

Abstract
As interest in biomass utilization has grown, the manipulation of lignin biosynthesis has received significant attention, such that recent work has demanded more robust lignin analytical methods. As the derivatization followed by reductive cleavage (DFRC) method is particularly effective for structurally characterizing natively acylated lignins, we used an array of synthetic β‐ether γ‐acylated model compounds to determine theoretical yields for all monolignol conjugates currently known to exist in lignin, and we synthesized a new set of deuterated analogs as internal standards for quantification using GC–MS/MS. Yields of the saturated ester conjugates ranged from 40 to 90 %, and NMR analysis revealed the presence of residual unsaturated conjugates in yields of 20 to 35 %. In contrast to traditional selected‐ion‐monitoring, we demonstrated the superior sensitivity and accuracy of multiple‐reaction‐monitoring detection methods, and further highlighted the inadequacy of traditional standards relative to isotopically labeled analogs.
Keywords: analytical methods, lignin, monolignol conjugates, redutive cleavage, synthetic methods
As a major component of biomass recalcitrant to depolymerization, lignin continues to impede efforts toward less energy‐intensive processes in the pulping and lignocellulosic biofuel industries.1, 2 Formed in a combinatorial fashion via consecutive oxidative radical‐coupling steps,3, 4 lignin contains a variety of phenylpropanoid subunits resulting from linking monomers to the growing polymer. β‐O‐4 ethers (hereafter termed simply β‐ethers), which account for ≈60 % of the interunit linkages, are among the most labile bonds in the polymer, but even they require harsh conditions and high temperatures to cleave.3, 5 In recent years, research efforts have focused on reducing the need for severe conditions through manipulation of the lignin biosynthetic pathway to incorporate novel monomers or upregulate production of more labile subunits (e.g., esters). Results have demonstrated that plants exhibit a high degree of metabolic plasticity for in vivo lignification,6, 7, 8, 9, 10 thus setting the stage for advantageous plant bioengineering. To keep up with these advances, the methodologies for characterizing lignin need to likewise evolve.11
For many years, NMR spectroscopy has offered excellent, non‐degradative structural information.12, 13, 14 However, although peak integration allows for reasonably accurate quantification in 1 D NMR, the more useful 2 D methods have serious quantitative limitations, especially for rapidly relaxing samples, and cannot provide these data on an absolute scale. Thioacidolysis has served as a powerful degradative tool,15, 16, 17 although it yields a complicated portfolio of products for analysis.18 For monolignol (ML) conjugate analysis, its utility has suffered from reaction conditions that neither leave esters intact nor cleave them completely;19 recent efforts have addressed this issue, as evidenced by the detection of labile ester conjugates in lignin from transgenic Arabidopsis thaliana.20
In unique fashion, derivatization followed by reductive cleavage (DFRC), a degradative method similar to thioacidolysis in that it cleaves β‐ether units in lignins to release analyzable monomers and dimers, uses reductive cleavage instead of hydrolysis to provide useful structural information.21, 22 DFRC offers several features: it selectively cleaves β‐ether bonds while retaining ester linkages, thus providing conclusive evidence of monolignol conjugate incorporation into lignins; a final acetylation step simplifies data interpretation by reducing the number of analytes; and two modified versions of the protocol use common reagents to distinguish between freephenolic and etherified units in the polymer, as well as natural γ‐acetates and those introduced by the method itself (Scheme 1). In recent years, DFRC has been an indispensable tool in confirming the incorporation of monolignol acetate (Ac),23, 24 p‐coumarate (pCA),25, 26 benzoate (BA),27 p‐hydroxybenzoate (pBA),27, 28 vanillate (VA),27 and ferulate (FA)9, 29, 30 conjugates into the lignin polymer.
Scheme 1.
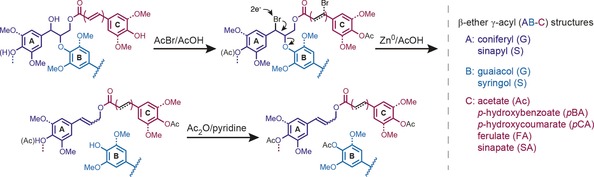
Mechanistic details for each step in the DFRC process. Acetylation of hydroxyl units and bromination of benzylic positions by acetyl bromide (AcBr)/acetic acid (AcOH) (derivatization) precede two‐electron reductive cleavage of β‐ethers and debromination of cinnamate‐derived moieties via zinc/acetic acid. In the final step the product mixture is acetylated using a mixture of acetic anhydride (Ac2O)/pyridine.
In spite of its usefulness, however, DFRC in its current form has some shortcomings. “Theoretical” yields based on synthetic models have diverged from those obtained using real plant samples.21, 31 Furthermore, reproducibility has presented a challenge, partly owing to the variety of internal standards that have been used over the years.9, 21, 32 Finally, the discovery of new conjugates in the plant cell wall has required improvements to the original method. Herein, we detail our reconstruction of the DFRC protocol to yield an accurate and reproducible method for the quantification of releasable monolignols and monolignol conjugates from plant biomass.
We report the reaction yields for β‐ether bond cleavage using a family of model compounds designed to represent the different β‐ether crosslinking permutations that exist between coniferyl alcohol, sinapyl alcohol, and the variously γ‐acylated monolignol conjugates (Scheme S1). The model compounds are abbreviated using an AB‐C nomenclature scheme, where “A” is either the coniferyl (G) or sinapyl (S) alcohol base unit, “B” is the β‐ether ring, either guaiacol (G) or syringol (S), and C is the γ‐acylating unit, acetate (Ac), p‐hydroxybenzoate (pBA), p‐coumarate (pCA), ferulate (FA), or sinapate (SA). As shown in Scheme 1, loss of the etherified “B” ring (either guaiacol or syringol moieties in our models) through reductive cleavage regenerates the vinyl group, whereas loss of bromide on the ester gives a saturated sidechain; this pattern is diagnostic for the cleavage of β‐ethers by the DFRC method (Figure 1). To quantitate these analytes more accurately, deuterated standards were synthesized. Deuteroacetylation has proven useful in this regard,33 but ketene loss upon electrospray ionization (ESI) and coelution with other products complicates mass spectrometric (MS) peak assignment of the standards versus the natural‐abundance isotopes (13C and 2H) of the lignin products. Therefore, in addition to deuteroacetylating the phenols and free γ‐hydroxyls, two to four additional deuterium labels were incorporated on either side of the ester (Scheme 2), providing multiple unique MS and multiple reaction monitoring (MRM) fragments between the isotopically labeled internal standards and the DFRC products. In conjunction with a natural abundance “dihydro” conjugate calibration curve, we used this new set of internal standards to quantify DFRC products.
Figure 1.
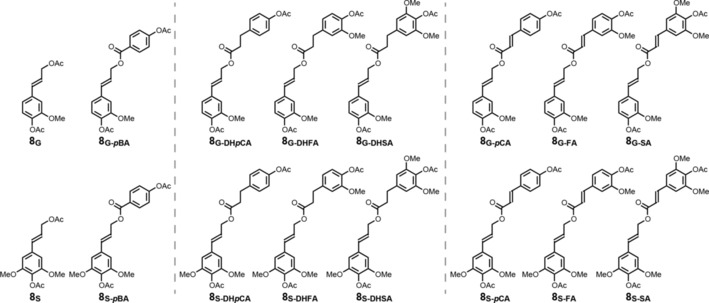
Characteristic products obtained after subjecting lignin (or model dimers, in this case) to DFRC. Top row contains coniferyl alcohol conjugates and the bottom row contains sinapyl alcohol conjugates.
Scheme 2.
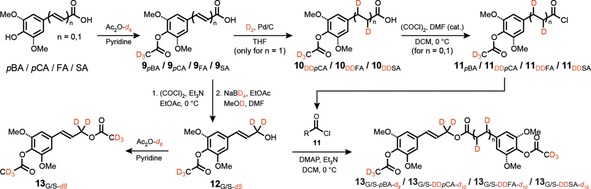
Synthesis of deuterated analogs used for quantitation of monolignols and monolignol conjugates. DD stands for “dideutero,” corresponding to where the hydroxycinnamate double bond has been deuterated to match the hydrogenated DFRC products. Natural‐abundance analogs were synthesized following an identical procedure using non‐deuterated reagents.
Additionally, we optimized each DFRC step to obtain the maximum detectable release of monolignol conjugates from extractive‐free corn stover, using p‐coumarate as a proxy for all conjugates. We obtained essentially the same conditions for bromination as in the original DFRC paper (2.5 h instead of 3 h)21 and we discovered that acetylation of the phenols in the product mixture, formed during the reductive cleavage step, occurs almost immediately after addition of the reagents (i.e., acetic anhydride and pyridine). The most important result gleaned from these efforts was our observation that zinc morphology had a significant impact on reaction yield. Zinc nanopowder (Sigma–Aldrich, P/N: 578 002, particle size <50 nm) greatly increased the yield over that of standard zinc dust for a given reduction time, while simultaneously decreasing variability between samples. As for differences between the rates of cleavage for MLs and ML conjugates, in both cases, zinc reductive cleavage was complete within 30 min; longer reduction times did not diminish the reaction yield (Figure S1 c).
Following optimization of each step, we have used synthetic model compounds to demonstrate that the DFRC procedure cleaves β‐ethers with approximately 80 % efficiency for monolignols and ML‐pBAs, and 40–50 % for ML‐pCAs, ML‐FAs, and ML‐SAs (sinapates), based solely upon quantification of the expected saturated conjugate, as discussed below (Figure 2). All compounds synthesized for the determination of “theoretical” DFRC reaction yields, for authentication of products, and for use as internal standards, along with details regarding reaction kinetics can be found in the Supporting Information. The detailed procedure can be found in the Experimental Section below, as well as in several recent papers that have applied this method to the analysis of monolignol conjugates.29, 30, 34
Figure 2.
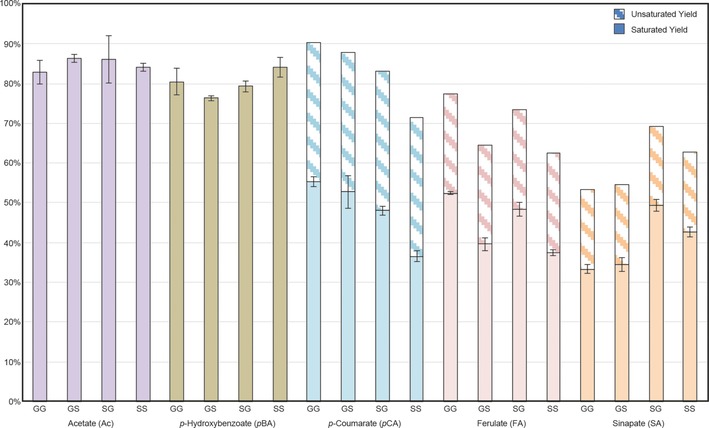
DFRC yields from β‐ether model compounds. Solid bars represent yields from compounds containing fully saturated esters, whereas hatched bars correspond to unsaturated cinnamate products, as calculated from the NMR‐determined ratio of unsaturated and saturated components.
In the course of our model compound studies, we found monolignol conjugate yields to be unusually low in comparison to general yields published in the original DFRC method paper, although monomer yields were as expected.35 As the data demonstrate (Figure 2), yield varied as a function of the moiety acylating the γ‐hydroxyl unit. In particular, when moving from the pBAs to the pCAs, yields dropped off markedly. Efforts to identify potential byproducts by GC–MS/MS were unsuccessful owing to poor peak resolution and MS responsiveness, but NMR analysis provided important insight; 1H spectra collected immediately following bromination and final acetylation showed residual vinyl peaks in both cases (albeit with different chemical shifts). Because the yield of ML‐pCAs obtained from corn stover varied little as a function of bromination time beyond 3 h (Figure S1 b), the presence of these residual vinyl peaks suggested that the ester had established an equilibrium between its brominated and unbrominated form, only the former of which would yield the expected saturated product.
To test this hypothesis, we ran DFRC on a larger scale (≈100 mg), using three permutations of model compound 7 (Figure S1): GG‐pCA, GG‐FA, and GG‐SA. Comparing the 1H NMR spectra of the product mixtures with those of the appropriate standards [e.g., G‐pCA and G‐DHpCA], we were able to quantify the ratio of unsaturated products relative to saturated ones by integrating the peaks corresponding to the α and β protons. We discovered that, as electron density increased on the aromatic ring of the γ‐acylating group owing to the presence of methoxyl units, the relative abundance of the unsaturated conjugate decreased, suggesting that the reaction equilibrium in the bromination step had moved more toward the brominated product. Indeed, others have demonstrated that bromination of electron‐rich vinyl groups proceeds rapidly and efficiently,36 and the observed trend in our own data set (≈35 % unsaturated conjugate arising from nonbrominated material for pCA, ≈25 % for FA, and ≈20 % for SA) further confirms this reactivity pattern. After incorporating quantitative data for the residual unsaturated conjugates based on our NMR‐determined ratios, the total yields for each conjugate are acceptable (Figure 2). Figures comparing the 1H NMR spectra of the crude DFRC reaction mixture with the appropriate standards can be found in the Supporting Information (Figures S2–S4).
In addition to optimizing yield and reaction kinetics, we also evaluated various internal standards and analytical methods for optimal performance. We compared chromatograms obtained from DFRC analysis of Alaskan cedar, poplar, and corn stover under scan mode (100–600 m/z) and MRM mode (Figure 3, Table 1). By using MRM technology, we observed significant reduction in background noise, thus allowing for greater accuracy in quantitation of low‐abundance monolignol conjugates (e.g., released ML‐DHFA esters). Yet, even with these improvements, the residual unsaturated conjugates (as described above), such as ML‐FA and ML‐SAs (the later of which has never been found in planta), were not detected in plant samples. Previous work has, however, successfully quantified both S‐DHpCA and S‐pCA by GC–MS in plants that contain ML‐pCAs;37 as such, our analysis of corn stover included quantitative data for both the saturated and unsaturated isomers (Figure 3). As a demonstration of the improvements offered by these new analytics, we then quantified several analytes using selected ion monitoring (SIM) and MRM methods and compared the results (Table 2). For abundant compounds (e.g., G in all species, S‐DHpCA in corn stover, and S‐pBA in poplar), we observed comparable performance between SIM and MRM scans. On average, we observed a difference of approximately 20 % between the methods for abundant monolignols and monolignol conjugates. The key advantage of MRM over SIM became apparent in the quantitation of low‐abundance monolignol conjugates (e.g., G‐pCA and G‐DHpCA in corn stover and G‐DHFA in poplar); whereas MRM allowed for accurate quantitation, these conjugates were not detectable by SIM.
Figure 3.
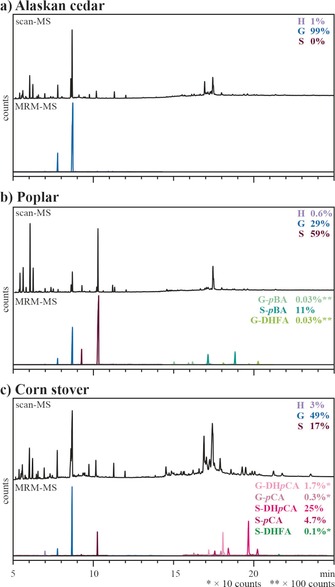
Comparison of chromatograms obtained under scan (100–600 m/z) and MRM acquisition mode for a) Alaskan cedar, b) poplar, and c) corn stover. In each case, the Figure displays the total ion chromatograms from the MS scan appears above the MRM traces of the transitions used for quantitation. The MRM traces were scaled according to their relative abundance as indicated on the right side of the chromatograms. Quantitative data can be found in Table 1.
Table 1.
Quantitative data of released target compounds as determined under SIM and MRM modes for Alaskan cedar, poplar, and corn stover. The corresponding GC–MS chromatograms are shown in Figure 3.
| Source | Method | Value [μmol (g whole cell wall)−1] | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| H‐OH/Ac | G‐OH/Ac | S‐OH/Ac | G‐pBA | S‐pBA | G‐pCA | S‐pCA | G‐FA | S‐FA | ||
| Alaskan cedar | SIM | 1.46±0.10 | 228.7±8.1 | |||||||
| MRM | 1.76±0.04 | 181.4±7.2 | ||||||||
| Poplar | SIM | 1.11±0.08 | 91.0±2.8 | 232.0±6.2 | ND | 14.9±1.3 | ND | |||
| MRM | 1.12±0.01 | 75.0±2.4 | 199.4±7.0 | 0.038±0.002 | 19.4±0.6 | 0.051±0.003 | ||||
| Corn stover | SIM | 1.79±0.07 | 31.9±0.6 | 8.4±0.4 | ND[a] | 11.6±0.0[a] | ND | |||
| MRM | 2.24±0.06 | 32.5±0.9 | 11.5±0.4 | 1.21±0.02[a] | 18.8±0.5[a] | 0.060±0.003 | ||||
[a] These values include quantitative data for both the saturated and unsaturated conjugates.
Table 2.
Heatmap depicting the relative suitability of internal standards for various DFRC analytes.[a]
| Analytes | Difference in value compared to the deuterated analog [%] | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| tetracosane | BPO | DEDF | H‐d 8 | G‐d 8 | S‐d 8 | G‐pBA‐d 8 | S‐pBA‐d 8 | G‐DD pCA‐d 10 | S‐DD pCA‐d 10 | G‐DDFA‐d 10 | S‐DDFA‐d 10 | |
| H‐OH/Ac | 30 | 47 | 43 | 0 | 2 | 2 | 17 | 40 | 22 | 26 | 24 | 56 |
| G‐OH/Ac | 35 | 41 | 145 | 4 | 0 | 4 | 91 | 146 | 95 | 124 | 141 | 158 |
| S‐OH/Ac | 31 | 45 | 124 | 4 | 2 | 0 | 63 | 102 | 73 | 88 | 98 | 114 |
| G‐pBA | 3517 | 4580 | 54 | 29 | 14 | 19 | 0 | 43 | 15 | 17 | 3 | 62 |
| S‐pBA | 53 | 60 | 64 | 50 | 37 | 41 | 30 | 0 | 40 | 18 | 32 | 13 |
| G‐DHpCA | 308 | 308 | 9 | 44 | 50 | 47 | 27 | 34 | 0 | 17 | 6 | 46 |
| S‐DHpCA | 11 | 25 | 7 | 25 | 35 | 32 | 9 | 14 | 14 | 0 | 9 | 24 |
| G‐DHFA | 2123 | 2378 | 39 | 26 | 4 | 2 | 4 | 47 | 12 | 20 | 0 | 66 |
| S‐DHFA | 2133 | 2351 | 4 | 12 | 71 | 67 | 17 | 11 | 36 | 24 | 30 | 0 |
[a] ▪=deuterated analogue, ▪=<10 %, ▪=10–25 %, ▪=25–50 %, ▪=>50 % difference.
To determine the reliability of various standards, we compared the accuracy of commercially available (or easily prepared) internal standards to isotopically labeled conjugates (using the latter as the “ideal” standard and basis for all calculations) to determine whether a less‐specialized standard could afford comparable performance. After calculating the percentage difference between the “ideal” conjugate and all others that were used in the course of this project, we created a heatmap as a visual aid for evaluating performance of a given analyte (Table 2). In almost every case, the traditional standards [tetracosane, 1,1‐bis‐(4‐hydroxyphenyl)ethane (BPO), which is detected as 1,1‐bis‐(4‐acetoxyphenyl)ethane (BPA), and diethyl 5,5′‐diferulate diacetate (DEDF)] showed poor performance. Furthermore, no single standard offered exceptional performance across the board, thus highlighting the differing MS response factors of each analyte and the necessity of using appropriate standards in MS methods.
Taken together, we contend that the various facets of our work have deconstructed a valuable but increasingly dated tool and fully rebuilt it into a robust method for lignin structural analysis. Yield data based on our expansive array of synthetic model compounds have offered insight into mechanistic details (e.g., the presence of unsaturated ester conjugates), as well as reasonable expectations for how much the method can release from biomass. Our use of isotopically labeled internal standards and cutting‐edge analytics have improved sensitivity such that we can detect cinnamate ester products present at low concentrations in the plant cell wall, which has major implications for plant breeding programs.9, 29, 30 As more researchers direct these programs toward lignin degradation and valorization, mature analytical methods will only grow in importance. By effectively redesigning DFRC from the ground up, we have given it the unique capability of detecting and quantifying low‐abundance monomers and monolignol conjugates, data which will provide critical insight into the chemical structure of engineered lignins and how best to utilize them.
Experimental Section
In detail, the optimized DFRC method (run on synthetic model compounds) is as follows. In a two‐dram vial containing a magnetic stir bar, the β‐ether model compound (1 mg) was dissolved in acetyl bromide‐acetic acid solution (1:4 v/v, 1.0 mL). The vial was sealed with a polytetrafluoroethylene (PTFE)‐lined cap and heated at 50 °C for 2.5 h, after which the solvent was removed under reduced pressure on a SpeedVac (Savant SPD131DDA concentrator with a RVT5105 refrigerated vapor trap and a OFP400 pump, Thermo Scientific, 50 °C, 25 min, 35 torr min−1, 1.0 torr). The resulting film was treated with absolute ethanol (0.5 mL), followed by removal of the solvent on the SpeedVac (50 °C, 30 min, 35 torr min−1, 1.0 torr). Ethanol quenches any residual acetyl bromide and, when running whole cell wall or lignin samples, the subsequent removal of ethanol in vacuo affords the brominated biomass as a film. In the next step, these brominated model compounds were dissolved in dioxane/acetic acid/water (5:4:1 v/v, 2 mL), and zinc nanopowder (50 mg) was charged to the vial with continuous stirring of the suspension at room temperature. After 15 min, the mixture was transferred to a separatory funnel containing saturated ammonium chloride (15 mL) and the corresponding deuterated internal standard (ISTD) using dichloromethane to rinse the reaction vessel (3×1 mL). The organic phase was separated, and the aqueous phase was extracted with dichloromethane (3×10 mL). The combined organic fractions were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. Reaction with pyridine/acetic anhydride (1:1 v/v, 3 mL) for 1 h assured full acetylation of the product. Removal of these solvents under reduced pressure yielded the final oil for analysis. Each sample was analyzed on a Shimadzu GCMS‐TQ8030 operating in MRM mode. The GC–MS/MS program and acquisition parameters are listed in the Supporting Information Tables S1–S3.
Conflict of interest
Patent submitted for deuterated monolignol conjugates.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was partially funded by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE‐FC02‐07ER64494 and DE‐SC0018409), and by Stanford's Global Climate and Energy Program (GCEP). We also thank the UW‐Madison Chemistry Department Mass Spectrometry Lab for acquisition of HRMS data. The purchase of the Thermo Q Exactive™ Plus in 2015 was funded by NIH Award 1S10 OD020022‐1 to the Department of Chemistry.
M. Regner, A. Bartuce, D. Padmakshan, J. Ralph, S. D. Karlen, ChemSusChem 2018, 11, 1600.
References
- 1. Chang M. C. Y., Curr. Opin. Chem. Biol. 2007, 11, 677. [DOI] [PubMed] [Google Scholar]
- 2. Himmel M. E., Ding S. Y., Johnson D. K., Adney W. S., Nimlos M. R., Brady J. W., Foust T. D., Science 2007, 315, 804. [DOI] [PubMed] [Google Scholar]
- 3. Boerjan W., Ralph J., Baucher M., Annu. Rev. Plant Biol. 2003, 54, 519. [DOI] [PubMed] [Google Scholar]
- 4. Ralph J., Lundquist K., Brunow G., Lu F., Kim H., Schatz P. F., Marita J. M., Hatfield R. D., Ralph S. A., Christensen J. H., Boerjan W., Phytochem. Rev. 2004, 3, 29. [Google Scholar]
- 5. Pandey M. P., Kim C. S., Chem. Eng. Technol. 2011, 34, 29. [Google Scholar]
- 6. Tobimatsu Y., Elumalai S., Grabber J. H., Davidson C. L., Pan X., Ralph J., ChemSusChem 2012, 5, 676. [DOI] [PubMed] [Google Scholar]
- 7. Vanholme R., Morreel K., Darrah C., Oyarce P., Grabber J. H., Ralph J., Boerjan W., New Phytol. 2012, 196, 978. [DOI] [PubMed] [Google Scholar]
- 8. Tobimatsu Y., Chen F., Nakashima J., Jackson L., Escamilla-Treviño L. L., Dixon R. A., Ralph J., Plant Cell 2013, 25, 2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilkerson C. G., Mansfield S. D., Lu F., Withers S., Park J.-Y., Karlen S. D., Gonzales-Vigil E., Padmakshan D., Unda F., Rencoret J., Ralph J., Science 2014, 344, 90. [DOI] [PubMed] [Google Scholar]
- 10. Lan W., Lu F., Regner M., Zhu Y., Rencoret J., Ralph S. A., Zakai U. I., Morreel K., Boerjan W., Ralph J., Plant Physiol. 2015, 167, 1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lupoi J. S., Singh S., Parthasarathi R., Simmons B. A., Henry R. J., Renewable Sustainable Energy Rev. 2015, 49, 871. [Google Scholar]
- 12. Kim H., Ralph J., Akiyama T., BioEnergy Res. 2008, 1, 56. [Google Scholar]
- 13. Kim H., Ralph J., Org. Biomol. Chem. 2010, 8, 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mansfield S. D., Kim H., Lu F., Ralph J., Nat. Protoc. 2012, 7, 1579. [DOI] [PubMed] [Google Scholar]
- 15. Lapierre C., Monties B., Rolando C., C. R. Acad. Sci. Ser. III 1984, 299, 441. [Google Scholar]
- 16. Lapierre C., Monties B., Rolando C., J. Wood Chem. Technol. 1985, 5, 277. [Google Scholar]
- 17. Rolando C., Monties B., Lapierre C. in Methods in Lignin Chemistry (Eds.: C. W. Dence, S. Y. Lin), Springer-Verlag, Berlin-Heidelberg, 1992, pp. 334–349. [Google Scholar]
- 18. Ralph J., Grabber J. H., Holzforschung 1996, 50, 425. [Google Scholar]
- 19. Lapierre C. in Lignin and lignans: Advances in chemistry (Eds.: C. Heitner, D. R. Dimmel, J. A. Schmidt), CRC Press, Taylor&Francis Group, New York, 2010, pp. 11–48. [Google Scholar]
- 20. Sibout R., Le Bris P., Legee F., Cezard L., Renault H., Lapierre C., Plant Physiol. 2016, 170, 1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lu F., Ralph J., J. Agric. Food Chem. 1997, 45, 2590. [Google Scholar]
- 22. Lu F., Ralph J. in Lignin: Structural Analysis, Applications in Biomaterials, and Ecological Significance (Ed.: F. Lu), Nova Science Publishers, Inc., Hauppauge, New York, USA, 2014, pp. 27–65. [Google Scholar]
- 23. Bunzel M., Heuermann B., Kim H., Ralph J., J. Agric. Food Chem. 2008, 56, 10368. [DOI] [PubMed] [Google Scholar]
- 24. Ralph J., Lu F., J. Agric. Food Chem. 1998, 46, 4616. [DOI] [PubMed] [Google Scholar]
- 25. Petrik D. L., Karlen S. D., Cass C. L., Padmakshan D., Lu F., Liu S., Le Bris P., Antelme S., Santoro N., Wilkerson C. G., Sibout R., Lapierre C., Ralph J., Sedbrook J. C., Plant J. 2014, 77, 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith R. A., Gonzales-Vigil E., Karlen S. D., Park J.-Y., Lu F., Wilkerson C. G., Samuels L., Mansfield S. D., Ralph J., Plant Physiol. 2015, 169, 2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou S., Runge T., Karlen S. D., Ralph J., Gonzales-Vigil E., Mansfield S. D., ChemSusChem 2017, 10, 3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu F., Karlen S. D., Regner M., Kim H., Ralph S. A., Sun R.-c., Kuroda K.-i., Augustin M. A., Mawson R., Sabarez H., Singh T., Jimenez-Monteon G., Hill S., Harris P. J., Boerjan W., Mansfield S. D., Ralph J., BioEnergy Res. 2015, 8, 934. [Google Scholar]
- 29. Karlen S. D., Zhang C., Peck M. L., Smith R. A., Padmakshan D., Helmich K. E., Free H. C. A., Lee S., Smith B. G., Lu F., Sedbrook J. C., Sibout R., Grabber J. H., Runge T. M., Mysore K. S., Harris P. J., Bartley L. E., Ralph J., Sci. Adv. 2016, 2, e1600393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith R. A., Cass C. L., Mazaheri M., Sekhon R. S., Heckwolf M., Kaeppler H., de Leon N., Mansfield S. D., Kaeppler S. M., Sedbrook J. C., Karlen S. D., Ralph J., Biotechnol. Biofuels 2017, 10, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Holtman K. M., Chang H. M., Jameel H., Kadla J. F., J. Agric. Food Chem. 2003, 51, 3535. [DOI] [PubMed] [Google Scholar]
- 32. Franke R., Hemm M. R., Denault J. W., Ruegger M. O., Humphreys J. M., Chapple C., Plant J. 2002, 30, 47. [DOI] [PubMed] [Google Scholar]
- 33. Schäfer J., Urbat F., Rund K., Bunzel M., J. Agric. Food Chem. 2015, 63, 2668. [DOI] [PubMed] [Google Scholar]
- 34. Karlen S. D., Smith R. A., Kim H., Padmakshan D., Bartuce A., Mobley J. K., Free H. C. A., Smith B. G., Harris P. J., Ralph J., Plant Physiol. 2017, 175, 1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu F., Ralph J., J. Agric. Food Chem. 1997, 45, 4655. [Google Scholar]
- 36. Fahey R. C., Schneide H.-J., J. Am. Chem. Soc. 1968, 90, 4429. [Google Scholar]
- 37. del Río J. C., Lino A. G., Colodette J. L., Lima C. F., Gutiérrez A., Martínez A. T., Lu F., Ralph J., Rencoret J., Biomass Bioenergy 2015, 81, 322. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary