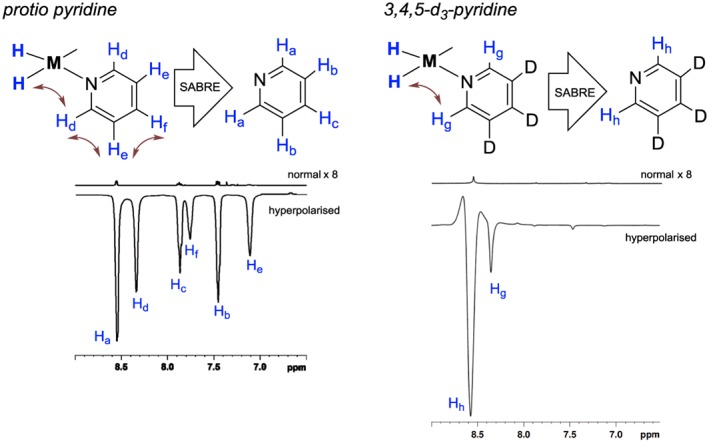
Left: two single scan 1H NMR spectra that illustrate pyridine (3) signals (attributed according to inset scheme) that result after SABRE (hyperpolarised) and when the polarization level matches that for thermodynamic equilibrium (normal). Right: two similar single scan 1H NMR spectra employing 3a. The normal traces are shown with a ×8 vertical expansion relative to the hyperpolarized trace
When the agent 3,4,5‐d 3‐pyridine (3a) was employed under identical conditions to those described for 3, the signal enhancement seen for the remaining ortho protons increases to 4,166 (6.7% polarization), shown in Table 1. However, upon moving to 2,4,6‐d 3‐pyridine (3b), the remaining meta protons exhibit a smaller gain associated with an enhancement of just 541 (0.9% polarization). Finally, 2,3,5,6‐d 4‐pyridine (3c) results in very little signal gain with its remaining proton showing a 163‐fold enhancement (0.51% polarization). These values are the average of three distinct measurements and are hence reproducible.
Table 1.
T 1 relaxation times and polarization levels achieved for pyridines 3 through SABRE in the specified solvent. Conditions: 20 mM substrate, 5 mM [IrCl(COD)IMes], activated with 3 bar p‐H2. o = ortho, m = meta, and p = para
| Substrate | Methanol‐d 4 | Ethanol‐d 6 | ||||
|---|---|---|---|---|---|---|
| Site T 1(no cat.)/s | Site T 1(with cat.)/s | Polarization level (%) | Site T 1(no cat.) / s | Site T 1(with cat.) /s | Polarization level (%) | |
(1).
|
o—28.1 m—26.2 p—33.8 |
o—1.5 m—5.2 p—5.5 |
o—4.3 ± 0.3 m—3.4 ± 0.1 p—5.4 ± 0.1 |
o—20.7 m—18.2 p—26.7 |
o—3.2 m—4.7 p—5.3 |
o—3.9 ± 0.2 m—1.8 ± 0.1 p—4.0 ± 0.1 |
(2).
|
o—51.2 | o—5.2 | o—5.8 ± 0.4 | o—72.4 | o—4.2 | o—6.7 ± 0.4 |
(3).
|
m—113.2 | m—46.7 | m—0.6 ± 0.1 | m—72.6 | m—13.8 | m—0.9 ± 0.1 |
(4).
|
p—84.7 | p—67.0 | p–0.54 ± 0.12 | p—57.5 | p—25.0 | p—0.51 ± 0.07 |
The lower polarization levels seen for 3b and 3c can be rationalized as the result of the fact that their protons are magnetically isolated from those of the hydride ligands in the iridium catalyst. Hence, smaller J‐couplings are involved in the polarization transfer step and less effective SABRE transfer results.37 By comparison, 3a features two ortho 1H nuclei that couple more strongly to the hydride ligands in the SABRE‐catalyst and they are therefore able to more readily receive magnetization. An analogous trend was found when methanol‐d 4 was used as the solvent, and in this case, a polarization level of 5.8% could be achieved with 3a.
The reduction in hydride‐substrate J‐coupling is somewhat offset by relaxation changes. At 400 MHz, in degassed ethanol‐d 6, protio‐pyridine (3) was found to exhibit T 1 relaxation times of between 18.2 and 26.7 s (Table 1). Its isotopologue 3a, however, has relaxation times for the two ortho protons of 72.4 s, whereas in 3b, the meta protons exhibit a 72.6 s value and in 3c, the para proton has a T1 of 57.5 s. The values determined in the 2H‐labelled isotopologues therefore reflect a dramatic improvement on those of the unlabelled 3. We note, however, that under the catalytic conditions used in SABRE, interactions with the iridium catalyst act to reduce the observed T 1 values. This is because the free and bound forms are in equilibrium and the observed value therefore reflects a weighted average of the two. For unlabelled pyridine, the new values lie between 3.2 and 5.3 s, whereas for 3a, 3b, and 3c, they are 4.2, 13.8, and 25.0 s, respectively. As percentages, the reductions caused by the presence of the catalyst in solution are therefore 94%, 81%, and 56%, respectively. This serves to demonstrate that the effect is most strongly manifest in the ortho protons, which exhibit the strongest spin‐spin coupling to the hydride ligands when bound to the iridium. Although 3b exhibits a better T 1 value for the meta position, it fails to receive strong magnetization through SABRE, a feature that is further demonstrated for the para proton of 3c. Again, analogous relaxation and polarization trends were observed in methanol‐d 4 solution. However, relaxation times for 3b and 3c were significantly extended in methanol‐d 4, compared to ethanol‐d 6.
Hence, for optimal SABRE activity we can conclude that it is desirable to locate a proton next to the catalyst binding site, where J HH is maximized. However, we tension this need with the fact that such an arrangement will also reduce the effective lifetime of the polarization under catalytic conditions. Clearly, the interplay between the T 1 of the proton site and the efficacy of polarization transfer must therefore be carefully considered when designing an optimized agent.
A series of hyperpolarized 13C measurements were then conducted using 3,4,5‐d 3‐pyridine (3a) in the presence of 1 for a 20::1 loading after SABRE at approximately 0.5 G. The resulting fully coupled single‐scan 13C hyperpolarized NMR spectrum is shown in Figure 3 alongside its thermally equilibrated 13C counterpart, which is a 2,048 scan average. A significant increase in the 13C resonances' signal‐to‐noise ratios is clearly observed in the hyperpolarized spectrum. Furthermore, the ortho peak (148 ppm) is observed as an antiphase doublet of doublets, whereas the meta (124 ppm) and para (137 ppm) peaks are split into antiphase triplets of doublets. This is consistent with the creation of I z S z terms, where the antiphase component is associated with the small, indirect J HC coupling. The meta and para signals are associated with the detection of a 13CD signal for the meta and para sites, which accounts for the in‐phase 1:1:1 splitting.
Figure 3.
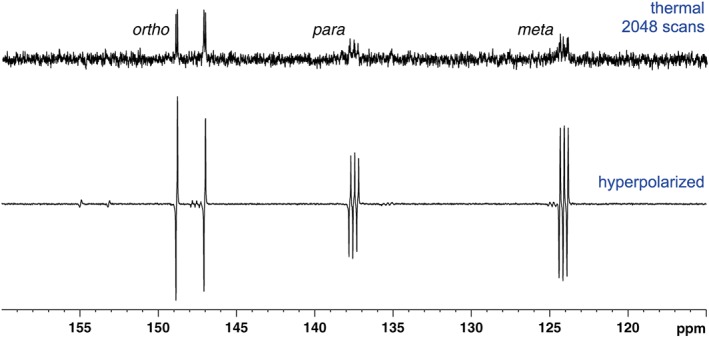
Top: Thermal 13C NMR spectrum of 3a (100 mM) and SABRE catalyst (5 mM) in methanol‐d 4 after 2,048 scans. Bottom: single‐scan hyperpolarized 13C NMR spectrum
When a similar experiment was recorded with concurrent 1H decoupling a much weaker signal was observed (Figure 4). This confirms that the dominant terms that are created under SABRE at 60 G are indeed I z S z based rather than S z. We expect that the levels of signal gain that are observed may be improved through the incorporation of a 15N label, which has recently been shown to reduce 13C polarization transfer losses due to quadrupolar relaxation.38
Figure 4.
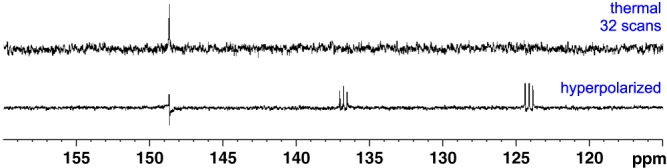
Top: thermal 13C{1H} NMR spectrum of 3a (100 mM) and SABRE catalyst (5 mM) in methanol‐d 4 over 32 scans. Bottom: single‐scan hyperpolarized 13C{1H} NMR spectrum
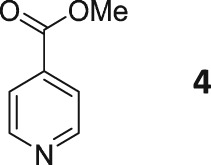
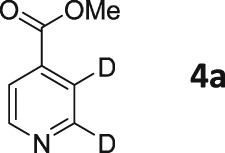
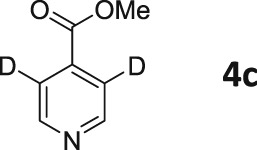
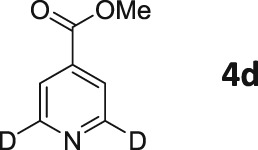
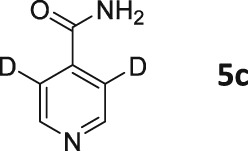
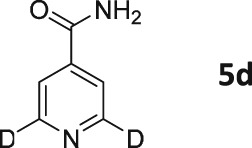
Following these observations, we turned our attention to the para‐substituted pyridine derivatives, methyl isonicotinate (4) and isonicotinamide (5). A range of doubly deuterated isotopologues of these compounds were synthesized according to Scheme 2, and the results of the related NMR studies are shown in Table 2. These substrates provide a range of 1H spin systems, and we expected them to exhibit markedly different hyperpolarization characteristics. This reflects the fact that 4a and 5a possess pairs of ortho and meta 1H nuclei that exhibit a mutual three‐bond coupling, in contrast, 4b and 5b contain two weakly coupled ortho and meta nuclei (5‐bond coupling), whereas 4c, 5c, 4d, and 5d contain pairs of equivalent ortho and meta nuclei, respectively.
Scheme 2.
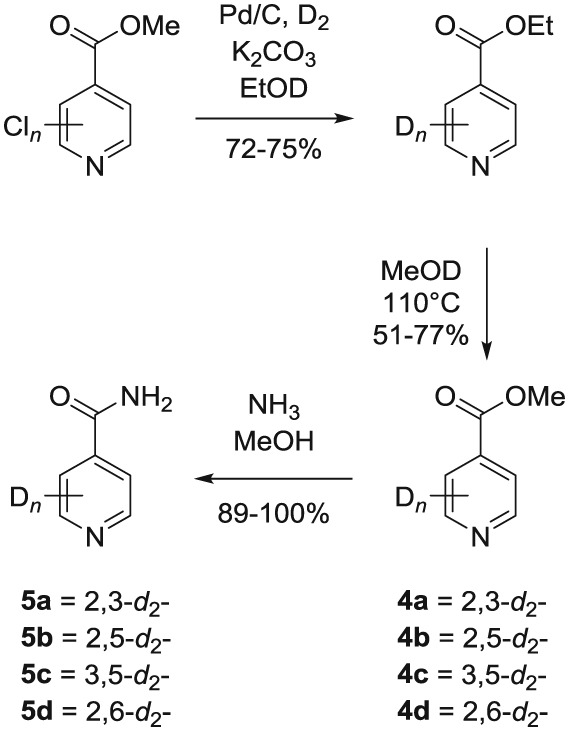
Synthetic route to the doubly 2H‐labelled methyl isonicotinates 4 and isonicotinamides 5
Table 2.
T 1 relaxation times (s) and polarization levels found for the methyl isonicotinates 4 and the isonicotinamides 5. Conditions: 20 mM substrate, 5 mM [IrCl(COD)IMes], activated with 3 bar p‐H2. o = ortho and m = meta
| Substrate | Methanol‐d 4 | ||
|---|---|---|---|
| Site T 1(no cat.)/s | Site T 1(with cat.)/s | Polarization level (%) | |
(5).
|
o—14.2 m—14.1 |
o—1.9 m—3.9 |
o—4.8 ± 0.1 m—1.0 ± 0.1 |
(6).
|
o—9.6 m—9.6 |
o—2.9 m—5.8 |
o—2.2 ± 0.1 m—1.4 ± 0.1 |
(7).
|
o—61.9 m—61.2 |
o—3.5 m—21.4 |
o—5.4 ± 0.4 m—5.6 ± 0.3 |
(8).
|
o—70.7 | o—4.1 | o—7.4 ± 0.3 |
(9).
|
m—72.8 | m—16.1 | m—7.4 ± 0.4 |
(10).
|
o—8.2 m—8.3 |
o—1.7 m—3.5 |
o—2.6 ± 0.2 m—0.2 ± 0.02 |
(11).
|
o—7.4 m—8.3 |
o—2.3 m—4.1 |
o—3.5 ± 0.3 m—0.4 ± 0.1 |
(12).
|
o—18.2 m—42.7 |
o—4.2 m—16.0 |
o—11.0 ± 0.5 m—11.1 ± 0.5 |
(13).
|
o—23.4 | o—2.9 | o—5.8 ± 0.7 |
(14).
|
m—47.2 | m—8.3 | m—4.2 ± 0.1 |
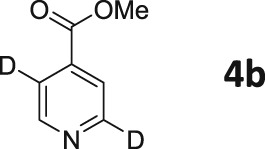
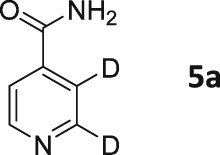
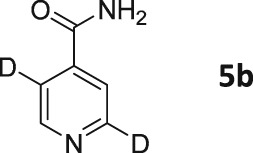
For these ester and amide substrates, deuteration at the 2‐ and 3‐positions (4a and 5a) resulted in limited changes in their relaxation times relative to their 1H counterpart and somewhat similar polarization levels as a result of SABRE were observed (Table 2). These results indicate that relaxation within these molecules is driven by interactions between the adjacent 1H nuclei. Analogues b–d, with more isolated 1H spin systems, might therefore be predicted to have higher T 1 values. In support of this, we found that deuteration at the 2‐ and 5‐positions (4b and 5b) does indeed result in significantly improved relaxation times and SABRE polarization levels (Figure 5). Now, the T 1 values are over 60 s for methyl 2,5‐d 2‐isonicotinate 4b, which compare to 14 s for protio 4. In the case of isonicotinamide 5b, the SABRE polarization levels improve from the original 2.6% and 0.2% levels for the ortho and meta positions of 5 (in methanol‐d 4), respectively, to 11.0% and 11.1%, respectively. In this case, the extension of the T 1 values determined for the meta proton in the presence of the SABRE catalyst greatly assists in increasing the observed polarization level of the proton at this position.
Figure 5.
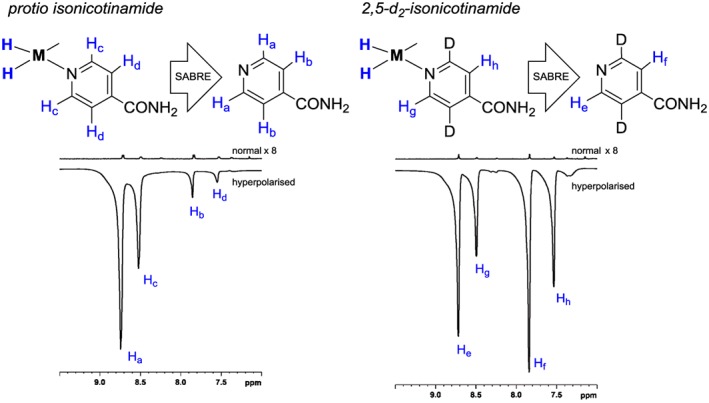
Left: single scan 1H NMR spectra for hyperpolarized and fully relaxed isonicotinamide 5 at 400 MHz. Right: corresponding single scan 1H NMR spectra of 5b. The normal traces are shown with a ×8 vertical expansion relative to the hyperpolarized traces
In general, compounds with only ortho 1H nuclei (4c and 5c) gave slightly improved levels of polarization and T 1 values that compare to those of the parent, but still suffer from short relaxation times in the presence of the SABRE catalyst. This is consistent with the results outlined earlier for 3a. Compounds 4d and 5d, with only meta 1H nuclei now give higher T 1 values, but as expected, the improvement in achieved polarization level is minimal, in accordance with the weaker J‐coupling that connects them to hydride ligands in the catalyst.
These trends confirm our earlier conclusions that substrates containing a 1H nucleus at the ortho position result in efficient polarization transfer and that 1H nuclei that are isolated from each other, and the hydride ligands of the metal catalyst, lead to increased relaxation times under SABRE. These changes contribute cooperatively to produce a strong, long‐lived hyperpolarized signal.
Related hyperpolarized 13C NMR experiments on the labelled isonicotinamides (5a–5d) showed that superior signal enhancements could be achieved with these substrates (Figure 6). With a 20‐fold concentration of substrate to catalyst and polarization transfer at approximately 0.5 gauss, compound 5a showed strong SABRE responses for the quaternary carbons, including those adjacent to the 2H labels. The remaining CH positions showed a much lower, but detectable signal enhancement, an effect which is predicted to be due to an increased rate of relaxation. Compound 5b produced a similar outcome; the higher levels of 1H polarization enabling increased magnetization transfer to be relayed into the most distant quaternary carbon. In the case of compound 5c, a clear SABRE signal was observed for all carbons on the aromatic ring, notably including that of the CH at the ortho position. The carbonyl carbon showed significantly smaller polarization, as only a weak 4 J CH coupling to the ortho proton is possible from that site for transfer. Analogue 5d gave very little 13C polarization, with only the carbonyl position being visible. It should be noted that in all cases, the dominant peaks appear in antiphase due to the observation of I z S z derived states.
Figure 6.
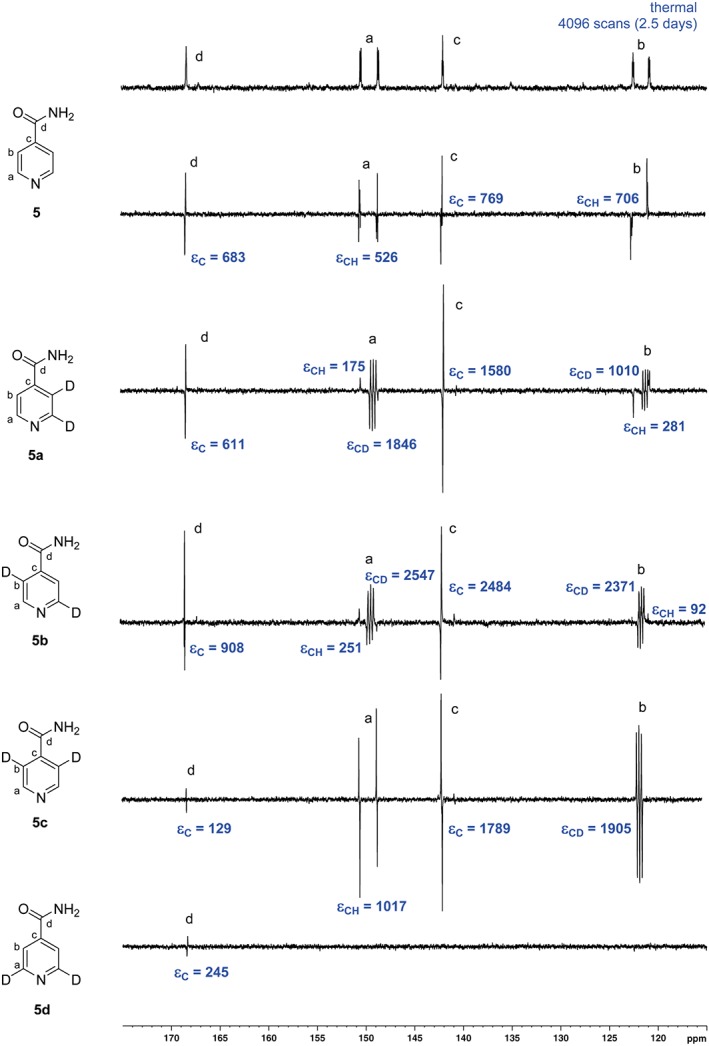
Series of hyperpolarized single‐scan 13C NMR spectra of 5–5d (100 mM) and [IrCl(COD)(IMes)] (5 mM) in methanol‐d 4 under p‐H2 after transfer at 0.5 G. Thermally polarized reference 13C NMR spectrum (top), acquired over 4,096 scans. ε = enhancement factor, fold gain
3. CONCLUSIONS
An investigation has been made into the effect of selectively incorporating 2H labels into a number of pyridine‐based substrates and the subsequent effect this change had on the observed hyperpolarization levels achieved through the SABRE process. We completed studies on three isotopologues of pyridine (3), four isotopologues of methyl isonicotinate (4), and four isotopologues of isonicotinamide (5). We find that harnessing hyperpolarization sites adjacent to the nitrogen centre of pyridine, which binds to the SABRE catalyst metal centre, is crucial for efficient polarization transfer through its stronger 4 J HH coupling to the parahydrogen‐derived metal hydride ligands. Furthermore, we confirm that the presence of spin‐isolated hyperpolarization sites in these agents both increases signal lifetime through reduced relaxation and allows the detection of strongly hyperpolarized 1H responses. These changes also facilitate the detection of strong 13C NMR signals, most notably for the corresponding quaternary and CD positions. These signals typically appear with anti‐phase character due their origin in a heteronuclear longitudinal two spin order term (1H─13C) involving an indirect coupling. Agent 5b produced the strongest carbonyl signal as a consequence of a 3 J HC coupling, which must lead to limited internal peak cancelation due to its small value. The 13CH signals of 5c are also dramatically stronger than those of 5a, 5b, and 5d in accordance with a predicted long relaxation time. The spin isolation of the C─13CONH2 groups in 5c and 5d though acts to reduce its detectability, although the signal for C─13CONH2 is strongly visible in 5a and 5b. Hence, we conclude that a 2H‐labelling strategy can be used to control not only 1H signal gains but also those of 13C. We expect that this strategy will enable improvement in polarization in molecules containing other heteronuclei, such as 15N, and work is ongoing to achieve this goal. Additionally, a detailed investigation into the mechanism of polarization transfer in molecules containing 2H nuclei would be of interest.
Supporting information
Data S1 Supporting Information
ACKNOWLEDGEMENT
We thank the Wellcome Trust (092506 and 098335) for funding.
Norcott P, Burns MJ, Rayner PJ, Mewis RE, Duckett SB. Using 2H labelling to improve the NMR detectability of pyridine and its derivatives by SABRE. Magn Reson Chem. 2018;56:663–671. https://doi.org/10.1002/mrc.4703
REFERENCES
- 1. Ardenkjær‐Larsen J. H., Fridlund B., Gram A., Hansson G., Hansson L., Lerche M. H., Servin R., Thaning M., Golman K., Increase in signal‐to‐noise ratio of > 10,000 times in liquid‐state NMR, Proc. Natl. Acad. Sci. U. S. A. 2003, 100(18), 10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Keshari K. R., Wilson D. M., Chemistry and biochemistry of 13C hyperpolarized magnetic resonance using dynamic nuclear polarization, Chem. Soc. Rev. 2014, 43(5), 1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ardenkjaer‐Larsen J. H., On the present and future of dissolution‐DNP, J. Magn. Reson. 2016, 264(Supplement C), 3. [DOI] [PubMed] [Google Scholar]
- 4. Bowers C. R., Weitekamp D. P., Transformation of symmetrization order to nuclear‐spin magnetization by chemical reaction and nuclear magnetic resonance, Phys. Rev. Lett. 1986, 57(21), 2645. [DOI] [PubMed] [Google Scholar]
- 5. Bowers C. R., Weitekamp D. P., Parahydrogen and synthesis allow dramatically enhanced nuclear alignment, J. Am. Chem. Soc. 1987, 109(18), 5541. [Google Scholar]
- 6. Golman K., Axelsson O., Jóhannesson H., Månsson S., Olofsson C., Petersson J. S., Parahydrogen‐induced polarization in imaging: Subsecond 13C angiography, Magn. Reson. Med. 2001, 46(1), 1. [DOI] [PubMed] [Google Scholar]
- 7. Adams R. W., Aguilar J. A., Atkinson K. D., Cowley M. J., Elliott P. I. P., Duckett S. B., Green G. G. R., Khazal I. G., López‐Serrano J., Williamson D. C., Reversible interactions with para‐hydrogen enhance NMR sensitivity by polarization transfer, Science 2009, 323(5922), 1708. [DOI] [PubMed] [Google Scholar]
- 8. Cowley M. J., Adams R. W., Atkinson K. D., Cockett M. C. R., Duckett S. B., Green G. G. R., Lohman J. A. B., Kerssebaum R., Kilgour D., Mewis R. E., Iridium N‐heterocyclic carbene complexes as efficient catalysts for magnetization transfer from para‐hydrogen, J. Am. Chem. Soc. 2011, 133(16), 6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lloyd L. S., Asghar A., Burns M. J., Charlton A., Coombes S., Cowley M. J., Dear G. J., Duckett S. B., Genov G. R., Green G. G. R., Highton L. A. R., Hooper A. J. J., Khan M., Khazal I. G., Lewis R. J., Mewis R. E., Roberts A. D., Ruddlesden A. J., Hyperpolarisation through reversible interactions with parahydrogen, Catal. Sci. Technol. 2014, 4(10), 3544. [Google Scholar]
- 10. Adams R. W., Duckett S. B., Green R. A., Williamson D. C., Green G. G. R., A theoretical basis for spontaneous polarization transfer in non‐hydrogenative parahydrogen‐induced polarization, J. Chem. Phys. 2009, 131(19), 194505. [DOI] [PubMed] [Google Scholar]
- 11. Barskiy D. A., Pravdivtsev A. N., Ivanov K. L., Kovtunov K. V., Koptyug I. V., A simple analytical model for signal amplification by reversible exchange (SABRE) process, PCCP 2016, 18(1), 89. [DOI] [PubMed] [Google Scholar]
- 12. Atkinson K. D., Cowley M. J., Elliott P. I. P., Duckett S. B., Green G. G. R., López‐Serrano J., Whitwood A. C., Spontaneous transfer of parahydrogen derived spin order to pyridine at low magnetic field, J. Am. Chem. Soc. 2009, 131(37), 13362. [DOI] [PubMed] [Google Scholar]
- 13. Hövener J.‐B., Schwaderlapp N., Borowiak R., Lickert T., Duckett S. B., Mewis R. E., Adams R. W., Burns M. J., Highton L. A. R., Green G. G. R., Olaru A., Hennig J., von Elverfeldt D., Toward biocompatible nuclear hyperpolarization using signal amplification by reversible exchange: Quantitative in situ spectroscopy and high‐field imaging, Anal. Chem. 2014, 86(3), 1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rayner P. J., Burns M. J., Olaru A. M., Norcott P., Fekete M., Green G. G. R., Highton L. A. R., Mewis R. E., Duckett S. B., Delivering strong 1H nuclear hyperpolarization levels and long magnetic lifetimes through signal amplification by reversible exchange, Proc. Natl. Acad. Sci. U. S. A. 2017, 114(16), E3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Atkinson K. D., Cowley M. J., Duckett S. B., Elliott P. I. P., Green G. G. R., López‐Serrano J., Khazal I. G., Whitwood A. C., Para‐hydrogen induced polarization without incorporation of para‐hydrogen into the Analyte, Inorg. Chem. 2009, 48(2), 663. [DOI] [PubMed] [Google Scholar]
- 16. Zeng H., Xu J., Gillen J., McMahon M. T., Artemov D., Tyburn J.‐M., Lohman J. A. B., Mewis R. E., Atkinson K. D., Green G. G. R., Duckett S. B., van Zijl P. C. M., Optimization of SABRE for polarization of the tuberculosis drugs pyrazinamide and isoniazid, J. Magn. Reson. 2013, 237, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roy S. S., Rayner P. J., Norcott P., Green G. G. R., Duckett S. B., Long‐lived states to sustain SABRE hyperpolarised magnetisation, PCCP 2016, 18(36), 24905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fekete M., Rayner P. J., Green G. G. R., Duckett S. B., Harnessing polarisation transfer to indazole and imidazole through signal amplification by reversible exchange to improve their NMR detectability, Magn. Reson. Chem. 2017, 55(10), 944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Truong M. L., Theis T., Coffey A. M., Shchepin R. V., Waddell K. W., Shi F., Goodson B. M., Warren W. S., Chekmenev E. Y., 15N hyperpolarization by reversible exchange using SABRE‐SHEATH, J. Phys. Chem. C 2015, 119(16), 8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pravdivtsev A. N., Yurkovskaya A. V., Vieth H.‐M., Ivanov K. L., RF‐SABRE: A way to continuous spin hyperpolarization at high magnetic fields, J. Phys. Chem. B 2015, 119(43), 13619. [DOI] [PubMed] [Google Scholar]
- 21. Dücker E. B., Kuhn L. T., Münnemann K., Griesinger C., Similarity of SABRE field dependence in chemically different substrates, J. Magn. Reson. 2012, 214, 159. [DOI] [PubMed] [Google Scholar]
- 22. Roy S. S., Norcott P., Rayner P. J., Green G. G. R., Duckett S. B., A Hyperpolarizable 1H magnetic resonance probe for signal detection 15 minutes after spin polarization storage, Angew. Chem. Int. Ed. 2016, 55(50), 15642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Appleby K. M., Mewis R. E., Olaru A. M., Green G. G. R., Fairlamb I. J. S., Duckett S. B., Investigating pyridazine and phthalazine exchange in a series of iridium complexes in order to define their role in the catalytic transfer of magnetisation from para‐hydrogen, Chem. Sci. 2015, 6(7), 3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Roy S. S., Norcott P., Rayner P. J., Green G. G. R., Duckett S. B., A simple route to strong carbon‐13 NMR signals detectable for several minutes, Chem. ‐ Eur. J. 2017, 23(44), 10496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Olaru A. M., Burt A., Rayner P. J., Hart S. J., Whitwood A. C., Green G. G. R., Duckett S. B., Using signal amplification by reversible exchange (SABRE) to hyperpolarise 119Sn and 29Si NMR nuclei, Chem. Commun. 2016, 52(100), 14482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gloggler S., Muller R., Colell J., Emondts M., Dabrowski M., Blumich B., Appelt S., Para‐hydrogen induced polarization of amino acids, peptides and deuterium‐hydrogen gas, PCCP 2011, 13(30), 13759. [DOI] [PubMed] [Google Scholar]
- 27. Shen K., Logan A. W. J., Colell J. F. P., Bae J., Ortiz G. X., Theis T., Warren W. S., Malcolmson S. J., Wang Q., Diazirines as potential molecular imaging tags: Probing the requirements for efficient and long‐lived SABRE‐induced hyperpolarization, Angew. Chem. Int. Ed. 2017, 56, 12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mewis R. E., Green R. A., Cockett M. C. R., Cowley M. J., Duckett S. B., Green G. G. R., John R. O., Rayner P. J., Williamson D. C., Strategies for the hyperpolarization of acetonitrile and related ligands by SABRE, J. Phys. Chem. B 2015, 119(4), 1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burns M. J., Rayner P. J., Green G. G. R., Highton L. A. R., Mewis R. E., Duckett S. B., Improving the hyperpolarization of 31P nuclei by synthetic design, The Journal of Physical Chemistry B 2015, 119(15), 5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pravdivtsev A. N., Yurkovskaya A. V., Zimmermann H., Vieth H.‐M., Ivanov K. L., Transfer of SABRE‐derived hyperpolarization to spin‐1/2 heteronuclei, RSC Adv. 2015, 5(78), 63615. [Google Scholar]
- 31. Pravdivtsev A. N., Yurkovskaya A. V., Vieth H.‐M., Ivanov K. L., Spin mixing at level anti‐crossings in the rotating frame makes high‐field SABRE feasible, PCCP 2014, 16(45), 24672. [DOI] [PubMed] [Google Scholar]
- 32. Holmes A. J., Rayner P. J., Cowley M. J., Green G. G. R., Whitwood A. C., Duckett S. B., The reaction of an iridium PNP complex with parahydrogen facilitates polarisation transfer without chemical change, Dalton Trans. 2015, 44(3), 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Norcott P., Rayner P. J., Green G. G. R., Duckett S. B., Achieving high 1H nuclear hyperpolarization levels with long lifetimes in a range of tuberculosis drug scaffolds, Chem. ‐ Eur. J. 2017, 23(67), 16990. [DOI] [PubMed] [Google Scholar]
- 34. Pavlik J. W., Laohhasurayotin S., Synthesis and spectroscopic properties of isomeric trideuterio‐ and tetradeuterio pyridines, J. Heterocycl. Chem. 2007, 44(6), 1485. [Google Scholar]
- 35. Pavlik J. W., Laohhasurayotin S., The photochemistry of 3,4,5‐trideuteriopyridine, Tetrahedron Lett. 2003, 44(44), 8109. [Google Scholar]
- 36. Kurhanewicz J., Vigneron D. B., Brindle K., Chekmenev E. Y., Comment A., Cunningham C. H., DeBerardinis R. J., Green G. G., Leach M. O., Rajan S. S., Rizi R. R., Ross B. D., Warren W. S., Malloy C. R., Analysis of cancer metabolism by imaging hyperpolarized nuclei: Prospects for translation to clinical research, Neoplasia 2011, 13(2), 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eshuis N., Aspers R. L. E. G., van Weerdenburg B. J. A., Feiters M. C., Rutjes F. P. J. T., Wijmenga S. S., Tessari M., Determination of long‐range scalar 1H–1H coupling constants responsible for polarization transfer in SABRE, J. Magn. Reson. 2016, 265(Supplement C), 59. [DOI] [PubMed] [Google Scholar]
- 38. Barskiy D. A., Shchepin R. V., Tanner C. P. N., Colell J. F. P., Goodson B. M., Theis T., Warren W. S., Chekmenev E. Y., The absence of Quadrupolar nuclei facilitates efficient 13C hyperpolarization via reversible exchange with Parahydrogen, ChemPhysChem 2017, 18(12), 1493. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Supporting Information